
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInner residues of macrothiolactone in autoinducer peptides I/IV circumvent spontaneous S-to-O acyl transfer to the upstream serine residue†
Masanobu
Nagano
 ,
Satoshi
Ishida
and
Hiroaki
Suga
*
,
Satoshi
Ishida
and
Hiroaki
Suga
*
Graduate School of Science, The University of Tokyo, 113-0033, Japan
First published on 26th January 2022
Abstract
Autoinducing peptides I and IV (AIP-I/IV) are naturally occurring cyclic thiodepsipeptides (CTPs) bearing a Ser–Thr–Cys–Asp/Tyr (STC[D/Y]) tetrapeptide motif, where the Cys thiol (HSC) in the side-chain is linked to the Met C-terminal carboxylic acid (MCOOH) to form 5-residue macrothiolactones,−SC(D/Y)FIMCO−. We have recently reported that CTPs containing SX1CX2 motifs spontaneously undergo macrolactonization to yield cyclic depsipeptides (CDPs) by an unprecedented rapid S-to-O acyl transfer to the upstream Ser hydroxyl group. Interestingly, even though the STC[D/Y] motif in AIP-I/IV is a member of the SX1CX2 motif family, it maintains the CTP form. This suggests that AIP-I/IV have a structural or chemical motive for avoiding such an S-to-O acyl transfer, thus retaining the CTP form intact. Here we have used genetic code reprogramming to ribosomally synthesize various AIP-I analogs and studied what the determinant is to control the formation of CTP vs. CDP products. The study revealed that a Gly substitution of the inner Asp/Tyr or Met residues in the thiolactone drastically alters the resistance to the promotion of the S-to-O acyl transfer, giving the corresponding CDP product. This suggests that the steric hindrances originating from the α-substituted sidechain in these two amino acids in the AIP-I/IV thiolactone likely play a critical role in controlling the resistance against macrolactone rearrangement to the upstream Ser residue.
Introduction
Naturally occurring peptides often possess unique macrocyclic structures and exhibit a broad spectrum of bioactivity, and thereby the discovery of such natural products has been an important mainstream in academia and the pharmaceutical industry.1,2 Among them, a class of macrocyclic thiodepsipeptides (CTPs) rarely exist in nature due to their chemical instability of the thiolactone bond.3 However, there are some examples of CTPs that exhibit bioactivities, e.g. thiocoraline4 and noshipeptide5 with anti-cancer and antibiotic activities.Among others, autoinducing peptides (AIPs) produced by Staphylococcus aureus are known as a family of CTPs composed of a 2–4-residue N-terminal exotail and a 5-residue thiolactone.6–9 When the AIPs are secreted from cells, they interact with the cognate membrane protein receptor, referred to as accessory gene regulatory C (AgrC), to activate downstream bacterial virulence signalling, which is regulated by the local density of the extracellular AIPs, referred to as quorum sensing “autoinducers”. AIPs are ribosomally expressed as a precursor linear form, referred to as AgrD, which is composed of leader, core, and AgrB-recognition domains (Fig. 1a). The AgrD is post-translationally thiolactonized by AgrB and further proteolyzed to the mature AIPs as side-chain-to-tail cyclized CTPs.10
 | ||
| Fig. 1 Ribosomal synthesis of cyclic thiodepsipeptides (CTPs) possessing an SX1CX2 motif and a downstream thioester. (a) Biosynthesis of autoinducing peptide I (AIP-I) from AgrD-I as a precursor peptide. (b) One-pot in vitro ribosomal cyclic depsipeptide (CDP) synthesis. In this case a CTP acts as an intermediate and is spontaneously converted to CDP via an S-to-O acyl transfer modulated by an SX1CX2 motif. N ≥ 1, depending on the X2 residue. | ||
We have recently discovered that when a SPCG (Ser–Pro–Cys–Gly) motif in peptides is S-acylated on the Cys's thiol side chain upon incubation with a thioacyl donor molecule, the acyl group spontaneously transfers to the Ser's hydroxyl side-chain via an S-to-O acyl transfer in a site-specific manner.11,12 This unprecedented chemistry led us to investigate in vitro ribosomal synthesis of side-chain-to-tail cyclized depsipeptides (CDPs) in one-pot, where a nascent linear peptide with a combination of an SPCG motif and a backbone thioacyl donor downstream macrothiolactonizes to form the corresponding CTP. This CTP spontaneously undergoes macrolactonization via S-to-O acyl transfer, yielding the corresponding CDP (Fig. 1b). During the above study, we were able to alleviate the SPCG to the SX1CX2 motif, where X1 and X2 represent nearly any amino acid, except Pro at X2. Most importantly, the chemistry allows us to access a broad range of CDPs in a nearly context-independent manner and with ring sizes consisting of 7–17 residues expanded from the parental 5–15 residue-ring sizes of the CTPs.
