
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOptimizing CSP1 analogs for modulating quorum sensing in Streptococcus pneumoniae with bulky, hydrophobic nonproteogenic amino acid substitutions†
Tahmina A.
Milly
a,
Alec R.
Buttner
b,
Naomi
Rieth
b,
Elizabeth
Hutnick
b,
Emilee R.
Engler
b,
Alexandra R.
Campanella
c,
Muralikrishna
Lella
a,
Michael A.
Bertucci
*c and
Yftah
Tal-Gan
 *a
*a
aDepartment of Chemistry, University of Nevada, Reno, 1664 North Virginia Street, Reno, Nevada 89557, USA. E-mail: ytalgan@unr.edu
bDepartment of Chemistry, Moravian University, 1200 Main St., Bethlehem, PA 18018, USA
cDepartment of Chemistry, Lafayette College, 701 Sullivan Rd., Easton, PA 18042, USA. E-mail: bertuccm@lafayette.edu
First published on 28th January 2022
Abstract
The prompt appearance of multiantibiotic-resistant bacteria necessitates finding alternative treatments that can attenuate bacterial infections while minimizing the rate of antibiotic resistance development. Streptococcus pneumoniae, a notorious human pathogen, is responsible for severe antibiotic-resistant infections. Its pathogenicity is influenced by a cell-density communication system, termed quorum sensing (QS). As a result, controlling QS through the development of peptide-based QS modulators may serve to attenuate pneumococcal infections. Herein, we set out to evaluate the impact of the introduction of bulkier, nonproteogenic side-chain residues on the hydrophobic binding face of CSP1 to optimize receptor-binding interactions in both of the S. pneumoniae specificity groups. Our results indicate that these substitutions optimize the peptide–protein binding interactions, yielding several pneumococcal QS modulators with high potency. Moreover, pharmacological evaluation of lead analogs revealed that the incorporation of nonproteogenic amino acids increased the peptides’ half-life towards enzymatic degradation while remaining nontoxic. Overall, our data convey key considerations for SAR using nonproteogenic amino acids, which provide analogs with better pharmacological properties.
Introduction
Bacteria coordinate their various physiological behaviors and control gene expression in response to changes in cell density by utilizing intercellular chemical signaling pathways in a process known as quorum sensing (QS).1 QS involves the detection of a small signaling molecule known as an autoinducer that is synthesized and then actively or passively secreted. In Gram-positive bacteria, the signals that mediate this self-propagating mechanism are generally peptides and, thus, are referred to as autoinducing peptides (AIPs).2 The AIPs are detected by a membrane-bound receptor, typically a histidine-kinase receptor, prompting a response when the AIP reaches a certain threshold concentration.1,3 This sensing enables bacterial cells to communicate with the other neighboring cells and establish different group-beneficial traits, including the production of virulence factors, sporulation, bioluminescence, root nodulation, swarming, biofilm formation, and competence development.4–7 Specifically, many human pathogenic species utilize QS to effectively attack their host and establish an infection. As such, QS has gained significant attention as an anti-pathogenic drug target for the development of novel therapeutics, especially considering the ongoing emergence of multidrug resistant bacterial pathogens.8–13 Interception of QS circuits’ signal-receptor interactions using signal molecule-based drugs would lead to an attenuation of bacterial pathogenicity rather than induction of cell death, thus limiting the potential for resistance development while still preventing many bacterial pathogenic traits.14,15Streptococcus pneumoniae, or pneumococcus, is a Gram-positive bacterium that populates the nasopharyngeal cavity and upper respiratory tract of humans. As an opportunistic pathogen, it is responsible for more than 1 million pneumococcal infections including bacteremia, sepsis, meningitis, and pneumonia in the United States alone.16,17 Moreover, recombinogenic pneumococcus is intrinsically resistant to several antibiotics such as vancomycin, linezolid, quinolones, and beta lactams.18–20 A major contributor to the development of antibiotic resistant strains is the ability of pneumococci to exchange intra- and inter-species genetic material with other neighboring species.13,20–22 Specifically, competent pneumococci have been shown to acquire antimicrobial resistance and virulence genes through transformation from its closely related species, Streptococcus mitis, which provides a significant advantage to pneumococci by enabling rapid evolution of the genome and capsular diversity.13,21,22 Therefore, the costs associated with pneumococcal infections as well as high-speed accumulation of multi-drug resistance by S. pneumoniae necessitates treating this pathogen using alternative approaches. In S. pneumoniae, the acquisition of antibiotic-resistance genes and pathogenicity is directly associated with the activation of the pheromone-responsive competence regulon, a conserved QS circuit (Fig. 1).23 Thus, this QS circuitry can be utilized as an excellent target for the design of anti-virulence drug leads to control pneumococcal infections.
