
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCurrent state of copper-based bimetallic materials for electrochemical CO2 reduction: a review
Otmane Zoubir a,
Lahoucine Atourki*a,
Hassan Ait Ahsaine*b and
Amal BaQaisc
a,
Lahoucine Atourki*a,
Hassan Ait Ahsaine*b and
Amal BaQaisc
aMANAPSE Lab, Faculty of Sciences, Mohammed V University in Rabat, Morocco. E-mail: l.atourki@um5r.ac.ma
bLaboratoire de Chimie Appliquée des Matériaux, Faculty of Sciences, Mohammed V University in Rabat, Morocco. E-mail: h.aitahsaine@um5r.ac.ma
cDepartment of Chemistry, College of Science, Princess Nourah bint Abdulrahman University, P.O. Box 84428, Riyadh 11671, Saudi Arabia
First published on 21st October 2022
Abstract
The increasing CO2 concentration in the atmosphere has caused profound environmental issues such as global warming. The use of CO2 as a feedstock to replace traditional fossil sources holds great promise to reduce CO2 emissions. The electrochemical conversion of CO2 has attracted much attention because it can be powered by renewable sources such as solar energy. In this review article, we provide insight into the important parameters when studying CO2RR and give a comprehensive review on the description of synthesis methods with electrocatalytic CO2 reduction over bimetallic copper-based materials. Due to the important bibliographic data on Cu bimetallic materials, we have limited this review to Sn, In, Pd, Zn and Ag. At the end of this review, challenges and perspectives for further upgrading have been included to briefly highlight the important future considerations of this rapidly growing technology.
1. Introduction
The intensification of human industrial activities has gradually disrupted environmental stability on earth, causing more CO2 production.1 The increasing CO2 concentration level in the atmosphere has resulted in severe problems such as the greenhouse effect, leading to global warming, melting glaciers, and more disastrous weather.2 Therefore, it is of critical importance to reduce atmospheric CO2 concentration. To this end, much effort has been devoted to define potential methods to mitigate CO2 emissions. An appealing solution is to use CO2 as the carbon source to produce value-added products such as carbon monoxide (CO), formic acid (HCOOH), formate (HCOO−), methanol (CH3OH), methane (CH4), ethylene (C2H4), ethanol (C2H5OH), as well as others by applying the renewable energy as energy input.3 A variety of alternative CO2 utilization approaches are being studied, including biological,4 thermochemical,5,6 photochemical7–10 and electrochemical methods.11–14 For thermochemical CO2 conversion via a reforming process, it requires not only high reaction temperatures and pressures but also an equal amount of hydrogen as the reducing agent, which is energetically hazardous for large-scale applications. For the photochemical process, the selectivity and production rate of the photocatalytic active systems are too low to be economically valid. In contrast, CO2 electrochemical reduction (CO2RR) appears to be one of the most promising technologies owing to its feasible operating conditions, scalability, and the increasing sources of green electricity with zero-CO2 emissions.15 However, challenges still exist such as slow electron transfer kinetics resulting in low exchange current densities, low energetic efficiency, and poor selectivity.16The development of efficient electrocatalysts plays a key role in the electrochemical reduction of CO2 in terms of activity and selectivity.17 Over the last few decades, enormous efforts have been devoted into investigating CO2 electrocatalysts for the electrochemical CO2 reduction reaction (CO2RR).2 The most commonly explored electrocatalysts for CO2 reduction can be divided into three groups: metallic; non-metallic; molecular catalysts. Polycrystalline monometallic catalysts offer unique merits such as simple structure, ease of handling, robustness, and other advantages, making them particularly appealing candidates for fundamental research.14 Based on the primary CO2 reduction product, four distinct groups of monometallic catalysts have been identified: (1) CO selective metals such as (Pd, Au, Zn, Ag, and Ga); (2) metals that mainly produce HCOOH (e.g., Hg, Pb, Cd, In, Sn, and Ti); (3) metals that form hydrocarbons such as CH4 and C2H4 (e.g., Cu); (4) metals that mainly produce hydrogen, H2 (Ni, Pt, Fe, and Ti). Among all the aforementioned monometallic CO2 electrocatalysts, copper (Cu) is the only metal that can deeply reduce CO2 to products such as hydrocarbons and alcohols with acceptable activity and efficiency.18 However, the selectivity of Cu toward certain products is typically poor.19 Recently, there has been growing interest in bimetallic catalysts for CO2 reduction; they represent new trends and opportunities in electrochemical CO2 conversion.20,21 Metals and metal oxide-based catalysts especially the Cu based electrocatalysts have been substantially reported as efficient CO2RR catalysts.22–24
They are easy to synthesize, relatively stable in working conditions. These materials do not require special preparation conditions compared to other molecular. Many remarkable contributions that have been reported using serious preparation control of structures in morphological tailoring,25 steering surfaces,23 Metal atom decoration,26 atomic scale structuration,27 guided alloying,28,29 electrolyte design,30 and strained surfaces.31 As robust as Cu based electrodes are, they still suffer from a reconstruction during electrolysis. This double-edged sword can degrade the efficiency of the catalysts with altering the well-defined Cu morphology and highly active sites,32,33 or reconstructs the generating unique Cu active sites with increased catalytic activity for specific products.34
The focus throughout this article will be on the use of copper-based bimetallic materials to reduce carbon dioxide into value-Added products. We have reviewed the Cu-M electrocatalysts (M: Sn, In, Pd, Zn and Ag) as they the most important reported electrocatalysts for CO2RR in the last decade. In this article, we will start by discussing the basic principles of CO2 electroreduction, including thermodynamics, reaction mechanism, and factors influencing CO2 electroreduction. Next, we will look at the electrochemical methods for CO2 electroreduction. Finally, copper-based bimetallic materials will be discussed in detail, including synthesis approaches and catalytic performance.
2. Fundamentals of electrocatalytic CO2 reduction
2.1 Thermodynamics of CO2 reduction
In thermodynamic, the Gibbs free energy (G) can be used to determine the maximum amount of reversible work that can be derived from any system at constant pressure (P) and temperature (T). The change in Gibbs energy ΔG can be used to measure the spontaneity of a specific reaction. If the Gibbs free energy decreases while the reaction proceeds, the reactants will spontaneously be converted into products. If G increases, the reaction will spontaneously proceed in the opposite direction, thereby making the starting materials.35In electrochemistry, Gibbs energy can be accurately controlled by applied potential as shown in equation (Qiao et al. 2016):36
| ΔG = −nFEcell | (1) |
This equation indicates the quantitative relationship between chemical energy and electrical energy in cell reactions (Table 1).
| Product | Gibbs free energy ΔG (kJ mol−1) |
|---|---|
| C | 0 |
| CO | −137.3 |
| CO2 | −394.4 |
| CH4 | −50.8 |
| C2H4 | −68.1 |
| CH3OH | −161.6 |
| H2O (steam) | −228.6 |
| H2O (water) | −237.2 |
| O2 | 0 |
| H2 | 0 |
2.2 Reaction mechanism
Researchers across the world have devoted tremendous effort to understand the mechanism for CO2 electrochemical reduction. Especially, why different metals generate different products. In this regard, they have proposed many pathways for electrochemical reduction (ERC), and continue to propose novel reaction pathways based on their understanding of CO2 reduction on metallic electrodes. However, since ERC is a surface phenomenon, there is no generalized reaction towards the products obtained. The mechanism must depend on the type and morphology of the catalyst.38,39 The electrochemical reduction of CO2 is a multi-step reaction process that may proceed via two to fourteen electron reaction pathways (Table 2) and yielding diverse reduction products. This multi-step reaction generally involves three major steps: (1) chemical adsorption of CO2 on the surface of the catalyst; (2) electron/proton transfer to separate C–O bonds and/or form C–H bonds; (3) desorption of products from the surface of electrocatalyst and diffusion into the electrolyte.40| Product | Reduction potentials of CO2 | E° (V) vs. SHE (pH 7) |
|---|---|---|
| Carbon monoxide | CO2 + 2e− + 2H+ → CO(g) + H2O | −0.53 |
| Hydrogen | 2e− + 2H+ → H2(g) | −0.42 |
| Formic acid | CO2 + 2e− + 2H+ → HCOOH(l) + H2O | −0.61 |
| Formaldehyde | CO2 + 4e− + 4H+ → CH2O(l) + H2O | −0.50 |
| Methanol | CO2 + 6e− + 6H+ → CH3OH(l) + H2O | −0.39 |
| Methane | CO2 + 8e− + 8H+ → CH4(g) + H2O | −0.25 |
| Ethylene | CO2 + 12e− + 12H+ → C2H4(g) + 4H2O | −0.38 |
| Ethane | CO2 + 14e− + 14H+ → C2H6(g) + 4H2O | −0.28 |
In a typical CO2 electrolyzer, anode and cathode are placed into two chambers separated with an ion-conducting membrane. At the anode, water is oxidized to form molecular oxygen, while CO2 is reduced to form carbon species at the cathode. The thermodynamic potential that allows the one-electron reduction of CO2 to form CO2− is −1.90 V vs. SHE in pH 7.0 aqueous solution, making the reaction uphill and unfavorable.41–43 The first step involves one-electron transfer to generate a key intermediate CO2−, which is the rate-limiting step in the reaction. In contrast, proton-assisted multi-electron transfer processes are more advantageous and take place almost instantaneously within the potential range of −0.2 V to −0.6 V relative to SHE, resulting in a variety of CO2 derivative products depending on the catalyst and electrolyte used.14,44 There are two possible pathways in which CO2− can be reduced after it has been formed (Fig. 1). The first one is based on the protonation of CO2− oxygen atom, thus forming *COOH, which is further reduced to CO and desorbs from the electrode surface.45,46 The second pathway is the protonation of CO2− carbon atoms to form HCOO* at high overpotential levels, which is further reduced to HCOO−.47
 | ||
| Fig. 1 Most possible pathways for electrocatalytic CO2RR on metal electrodes in aqueous solutions. Reproduced with permission from ref. 14. Copyright ©1994, Elsevier. | ||
The types and the number of target products are controlled by tuning the binding energy of the adsorbed intermediate such as (CO*, COOH*, CHO*, COH*).48–50 For example, when the interaction between the surface of the electrocatalysts and the reduction intermediates is not strong enough, CO and HCOO− are the main reduction products. Because in the case of weak binding strength C–O bond does not dissociate. However, the electrocatalysts that bind CO* strongly produced a limited amount of CO and HCOOH because CO* intermediate will stick much longer on the surface of the catalyst, thus will be further reduced to other products.51 Most CO2 reduction electrocatalysts produce mainly CO and HCOO− products. However, only a few electrocatalysts, such as Cu, can further reduce CO to alcohols and hydrocarbons at low overpotentials.18,52,53 The basic explanation for its potential to generate products apart from CO is that Cu binds *CO neither too weakly nor too strongly.19
3. Copper-based bimetallic materials