Among the known S. aureus AIPs, we have noticed that AIP-I and AIP-IV contain an STC(D/Y) motif belonging to the SX1CX2 motif family (Fig. 1a). Therefore, it is possible that the occurrence of a similar S-to-O acyl transfer reaction in AIP-I/IV rearranges the CTP to a “chemically more stable” CDP. However, previous studies on AIP-I and their mutants including insights into their structure and activity relationship (SAR) did not report such an S-to-O acyl transfer event.13–15 This fact has motivated us to investigate AIP-I/IV using our one-pot ribosomal CTP/CDP synthesis approach to understand the nature of suppression of the S-to-O acyl transfer and why the CTP form can be maintained.
Results and discussion
First, we conducted a one-pot in vitro ribosomal synthesis of AIP-I using our genetic code reprogramming approach in place of chemical16 or intein-mediated synthesis.17 For this purpose, a mRNA template (mR1) for expression of an AIP-I analog, referred to as AIP-Ia, was designed (Fig. 2a and b). In this molecule, the thiolactone (C8–M12), STCD motif (S6–D9), and Y5 were kept as the same residues as wildtype AIP-I, whereas the N-terminal peptide sequence at the upstream position of Y5 was replaced with a short peptide (fM1–G4) to facilitate peptide analysis by MALDI-TOF MS (Fig. 2b). A thioester was directly introduced downstream by reassigning the Trp codon (UGG) with α-thio-p-chloro-phenyl lactic acid (HSFCl13) using a Trp-deficient FIT (flexible in vitro translation) system,18,19 which gives an alternative route of enzymatic thioesterificaion using AgrB or intein, and the sequence was terminated using a following short peptide (YRA) (Fig. 2a). As expected, the expression of pre-AIP-Ia with the HSFCl13 reassignment afforded a single peak of the peptide corresponding to AIP-Ia by MALDI-TOF MS (Fig. 2c, [M + H]+ = 1390.72 m/z observed), and no precursor linear peptide, pre-AIP-Ia ([M + H]+ = 1996.75 m/z), was detected, indicating that one-pot synthesis of AIP-Ia was achieved.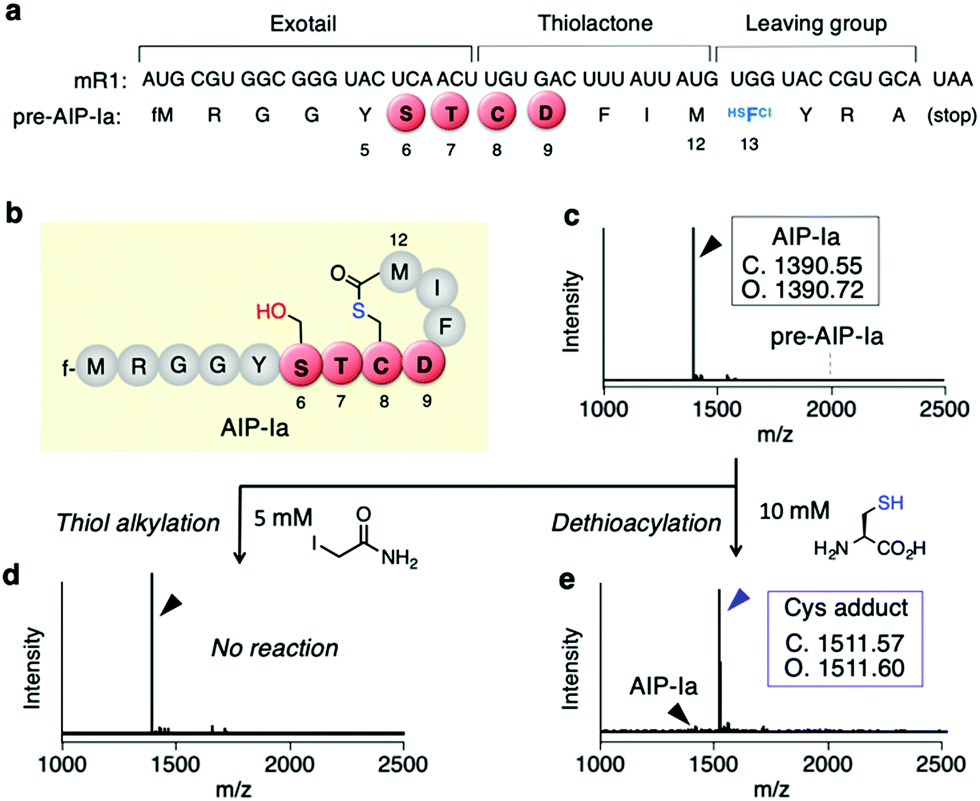 | ||
| Fig. 2 One-pot in vitro ribosomal synthesis of the AIP-I analog (AIP-Ia) without post-translational enzymes. (a) mRNA (mR1) and precursor peptide sequence (pre-AIP-Ia) used for in vitro CTP/CDP synthesis. 3’-Acylated tRNA, HSFCl–tRNAEnAsn#3CCA, was in vitro-prepared using a flexizyme in advance, and then used for translation in the Trp-deficient FIT system to generate a backbone thioester. f, formyl group. (b) Peptide sequence of AIP-Ia as a product. (c) MALDI-TOF MS spectrum of a crude translation product after 30 min translation. (d) Alkylation of the translation product by IAA. (e) Dethioacylation of thiolactone by addition of cysteine. | ||