 | ||
| Fig. 1 S. pneumoniae CSP-mediated QS circuit. The pre-CSP peptide, ComC, is being processed and secreted by the ComAB transporter as the mature CSP signal. At high concentration, CSP can effectively bind and activate a transmembrane histidine kinase receptor, ComD, which, after being activated, transfers a phosphate group to its cognate response regulator, ComE. Phosphorylated ComE then triggers the transcription of numerous genes, including the effector molecule of the circuitry, ComX, which regulates QS-mediated phenotypes. | ||
In S. pneumoniae, the competence regulon is triggered by a 17-amino acid AIP termed the competence stimulating peptide (CSP, Fig. 1).5 With the help of a proteolytic ATP binding cassette (ABC) transporter, (ComAB, Fig. 1), the CSP pro-peptide, ComC, is processed and the mature signaling molecule, CSP, is exported out of the cell.5 Upon reaching a threshold concentration, CSP can effectively bind and activate a membrane-bound histidine kinase receptor, ComD, resulting in phosphorylation of the response regulator, ComE.23–25 Phosphorylated ComE then acts as a transcription factor and initiates the transcription of the comAB and comCDE genes, resulting in autoinduction of the QS circuitry. ComE also initiates the transcription of the gene for the effector molecule of the QS circuit, the alternative sigma factor, ComX, which controls different QS-regulated phenotypes.25–27 The majority of S. pneumoniae strains can be divided into two main pherotypes or specificity groups based on the AIP they produce (CSP1 or CSP2, Fig. 1), along with their own specific ComD receptor (ComD1 or ComD2, respectively).28 These two peptide pheromones share approximately 50% sequence similarity, with most of the variation occurring amongst hydrophobic residues in the central region of the pheromone, allowing them to confer high selectivity toward their respective cognate receptors.28–31
The pneumococcal competence regulon communication pathway can be modulated through impediment of the peptide–receptor interaction by using synthetic AIP analogs. To improve the potency and pharmacological properties of CSP-based QS modulators, Yang et al. previously performed a systematic structure–activity relationship (SAR) analysis of the native CSP1 signal.29,30 The results of their studies suggested that an α-helix is the bioactive conformation of CSP1, and that the hydrophobic side of the helix plays a crucial role in the binding of CSP1 to ComD1. Specifically, hydrophobic residues in positions 4, 7, 8, 11 and 12 of the CSP1 sequence form a hydrophobic patch that spans two full helical turns and stabilizes CSP1–ComD1 binding. These results suggest that any structural change affecting this hydrophobic patch will alter the CSP1–receptor binding interaction.30 In a previous study, we incorporated highly conservative point mutations to the hydrophobic side-chain residues in these positions of the CSP1 sequence using both proteogenic and nonproteogenic amino acids.32 Information gained from this work suggested that the side-chain residues do not fully occupy the hydrophobic binding pockets and, thus, the CSP1–ComD1 binding interactions could be further optimized utilizing elongated side-chain residues. We advanced these results by incorporating multiple mutations containing several of the nonproteogenic amino acids in the CSP1 sequence.33 The findings from this study provided several important structural insights, specifically the preference of ComD1 for linear, hydrophobic, nonproteogenic amino acids. Combined, the results obtained from these two studies revealed strong potential for even further optimization of the binding interaction between CSP1 and ComD1. In this work, we focused on assessing the hydrophobic pockets within the ComD1 receptor through the introduction of bulkier and more hydrophobic nonproteogenic amino acids, namely a non-natural Phe-derivative, cyclohexylalanine (Cha), and an extended aliphatic hydrophobic residue, homoleucine (HLeu), in key hydrophobic positions (4, 7, 8, 11, and 12). To this end, we rationally designed and chemically synthesized a library of singly and multiply mutated CSP1 peptides intended to develop novel CSP-based QS modulators with enhanced activities against both pneumococcal ComD receptors. Our analysis revealed several nanomolar- and picomolar-range ComD1 and ComD2 activators containing these two nonproteogenic amino acids, suggesting that size and hydrophobicity, rather than the aromaticity of the amino acid side chains, dictate the stabilization of the binding interaction. Our next goal was to construct potent inhibitory peptides of the S. pneumoniae competence regulon by combining the lead CSP1 analogs with a previously characterized E1A substitution.8 Indeed, through this analysis, we were able to develop several low nanomolar-range ComD1 inhibitors. Moreover, we evaluated the pharmacological properties of lead analogs and observed a significant increase in stability towards enzymatic degradation while maintaining low toxicity. In addition to yielding a series of new QS activators and inhibitors, our results provide valuable information regarding the ComD1 hydrophobic binding pockets and key SAR knowledge of the CSP1 pheromone. This information can be utilized for the rational design of highly potent, pharmacologically stable CSP-based QS modulators with therapeutic potential.