In literature, there is numerous method used to deposit a layer of metal onto another metal, ranging from physical deposition methods:55 electron beam evaporation, pulsed layer deposition, sputtering to the chemical deposition methods:56,57 chemical vapor deposition, atomic layer deposition; some of these methods are expensive due to the need of vacuum system and others lack safety. However, electrochemical methods are generally simple and low-cost.58 The electrodeposition is one of the major electrochemical techniques used to produce inorganic electrodes with more complex compositions.59 It provides the main advantage of monitoring and controlling the thickness of the samples by simply varying the deposition current/potential.60 This technique can be applied to various types of materials including oxides, phosphates, chalcogenides, and metals (e.g., monometallic, bimetallic, ternary compounds). Due to its low cost and easy scalability, electrodeposition is particularly appealing for producing catalyst electrodes for use in solar fuel production.59 In this technique, the cations are reduced in the cathode, and the reactive metal anodes are dissolved and re-deposited to load onto the surface of cathode.Copper, which is the magical element for CO2 electrocatalysis giving a variety of C2 to C3 chemicals, holds a unique interest in the CO2 electrocatalysis community. This uniqueness of Cu may lie in its moderate binding energy for some key intermediates, such as adsorbed hydrogen *H and adsorbed *CO,61 which favors the protonation of *CO into deeper reduction products. The Cu surfaces can be modulated, tuned and controlled on various supports which leads to different reactions tracks and pathways encompassing intermediates, *CO dimerization, C–C coupling, C1–C2 coupling etc. Some authors have highlighted that the coalescence, fragmentation and aggregation of copper nanoparticles might cause a stability issues in Cu based electrocatalysts and others suggested that many factors are affecting the CO2RR efficiency of Cu catalysts.54,62 It is well known that properties of bimetallic catalysts are significantly different from their monometallic counterparts because of the change in the electronic structures. Additionally, the bimetallic configuration provides new active sites, thus optimizing the binding strengths between intermediates and active sites.63,64
3.1 Cu–Sn bimetallic materials
Wang et al.65 have reported the unexplored phase and structural engineering of Cu/Sn catalysts for enhancing CO2 reduction reaction by simple thermal annealing of CuSn core–shell NPs in controlled conditions. Cu–SnO2 catalysts were engineered with three distinct structures: the CuO/hollow SnO2 heterostructure of CuSn NPs/C-A, the Cu41Sn11@SnO2 core–shell structure of CuSn NPs/C–H and the Cu NPs/hollow SnO2 Janus structure of CuSn NPs/C-AH. The electrocatalytic performance results showed that CO was the main product of the CO2 reduction reaction under the potential of −0.7 V vs. RHE. The CuSn NPs/C-A catalyst achieved (70.1% FECO) and CuSn NPs/C-AH (45.1% FECO), which outperformed CuSn NPs/C–H (20.1% FECO), Cu NPs/C (13.5% FECO), and SnO2 NSL/C 8.4% FECO). Furthermore, at the same potential (−0.7 V vs. RHE) the CuSn NPs/C-A exhibited the highest current density of CO approximately (1.66 mA cm−2), which surpassed the CuSn NPs/C-AH (0.86 mA cm−2) and Cu NPs/C (0.28 mA cm−2) and was far better than the CuSn NPs/C–H (0.21 mA cm−2) and SnO2 NSL/C (0.05 mA cm−2). It is clear that there was a selectivity transformation from CO to HCOOH when the electrolysis potential is changed from −0.7 V vs. RHE to −1.0 V vs. RHE. For all catalysts except Cu NPs/C, the main product of CO2 reduction reaction was HCOOH. By contrast, CuSn NPs/C-A still exhibited the highest FEHCOOH of 71.5%. Moreover, at −1.0 V vs. RHE, CuSn NPs/C-A reached the maximum partial current density of HCOOH around (12.6 mA cm−2), which was higher than the values of the CuSn NPs/C-AH (mA cm−2 Cu NPs/C–H (10.5 mA cm−2), Cu NPs/C, and SnO2 NSL/C. In addition, the investigation of the stability using chronoamperometry at −0.7 V vs. RHE indicated that CuSn NPs/C-A, CuSn NPs/C-AH, CuSn NPs/C–H catalyst exhibited stable current densities in the span of 10 h. Ju et al.66 demonstrated the importance of using CO2 gaseous as a feedstock to enhance CO2 mass transport and achieve high CO partial current density. In this regard, the authors employed Sn/Cu-nanofiber electrodes as freestanding gas diffusing electrodes GDEs. They fabricated electrospun polyvinylidene fluoride (PVDF) nanofibers with uniform Cu coating and then used electrochemical underpotential deposition (UPD) to decorate the surface of Cu with Sn. The faradaic efficiencies for CO (Fig. 2) were above 80% at potential ≤ −0.9 V and the partial current densities for CO exhibited 104 mA cm−2 at −1.2 V. However, at potentials ≤ −1.0 V a sparse amount of C2H4 less than 8.2% has been detected with current densities lower than 10 mA cm−2 (Fig. 2).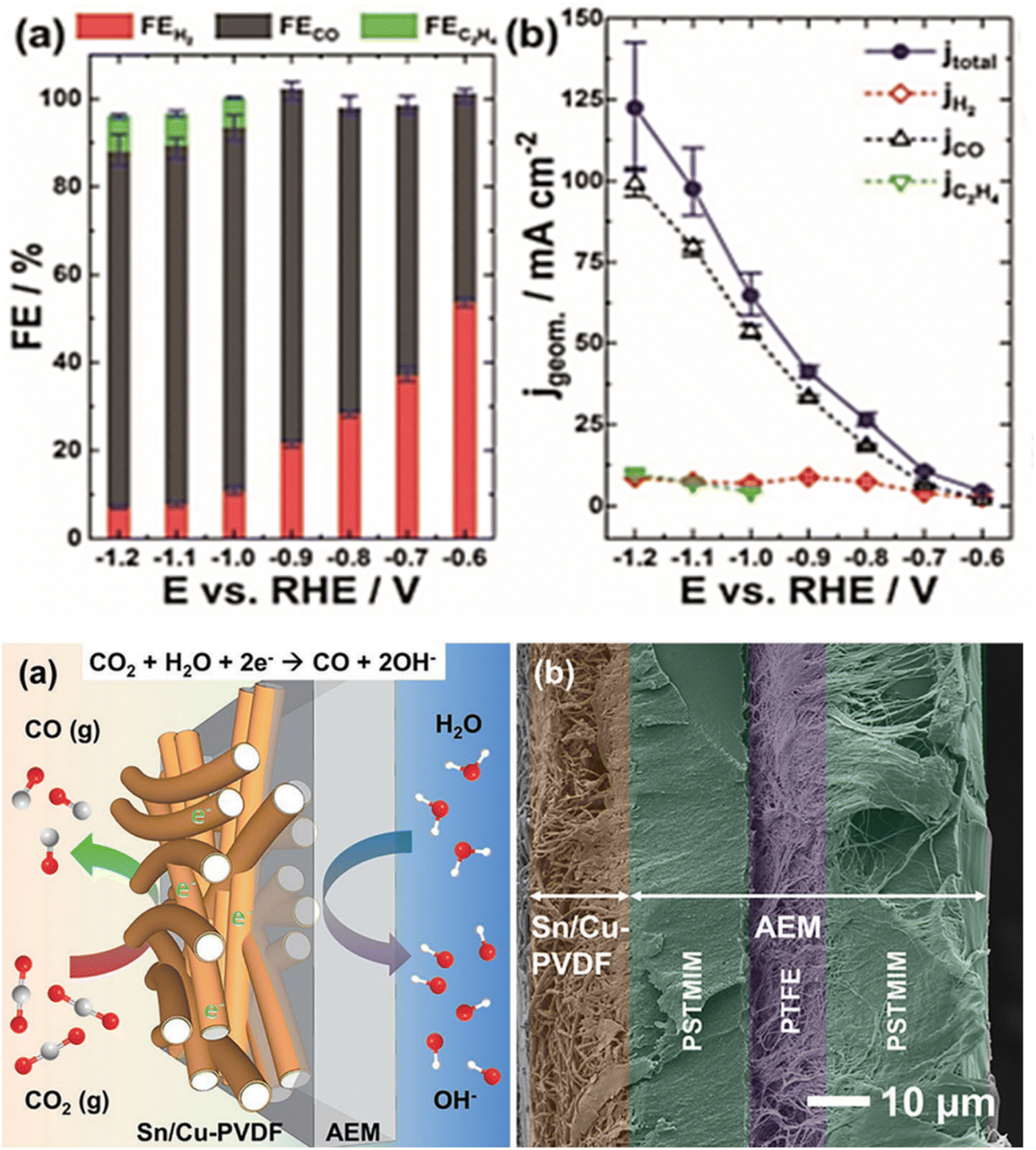 | ||
| Fig. 2 (Up): (a) Faradaic efficiencies, (b) current densities for electrochemical CO2 reduction at a Sn/Cu-PVDF GDE at (−0.6 to −1.2 V vs. RHE). (Bottom): (a) Scheme and (b) cross-sectional SEM image of a Sn/Cu-PVDF/AEM assembly used for electrocatalytic reduction of gaseous CO2. Reproduced with permission.66 Copyright ©2019, Wiley-VCH. | ||
Furthermore, the Sn/Cu-PVDF GDE maintained long-term stability of 135 h at −1.0 V with the average CO FE (84.9%), while the average FEs for H2 and C2H4 were 10.6% and 4.4%, respectively. In addition, it has been shown that the relatively high permeability of Sn/Cu nanofiber GDEs was essential for ensuring high CO2 to CO conversion rates and inhibiting the hydrogen evolution reaction. Jiang et al.67 provided a method for producing a high-performance bimetallic catalyst using an electronically regulated Cu in a heterogeneous bimetallic catalyst of Sn nanoparticles. The performance of the BM Sn–Cu catalyst was investigated under a wide potential range (from −0.75 V to −1.15 V vs. RHE). The BM Sn–Cu catalyst generated HCOO− as the primary product during CO2 electrochemical reduction. As shown in (Fig. 3a) the BM Sn–Cu electrode demonstrated the higher faradaic efficiency toward HCOO− about 92% at −0.95 V vs. RHE, while the total faradaic efficiency of CO and H2 on the BM Sn–Cu electrode was less than 10% (Fig. 3b). The authors suggested that Sn plays a key in the enhanced catalytic performance of BM Sn–Cu catalyst toward HCOO−. The electrocatalytic activity of Cu polished, BM Sn–Cu, and M Sn electrodes was examined under potentiostatic conditions in potential ranges from 0 to −1.2 V vs. RHE. The current density measurements (Fig. 3c) showed that among all electrodes, BM Sn–Cu electrode exhibited a higher HCOO− partial current density of 10.8 mA cm−2 at −0.95 V vs. RHE. This finding confirms that the embedded structure of BM Sn–Cu electrode can provide more active sites for CO2 reduction. The DFT calculations were performed to understand the electronic structure of BM Sn–Cu catalyst for CO2 electrochemical reduction. It was found that regulating the electronic structure of the catalyst promotes the formation of the HCOO* intermediate. Fig. 3d illustrates the proposed mechanism reaction for CO2 electrochemical reduction to HCOO− on the BM Sn–Cu electrode.
 | ||
| Fig. 3 (a) Faradaic efficiencies for HCOO− at potential range from −0.75 V to −1.15 V vs. RHE. (b) CO and H2 faradaic efficiencies on the polished Cu, M Sn, BM Sn–Cu at potential range from −0.75 V to −1.15 V vs. RHE in saturated 0.1 M KHCO3. (c) The partial current density of HCOO− on the polished Cu, M Sn, BM Sn–Cu, M Sn, BM Sn–Cu. (d) Proposed reaction mechanism for CO2 electrochemical reduction to HCOO− of BM Sn–Cu electrode. Reproduced with permission.67 Copyright ©2019, Elsevier. | ||
Ye et al.68 developed a new approach for designing the Cu–Sn alloy catalyst toward the electroreduction of CO2 to HCOO− through a modified electrodeposition method. For the electrocatalytic performances, the Cu–Sn catalyst was evaluated under a wide potential range (from −0.7 V to −1.2 V vs. RHE). The Cu–Sn catalyst produced HCOO− as the main product during CO2 electrochemical reduction. The catalyst reduced the faradaic efficiency for H2 evolution from (>90% at −0.75 V vs. RHE) to (less than 10% at −1.14 V vs. RHE) and improved the HCOO− faradaic efficiency reaching 82.3% at −1.14 V vs. RHE. In contrast, Sn on CFC catalyst exhibited HCOO− faradaic efficiency of greater than 55% at −1.14 V vs. RHE while few amounts of H2 and CO were also obtained. The authors attributed the improved performance of Sn–Cu alloy catalyst towards HCOO− production to the stepped (211) surface of the Cu–Sn alloy. The HCOO− mass activity of Cu–Sn alloy catalyst increased along with increasing the applied potential reaching the maximum (1490.6 mA mg−1) at −1.14 V vs. RHE. However, the HCOO− mass activity of the Cu–Sn catalyst decreased with the increase of the deposition time. The result showed that the highest mass activity was achieved at a deposition time of 0.01 s. Moreover, the authors investigated the correlation between CO2RR performance and Sn deposition time. To this end, the results demonstrated that the faradaic efficiency of HCOO− remained stable at higher than 82% under various electrodeposition times. Dong et al.69 demonstrated the effect of the local corrosion phenomenon of Cu–Sn catalysts on the catalytic selectivity of CO2 reduction via the pattering of the Sn layer on Cu foil. They found that the thickness of Sn patterns has a significant impact on the surface composition of Cu and Sn. In the Cu–Sn catalyst preparation, the Cu foil was electrochemically polished in the H3PO4 solution for 180 s by applying an anodic potential of 2.5 V versus the IrOx counter electrode. Then, the Cu foil was cleaned with deionized water and dried using N2 blowing.
For the photolithography process, the photoresist was spin-coated for 40 s at 6000 rpm and softly baked for 60 s at 110 °C. The photoresist-coated sample was moved to a mask aligner, and then the samples were contacted with a patterned photomask. The prepared Cu–Sn catalyst was sealed with epoxy except for the active area of the electrode (0.283 cm2). In order to investigate catalytic performances, the authors prepared four samples denoted as Cu foil, Cu/Sn 3 nm, Cu/p-Sn 3 nm, and Cu/p-Sn 20 nm and they evaluated them at different applied potentials from −0.6 V to −1.2 V vs. RHE. The Cu foil obtained a faradaic efficiency of greater than 50% at all potentials and began to produce CH4 at high potentials (−1.2 VRHE). In contrast, constructing a thick Sn layer 3 nm on Cu foil suppressed the H2 evolution (FEH2 < 5.5%) and increased the selectivity towards HCOOH (FEHCOOH > 80%) at high potentials (from −0.8 VRHE to −1.2 VRHE). The Cu/p-S 3 nm exhibited unique selectivity towards CO, the faradaic efficiency reached the highest value (FECO = 58.1%) at −1.0 V vs. RHE. At a potential of −1.2 VRHE the FECO of Cu/p-Sn catalyst decreased and FECH4 increased. On the other hand, Cu/p-Sn (20 nm) showed an increased selectivity towards HCOOH about 61.6% at −1.2 VRHE. Furthermore, the authors investigated the effect of the pattering of Sn (tSn) on the catalytic selectivity of bimetallic Cu–Sn catalyst. They measured the faradaic efficiencies of Cu/Sn and Cu/p-Sn for various tSn values. The Cu/Sn catalyst with a thin Sn layer (tSn = 1 nm) exhibited a high CO FE of 86.1% at −1.0 VRHE. However, the Cu/Sn catalyst with (tSn > 2 nm) showed a high FEHCOOH of greater than 80% at −1.0 VRHE. In comparison to Cu/Sn, the Cu/p-Sn catalyst with thin Sn patterns (between 2 nm to 4 nm) exhibited a high FECO of greater than 51.2%. However, increasing Sn patterns (tSn > 5 nm) resulted in a decrease in CO FE (40%) and an increased in HCOOH FE to 59.7%. Hu et al.70 synthesized Cu–Sn core–shell nanowire array catalysts based on 3-D macroporous nickel (Ni) foams through two-step deposition annealing and electroreduction treatment. The electrochemical characterizations for CO2RR revealed that constructing a Sn shell on Cu nanowires reduced faradaic efficiencies for H2 from 55.7% to 10.1% and HCOOH formation from 12.3% to 2% and improved CO faradaic efficiency reaching 90% instead of 32% at an applied potential of −0.8 V vs. RHE. The authors attributed the improved performance to the suppressed adsorption of H* and the ordered arrangement of Sn and Cu atoms. Moreover, the authors investigated the correlation between CO2RR performance and Sn deposition time. The results showed that the efficiency of CO increased with increasing the deposition time reaching the maximum at 10 s and then decreased with time while HCOOH, H2 efficiencies are on an opposite trend. Furthermore, the effect of applied potential of c/s Cu–Sn NW foam was also investigated, indicating that the production selectivity toward CO remained stable between −0.6 to −1.2 V with a faradaic efficiency higher than 90% and, the total current density reached −13.2 and −19.3 mA cm−2 between −0.8 to −1.2 V vs. RHE. In terms of stability, the Cu–Sn core/shell nanowire array catalyst exhibited good stability. After 8 cycles of use, the catalyst achieved stable current densities of 2 h for each cycle (Fig. 4). According to the authors, good stability could be ascribed to the formation of Cu6Sn5 in the interface.