Since conversion of CTP to CDP via the S-to-O acyl transfer would not result in any mass difference, in order to validate the exact form of AIP-Ia we performed post-translational modification on the peptide product in two ways. First, we treated the peptide with iodoacetamide (IAA) that could alkylate a free Cys residue present in the peptide sequence. If the thiolactone was formed, such a free Cys residue could be absent in the peptide and thereby no alkylation should occur. As expected, no alkylated product was observed by MALDI-TOF analysis, suggesting that the thiol group on Cys8 was used to form an S-acyl isopeptide bond (Fig. 2d). Second, the CTP form of AIP-Ia was confirmed by dethioacylation using 10 mM of Cys. If the peptide product includes a thioester bond, Cys cleaves the bond in a manner of native chemical ligation to give the Cys adduct at the C-terminal via ring-opening.20 In fact, the addition of Cys to the translated product promoted a mass shift by 121 Da, indicating that the corresponding Cys adduct was produced (Fig. 2e). These studies have solidly validated that the AIP-Ia has the desired thiolactone form, which does not undergo the S-to-O acyl transfer to the side chain of S6 even though it has the STCD motif.
To determine which residues are important for maintaining the CTP form, we first conducted mutations of T7 being the upstream residue adjacent to Cys8. Thus, the T7 residue (X1 in SX1CX2) in the exotail part of AIP-Ia was substituted by 8 distinct amino acids (X1 = A, Y, K, N, Q, E, G, and P in Fig. 3a and Table S1a, ESI†). The respective translated products and IAA-labelled products were analyzed by MALDI-TOF MS. The mass value in each peptide corresponded to the CTP form regardless of the residue at the T7 position (Fig. S1, ESI†). For the D9 residue (X2) in the thiolactone ring, it was mutated to 7 distinct amino acids (X2 = A, Y, K, N, Q, E, and G in Fig. 3b and Table S1a, ESI†). The mutations A, Y, K, N, Q, and E also retained the CTP form (Fig. S2, ESI†). Importantly, the D9Y mutant corresponds to the naturally occurring AIP-IV thiolactone, meaning that the S-to-O acyl transfer does not take place in both AIPs. In contrast, for the D9G mutant (X2 = G), the post-translational alkylation gave a mass shifted by 57 Da, corresponding to the formation of alkylated CDP (D9G–CDP–AA in Fig. 3c and d). This implies that the single substitution of D9G in the thiolactone of AIP-Ia dramatically promotes the S-to-O acyl transfer.