Results and discussion
Design and synthesis of nonproteogenic CSP1 analogs
Designing optimized peptide ligands that interact with receptor proteins involves the enhancement of individual peptide-protein binding interactions.34 For instance, an unoccupied binding site within the protein due to the steric limits of a proteogenic amino acid side chain could be optimized through substitution of a bulkier amino acid side chain (Fig. 2). Contrary, when there is an unfavorable steric clash within the binding pocket due to the presence of larger amino acid side chain, the binding interactions could be optimized through utilization of a smaller amino acid side chain.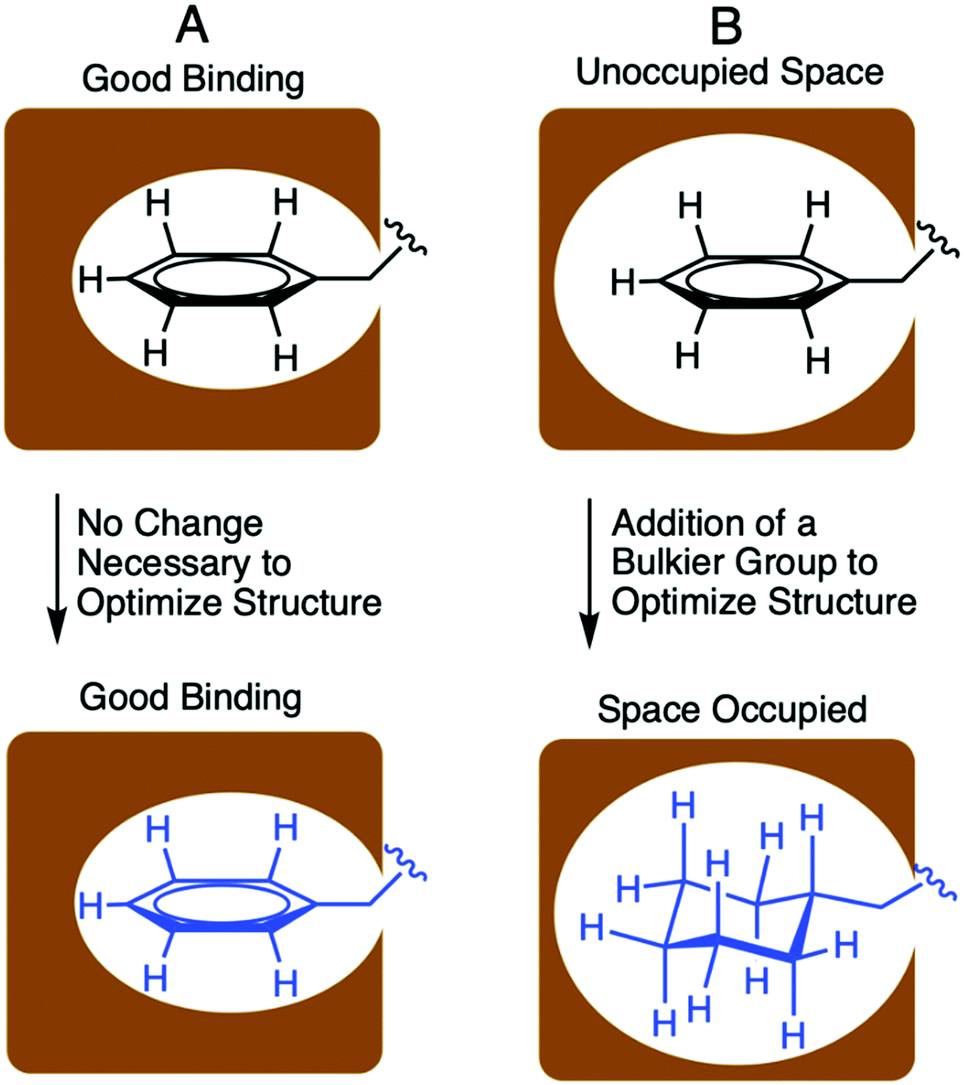 | ||
| Fig. 2 Portrayal of optimized and unoptimized protein–peptide binding interactions. (A) represents an optimized binding interaction where the side chain residue fills the binding pocket entirely. (B) represents an unoptimized binding interactions due to limited contacts of small side chain with the binding pocket. This interaction was improved through incorporation of a bulkier, nonproteogenic side chain. | ||
In the context of S. pneumoniae CSP1, our previous investigations of the CSP1–ComD1 binding interaction revealed that positions 4, 7, 8, 11, and 12 in CSP1 have unoccupied space within the receptor binding site.32,33 We aimed to assess the steric limit of the CSP1–ComD1 interaction by utilizing bulkier, hydrophobic nonproteogenic substituents. To this end, aliphatic and aromatic hydrophobic residues (Leu, Ile or Phe) in positions 4, 7, 8, 11, and 12 were substituted with the nonproteogenic amino acids, cyclohexylalanine (Cha) or homoleucine (HLeu) (Fig. 3).