 | ||
| Fig. 4 (a) Effect of applied potential on product distribution and current density of c/s Cu Sn NW@foam for CO2RR. (b) Stability of c/s Cu Sn NW@foam catalyst for CO2RR by re-repeated use. Reproduced with permission.70 Copyright ©2018, Wiley-VCH. | ||
Sarfraz et al.71 prepared a highly CO selective catalyst by the electrodeposition of Sn on the surface of oxide-derived copper (OD-Cu). To elucidate the catalytic performance the authors prepared three samples: Sn deposited on OD-Cu denoted as (Cu–Sn), OD-Cu, and Sn deposited on Sn. The CO2RR electrocatalytic showed that Cu–Sn exhibited a CO faradaic efficiency of greater than 90% over a wide potential range (from −0.5 to −0.8 V vs. RHE), while only trace of HCOOH and H2 were detected. Moreover, the Cu–Sn catalyst maintained long-term stability at least 14 h. The CO faradaic efficiency of OD-Cu catalyst increased with increasing the potential and reached a maximum (≈63%) at −0.6 V vs. RHE and then drop to 32% at −0.8 V vs. RHE. In comparison, HCOOH faradaic efficiency increased along with increasing overpotential, reaching 45% FE at −0.8 V vs. RHE. On the other hand, Sn deposited on Sn electrode showed completely H2 production. In addition, the authors have elucidated the effect of Sn deposition amount on the pre-reduced OD-Cu. The optimal amount of Sn was obtained at 3.9 μmol cm−2, achieving selectivity of 90% CO FE and 5% HCOOH FE at −0.6 V vs. RHE; however, the excess of Sn deposited on OD-Cu favored H2 generation. In order to investigate the catalytic performance of core–shell Cu–SnO2 nanoparticles, Li et al.72 synthesized two types of CuSnO2 nanoparticles with the same core radius (7 nm) but different SnO2 shell thicknesses (0.8 nm and 1.8 nm). They found that the thickness of Sn shell has a significant effect on product selectivity. The nanoparticles with thinner SnO2 shell 0.8 nm (Fig. 5a) reached a maximum FE of 93% toward CO at −0.7 V vs. RHE with a current density of 4.6 mA cm−2. The nanoparticles with thicker SnO2 shell 1.8 nm (Fig. 5b) could produce 85% HCOO− at −0.9 V vs. RHE and less than 1% of CO formation. Based on density functional theory (DFT) calculations, the high selectivity of 0.8 nm SnO2 shell toward CO was ascribed to the large compressive strain on the surface (10%), Cu doping on 0.8 nm SnO2, and the less negative overpotential for CO (−1.87 V vs. RHE) compared to HCOO− (−2.21 V vs. RHE). To investigate the activity and the selectivity of Sn–Cu catalysis toward CO2 reduction. Li et al.73 engineered Cu/SnO2 electrode by electrodepositing SnO2 films on porous copper foam followed by electrochemical pre-reduction. The same procedure was used to fabricate the A-Cu/SnO2 electrode, but with an additional annealing step. The Cu foil was mechanically polished with 200-grade sandpaper, electropolished in 85% of phosphoric acid, and washed with acetone and deionized water. Epoxy resin was used to encapsulate the back of Cu foil. The Cu foam was deposited on the pretreated Cu foil at a current density of −3 A cm−2 for 15 s in a plating bath containing (0.2 M CuSO4, 1.5 M H2SO4). The Sn foil (2 cm × 2 cm) was electrodeposited on Cu foam electrode via electrodeposition method using a constant potential of −0.3 V while varying deposition time (5, 15, 30, 45, and 60 min) in the electrolyte consisting of (0.02 M SnCl4, 0.1 M NaNO3, 0.075 M HNO3). The electrochemical pre-reduction was carried out in a CO2- saturated 0.1 M KCO3 at −0.5 V vs. RHE for 1 h. The resultant electrode was labeled as Cu/SnO2 electrode. A-Cu/SnO2 electrode was obtained by applying an additional annealing step (200 °C for 6 h in muffle furnace) between electrodepositing and pre-reduction processes (Fig. 5).
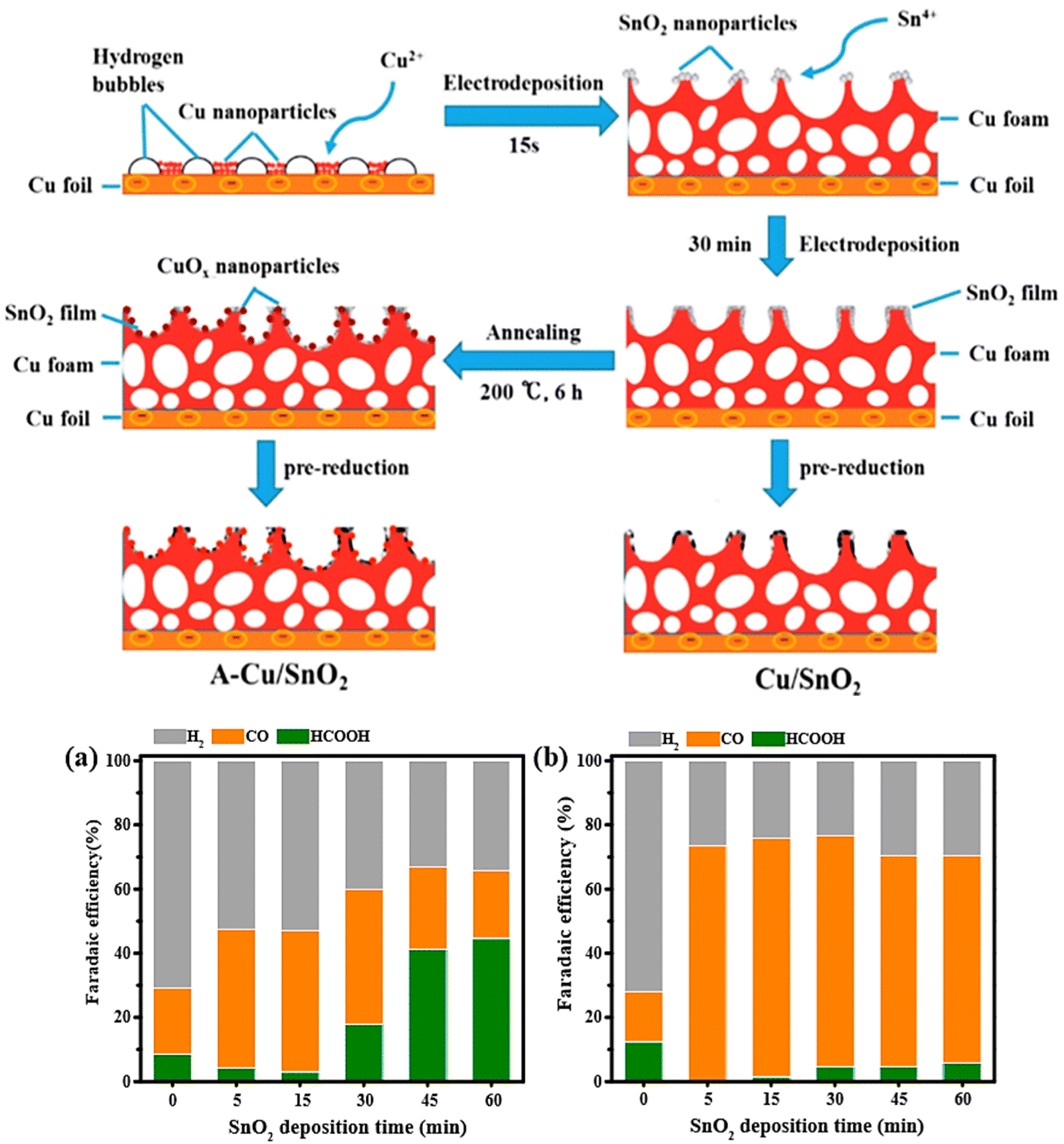 | ||
| Fig. 5 (Up) Schematic drawing of the fabrication of Cu/SnO2 and A-Cu/SnO2. (Bottom) Comparison of faradaic efficiencies of H2, CO, and HCOOH for (a) Cu/SnO2 and (b) A-Cu/SnO2 electrodes with different deposition times at −1.0 V vs. RHE in CO2-saturated 0.1 M KHCO3.73 | ||
For the electrocatalytic performance, Cu/SnO2 and A-Cu/SnO2 were evaluated under a wide potential range (from −0.8 V to −1.2 V vs. RHE). The Cu/SnO2 catalyst achieved 75% of H2 FE and 25% of CO FE at −0.8 V vs. RHE. In contrast, A-Cu/SnO2 exhibited a higher selectivity reaching nearly 60% FE at −0.8 V vs. RHE and a significant decrease of FE of H2 approximately 40% at −0.8 V. This comparison indicated that A-Cu/SnO2 has not only higher selectivity toward CO than Cu/SnO2, but also higher CO2 reduction activity. Furthermore, the authors investigated the effect of SnO2 deposition time on Cu foam for both electrodes Cu/SnO2 and A-CuSnO2 at −1.0 V vs. RHE. In the case of Cu/SnO2 electrodes, the efficiency of HCOOH increased with increasing the deposition time reaching the maximum at 60 min, while H2 efficiency decreased along with increasing the deposition time. In contrast, CO FE achieved a higher level at 30 min and then decreased dramatically at 45 min of SnO2 deposition time, and has since continued to decline. For A-Cu/SnO2 with different deposition times of SnO2, the production selectivity remained almost constant at greater than 70% FE for CO, and the least formation product was for HCOOH with FE (<5%), with a remarkable suppression of hydrogen evolution reaction. In terms of stability, A-Cu/SnO2 catalyst with a deposited time of 15 min exhibited long-term stability of about 10 h. The selectivity for CO was maintained around 75% during the 10 h electrolysis, and the total current density was kept well at −8.5 mA cm−2.
This class of bimetallic is very interesting as it can be tuned by different synthesis method to prepare active electrode for CO2RR. However, one of the main drawbacks of these Cu–Sn materials is their shortcoming to further reduce CO2 to multicarbon hydrocarbons and other oxygenates value added chemicals. Accordingly, these materials do not offer a promising way to obtain a clean renewable fuel of high energy density and close the carbon loop cycle. The main products of Cu–Sn are carbon monoxide and formic acid or it can be tuned from one to another by varying the Cu/Sn composition.74 This is barrier towards viable utilization of these materials for CO2 valorization to C2 and oxygenates.
3.2 Cu–In bimetallic materials
Luo et al.75 synthesized Cu–In catalyst by simply depositing In3+ on Cu(OH)2 NWs followed with mild oxidation and in situ electroreduction treatments. The Cu(OH)2 nanowires were synthesized via simple chemical oxidation. For this purpose, Cu foil (1 cm × 1 cm) was cleaned with water and sonicated in acetone for 5 min. Then, it was further etched in a solution containing 2 M of HNO3 for 5 min to remove surface impurities. After immersing the Cu foil in a solution consisting of (2.5 M NaOH, 0.125 M (NH4)2 S2O8) for 10 min to grow Cu(OH)2 NWs, the color of the solution changed from reddish to dark cyan, indicating the formation of Cu(OH)2 NWs.76 Tin (In) was deposited by dipping Cu foil into a solution containing InCl3 for 30 s. The catalyst was obtained by in situ electroreduction before the CO2 electroreduction experiments.The electrocatalytic performance showed that the CO faradaic efficiency of CuNWs has increased gradually reaching the maximum (45%) at −0.6 V before dropping significantly to 16% at −1.0 V. HCOOH selectivity increased steadily along with decreasing the applied potential reaching the highest FE at −0.9 V and then decreased gradually at −1.0 V. While H2 exhibited a high FE > 50% at a potential between (−0.4 to −0.5 V).
In contrast, the CuIn20 catalyst produced CO as a main product throughout the entire examined potential range. Thus, CO FE of CuIn20 reached almost 95% in the range of −0.6 to −0.8 V.
The current density measurements of Cu NWs and CuIn20 under potentiostatic conditions showed that the CO partial current density of CuIn20 was 5 times higher than that of Cu NWs catalyst. The stability test of CuIn20 and Cu NWs was carried out at an applied potential of −0.6 V. Initially, Cu NWs catalyst exhibited 50% CO FE and dropped to 32% after 12 h of operation. Whereas, the CuIn20 catalyst exhibited a stable current density of nearly 1.7 mA cm−2 and maintained an average (90%) of CO FE in the span of 60 h. According to the authors, the long-term stability of CuIn20 catalyst could be ascribed to its low sensitivity to metal impurities.
The role of the In deposition amount on the CO2RR was studied by immersing Cu NWs in various concentrations of InCl3 solution. The result showed that the current density and CO faradaic efficiency depended strongly on the In amount. It was found that CO FE increased with increasing In amount (0.52 μmol cm−2) reaching a maximum of approximately 93% for CuIn20 catalyst and then decreased for a higher amount of In.