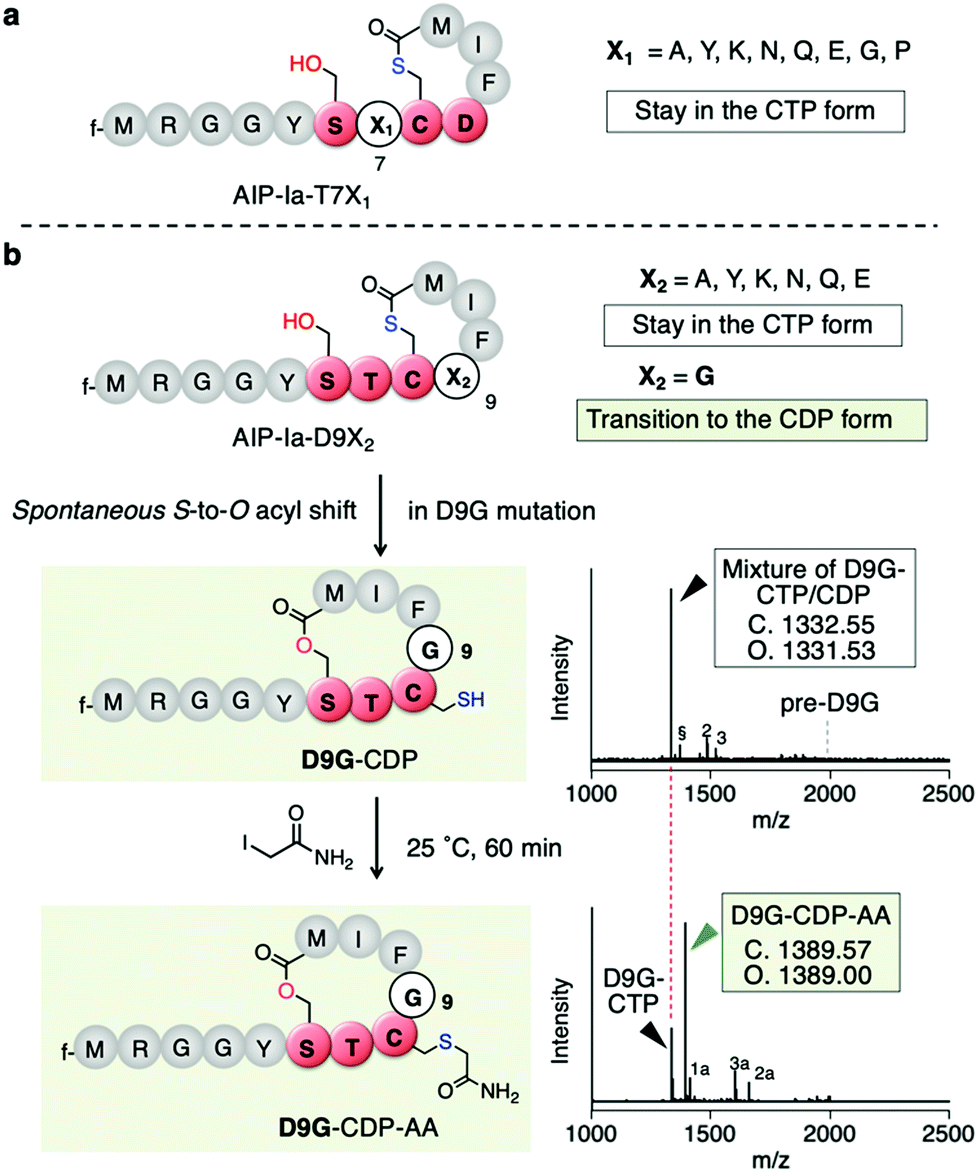 | ||
| Fig. 3 Mutation study of the X1 and X2 positions of the SX1CX2 motif. (a) Mutation study of AIP-Ia at Thr7 to an X1 residue. All the resulting translation products maintained the CTP form. (b) Mutation study at Asp9 to an X2 residue. Only mutation to Gly facilitated S-to-O acyl transfer. The second bottom shows a structure of D9G-CDP and its MALDI-TOF MS analysis of the crude translation products, which was a mixture of CTP and CDP forms. pre-D9G, a linear precursor peptide with a D9G mutation. Potassium adduct (§), DTT adduct (2), and Cys adduct (3) were also observed. 2 and 3 are attributed to an in vitro translation system (see ref. 11). The bottom is a structure of alkylated D9G-CDP by IAA and its MALDI-TOF MS analysis. f, formyl group; 1a, alkylated hydrolyzed peptide; 2a, tri-alkylated DTT adduct; 3a, bis-alkylated Cys adduct. | ||
To determine the ratio of CDP/CTP form in the D9G mutant, the translated product was semi-quantitatively analyzed by LC-MS. A positive control, AIP-Ia, was subjected to LC-MS, and then the extracted ion chromatogram (EIC) was evaluated. We observed no separated peak and no alkylated product using IAA (Fig. S3a, ESI†). On the other hand, when the same experiment was performed on D9G–CTP, the EIC chromatogram gave two peaks of 19% (11.0 min) and 81% (11.6 min), which were assigned to a D9G–CDP and a D9G–CTP, respectively (Fig. S3b, ESI†). Extending the translation time from 30 min to 60 min increased the CDP product from 19% to 45%, suggesting that the S-to-O acyl transfer in D9G–CTP slowly proceeded. The IAA alkylation of D9G–CDP (the peak observed at 11.0 min) afforded a fraction of D9G–CDP–AA (a peak observed at 10.5 min) in an approximately 54% yield. These results confirmed the occurrence of the S-to-O acyl transfer of the D9G mutant.