 | ||
| Fig. 3 Conservative point mutations of CSP1 performed in this study. The CSP1 sequence is presented using the one letter amino acid code. Residues in red were replaced by the green residues. HLeu, homoleucine; Cha, cyclohexylalanine. | ||
Utilization of Cha substitutions in positions 7, 8 and 11 allowed us to increase the side chain hydrophobic surface area, while minimizing alterations to chain length and polarity. This mutation also removes the aromaticity of the Phe residue, abolishing forces governed by π electrons, such as π–π stacking. In parallel, we employed a series of HLeu substitutions intended to explore the spatial extremes of each position, reaching the upper limit of carbon chain length with a nonproteogenic amino acid substitution. At positions 7, 8, and 11, HLeu substitution similarly provided an opportunity to explore the effects of converting aromatic side chains to aliphatic residues. Overall, by systematically substituting HLeu and Cha at these five positions (4, 7, 8, 11, and 12), we created a library of analogs that explores the effects of interconverting cyclic and aliphatic residue sidechains, while also assessing the effects of size, hydrophobicity, and aromaticity. The analogs were built using standard solid-phase peptide synthesis (SPPS) protocols on Wang resin,35 followed by purification using semi-preparative RP-HPLC to >95% purity and their identity confirmed by mass spectrometry (for full details see the ESI†).
Nonproteogenic singly substituted CSP1 analogs
To evaluate the ability of the CSP1 analogs to modulate the activity of the pneumococcal ComD receptors (both ComD1 and ComD2), we utilized a β-galactosidase cell-based bacterial reporter assay with the two previously constructed reporter strains, D39pcomx::lacZ and TIGR4pcomx::lacZ.8 These are two wild type strains capable of producing their native CSPs and carry the lacZ gene under the control of the comX promoter. Activation of the ComD receptors can therefore be assessed by measuring β-gal activity. All the CSP1 analogs were initially screened for their ability to modulate both ComD receptors at high analog concentration, followed by the determination of their EC50/IC50 values through dose–response curves (see the ESI† for initial screening and dose–response curves).Biological evaluation of the singly substituted CSP1 analogs revealed that substitution of either HLeu or Cha at all but the 11th position resulted in comparable or higher potency against the ComD1 receptor relative to the native CSP1. Each substitution also maintained activity against the ComD2 receptor (see Table 1). The data from single, nonproteogenic substitutions at L4 revealed that increased size and hydrophobicity does not lead to a significant change in activity compared to the native side chain at this position. The EC50 values are comparable between the native CSP1, L4Cha, and L4HLeu against both the ComD1 and ComD2 receptors (about 2-fold reduction and 2-fold increase in potency against ComD1 and ComD2, respectively). Regarding the 12th position, substitution of Cha or HLeu for Ile resulted in two potent ComD1 activators, I12Cha and I12HLeu, exhibiting EC50 values of 3.1 and 5.1 nM, respectively (Table 1). These data support the previous observation that the binding pocket of the 12th residue in the ComD1 receptor is not fully occupied by Ile and thus the CSP1–ComD1 binding interaction can be optimized utilizing bulkier hydrophobic side chain residues at this position.32 Contrary to the ComD1 receptor, these two mutations were not as well tolerated against the ComD2 receptor, resulting in a reduction in potency compared to CSP1 (EC50 values of I12Cha and I12HLeu are >1000 nM and 870 nM, respectively, Table 1). This suggests that the introduction of bulky side chains in the ComD2 binding pocket for this position is not as permissible.