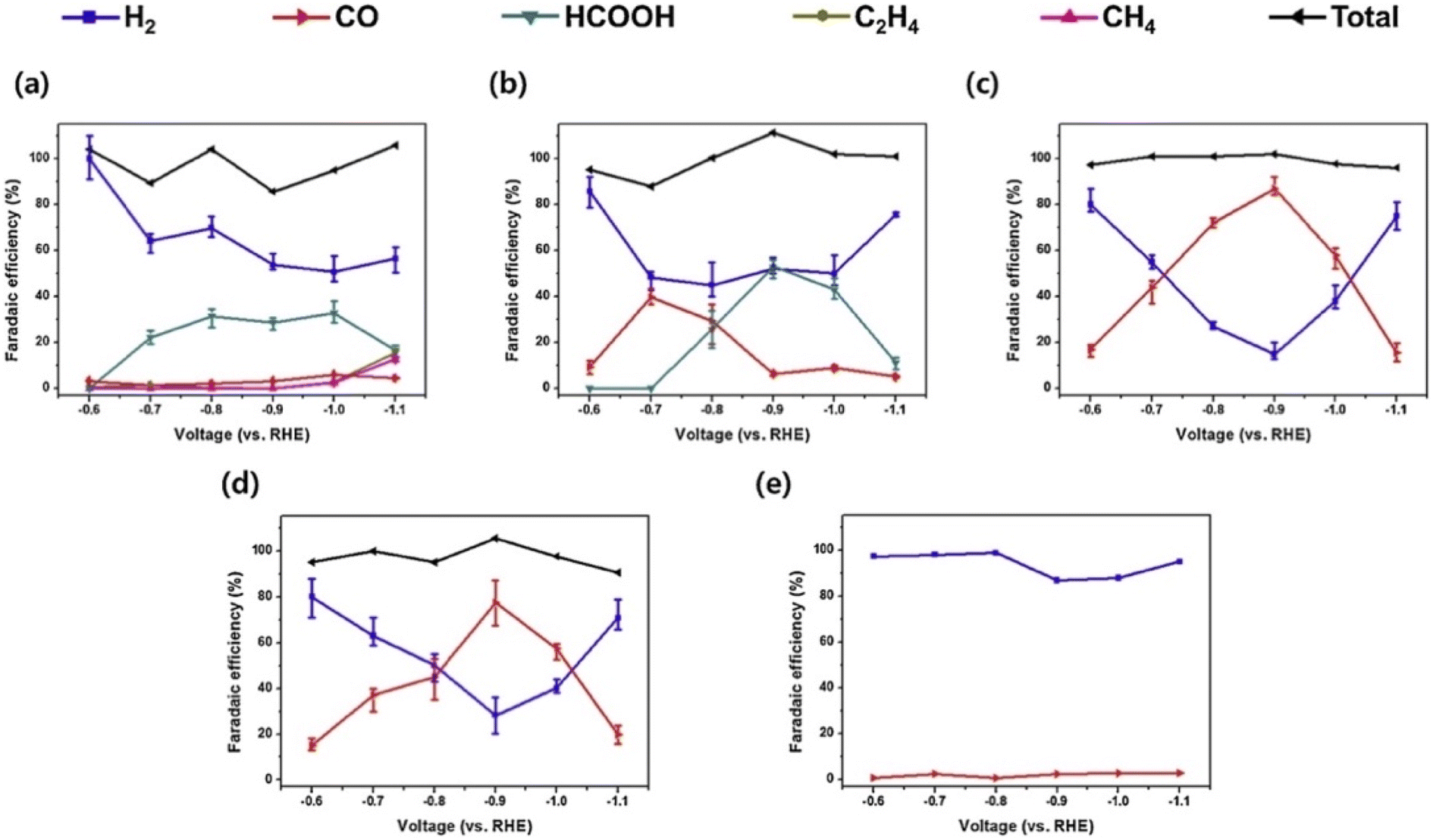
Rasul et al.77 developed a highly selective Cu–In electrocatalyst for the electrochemical reduction of CO2 to CO. The Cu foil (1 cm × 3 cm) was cleaned in 1 M HCl for several seconds, rinsed with Milli-Q water, and dried at room temperature. The foil was further dried with Kimwipes to avoid partial oxidation of the electrode. OD-Cu electrode was obtained by thermally oxidizing Cu metal sheet at 500 °C for 2 h under static air in a muffle furnace. Afterward, Cu–In electrode was obtained through the in situ electrochemical reduction of the OD-Cu electrode in a solution consisting of (0.05 M InSO4, 0.4 M citric acid) at a current density of −10 mA for 90 min.
The electrocatalytic performance of In–Cu and OD-Cu catalysts was investigated under a wide potential range potential from (−0.3 to −0.7 V vs. RHE). The results indicated that OD-Cu catalysts (Fig. 6b) produced mainly H2 at −0.3 V. However, CO and HCOOH selectivity increased along with increasing the applied potential and reached a maximum of 40% and 30%, respectively, at −0.6 V vs. RHE. In contrast, Cu–In electrode (Fig. 6a) exhibited a CO selectivity of 23% and a negligible H2 level at −0.3 V vs. RHE. The conversion of CO2 to CO increased with more negative polarization of the electrode, reaching a maximum faradaic efficiency of 95% at −0.7 V vs. RHE.
 | ||
| Fig. 6 (a) The current density of OD-Cu and Cu–In, (b) faradaic efficiencies of OD-Cu, (c) faradaic efficiencies of Cu–In, (d) test stability for the Cu–In catalyst at −0.6 V vs. RHE in 0.1 M KHCO3/CO2. Reproduced with permission.77 Copyright ©2015, Wiley-VCH. | ||
Cu–In and OD-Cu catalysts (Fig. 6c) exhibited similar values for total current density in the same electrochemical conditions. On the other hand, the stability of Cu–In catalyst (Fig. 6d) was evaluated at −0.6 V in 0.1 M KHCO3, indicating good stability for 7 h with 85% CO FE.
3.3 Cu–Pd bimetallic materials
Palladium is as an intriguing catalyst owing to its low barrier toward H2 adsorption and ease of formation of the metal hydride (PdHx).78–81 It is regarded as a promising candidate for catalyzing CO2 hydrogenation reactions and electrochemical CO2 reduction reactions because it facilitates the hydrogenation and gives mainly CO in CO2RR due to the hydride formation, which inhibits *CO binding and affects selectivity toward *COH/*CHO for further CO2RR C2 byproducts.48,82For CO2RR, Mun et al.,83 have prepared monodisperse Cu–Pd nanoparticles (NPs) with various compositions via a colloidal method. Different products were obtained for when comparing with bulk copper catalyst. It was observed that alloying Cu to Pd suppresses hydrocarbon evolution and leads to CO production with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Cu–Pd being the best electroactive catalyst giving 87% CO at −0.9 V vs. RHE (Fig. 7). The DFT results indicate that alloying Pd to Cu enhances the energy barrier of the CO protonation step, which is the potential-determining step for hydrocarbon production, resulting in a change in CO2RR selectivity: Pd alloying causes H* to be more firmly adsorbed on CuPd(211) than on Cu(211), and the energy binding of adsorbed H* on Cu–Pd alloy is much lower energy than that of Cu(211), indicating that the HER is suppressed. The limiting potential of the favored CO* protonation step is higher on CuPd(211) (−1.03 V) than on Cu(211) (−0.60 V) and as reported elsewhere50,82,84 the HCO* formation is much more favored than COH* formation on Cu(211), and on CuPd(211). This indicates the hydrocarbon suppression on Cu–Pd alloy.
:1 Cu–Pd being the best electroactive catalyst giving 87% CO at −0.9 V vs. RHE (Fig. 7). The DFT results indicate that alloying Pd to Cu enhances the energy barrier of the CO protonation step, which is the potential-determining step for hydrocarbon production, resulting in a change in CO2RR selectivity: Pd alloying causes H* to be more firmly adsorbed on CuPd(211) than on Cu(211), and the energy binding of adsorbed H* on Cu–Pd alloy is much lower energy than that of Cu(211), indicating that the HER is suppressed. The limiting potential of the favored CO* protonation step is higher on CuPd(211) (−1.03 V) than on Cu(211) (−0.60 V) and as reported elsewhere50,82,84 the HCO* formation is much more favored than COH* formation on Cu(211), and on CuPd(211). This indicates the hydrocarbon suppression on Cu–Pd alloy.
 | ||
| Fig. 7 Faradaic efficiencies of the Cu–Pd NP/C catalysts in 0.1 M KHCO3 solution: (a) Cu NP/C, (b) Cu3Pd NP/C, (c) CuPd NP/C, (d) CuPd3 NP/C, and (e) Pd NP/C. Reproduced with permission.84 Copyright ©2019, Elsevier. | ||
Recently, Xie and coworkers85 have prepared Cu–Pd heterostructures via dual potential electrochemical deposition which promotes CO2 reduction and stabilizes CO* to preferable CH4 formation. A small amount of HCOOH and C2H4 were observed. The aforementioned heterostructure exhibited a higher CO2-to-CH4 selectivity of 32% at high potentials of −1.2 and −1.25 V vs. RHE compared to the bare Cu and Pd metals. Using the DFT calculations, it was determined that adsorbed CO is the key intermediate for CH4 and C2H4 production which was in agreement with previous reports.86,87
A pyridine derivative abbreviated PYD was entrapped in Cu–Pd alloy to prepare and organically PYD@Cu–Pd composite for the CO2RR to combine two active sites for alcohols electrosynthesis in a single catalyst. Cu nanoparticles (NPs), Pd NPs, Cu–Pd alloy, PYD@Cu and PYD@Pd composite were also prepared and tested as control experiments.88 The PYD@Cu–Pd gives 26% and C2H5OH with 12% faradaic efficiencies at −0.04 and −0.64 V vs. RHE, respectively. Remarkably, the PYD@Pd and Cu–Pd yielded respectively 35% faradaic efficiency of CH3OH at −0.04 V vs. RHE and 11% of C2H5OH at −0.64 V vs. RHE. This means that the PYD@Cu–Pd catalyses CO2 to both CH3OH and C2H5OH l using two active regions: Cu–Pd (For C2H5OH) and pyridine ring of PYD@Pd to PYD H* (CH3OH).89 Other control catalysts did not lead to alcohol formation. Longer stability screening for the PYD@Cu–Pd composite at constant cathodic potential of −0.04 and −0.64 V vs. RHE was reported for 14 h. The current density remained constant for both potential values, reaching 21 mA cm−2 and 5 mA cm−2 for CH3OH and C2H5OH production, respectively, throughout the test. Moreover, the CH3OH FE could maintain around 25% for 15 times repeated electrolysis and a small drop on the faradaic efficiency to C2H5OH was observed throughout the 15 cycles. This work shows the importance of the synergic effect of gathering organic catalyst and metal alloy in a very engineered fashion to put in work two alcohol sites for the CO2RR.
Whereas, Wang and coworkers90 have investigated the CuPd alloy with Pd rich surface supported on carbon for the CO2RR to HCOO− with 2 electron transfer. The prepared Cu20P820/C catalyst using adsorbate-induced surface segregation method shows a modest faradaic efficiency for the HCOO− electrosynthesis of 7.37%. When the same catalyst composition was prepared using by the NaH2PO2 reduction solution method instead of the H2 thermal reduction process, the faradaic efficiency for HCOO− production increased to 60% at −0.75 V vs. Ag/AgCl. The authors have confirmed using ex situ XPS that the catalytic activity of the catalyst can be attributed to the lower bonding strength of H* (adsorbed hydrogen) on the Cu20Pd80/C surface, which is promoted by the ligand effect between Cu and Pd as confirmed by the opposite band energy shift on Pd 3d5/2 and Cu 2p3/2 as well as the upshift of the d-band center for Cu20Pd80/C NPs. Then, the lower bonding strength of hydride on the surface catalyst would reduce the activation energy barrier of the catalyst to transfer hydrogen from its surface to CO2 and to form the HCOO* intermediate. Long-term chronoamperometry of 140 h have led to the CO catalyst poisoning and deactivation.
Carbon monoxide electroformation from CO2RR was reported by Chen et al.,91 using bimetallic Cu–Pd nanoalloys with different compositions and morphologies: spherical (noted as S) Cu–Pd nanoalloys with Cu/Pd molar ratio of 1/0.3 have the highest faradaic efficiency toward CO conversion (93%), while the dendritic Cu–Pd nanoalloys (noted as D) have the highest faradaic efficiency for H2 (65.2%) via hydrogen evolution reaction at a polarized potential of −0.87 V vs. RHE (Fig. 8). While the concave nanocubes of Cu–Pd–C (noted as C) shows much lower CO FE. This work has shed light on the effect of morphologies on CO2RR electroconversion, this will pave the way towards to catalysis by design and morphology dependent activity.