Since the D9G mutation in the AIP-Ia thiolactone promoted the spontaneous rearrangement from CTP to CDP, we next performed Gly scanning of other amino residues in the thiolactone ring (F10, I11, and M12 in Table S1b, ESI†). Neither the F10G nor I11G mutation induced the conversion of CTP to CDP (Fig. S4a and b, ESI†). On the other hand, a fraction of the M12G mutant rearranged to CDP (M12G–CDP–AA in Fig. 4a and b, Fig. S4c, ESI†). The double G mutant at the 9th and 12th positions (D9G and M12G) underwent a major conversion to the CDP–AA (Fig. 4a and b, Fig. S4d, ESI†). More drastic replacement of the DFIM thiolactone sequence with GGSG (9–12GS) gave a nearly complete conversion to the CDP form (Fig. 4a and b, Fig. S4e, ESI†), which was consistent with our previous report.11 Note that the double G mutant at the 10th and 11th positions (F10G and I11G) completely left the CTP intact (Fig. S4f and Table S1b, ESI†), indicating that D9 and M12 are the critical determinants for making the thiolactone resistant to the rearrangement. Likely, G mutation at the 9th or 12th position in the AIP-Ia alleviates the conformational rigidity of the thiolactone ring and reduces the steric hindrances around the carbonyl group of the thioester, allowing for the nucleophilic attack of the hydroxyl group of S6 on the thioester bond and the formation of CDP via the S-to-O acyl transfer.
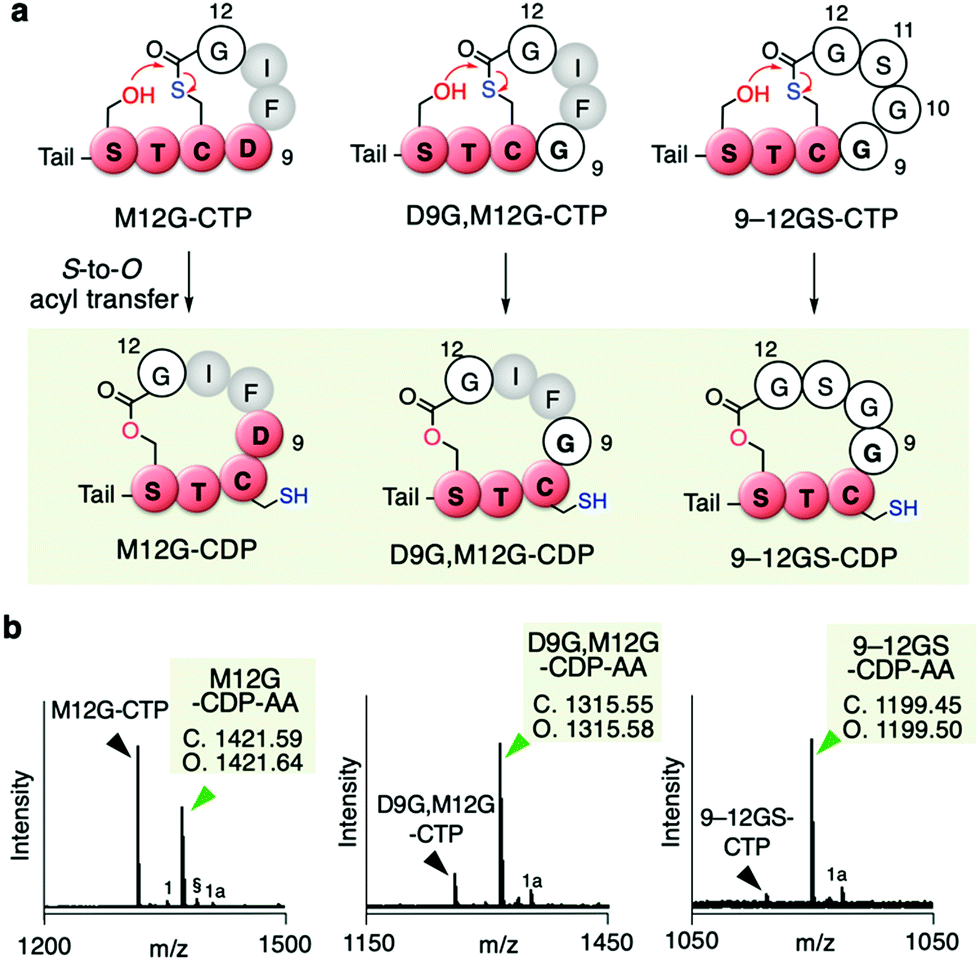 | ||
| Fig. 4 Mutation of inner residues of the AIP-Ia thiolactone to Gly residue(s). (a) Spontaneous rearrangement of AIP-Ia mutants to the corresponding CDPs during the in vitro translation. The precursor peptides were expressed in the Trp-deficient FIT system. Tail; N-terminal exotail tail sequence (fMRGGY). (b) MALDI-TOF MS analysis of the post-translationally alkylated product by IAA. §, potassium adduct; 1, hydrolyzed peptide; 1a, alkylated hydrolyzed peptide (ref. 11). | ||
To this end, we further tested if the ring expansion of the 5-residue thiolactone could induce the S-to-O acyl transfer upon keeping these two key residues, D9 and M12(+X) in the ring. We thus prepared 6, 7, and 8-residue ring CTPs, where more structurally relaxed peptide sequences consisting of Gly and Ser residues were introduced between Asp9 and Met12 in the thiolactone ring (Fig. 5a and b, Fig. S5, Table S1c, ESI†). Interestingly, the S-to-O acyl transfer in the 6- and 7-residue ring CTPs did not occur, whereas it did occur in the 8-residue ring CTP to form a 10-residue ring CDP in approximately half of the fraction. This result suggests that D9 and M12(+2) residues in up to 7-residue thiolactone rings prohibit the S-to-O acyl transfer to the upstream Ser residue in the STCD motif, but further ring expansion very likely loosens the ring constraints to facilitate the S-to-O acyl transfer, affording the corresponding CDP.