| Peptide name | Peptide Sequence | ComD1 | ComD2 | ||
|---|---|---|---|---|---|
| EC50b (nM) | 95% CIc | EC50b (nM) | 95% CIc | ||
| a See the Experimental details for methods and the ESI for plots of agonism dose response curves. b EC50 values were determined by testing peptides over a range of concentrations. c 95% confidence interval. d Data from ref. 29. | |||||
| CSP1d | EMRLSKFFRDFILQRKK | 10 | 6.3–17 | 530 | 500–560 |
| CSP1-L4Cha | EMR(Cha)SKFFRDFILQRKK | 17 | 12–23 | 590 | 300–1200 |
| CSP1-L4HLeu | EMR(HLeu)SKFFRDFILQRKK | 11 | 7.0–17 | 220 | 130–360 |
| CSP1-F7Cha | EMRLSK(Cha)FRDFILQRKK | 1.5 | 0.95–2.3 | 780 | 550–1100 |
| CSP1-F7HLeu | EMRLSK(HLeu)FRDFILQRKK | 0.82 | 0.77–0.87 | 75 | 35–160 |
| CSP1-F8Cha | EMRLSKF(Cha)RDFILQRKK | 4.8 | 2.7–8.3 | 490 | 230–1100 |
| CSP1-F8HLeu | EMRLSKF(HLeu)RDFILQRKK | 3.3 | 2.0–5.2 | 520 | 390–710 |
| CSP1-F11Cha | EMRLSKFFRD(Cha)ILQRKK | 110 | 67–200 | 210 | 110–420 |
| CSP1-F11HLeu | EMRLSKFFRD(HLeu)ILQRKK | 67 | 32–140 | 540 | 300–980 |
| CSP1-I12Cha | EMRLSKFFRDF(Cha)LQRKK | 3.1 | 2.0–5.0 | >1000 | – |
| CSP1-I12HLeu | EMRLSKFFRDF(HLeu)LQRKK | 5.1 | 5.0–5.3 | 870 | 800–950 |
According to data from previous structural studies of CSP1, the positions bearing Phe (7, 8, and 11) are located on one side of the helix and mostly occupy the hydrophobic binding site within the ComD1 receptor.30 The Phe side chains were assumed to effectively interact with the ComD1 binding pocket through both hydrophobic and aromatic interactions. The results of our singly substituted CSP1 analogs revealed that substitution of Cha or HLeu for Phe at positions 7 and 8 resulted in more potent analogs against ComD1. With the exception of F7Cha, all the resulting analogs at these positions exhibited enhanced or comparable activity against ComD2 compared to CSP1 (Table 1). These results suggest that these sites in the ComD1 binding pocket may be larger than originally thought. Specifically, the activation data of single mutations revealed that the F7HLeu derivative is the most potent ComD1 agonist reported to date and a highly potent ComD2 agonist (>12-fold increase in potency against ComD1, EC50 = 0.82 nM; and >7-fold increase in potency against ComD2, EC50 = 75 nM; Table 1). As observed with F7, the F8 position favors both the Cha and the HLeu substitutions, affording modestly improved ComD1 activators (more than 2-fold increase in potency) and exhibiting similar activity to CSP1 against the ComD2 receptor. This result is consistent with our previous observation, suggesting that the binding pocket for the eighth residue can accommodate elongated hydrophobic side chains.31 On the contrary, an opposite trend was observed for the 11th residue. An 11-fold (F11Cha) and ∼7-fold (F11HLeu) reduction in potency against ComD1 was observed, depicting that this position is either more spatially restricted than the other positions or that the aromaticity of the Phe side-chain residue plays a critical role at this site on the receptor.