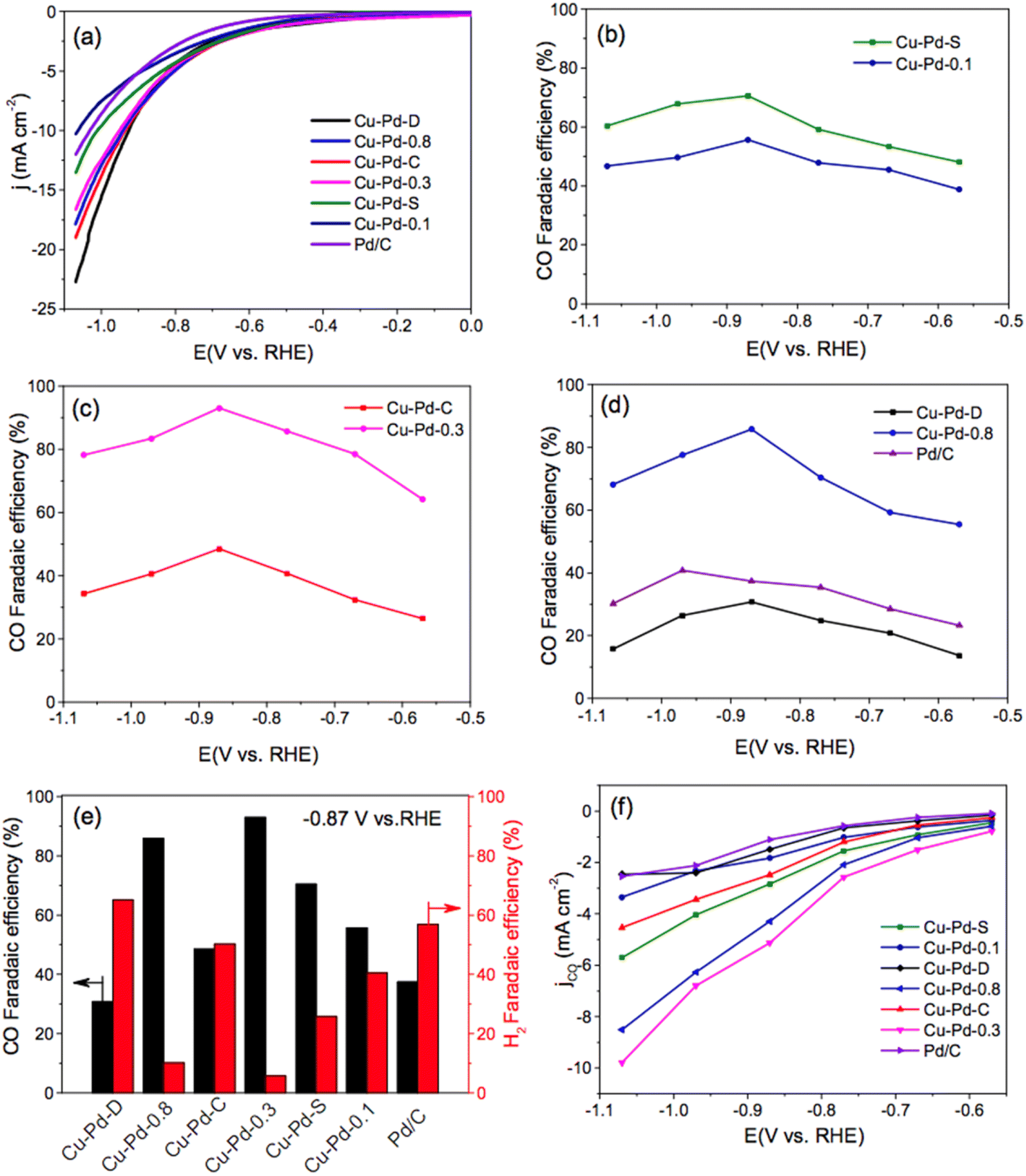 | ||
| Fig. 8 LSV curves (a) of bimetallic Cu–Pd–S, Cu–Pd-0.1, Cu–Pd–C, Cu–Pd-0.3, Cu–Pd-D, Cu–Pd-0.8 nanoalloys, commercial Pd/C catalyst in CO2 saturated 0.5 M KHCO3 solution with a scan sweep of 10 mV s−1, the CO faradaic efficiency at different applied potentials (b–d), the CO and H2 faradaic efficiency at a fixed potential of −0.87 V (e), and CO partial current density of these nanoalloys (f). Reproduced with permission.91 Copyright ©2018, American Chemical Society. | ||
C2 byproducts can be also formed through direct CO2RR. For instance, Feng and coworkers92 have prepared Cu–Pd catalysts using simple electrodeposition method on carbon paper. The electrochemical CO2RR performance of the Cu–Pd/CP outperforms the bare Cu/CP and Pd/CP leading to higher current density suggesting favorable bonding and CO2 activation. The faradaic efficiency of C2H4 reached 45.23%, at −1.2 V vs. RHE, whereas, Cu/CP recorded only 30% and no C2H4 on Pd/Pd catalyst. This reported C2H4 efficiency outperformed the state-of-the art metal catalysts reported elsewhere.93–96 Ma et al.97 investigated the effect of geometric arrangement on the Cu–Pd activity and selectivity. To this end, the authors synthesized different bimetallic Pd–Cu catalysts with disordered, ordered, and phase-separated atomic arrangements.
The electrocatalytic performance of CuPd bimetallic nanoparticles with distinct geometric arrangements exhibited different selectivity toward C1 and C2 products. For the ordered CuPd nanoparticles the faradaic efficiency toward C1 products (mainly CO) achieved 80%, while C2 products exhibited the lowest FE < 5%. However, the disordered CuPd nanoparticles reached a higher FE for CH4 the ordered or phase-separated nanoparticles. In contrast, the phase-separated exhibited the highest FE 50% toward C2 products (primarily C2H4). This could be due to the fact that the binding *CO in the phase-separated is less affected by the segregation of the two sites. Regarding the current density measurements, the phase-separated sample exhibited the highest total current density of 370 mA cm−2, while the ordered sample achieved the lowest total current density. Furthermore, the authors investigated the correlation between the catalyst composition and its activity and selectivity. Therefore, they prepared two samples with different (Cu:Pd) ratios for the disordered structure: Cu3Pd and CuPd3. The electrochemical characterization showed that the CH4 FEs for disordered CuPd were higher than that for both Cu3Pd and CuPd3. In addition, the FEs of C2 products increased along with increasing Cu concentration.
Zhang et al.98 reported an electrodeposition strategy for designing highly dispersed, ultrafine PdCu catalysts. PdCu catalyst exhibited a greater than two-fold enhancement in FE for CO2 reduction to CH4 as compared to the Cu catalyst. The maximum FE for CH4 was 33% at −1.8 V (vs. Ag/AgNO3). They indicated that the improvement was originated from a synergistic reaction between the Cu–CO and Pd–H sites during electrochemical CO2 reduction. The onset of CO2 reduction to CH4 at the PdCu catalyst appeared at ∼400 mV and ∼200 mV more positive than at the Pd and Cu catalysts, respectively.
Chen et al.99 prepared nanostructured Cu2O-derived Cu catalyst and PdCl2 for CO2 to C2H6 conversion. The Cu2O-derived Cu catalyst was prepared by the electrochemical deposition on Cu discs. When the addition of PdCl2 was added into the catholyte during the CO2 reduction at −1.0 V (vs. RHE), the C2H6 formation could be achieved with an FE of 30.1% at the same potential. According to their mechanistic studies, C2H4 was first produced from CO2 reduction at the Cu sites. The hydrogenation occurred with the help of adsorbed PdClx to produce C2H6. The efficient conversion of C2H4 to C2H6 required both copper and PdClx sites. However, adding of other palladium-based particles to the electrolyte, such as Pd0, PdO, or Pd–Al2O3, did not reach the same conversion efficiency.
Zhu et al.100 employed thermal reduction treatment and in situ growth to prepare the CuPd catalyst. According to the electrocatalytic results, the FE of CuPd(100) catalyst achieved 50.3% at −1.4 V vs. RHE, which was higher than that of the Cu catalysts (23.6%) at the same potential. The CuPd(100) catalyst also had the highest electrochemically active surface area of 7.04 × 10−3 mF cm−2. These results were consistent with the DFT prediction, implying that CuPd(100) could provide enough CO* for the C–C coupling by lowering the energy barrier of the CO2* hydrogenation step.
Lai et al.101 used a pulse program consisting of an open-circuit voltage (OCV) and a cathodic potential to investigate the effect of spontaneous reoxidation on the catalytic performance of Cu–Pd bimetallic catalysts. The results showed that the simultaneous presence of Cu and Pd was critical for achieving high CO FE for the bimetallic catalysts. Among all Cu–Pd catalysts, the Cu5Pd5 reached the maximum FE of 88% with the partial current density of −132 mA h g−1 at an applied cathodic potential of −0.7 V for 210 s and anodic potential at OCV for 450 s. The DFT calculation suggested that such efficient CO2 conversion to CO resulted from the reduced *CO binding strength.
Li et al.102 constructed a novel Cu–Pd catalyst Cu@PIL@Pd by impregnating PdCl2 into the Cu@PIL with Cl-as the anion. It was found that the Cu@PIL@Pd-2 catalyst exhibited the highest FE toward C2+ of 68.7% with a high partial current density of 178.3 mA cm−2 at a cathodic potential of −1.01 V vs. RHE. The catalyst also showed the highest CH4 FE of 42.5% with a partial current density of 172.8 mA cm−2 at −1.24 V vs. RHE. The authors attributed the high selectivity of the Cu@PIL@Pd-2 catalyst toward C2+ products to the synergy effect between the adjacent Pd and Cu sites. The Cu was found to promote the CO generation on the Pd sites, while the abundant Cu–Pd interfaces promoted the *CO spillover from Pd sites to adjacent Cu sites, promoting the C–C coupling reaction.
Shen et al.103 investigated CO electroreduction on a series of Cu–Pd catalysts prepared with different compositions: Cu70Pd30, Cu49Pd51, and Cu23Pd77. The results showed that the Cu70Pd30 catalyst produced acetate with a FE of >14% at −0.5 V vs. RHE and ethanol with a FE of 29% at −1.2 V vs. RHE. For the Pd-rich Cu23Pd77 catalyst, H2 was the main product with a FE of >40% throughout the investigated potential range. In contrast, the Cu49Pd51 achieved a high selectivity toward acetate with a FE of >65% at −1.2 V vs. RHE. The acetate selectivity of Cu49Pd51 in the alkaline electrolyte was explained by the asymmetrical C–C coupling mechanism via *CO–*CHO coupling.
3.4 Cu–Zn bimetallic materials
Zinc (Zn) is environmentally friendly and less expensive as compared to other co-catalysts such as Ag, Pd, and Au.104 Zn has attracted more attention due to its ability to reduce CO2 to CO, HCOOH, and syngas at different overpotential.105,106Because hydrogen evolution reaction occurs more slowly on Zn electrodes than on Cu electrodes,107 forming bimetallic Cu–Zn catalyst is one approach to enhance the electrocatalytic activity of Cu electrode toward CO2 reduction and promotes multicarbon products. Ren et al.108 prepared a series of oxide-derived CuxZn catalysts to promote the reduction of CO2 to C2H5OH. They revealed that introducing a co-catalyst improved the selectivity and the efficiency of CO2 reduction.
CuxZn oxide films were prepared by dissolving 0.3 M CuSO4 and 2.3 M lactic acid in ultrapure water. NaOH was added to the solution to adjust the pH to 12, 10.6, or 9. Then 10 mM of ZnCl2 was added to the solution. The oxide films CuxZn were deposited onto Cu disks using a current density of −0.92 mA cm−2 for 10 min. During the deposition process, the electrolyte was kept at 960 °C and stirred at 300 rpm. The CuxZn catalysts exhibited distinct selectivities toward C2H4 and C2H5OH formation. The faradaic efficiency of CuxZn catalysts toward C2H5OH increased along with increasing the amount of Zn, achieving the maximum FE of 29.1% on Cu4Zn at −1.05 V vs. RHE. On the contrary, the faradaic efficiency toward C2H4 decreased along with increasing Zn amount, achieving the lowest FE of 4.1% on Cu2Zn.
At −1.05 V vs. RHE, Cu4Zn catalyst reached the highest partial current density of C2H5OH approximately −8.2 mA cm−2, Cu10Zn catalyst achieved a partial current density of almost −4.0 mA cm−2 toward C2H5OH. Whereas Cu2Zn exhibited the lowest partial current density of almost −2.3 mA cm−2 toward C2H5OH. Furthermore, Cu4Zn catalyst maintained remarkable stability toward C2H5OH with a faradaic efficiency of 29.1% in the span of 5 h. However, the FE of C2H5OH decreased to 25% for 10 h electrolysis. The authors proposed a mechanism to elucidate the electroreduction of CO2 to C2H5OH. In the first step, CO2 is reduced to *CO at Cu or Zn sites and is then reduced to CHO or CHx (x = 1–3) intermediates. Because of the weak adsorption of CO on Zn sites, desorbed CO could diffuse and overflow on the Cu sites. This CO could insert between Cu sites and *CH2 to form *COH2 which is further reduced to acetaldehyde and finally C2H5OH.
Keerthiga and Chetty109 used a simple electrodeposition approach to design a hierarchically structured Zn electrocatalyst on Cu electrode. They prepared two samples Cu/Zn-A (in electrolytic bath containing 0.6 M of sodium zincate) and Cu/Zn-B (in electrolytic bath containing 6 M of sodium zincate). For the electrocatalytic performances, the prepared catalysts were evaluated under a wide potential range (from −0.7 V to −1.8 V vs. NHE). The Cu/Zn-B catalyst produced a maximum CH4 formation of 52%, C2H6 formation of 24%, and H2 formation of 8.7% at a potential of −1.6 V vs. NHE. Furthermore, the Cu/Zn-B catalyst exhibited a higher current density of 0.70 mA cm−2 for CH4 at a potential of −1.6 V vs. NHE compared to the Cu (0.14 mA cm−2) and Zn (0.01 mA cm−2) electrodes. However, Cu/Zn-A catalyst produced mainly H2, with only minor hydrocarbon formation. The results suggested that Zn deposition on Cu reduced hydrogen evolution and favored the addition of protons to form CH4. The porous nature of hierarchically structured Zn could allow the diffusion of CO2 through the Cu/Zn interface for the reduction while maintaining high catalytic activity due to the of the large electrode area of the hierarchical structure reducing the surface poisoning.
Su et al.110 demonstrated the catalytic synergies of Cu and Zn to enhance the CO2 electroreduction towards liquid C2 products using hierarchically porous Cu/Zn catalysts.
The bimetallic catalysts were prepared based on an interfacial self-assembly method, in which the alloy composition of the catalysts can be adjusted by varying the ratios of metal precursors. The electrocatalytic performance results showed that hierarchically macroporous–mesoporous (HMMP) Cu5Zn8 demonstrated a high C2H5OH selectivity of 46.6% with C2H5OH current density of −2.3 mA cm−2 at −0.8 V vs. RHE. The Cu5Zn8 electrocatalyst maintained an excellent stability of 11 h towards CO2 electroreduction. Grätzel and coworkers111 prepared CuZn bimetallic catalyst by applying 100 atomic layer deposition cycles of ZnO precursor on CuO nanowires. The obtained CuZn bimetallic catalysts exhibited 32% FE for C2H5OH production and C2H5OH current density of −10.5 mA cm−2 at −1.15 V vs. RHE. Based on their understanding, the authors proposed a mechanism of C2H5OH formation on Cu and CuZn catalysts (Fig. 9). On Cu catalyst, C2H4 and C2H5OH are produced through the dimerization of two adsorbed CO, resulting in the formation of C2H4. On CuZn catalyst, the CO produced on Zn sites tends to be released into the electrolyte due to the low binding energy between CO and Zn. At low overpotential, the formation of *CH3 intermediate is minimal, and most of CO is released as CO gas. At higher overpotential, the *CH3 intermediate is produced in large amounts and combines with the CO present near Cu sites to form C2H5OH.