 | ||
| Fig. 5 One-pot ribosomal synthesis of AIP-Ia with various ring-sizes. (a) The corresponding precursor peptides were expressed in the Trp-deficient FIT system and the CTPs consisting of 6–8 ring sizes were analyzed by MALDI-TOF MS. Tail; N-terminal exotail tail sequence (fMRGGY). (b) MALDI-TOF MS analysis of post-translationally alkylated product by IAA. 1a, alkylated hydrolyzed product. | ||
Conclusions
We recently reported that CTPs containing an SX1CX2 motif spontaneously undergo macrolactonization via rapid S-to-O acyl transfer, yielding the corresponding CDPs. Though the naturally occurring autoinducing CTPs, AIP-I and IV, contain STCD and STCY motifs (X1 = T and X2 = D or Y in the SX1CX2 motif), respectively, we experimentally confirmed that the S-to-O acyl transfer does not take place in an AIP-I analog, leaving the CTP form intact. Taking advantage of the FIT system with genetic code reprogramming, we have prepared various mutants for the outer and inner residues of the thiolactone ring, revealing that a Gly substitution of the Asp/Tyr residue in the STC(D/Y) motif and a C-terminal Met residue (whose carboxyl group forms the thioester bond) dramatically alter the resistance to the S-to-O acyl transfer, rearranging into the CDP form. Since mutations from the Asp/Tyr residue to arbitrary amino acids other than Gly do not alter the resistance, the steric hindrances originating from the α-substituted sidechain likely play a critical role in controlling the reactivity of the thioester bond to the hydroxyl group of the upstream Ser. Moreover, the expansion of the thiolactone ring from 5 residues to 6 or 7 residues still maintains the CTP form as far as the aforementioned critical residues are kept, whereas that to 8 residues promotes the S-to-O acyl transfer, yielding the corresponding CDP consisting of the 10-residue lactone ring. We propose that not only the steric hindrance by the inner residues adjacent to the thioester bond in the thiolactone but also the ring constraints in the AIP-I/IV effectively prevent the undesired ring-expansion21 to CDP. Likely, the naturally occurring AIP-I/IV could have evolved to maintain the CTP scaffold by taking smaller ring-size and placing appropriate residues in the thiolactone to make the necessary interaction with the cognate receptor, AgrC.So far, more than 40 AIPs with a thiolactone have been reported (Table S2, ESI†),9,22 and among them 8 AIPs have the SX1CX2 motif in addition to the S. aureus AIP-I/IV described in this study. However, none of them has a Gly residue at the critical positions near the thioester bond, implying that these naturally occurring AIPs are also resistant to the S-to-O acyl transfer, i.e., they maintain the CTP form. This information is useful to predict the naturally occurring thiolactone structure of AIPs,23–25 giving guidance for the study of the structure–activity relationship in quorum sensing modulators.13,26
Experimental
Oligonucleotides
The oligonucleotides used in this study are listed in Tables S2 and S3 (ESI†). Reverse primers for assembling tRNA templates were modified with 2′-O-methylation (G(me)) at the second last nucleotide of the 5′ termini to suppress non-templated nucleotide addition in in vitro transcription with T7 polymerase.27 Standard oligonucleotides were purchased from Eurofins Genomics K.K. (Japan) and methylated oligos were purchased from Gene Design Inc. (Japan).Preparation of flexizymes and tRNA
Flexizymes (eFx) and tRNAPro1E2CCA were prepared by run-off in vitro transcription with T7 RNA polymerase as previously described.18,28Thioacylation of tRNA with HSPhe4Cl by flexizymes