Nonproteogenic poly substituted CSP1 analogs
The single mutant library revealed several potent CSP-based pneumococcal QS activators. Therefore, we sought to utilize the recently acquired SAR insight to rationally design more potent QS modulators. To this end, a library of poly-substituted peptides was synthesized to examine potential synergistic effects on ComD activation. These mutations focused on positions 7, 8, and 12, based on the results of the singly substituted analogs (Table 2). Biological evaluation revealed that all the doubly substituted peptides possess EC50 values against ComD1 at or below the EC50 value of the native CSP1 signal. Our results revealed that a combination of two Cha substitutions at positions 7 and 12 yielded the most potent pan-group activator in our library (F7Cha/I12Cha, EC50 = 0.97 nM against ComD1 and 70 nM against ComD2; Table 2). This dual-modified analog displayed a cumulative effect, as the recorded activation was greater than that of the single replacement analogs (compare the EC50 values of F7Cha and I12Cha against the ComD1 and ComD2 receptors in Table 1 with the EC50 values of F7Cha/I12Cha against the ComD1 and ComD2 receptors in Table 2). This result suggests that the increased hydrophobicity and steric bulk in both the 7th and 12th positions, brought upon by the introduction of the Cha residues, allows for optimized interactions in both binding pockets. Likewise, HLeu was incorporated into several doubly substituted peptides. This resulted in another potent pan-group activator formed by combining Cha and HLeu substitutions at the 7th and 12th positions, respectively (F7Cha/I12HLeu), which exhibited a 3- to 4-fold increase in potency against both ComD receptors. While some doubly substituted analogs exhibited increased potency relative to CSP1 against both pneumococcal receptors, most resulted in similar activities to CSP1 against ComD2, supporting our previous observation that there are different binding requirements for the ComD1 and ComD2 receptors (Table 2).32| Peptide name | Peptide sequence | ComD1 | ComD2 | ||
|---|---|---|---|---|---|
| EC50b (nM) | 95% CIc | EC50b (nM) | 95% CIc | ||
| a See the Experimental details for methods and the ESI for plots of agonism dose response curves. b EC50 values were determined by testing peptides over a range of concentrations. c 95% confidence interval. d Data from ref. 29. | |||||
| CSP1d | EMRLSKFFRDFILQRKK | 10 | 6.3–17 | 530 | 500–560 |
| CSP1-F7Cha/F8Cha | EMRLSK(Cha)(Cha)RDFILQRKK | 2.2 | 1.4–3.5 | 590 | 330–1000 |
| CSP1-F7Cha/F8HLeu | EMRLSK(Cha)(HLeu)RDFILQRKK | 2.5 | 1.5–4.4 | 550 | 280–1100 |
| CSP1-F7HLeu/F8Cha | EMRLSK(HLeu)(Cha)RDFILQRKK | 4.8 | 2.9–8.1 | 340 | 190–640 |
| CSP1-F7HLeu/F8HLeu | EMRLSK(HLeu)(HLeu)RDFILQRKK | 7.2 | 3.4–15 | 710 | 530–940 |
| CSP1-F7Cha/I12Cha | EMRLSK(Cha)FRDF(Cha)LQRKK | 0.97 | 0.44–2.2 | 70 | 41–120 |
| CSP1-F7Cha/I12HLeu | EMRLSK(Cha)FRDF(HLeu)LQRKK | 3.0 | 1.4–6.4 | 140 | 68–300 |
| CSP1-F7HLeu/I12Cha | EMRLSK(HLeu)FRDF(Cha)LQRKK | 3.4 | 2.1–5.4 | 320 | 190–540 |
| CSP1-F7HLeu/I12HLeu | EMRLSK(HLeu)FRDF(HLeu)LQRKK | 1.2 | 0.80–1.8 | 350 | 180–680 |
| CSP1-F8Cha/I12Cha | EMRLSKF(Cha)RDF(Cha)LQRKK | 8.4 | 7.9–9.0 | 840 | 730–980 |
| CSP1-F8Cha/I12HLeu | EMRLSKF(Cha)RDF(HLeu)LQRKK | 5.4 | 3.7–7.8 | 380 | 250–600 |
| CSP1-F8HLeu/I12Cha | EMRLSKF(HLeu)RDF(Cha)LQRKK | 10 | 5.2–21 | 680 | 560–840 |
| CSP1-F8HLeu/I12HLeu | EMRLSKF(HLeu)RDF(HLeu)LQRKK | 3.3 | 2.7–4.0 | 400 | 230–720 |
E1A substituted CSP1 analogs