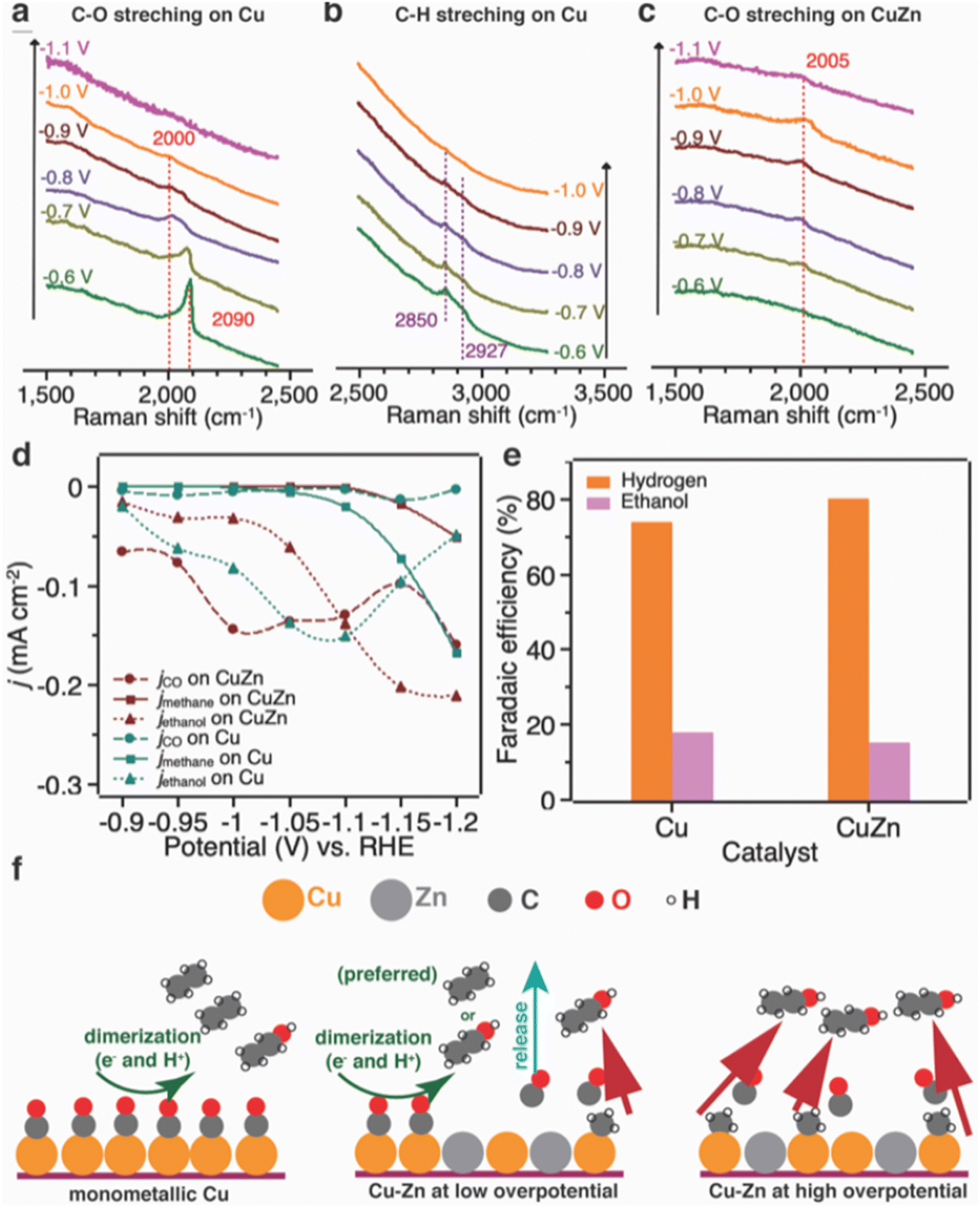 | ||
| Fig. 9 Operando Raman spectrum of (a and b) Cu catalyst and (c) CuZn catalyst at different potentials in 0.1 M KHCO3 under laser radiation at 785 nm; (d) partial current density (normalized against the electrochemical surface area) of C2H5OH, methane and carbon monoxide as a function of applied potential; (e) electrochemical reduction of acetaldehyde on Cu and CuZn catalyst; and (f) proposed mechanism of C2H5OH formation. Reproduced with permission.111 Copyright ©2019, Wiley-VCH. | ||
Dongare et al.112 engineered oxide derived CuO–ZnOx bimetallic catalysts via a simple co-precipitation method followed by an air atmosphere calcination step. The loading percentage of Cu to Zn in the electrocatalysts was adjusted by varying the precursor concentrations from x = 5 to 20 wt%. The performance of the CuO–ZnOx electrocatalysts was investigated under a wide potential range from −0.4 V to −1 V vs. RHE. The CuO–ZnOx electrocatalysts demonstrated different FE for HCOOH, C2H5OH, CH3OH, and n-propanol depending on the precursor concentrations. The CuO electrode showed low selectivity for C2H5OH about 7.10% of FE. In contrast, incorporating ZnO into CuO improved C2H5OH selectivity while suppressing the formation of HCOOH. Interestingly, CuO–ZnO10 electrode showed the highest faradaic efficiency of 22.27% for C2H5OH at −0.8 V vs. RHE. While, further increase in Zn concentration decreased the FE of C2H5OH to approximately 16.41% for CuO–ZnO20 electrode. Similar trend was found in the case of n-propanol and CH3OH. The FE of n-propanol increased from 4.5% to 10.23% using CuO and CuO–ZnO10 respectively at −0.6 V vs. RHE, and the FE of CH3OH increased from 7.44 to 15.02% on CuO and CuO–ZnO10 electrodes at −0.6 V vs. RHE. The authors claimed that the balanced CuO and ZnO percentage for maximum C–C coupling was responsible for the improved selectivity. The CuO–ZnO10 electrode gave a balanced CO supply, resulting in increased CO dimerization. The ZnO provided additional CO to increase the local CO concentration on the Cu surface and as a result, *CO surface coverage, resulting in a faster conversion of CO-to-C2H5OH products. Furthermore, increasing Zn concentration in the CuO–ZnOx electrode beyond 10% results in as oversupply of CO, which lowered the *CO dimerization.
The stability and long-term durability of CuO–ZnO10 electrode was also investigated. During 12 hours of operation, the CuO–ZnO10 electrode maintained a total current density of −3.75 mA cm−2 with no change in the electrode morphology. The reproducibility experiments on CuO–ZnO10 electrode revealed that the selectivity of C2H5OH was maintained for first three cycles but decreased for subsequent cycles.
3.5 Cu–Ag bimetallic materials
Hoang et al.113 aimed to enhance CO2 selectivity towards C2 products such as C2H4 and C2H5OH. For this purpose, Cu–Ag catalysts were electrodeposited at a constant current density of 4 mA cm−2 in a plating bath containing 0.1 M CuSO4·5H2O + 3.5 diamino-1,2,4 triazole (DAT), with or without 1 mM Ag2SO4, at pH = 1.5 adjusted by using H2SO4. (1 cm × 2.5 cm) of carbon paper was activated by immersing it in concentrated HNO3 for 60 min and used as a gas diffusion electrode before being electrodeposited with 2 C cm−2 of CuAg. The catalytic activity and selectivity of Cu and CuAg catalysts during CO2 electroreduction were evaluated in a flow electrolyzer. CO, C2H4, and C2H5OH were the major products using Cu-wire 0% Ag named (Cu-wire), CuAg-wire (6% Ag) electrodeposited with DAT named (CuAg-wire), and CuAg-wire (6% Ag) electrodeposited without DAT named (CuAg-poly).The electrocatalytic results showed that the catalysts exhibited different activities and product selectivities in 1 M KOH electrolyte. The CuAg-poly catalyst exhibited the lowest faradaic efficiency nearly 30% toward C2H4 and 20% toward C2H5OH at −0.7 V vs. RHE. Furthermore, CuAg-poly catalyst exhibited a low current density approximately −20 mA cm−2 toward CO, C2H4, and C2H5OH. Cu-wire catalyst achieved high faradaic efficiency as well as high current density toward C2 products. The faradaic efficiency for C2H4 increased along with increasing the potential reaching the maximum (40%) at low potential (−0.5 V vs. RHE), and C2H4 current density reached −100 mA cm−2 at −0.7 V vs. RHE. However, for the C2H5OH product, Cu-wire exhibited a faradaic efficiency of 20% at −0.5 V vs. RHE and a current density of −75 mA cm−2 at −0.7 V vs. RHE. In contrast, CuAg-wire catalyst achieved both high FE (60%) toward C2H4 and high C2H4 current density about- 180 mA cm−2 at −0.7 V vs. RHE. This high FE of C2H4 could be ascribed to the high pH solvent. Huang et al.114 have prepared Ag–Cu nanodimers for CO2-to-CO reduction, authors performed a synthesis Ag–Cu with different compositions. The catalytic activity enabled by the addition of Ag to Cu in the form of segregated nanodomains within the same catalyst accounts for a 3.4-fold increase in faradaic efficiency for C2H4 and 2-fold increase in partial current density for CO2 reduction when compared to the pure Cu counterpart. The FE of C2H4 electrosynthesis reached around 40% at −1.1 V vs. RHE (Fig. 10).
 | ||
| Fig. 10 (a–c) FEs for each gaseous product and for the main liquid product over (a) Ag NPs, (b) Cu NPs, and (c) Ag1–Cu1.1 NDs. (d and e) Total current density (d) and partial current density for the electroreduction of CO2 (e) over Ag NPs, Cu NPs, and Ag1–Cu1.1 NDs. (f) CO and C2H4 mass activity of Ag NPs, Cu NPs and Ag1–Cu1.1 NDs at −1.1 V vs. RHE. Reproduced with permission.114 Copyright ©2018, American Chemical Society. | ||
The Cu–Ag alloy was also used by Wang et al.115 as an interfacial catalyst for the electrochemical reduction of CO2 to C2H4. The faradaic efficiency for C2H4 formation reached over 42%, which is more than twice as high as that of pure Cu catalyst at −1.1 V vs. RHE. The Cu–Ag catalyst remains stable and produce steady state C2H4 for 30 h electrolysis. The mechanism formation C2H4 was explained by the steps as follows: (i) Ag nanoparticles capture CO2, (ii) CO2 molecule accepts one proton and one electron to form *COOH intermediate on Ag nanoparticle surface, (iii) *COOH accepts one proton and one electron to form *CO intermediate on Ag nanoparticle surface. (iv) Transfer of CO intermediate from Ag nanoparticle to Cu side via the Ag/Cu interface and finally (v) C2H4 is formed by coupling two CO molecules.
Wang et al.116 synthesized Cu–Ag bimetallic nanowire arrays as catalysts in three steps. First, thermal oxidation was used in ambient air to grow CuO/Cu2O nanowires on copper mesh. Then, in the presence of H2, thermal reduction was carried out to obtain Cu nanowires. Finally, the Cu–Ag bimetallic nanowires were produced via galvanic replacement between Cu nanowires and the Ag+ precursor. This catalyst leads to a reduction of CO2 to a multitude of C1 and C2 byproducts with HCOO− being the highest in faradaic efficiency at −0.6, −0.7 and −0.8 V vs. RHE. Interestingly, at −0.8 V CH3OH, C2H5OH and propanol were observed with small faradaic efficiency (around 8%).
Meyer's group117 have succeeded to reduce CO2 to acetate using monodispersed, ultrasmall Ag and Cu bimetallic nanoparticles on electrochemically polymerized poly-Fe(vbpy)3(PF6)2 films. The authors showed that the 8 electrons transfer to produce acetate can be obtained using a ratio variation of Ag/Cu. The maximum faradaic efficiency for acetate production reached 21.2% at −1.33 V vs. RHE. The presence of Ag on the surface of the cluster pair Cu2–Ag3 plays a crucial role in maximizing acetate production.
Whereas, Jaramillo's group118 have prepared CuAg thin films using nonequilibrium Cu/Ag alloying by physical vapor deposition technique. The prepared thin films prepared were tested for CO2 electroreduction, the faradaic efficiencies of different 2 electrons byproducts including H2, CO and HCOOH and further reduced (>2e−) hydrocarbon, alcohol, and carbonyl CO2RR products were analyzed. The films containing Cu-rich and Ag-rich phases with substantial nonequilibrium interphase miscibility demonstrated that Ag miscibility into Cu is an effective strategy to promote the electrocatalytic CO2R selectivity and activity toward liquid carbonyl products. This was attributed to lower surface binding energies of oxygen-containing intermediate species. The catalytic activity of CuAg versus Cu toward more reduced products was decreased, while the activity toward hydrocarbons and HER was significantly suppressed. The decreasing activity and selectivity to hydrocarbons and suppressing hydrogen was explained to be due to the Ag doping which weakens the Cu binding energy of *H species.