As previously reported,19 2 μL of 250 μM tRNA, 2 μL of 250 μM flexizyme and 2 μL of 0.5 M bicine-KOH buffer [pH 9.0] and 6 μL of deionized water were mixed and heated to 95 °C for 2 min and cooled to RT over 5 min. 4 μL of 3 M MgCl2 was added and the mixture was transferred to an ice-bath. Finally, 4 μL of 20 mM pre-reduced HSPhe4Cl–CME ester by TCEP·HCl aq. was added and the mixture was incubated on ice for 6 h. The reaction was quenched by adding 80 μL of 300 mM NaOAc [pH 5.2]. Nucleotides were precipitated by the addition of 200 μL ethanol followed by centrifugation. The pellet was rinsed with 60 μL of 0.1 M NaOAc [pH 5.2] in 70% ethanol twice and once with 25 μL of 70% ethanol. This reaction yields 500 pmol of crude aminoacylated tRNA.In vitro expression of AIP-Ia and its analogs by the Trp-deficient FIT system
A 5 μL in vitro translation was prepared by mixing a DNA template, 0.5 mM Trp-deficient 19 amino acids mix, 50 μM of pre-loaded HSPhe4Cl–tRNAPro1E2CCA, and in-house PURE system29 and incubated at 37 °C for 30 min.11 The crude reaction mixture was desalted using solid-phase extraction (SPE) C-tip (Nikkyo Technos) and eluted with 80% acetonitrile, 0.5% acetic acid, half saturated with the matrix (R)-cyano-4-hydroxycinnamic acid (Bruker), and spotted on a MALDI Ground Steel 384 Target plate. MALDI-TOF-MS measurement was performed under linear or reflector positive mode using an UltrafleXtreme (Bruker) with external calibration (Peptide Calibration Standard II, Bruker).Thiol capping in peptides by IAA for MS analysis
To 5 μL of in vitro translated AIP-Ia analogs, 0.55 μL of 50 mM IAA (#A39271, Thermo Fisher) in ultra-pure water and 0.5 μL of 20 mM TCEP were added and incubated for 1 hour at 25 °C. The reaction mixture was desalted and analyzed by MALDI-TOF MS.Dethioacylation
As previously reported,11 to 5 μL of in vitro translated AIP-Ia analogs, 0.55 μL of 100 mM cysteine was added and incubated for 40 min at 25 °C. The reaction mixture was desalted and analyzed by MALDI-TOF MS.LC-MS analysis of AIP-I analogs
To an in vitro-expressed crude product was added cold MeOH and the resulting solution was held on ice for 5 min. Precipitates were then spun down at 4 °C and to the supernatant was further added 10-fold volume of 1% TFA. After spinning down, the supernatant was used as a sample for LC-MS. A Waters Xevo G2-XS QTof instrument equipped with an Acquity I-Class UPLC system was used for LC-MS analysis. HPLC was done on an Acquity UPLC Peptide BEH C18 column (dimensions: 150 × 2.1 mm; pore size: 300 Å; particle size: 1.7 μm) using 0.1% (v/v) formic acid in water (solvent A) and 0.1% (v/v) formic acid in acetonitrile (solvent B) as a mobile phase. Analysis was performed at 60 °C and 0.3 mL min−1 flow rate running the following gradient: 1% B for 1.5 min; 1–61% B over 16.5 min; 95% B for 1 min; 1% B for 2 min (total run time: 19.5 min). MS analysis was done in a positive polarity/high-sensitivity mode with a 0.3 s scan time. Capillary voltage was set to 700 V; the electrospray ionization source and desolvation temperatures were 120 °C and 400 °C, respectively. Manufacturer-supplied leucine enkephalin was used as a lockspray standard for continuous mass axis referencing and the recommended lockspray setup procedure was performed prior to every run. LC-MS data were analyzed using MassLynx v.4.1.Author contributions
M. N. conceived the study. M. N. and S. I. planned and performed the experiments. M. N., S. I. and H. S. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the Japan Society for the Promotion of Science (JSPS) Grant-in-Aid for Scientific Research and Specially Promoted Research JP20H05618 to H.S. and JSPS Grants-in-Aid for Research Fellows 253534 to M.N., and JP16J04031 to S. I.Notes and references
- C. Morrison, Nat. Rev. Drug Discovery, 2018, 17, 531–533 CrossRef CAS PubMed.
- A. A. Vinogradov, Y. Yin and H. Suga, J. Am. Chem. Soc., 2019, 141, 4167–4181 CrossRef CAS PubMed.
- I. Avan, C. Dennis Hall and A. R. Katritzky, Chem. Soc. Rev., 2014, 43, 3575–3594 RSC.
- E. Erba, D. Bergamaschi, S. Ronzoni, M. Faretta, S. Taverna, M. Bonfanti, C. V. Catapano, G. Faircloth, J. Jimeno and M. D’Incalci, Br. J. Cancer, 1999, 80, 971–980 CrossRef CAS PubMed.