3.6 Summary of the performance of Cu-based catalysts for CO2RR
| Catalyst | Conditions | Highlighted product with FE |
|---|---|---|
| CuSn core–shell NPs:65 | 0.1 M KHCO3, -0.7 V vs. RHE | |
| CuSn NPs/c-A | CO (70.1%) | |
| CuSn NPs/c-AH | CO (45.1%) | |
| CuSn NPs/c-H | CO (20.1%) | |
| Sn/Cu-PVDF electrode66 | 0.1 M KHCO3, −0.9 V vs. RHE | CO (80%) |
| Cu–Sn core shell nanowire70 | 0.5 M KHCO3, −0.8 V vs. RHE | CO (90%) |
| Sn/OD-Cu71 | 0.1 M KHCO3, −0.6 V vs. RHE | CO (90%), HCOO− (5%) |
| Core–shell Cu–SnO2 NPs72 | 0.5 M KHCO3 | |
| 0.8 nm thick | −0.7 V vs. REH | CO (93%) |
| 1.8 nm thick | −0.9 V vs. RHE | HCOO− (85%) |
| A-Cu/SnO2 (ref. 73) | 0.1 M KHCO3, −1.0 V vs. RHE | CO (70%) |
| CuIn20 (ref. 75) | 0.1 M KHCO3, −0.6 V vs. RHE | CO (95%) |
| Cu–In electrode77 | 0.1 M KHCO3, −0.7 V vs. RHE | CO (95%) |
| Cu0.75In0.25 | 0.5 M NaHCO3, −0.7 V vs. RHE | CO (81%) |
| Cu4Zn119 | 0.1 M KHCO3, −1.05 V vs. RHE | C2H5OH (29.1%) |
| Cu5Zn8 (ref. 120) | 0.05 M KHCO3, −1.5 V vs. RHE | CO (80%) |
| Cu94Ag6 (ref. 113) | 1 M KOH, −0.7 V vs. RHE | C2H4 (60%), C2H5OH5 (25%) |
| Cu16Ag84 (ref. 121) | 0.5 M KHCO3, −0.83 V vs. RHE | CO (45%) |
| Ag57Cu43 (ref. 121) | 0.5 M KHCO3, −1.5 V vs. RHE | CO (40%) |
| Ag–Cu core–shell (surface Ag:Cu = 1:0.6) |
0.1 M KHCO3, −1.06 V vs. RHE | CO (5%), CH4 (18%), C2H4 (25%) |
| Ag–Cu core–shell122 (surface Ag:Cu = 1:7.63) |
0.1 M KHCO3, −1.06 V vs. RHE | CO (82%) |
| Cu38Cd62 (ref. 123) | 0.05 M KHCO3, −1.12 V vs. RHE | CO (43%), HCOO− (10%) |
| Ordered CuPd97 | 1 M KOH, −0.55 V vs. RHE | CO (80%) |
| Phase separated CuPd97 | 1 M KOH, −0.75 V vs. RHE | CO (17%), C2H4 (48%), C2H5OH (15%) |
| Disordered CuPd97 | 1 M KOH, −0.60 V vs. RHE | CO (60%), CH4 (1%), C2H4 (4%), C2H5OH (2%) |
| Pd85Cu15 (ref. 124) | 0.1 M KHCO3, −0.89 V vs. RHE | CO (86%) |
| o-AuCu125 | 0.1 M KHCO3, −0.77 V vs. RHE | CO (80%) |
| Cu@Au126 | 0.5 M KHCO3, −0.65 V vs. RHE | CO (30%) |
4. Challenges and prospects
Over the next few decades, fossil fuels may remain the main energy source. Mitigating the impact of waste CO2 emissions is still a key issue in modern society. Electrocatalytic CO2 reduction provides an intriguing method for reducing CO2, by which CO2 as a feedstock can be converted into fuel or value-added chemicals. Copper is a magic element that can give various CO2RR byproducts. Controlling, tuning and modulating Cu surfaces and Cu-based bimetallic materials is necessary to achieve stable multicarbon (i.e. targeted product) production. Regardless of structural control, fundamental understanding of electrolyte engineering, cell design, pressure, temperature should be also elucidated to give the working state of Cu electrodes. In this review, we have discussed the mechanism of CO2 electrochemical reduction on various copper-based catalytic materials and how certain factors determine the product distribution and its selectivity including synthesis method, modification, surface morphology and structure. Copper-based catalyst materials currently occupy an important position in electrolysis due to their exceptional properties. Efficient and robust copper-based materials have been successfully prepared through enhanced synthetic strategies. These efforts embody the most important areas to understand the basics of CO2 reduction and contribute positively to the development of new bimetallic catalysts. Regardless of the important steps that have been made in this direction and the encouraging research labor and production, we still feel that these following prospects should be taken into account:• Develop other or existing chemical synthesis and modification technique that requires a straightforward design of the active sites in the general catalysis by design procedure. Thusly, more exploration of the modification steps should be explored.
• Explore and investigate other metals as they can give a better energy efficiencies and selectivity, for instance Sultan and coworkers have developed Al2CuO4 nanosheets uniformly covered with CuO nanoparticles giving high FE for ethylene (82.4%) and remarkable stability in reaction condition to 100 h; this material achieved high current density of 421 mA cm−2 in a flow cell.127
• In depth-understanding of CO2RR mechanisms is still a challenging task due to the evolution of the active site and the reconstruction of Cu materials under reaction conditions.
• Apply advanced operando techniques and theoretical calculation methods are highly recommended to probe the oxidation state, the CO2 activation, and possible reaction intermediates of the electrochemical synthesized Cu-based catalysts. This will help to reorient and guide synthesis researchers to efficiently prepare a desired active site for a given CO2RR byproduct as well as understanding the reaction mechanism and provide further guidance for the catalyst development.
• Tremendous efforts should be directed towards the design and construction of larger CO2 electrolyzer pilots. It should be noted that the high currents of the electrolyzer as well as the overall energy efficiency are key requirements for further commercialization of this technology.
It is trustworthy to note that the catalytic activity and stability of the reported electrocatalysts are still far away from the industrialization that usually requires long-term stability of more than 1000 h at a high current density of over 200 mA cm−2.
Author contributions
Otmane Zoubir: writing the draft, writing – review & editing, literature analysis. Lahoucine Atourki: writing – review & editing; project administration, supervision and validation. Hassan Ait Ahsaine: writing the draft, project administration; supervision and validation. Amal BaQais: writing – review & editing.Conflicts of interest
The author declares that he has no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
Authors thank all the involved institutions.References
- N. S. Spinner, J. A. Vega and W. E. Mustain, Catal. Sci. Technol., 2012, 2, 19–28 RSC.
- W. Zhu, B. M. Tackett, J. G. Chen and F. Jiao, Top. Curr. Chem., 2018, 376, 41 CrossRef PubMed.
- N. Uemoto, M. Furukawa, I. Tateishi, H. Katsumata and S. Kaneco, ChemEngineering, 2019, 3, 15 CrossRef CAS.
- M. Mondal, S. Khanra, O. N. Tiwari, K. Gayen and G. N. Halder, Environ. Prog. Sustainable Energy, 2016, 35, 1605–1615 CrossRef CAS.
- W. C. Chueh and S. M. Haile, ChemSusChem, 2009, 2, 735–739 CrossRef CAS PubMed.
- M. Mikkelsen, M. Jørgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43–81 RSC.
- S. Bai, H. Qiu, M. Song, G. He, F. Wang, Y. Liu and L. Guo, eScience, 2022, 2, 428–437 CrossRef.
- C. Liu, J. J. Gallagher, K. K. Sakimoto, E. M. Nichols, C. J. Chang, M. C. Chang and P. Yang, Nano Lett., 2015, 15, 3634–3639 CrossRef CAS PubMed.
- M. Schreier, J. Luo, P. Gao, T. Moehl, M. T. Mayer and M. Grätzel, J. Am. Chem. Soc., 2016, 138, 1938–1946 CrossRef CAS PubMed.
- X. Chang, T. Wang and J. Gong, Energy Environ. Sci., 2016, 9, 2177–2196 RSC.
- Z. Cao, D. Kim, D. Hong, Y. Yu, J. Xu, S. Lin, X. Wen, E. M. Nichols, K. Jeong and J. A. Reimer, J. Am. Chem. Soc., 2016, 138, 8120–8125 CrossRef CAS PubMed.
- A. S. Hall, Y. Yoon, A. Wuttig and Y. Surendranath, J. Am. Chem. Soc., 2015, 137, 14834–14837 CrossRef CAS PubMed.
- M. Liu, Y. Pang, B. Zhang, P. De Luna, O. Voznyy, J. Xu, X. Zheng, C. T. Dinh, F. Fan and C. Cao, Nature, 2016, 537, 382–386 CrossRef CAS PubMed.
- Q. Lu and F. Jiao, Nano Energy, 2016, 29, 439–456 CrossRef CAS.
- Q. Lu, J. Rosen, Y. Zhou, G. S. Hutchings, Y. C. Kimmel, J. G. Chen and F. Jiao, Nat. Commun., 2014, 5, 1–6 Search PubMed.
- Z. Sun, T. Ma, H. Tao, Q. Fan and B. Han, Chem, 2017, 3, 560–587 CAS.
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS PubMed.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC.
- A. Vasileff, C. Xu, Y. Jiao, Y. Zheng and S.-Z. Qiao, Chem, 2018, 4, 1809–1831 CAS.
- F. Chang, M. Xiao, R. Miao, Y. Liu, M. Ren, Z. Jia, D. Han, Y. Yuan, Z. Bai and L. Yang, Electrochem. Energy Rev., 2022, 5, 1–35 Search PubMed.
- D. Karapinar, C. E. Creissen, J. G. Rivera de la Cruz, M. W. Schreiber and M. Fontecave, ACS Energy Lett., 2021, 6, 694–706 CrossRef CAS.
- B. Yang, K. Liu, H. Li, C. Liu, J. Fu, H. Li, J. E. Huang, P. Ou, T. Alkayyali and C. Cai, J. Am. Chem. Soc., 2022, 144, 3039–3049 CrossRef CAS PubMed.
- Z. Han, D. Han, Z. Chen, J. Gao, G. Jiang, X. Wang, S. Lyu, Y. Guo, C. Geng and L. Yin, Nat. Commun., 2022, 13, 1–10 Search PubMed.
- Y. Jia, F. Li, K. Fan and L. Sun, Adv. Powder Mater., 2022, 1, 100012 CrossRef.
- B. Zhang, Y. Jiang, M. Gao, T. Ma, W. Sun and H. Pan, Nano Energy, 2021, 80, 105504 CrossRef CAS.
- Y. F. Nishimura, H.-J. Peng, S. Nitopi, M. Bajdich, L. Wang, C. G. Morales-Guio, F. Abild-Pedersen, T. F. Jaramillo and C. Hahn, ACS Appl. Mater. Interfaces, 2021, 13, 52044–52054 CrossRef CAS PubMed.
- S. Gao, Y. Lin, X. Jiao, Y. Sun, Q. Luo, W. Zhang, D. Li, J. Yang and Y. Xie, Nature, 2016, 529, 68–71 CrossRef CAS PubMed.
- X. Zheng, Y. Ji, J. Tang, J. Wang, B. Liu, H.-G. Steinrück, K. Lim, Y. Li, M. F. Toney and K. Chan, Nat. Catal., 2019, 2, 55–61 CrossRef CAS.
- H. A. Hansen, C. Shi, A. C. Lausche, A. A. Peterson and J. K. Nørskov, Phys. Chem. Chem. Phys., 2016, 18, 9194–9201 RSC.
- D. Gao, I. T. McCrum, S. Deo, Y.-W. Choi, F. Scholten, W. Wan, J. G. Chen, M. J. Janik and B. Roldan Cuenya, ACS Catal., 2018, 8, 10012–10020 CrossRef CAS.
- E. L. Clark, C. Hahn, T. F. Jaramillo and A. T. Bell, J. Am. Chem. Soc., 2017, 139, 15848–15857 CrossRef CAS PubMed.
- Y. Li, F. Cui, M. B. Ross, D. Kim, Y. Sun and P. Yang, Nano Lett., 2017, 17, 1312–1317 CrossRef CAS PubMed.
- P. Grosse, D. Gao, F. Scholten, I. Sinev, H. Mistry and B. Roldan Cuenya, Angew. Chem., Int. Ed., 2018, 57, 6192–6197 CrossRef CAS PubMed.
- Z. Weng, Y. Wu, M. Wang, J. Jiang, K. Yang, S. Huo, X.-F. Wang, Q. Ma, G. W. Brudvig and V. S. Batista, Nat. Commun., 2018, 9, 1–9 CrossRef CAS PubMed.
- P. W. Atkins, J. De Paula and P. W. Atkins, Physical Chemistry, W.H. Freeman, New York, 2006 Search PubMed.
- J. Qiao, Y. Liu and J. Zhang, Electrochemical reduction of carbon dioxide. Fundamentals and technologies, CRC Press, 2016 Search PubMed.
- P. Basu, Chapter 5-Gasification Theory and Modeling of Gasifiers' in Biomass Gasification and Pyrolysis, Academic Press, Boston, 2010 Search PubMed.
- H. Hashiba, H. K. Sato, S. Yotsuhashi, K. Fujii, M. Sugiyama and Y. Nakano, Sustainable Energy Fuels, 2017, 1, 1734–1739 RSC.
- W. J. Durand, A. A. Peterson, F. Studt, F. Abild-Pedersen and J. K. Nørskov, Surf. Sci., 2011, 605, 1354–1359 CrossRef CAS.
- B. Khezri, A. C. Fisher and M. Pumera, J. Mater. Chem. A, 2017, 5, 8230–8246 RSC.
- A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. DuBois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille and P. J. Kenis, Chem. Rev., 2013, 113, 6621–6658 CrossRef CAS PubMed.
- L. Torrente-Murciano, D. Mattia, M. D. Jones and P. K. Plucinski, J. CO2 Util., 2014, 6, 34–39 CrossRef CAS.
- J. Wu, P. P. Sharma, B. H. Harris and X.-D. Zhou, J. Power Sources, 2014, 258, 189–194 CrossRef CAS.
- M. A. Tekalgne, H. H. Do, A. Hasani, Q. Van Le, H. W. Jang, S. H. Ahn and S. Y. Kim, Mater. Today Adv., 2020, 5, 100038 CrossRef.
- F. Calle-Vallejo and M. T. Koper, Angew. Chem., 2013, 125, 7423–7426 CrossRef.
- J. H. Montoya, C. Shi, K. Chan and J. K. Nørskov, J. Phys. Chem. Lett., 2015, 6, 2032–2037 CrossRef CAS PubMed.
- J. Wu, Y. Huang, W. Ye and Y. Li, Adv. Sci., 2017, 4, 1700194 CrossRef PubMed.
- X. Nie, W. Luo, M. J. Janik and A. Asthagiri, J. Catal., 2014, 312, 108–122 CrossRef CAS.
- Z. W. Ulissi, M. T. Tang, J. Xiao, X. Liu, D. A. Torelli, M. Karamad, K. Cummins, C. Hahn, N. S. Lewis and T. F. Jaramillo, ACS Catal., 2017, 7, 6600–6608 CrossRef CAS.
- H. A. Hansen, C. Shi, A. C. Lausche, A. A. Peterson and J. K. Nørskov, Phys. Chem. Chem. Phys., 2016, 18, 9194–9201 RSC.
- M. K. Birhanu, M.-C. Tsai, A. W. Kahsay, C.-T. Chen, T. S. Zeleke, K. B. Ibrahim, C.-J. Huang, W.-N. Su and B.-J. Hwang, Adv. Mater. Interfaces, 2018, 5, 1800919 CrossRef.
- Y. Hori, K. Kikuchi and S. Suzuki, Chem. Lett., 1985, 14, 1695–1698 CrossRef.
- Y. Hori, I. Takahashi, O. Koga and N. Hoshi, J. Phys. Chem. B, 2002, 106, 15–17 CrossRef CAS.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. Stephens, K. Chan and C. Hahn, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- Handbook of Deposition Technologies for Films and Coatings, ed. P. M. Martin, William Andrew Publishing, Boston, 3rd edn, 2010, pp. 1–31 Search PubMed.
- M. J. Hampden-Smith and T. T. Kodas, Chem. Vap. Deposition, 1995, 1, 39–48 CrossRef CAS.
- J. E. Crowell, J. Vac. Sci. Technol., A, 2003, 21, S88–S95 CrossRef CAS.
- I. M. Dharmadasa and J. Haigh, J. Electrochem. Soc., 2006, 153, G47 CrossRef CAS.
- G. V. Govindaraju, G. P. Wheeler, D. Lee and K.-S. Choi, Chem. Mater., 2017, 29, 355–370 CrossRef CAS.
- T. S. N. S. Narayanan, I.-S. Park and M.-H. Lee, in Surface Modification of Magnesium and its Alloys for Biomedical Applications, ed. T. S. N. S. Narayanan, I.-S. Park and M.-H. Lee, Woodhead Publishing, Oxford, 2015, pp. 29–87 Search PubMed.
- A. Bagger, W. Ju, A. S. Varela, P. Strasser and J. Rossmeisl, ChemPhysChem, 2017, 18, 3266–3273 CrossRef CAS PubMed.
- H. Tabassum, X. Yang, R. Zou and G. Wu, Chem. Catal., 2022, 2, 1561–1593 CrossRef.
- M. Sankar, N. Dimitratos, P. J. Miedziak, P. P. Wells, C. J. Kiely and G. J. Hutchings, Chem. Soc. Rev., 2012, 41, 8099–8139 RSC.
- A. Wang, X. Y. Liu, C.-Y. Mou and T. Zhang, J. Catal., 2013, 308, 258–271 CrossRef CAS.
- P. Wang, M. Qiao, Q. Shao, Y. Pi, X. Zhu, Y. Li and X. Huang, Nat. Commun., 2018, 9, 4933 CrossRef PubMed.
- W. Ju, F. Jiang, H. Ma, Z. Pan, Y. Zhao, F. Pagani, D. Rentsch, J. Wang and C. Battaglia, Adv. Energy Mater., 2019, 9, 1901514 CrossRef.
- X. Jiang, X. Wang, Z. Liu, Q. Wang, X. Xiao, H. Pan, M. Li, J. Wang, Y. Shao, Z. Peng, Y. Shen and M. Wang, Appl. Catal., B, 2019, 259, 118040 CrossRef CAS.
- K. Ye, A. Cao, J. Shao, G. Wang, R. Si, N. Ta, J. Xiao and G. Wang, Sci. Bull., 2020, 65, 711–719 CrossRef CAS.
- W. J. Dong, J. W. Lim, D. M. Hong, J. Y. Park, W. S. Cho, S. Baek, C. J. Yoo, W. Kim and J.-L. Lee, ACS Appl. Energy Mater., 2020, 3, 10568–10577 CrossRef CAS.
- H. Hu, Y. Wang, N. Du, Y. Sun, Y. Tang, Q. Hu, P. Wan, L. Dai, A. C. Fisher and X. J. Yang, ChemElectroChem, 2018, 5, 3854–3858 CrossRef CAS.
- S. Sarfraz, A. T. Garcia-Esparza, A. Jedidi, L. Cavallo and K. Takanabe, ACS Catal., 2016, 6, 2842–2851 CrossRef CAS.
- Q. Li, J. Fu, W. Zhu, Z. Chen, B. Shen, L. Wu, Z. Xi, T. Wang, G. Lu and J. Zhu, J. Am. Chem. Soc., 2017, 139, 4290–4293 CrossRef CAS PubMed.
- Q. Li, M. Li, S. Zhang, X. Liu, X. Zhu, Q. Ge and H. Wang, Catalysts, 2019, 9, 476 CrossRef CAS.
- M. Zhang, Z. Zhang, Z. Zhao, H. Huang, D. H. Anjum, D. Wang, J. He and K.-W. Huang, ACS Catal., 2021, 11, 11103–11108 CrossRef CAS.
- W. Luo, W. Xie, R. Mutschler, E. Oveisi, G. L. De Gregorio, R. Buonsanti and A. Züttel, ACS Catal., 2018, 8, 6571–6581 CrossRef CAS.
- W. Zhang, X. Wen, S. Yang, Y. Berta and Z. L. Wang, Adv. Mater., 2003, 15, 822–825 CrossRef CAS.
- S. Rasul, D. H. Anjum, A. Jedidi, Y. Minenkov, L. Cavallo and K. Takanabe, Angew. Chem., Int. Ed., 2015, 54, 2146–2150 CrossRef CAS PubMed.
- H. Conrad, G. Ertl and E. E. Latta, Surf. Sci., 1974, 41, 435–446 CrossRef.
- L. L. Jewell and B. H. Davis, Appl. Catal., A, 2006, 310, 1–15 CrossRef CAS.
- G. Kyriakou, M. B. Boucher, A. D. Jewell, E. A. Lewis, T. J. Lawton, A. E. Baber, H. L. Tierney, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Science, 2012, 335, 1209–1212 CrossRef CAS PubMed.
- S. Bai, Q. Shao, P. Wang, Q. Dai, X. Wang and X. Huang, J. Am. Chem. Soc., 2017, 139, 6827–6830 CrossRef CAS PubMed.
- A. A. Peterson and J. K. Nørskov, J. Phys. Chem. Lett., 2012, 3, 251–258 CrossRef CAS.
- Y. Mun, S. Lee, A. Cho, S. Kim, J. W. Han and J. Lee, Appl. Catal., B, 2019, 246, 82–88 CrossRef CAS.
- T. Adit Maark and B. R. K. Nanda, J. Phys. Chem. C, 2016, 120, 8781–8789 CrossRef CAS.
- J.-F. Xie, J.-J. Chen, Y.-X. Huang, X. Zhang, W.-K. Wang, G.-X. Huang and H.-Q. Yu, Appl. Catal., B, 2020, 270, 118864 CrossRef CAS.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311–1315 RSC.
- H.-P. Yang, S. Qin, Y.-N. Yue, L. Liu, H. Wang and J.-X. Lu, Catal. Sci. Technol., 2016, 6, 6490–6494 RSC.
- E. Barton Cole, P. S. Lakkaraju, D. M. Rampulla, A. J. Morris, E. Abelev and A. B. Bocarsly, J. Am. Chem. Soc., 2010, 132, 11539–11551 CrossRef CAS PubMed.
- W. J. Wang, S. Hwang, T. Kim, S. Ha and L. Scudiero, Electrochim. Acta, 2021, 387, 138531 CrossRef CAS.
- D. Chen, Q. Yao, P. Cui, H. Liu, J. Xie and J. Yang, ACS Appl. Energy Mater., 2018, 1, 883–890 CrossRef CAS.
- R. Feng, Q. Zhu, M. Chu, S. Jia, J. Zhai, H. Wu, P. Wu and B. Han, Green Chem., 2020, 22, 7560–7565 RSC.
- S. Jia, Q. Zhu, H. Wu, M. Chu, S. Han, R. Feng, J. Tu, J. Zhai and B. Han, Chin. J. Catal., 2020, 41, 1091–1098 CrossRef CAS.
- O. A. Baturina, Q. Lu, M. A. Padilla, L. Xin, W. Li, A. Serov, K. Artyushkova, P. Atanassov, F. Xu and A. Epshteyn, ACS Catal., 2014, 4, 3682–3695 CrossRef CAS.
- L. Hou, J. Han, C. Wang, Y. Zhang, Y. Wang, Z. Bai, Y. Gu, Y. Gao and X. Yan, Inorg. Chem. Front., 2020, 7, 2097–2106 RSC.
- Y. Chen, Z. Fan, J. Wang, C. Ling, W. Niu, Z. Huang, G. Liu, B. Chen, Z. Lai and X. Liu, J. Am. Chem. Soc., 2020, 142, 12760–12766 CrossRef CAS PubMed.
- S. Ma, M. Sadakiyo, M. Heima, R. Luo, R. T. Haasch, J. I. Gold, M. Yamauchi and P. J. A. Kenis, J. Am. Chem. Soc., 2017, 139, 47–50 CrossRef CAS PubMed.
- S. Zhang, P. Kang, M. Bakir, A. M. Lapides, C. J. Dares and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 15809–15814 CrossRef CAS PubMed.
- C. S. Chen, J. H. Wan and B. S. Yeo, J. Phys. Chem. C, 2015, 119, 26875–26882 CrossRef CAS.
- L. Zhu, Y. Lin, K. Liu, E. Cortés, H. Li, J. Hu, A. Yamaguchi, X. Liu, M. Miyauchi and J. Fu, Chin. J. Catal., 2021, 42, 1500–1508 CrossRef CAS.
- Y.-A. Lai, Y.-C. Chu, C.-J. Chang, K.-H. Chen, Y.-C. Hsiao, C.-C. Chang, M.-Y. Liao and H. M. Chen, Adv. Energy Sustain. Res., 2022, 2200075 CrossRef.
- X.-Q. Li, G.-Y. Duan, X.-X. Yang, L.-J. Han and B.-H. Xu, Fundam. Res., 2022 DOI:10.1016/j.fmre.2021.12.009.
- H. Shen, Y. Wang, T. Chakraborty, G. Zhou, C. Wang, X. Fu, Y. Wang, J. Zhang, C. Li and F. Xu, ACS Catal., 2022, 12, 5275–5283 CrossRef CAS.
- Z. Li, R. M. Yadav, L. Sun, T. Zhang, J. Zhang, P. M. Ajayan and J. Wu, Appl. Catal., A, 2020, 606, 117829 CrossRef CAS.
- F. Quan, D. Zhong, H. Song, F. Jia and L. Zhang, J. Mater. Chem. A, 2015, 3, 16409–16413 RSC.
- B. Qin, Y. Li, H. Fu, H. Wang, S. Chen, Z. Liu and F. Peng, ACS Appl. Mater. Interfaces, 2018, 10, 20530–20539 CrossRef CAS PubMed.
- J. Rosen, G. S. Hutchings, Q. Lu, R. V. Forest, A. Moore and F. Jiao, ACS Catal., 2015, 5, 4586–4591 CrossRef CAS.
- D. Ren, B. S.-H. Ang and B. S. Yeo, ACS Catal., 2016, 6, 8239–8247 CrossRef CAS.
- G. Keerthiga and R. Chetty, J. Electrochem. Soc., 2017, 164, H164 CrossRef CAS.
- X. Su, Y. Sun, L. Jin, L. Zhang, Y. Yang, P. Kerns, B. Liu, S. Li and J. He, Appl. Catal., B, 2020, 269, 118800 CrossRef CAS.
- D. Ren, J. Gao, L. Pan, Z. Wang, J. Luo, S. M. Zakeeruddin, A. Hagfeldt and M. Grätzel, Angew. Chem., Int. Ed., 2019, 58, 15036–15040 CrossRef CAS PubMed.
- S. Dongare, N. Singh, H. Bhunia and P. K. Bajpai, Electrochim. Acta, 2021, 392, 138988 CrossRef CAS.
- T. T. Hoang, S. Verma, S. Ma, T. T. Fister, J. Timoshenko, A. I. Frenkel, P. J. Kenis and A. A. Gewirth, J. Am. Chem. Soc., 2018, 140, 5791–5797 CrossRef CAS PubMed.
- J. Huang, M. Mensi, E. Oveisi, V. Mantella and R. Buonsanti, J. Am. Chem. Soc., 2019, 141, 2490–2499 CrossRef CAS PubMed.
- J. Wang, Z. Li, C. Dong, Y. Feng, J. Yang, H. Liu and X. Du, ACS Appl. Mater. Interfaces, 2019, 11, 2763–2767 CrossRef CAS PubMed.
- Y. Wang, C. Niu and Y. Zhu, Nanomaterials, 2019, 9, 173 CrossRef CAS PubMed.
- Y. Wang, D. Wang, C. J. Dares, S. L. Marquard, M. V. Sheridan and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 278–283 CrossRef CAS PubMed.
- D. Higgins, A. T. Landers, Y. Ji, S. Nitopi, C. G. Morales-Guio, L. Wang, K. Chan, C. Hahn and T. F. Jaramillo, ACS Energy Lett., 2018, 3, 2947–2955 CrossRef CAS.
- D. Ren, J. Fong and B. S. Yeo, Nat. Commun., 2018, 9, 925 CrossRef PubMed.
- A. Katoh, H. Uchida, M. Shibata and M. Watanabe, J. Electrochem. Soc., 1994, 141, 2054 CrossRef CAS.
- J. Choi, M. J. Kim, S. H. Ahn, I. Choi, J. H. Jang, Y. S. Ham, J. J. Kim and S.-K. Kim, Chem. Eng. J., 2016, 299, 37–44 CrossRef CAS.
- Z. Chang, S. Huo, W. Zhang, J. Fang and H. Wang, J. Phys. Chem. C, 2017, 121, 11368–11379 CrossRef CAS.
- M. Watanabe, M. Shibata, A. Kato, M. Azuma and T. Sakata, J. Electrochem. Soc., 1991, 138, 3382 CrossRef CAS.
- Z. Yin, D. Gao, S. Yao, B. Zhao, F. Cai, L. Lin, P. Tang, P. Zhai, G. Wang and D. Ma, Nano Energy, 2016, 27, 35–43 CrossRef CAS.
- D. Kim, C. Xie, N. Becknell, Y. Yu, M. Karamad, K. Chan, E. J. Crumlin, J. K. Nørskov and P. Yang, J. Am. Chem. Soc., 2017, 139, 8329–8336 CrossRef CAS PubMed.
- K. Chen, X. Zhang, T. Williams, L. Bourgeois and D. R. MacFarlane, Electrochim. Acta, 2017, 239, 84–89 CrossRef CAS.
- S. Sultan, H. Lee, S. Park, M. M. Kim, A. Yoon, H. Choi, T.-H. Kong, Y.-J. Koe, H.-S. Oh and Z. Lee, Energy Environ. Sci., 2022, 15, 2397–2409 RSC.
| This journal is © The Royal Society of Chemistry 2022 |