- N. M. Haste, W. Thienphrapa, D. N. Tran, S. Loesgen, P. Sun, S. J. Nam, P. R. Jensen, W. Fenical, G. Sakoulas, V. Nizet and M. E. Hensler, J. Antibiot., 2012, 65, 593–598 CrossRef CAS PubMed.
- G. Ji, R. Beavis and R. P. Novick, Science, 1997, 276, 2027–2030 CrossRef CAS PubMed.
- R. P. Novick and E. Geisinger, Annu. Rev. Genet., 2008, 42, 541–564 CrossRef CAS PubMed.
- B. Wang and T. W. Muir, Cell Chem. Biol., 2016, 23, 214–224 CrossRef CAS PubMed.
- M. Thoendel, J. S. Kavanaugh, C. E. Flack and A. R. Horswill, Chem. Rev., 2011, 111, 117–151 CrossRef CAS PubMed.
- R. Qiu, W. Pei, L. Zhang, J. Lin and G. Ji, J. Biol. Chem., 2005, 280, 16695–16704 CrossRef CAS PubMed.
- M. Nagano, Y. Huang, R. Obexer and H. Suga, J. Am. Chem. Soc., 2021, 143, 4741–4750 CrossRef CAS PubMed.
- A. A. Vinogradov, M. Nagano, Y. Goto and H. Suga, J. Am. Chem. Soc., 2021, 143, 13358–13369 CrossRef PubMed.
- C. P. Gordon, P. Williams and W. C. Chan, J. Med. Chem., 2013, 56, 1389–1404 CrossRef CAS PubMed.
- C. P. Gordon, S. D. Olson, J. L. Lister, J. S. Kavanaugh and A. R. Horswill, J. Med. Chem., 2016, 59, 8879–8888 CrossRef CAS PubMed.
- G. J. Lyon, J. S. Wright, T. W. Muir and R. P. Novick, Biochemistry, 2002, 41, 10095–10104 CrossRef CAS PubMed.
- C. P. Gordon, Org. Biomol. Chem., 2020, 18, 379–390 RSC.
- C. L. Malone, B. R. Boles and A. R. Horswill, Appl. Environ. Microbiol., 2007, 73, 6036–6044 CrossRef CAS PubMed.
- Y. Goto, T. Katoh and H. Suga, Nat. Protoc., 2011, 6, 779–790 CrossRef CAS PubMed.
- R. Takatsuji, K. Shinbara, T. Katoh, Y. Goto, T. Passioura, R. Yajima, Y. Komatsu and H. Suga, J. Am. Chem. Soc., 2019, 141, 2279–2287 CrossRef CAS PubMed.
- P. Dawson, T. Muir, I. Clark-Lewis and S. Kent, Science, 1994, 266, 776–779 CrossRef CAS PubMed.
- J. G. Johnson, B. Wang, G. T. Debelouchina, R. P. Novick and T. W. Muir, ChemBioChem, 2015, 16, 1093–1100 CrossRef CAS PubMed.
- D. N. Mcbrayer, D. Cameron, Y. Tal-gan and C. Cameron, Org. Biomol. Chem., 2020, 18, 7273–7290 RSC.
- B. H. Gless, M. S. Bojer, P. Peng, M. Baldry, H. Ingmer and C. A. Olsen, Nat. Chem., 2019, 11, 463–469 CrossRef CAS PubMed.
- B. H. Gless, B. S. Bejder, F. Monda, M. S. Bojer, H. Ingmer and C. A. Olsen, J. Am. Chem. Soc., 2021, 143, 10514–10518 CrossRef CAS PubMed.
- E. M. Molloy, M. Dell, V. G. Hänsch, K. L. Dunbar, R. Feldmann, A. Oberheide, L. Seyfarth, J. Kumpfmüller, T. Horch, H. D. Arndt and C. Hertweck, Angew. Chem., Int. Ed., 2021, 60, 10670–10679 CrossRef CAS PubMed.
- T. Yang, Y. Tal-gan, A. E. Paharik, A. R. Horswill and H. E. Blackwell, ACS Chem. Biol., 2016, 11, 1982–1991 CrossRef CAS PubMed.
- C. Kao, M. Zheng and S. Rüdisser, RNA, 1999, 5, 1268–1272 CrossRef CAS PubMed.
- T. Katoh, Y. Iwane and H. Suga, Nucleic Acids Res., 2017, 45, 12601–12610 CrossRef CAS PubMed.
- Y. Goto, T. Katoh and H. Suga, Nat. Protoc., 2011, 6, 779–790 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cb00225b |
| This journal is © The Royal Society of Chemistry 2022 |