Previously, Zhu et al. reported that the replacement of glutamic acid at position 1 with alanine in CSP1 resulted in an analog that exhibits competitive inhibition against ComD1.8 Having identified several pan-group QS activators in both our single and double mutant libraries, we set out to evaluate whether these pan-group activators could be converted into pan-group QS inhibitors by applying the same key modification. For this analysis, we chose the most potent pan-group activators in both the single and double mutant libraries and incorporated the E1A modification to afford a library of eight E1A-containing analogs. This library included six singly substituted CSP1 analogs containing the Cha or HLeu substitutions at positions 7, 8 or 12, as well as the two most potent doubly substituted pan-group activators (F7Cha/I12Cha and F7Cha/I12HLeu). Biological evaluation revealed that all the resultant analogs can only effectively inhibit the ComD1 receptor (Table 3 and Fig. S-4 and S-8, ESI†). The lack of inhibitory activity against the ComD2 receptor highlights the strict and different requirements for receptor inhibition compared to receptor activation. These results are consistent with a previous study demonstrating that direct conversion of pan-group activators into pan-group inhibitors requires further modification rather than just a single substitution.36| Peptide name | Peptide sequence | ComD1 | |
|---|---|---|---|
| IC50 (nM)b | 95% CIc | ||
| a See the Experimental details for methods and the ESI for plots of antagonism dose response curves. b IC50 values were determined by testing peptides over a range of concentrations. c 95% confidence interval. d Data from ref. 29. | |||
| CSP1-E1Ad | AMRLSKFFRDFILQRKK | 86 | 51–150 |
| CSP1-E1A/F7Cha | AMRLSK(Cha)FRDFILQRKK | 36 | 16–79 |
| CSP1-E1A/F7HLeu | AMRLSK(HLeu)FRDFILQRKK | 72 | 42–120 |
| CSP1-E1A/F8Cha | AMRLSKF(Cha)RDFILQRKK | 210 | 95–470 |
| CSP1-E1A/F8HLeu | AMRLSKF(HLeu)RDFILQRKK | 340 | 200–580 |
| CSP1-E1A/I12Cha | AMRLSKFFRDF(Cha)LQRKK | 590 | 380–910 |
| CSP1-E1A/I12HLeu | AMRLSKFFRDF(HLeu)LQRKK | 41 | 29–56 |
| CSP1-E1A/F7Cha/I12Cha | AMRLSK(Cha)FRDF(Cha)LQRKK | 72 | 39–140 |
| CSP1-E1A/F7Cha/I12HLeu | AMRLSK(Cha)FRDF(HLeu)LQRKK | 57 | 46–72 |
Comparing the bioactivities of the resulting single-substitution inhibitors reveals that there is no correlation between EC50 and IC50 values. However, several novel ComD1 inhibitors with low nanomolar potency were discovered. E1A/F7Cha (IC50 = 36 nM), E1A/I12HLeu (IC50 = 41 nM), and E1A/F7HLeu (IC50 = 72 nM) displayed higher inhibitory potency against the ComD1 receptor compared to the E1A substitution alone (IC50 = 86 nM). The remaining single-substituted analogs displayed inhibitory activity but were less potent (IC50 > 86 nM) (Table 3). Although single substitution of HLeu or Cha at the 12th position yielded two potent ComD1 activators (Table 1), only the I12HLeu combined with the E1A modification generated a more potent ComD1 inhibitor. Conversely, the I12Cha substitution combined with the E1A substitution yielded a ComD1 inhibitor with ∼7-fold reduced activity, exhibiting the sensitivity of the 12th position for ComD1 inhibition. This sensitivity is further underscored by data garnered after introducing the E1A substitution into our doubly mutated activators, F7Cha/I12Cha and F7Cha/I12HLeu. E1A/F7Cha/I12Cha displayed an 8-fold increase in potency relative to E1A/I12Cha, but was only half as potent as E1A/F7Cha. Similarly, E1A/F7Cha/I12HLeu was a less potent inhibitor than its doubly substituted precursors, E1A/F7Cha and E1A/I12HLeu. Thus, select substitutions of a single bulky, nonproteogenic amino acid in combination with the E1A modification proved to be more effective in harnessing inhibitory activity than combining multiple substitutions.
Pharmacological evaluation of lead CSP1 analogs
Our next goal was to evaluate the impact the non-proteogenic substitutions have on key pharmacological properties, i.e., metabolic stability and toxicity. To this end, we first evaluated the stability of CSP1, CSP1-F7Cha/I12Cha (lead activator), and CSP1-E1A/F7Cha (lead inhibitor) towards trypsin and chymotrypsin degradation. Our results indicate that CSP1 is highly susceptible to enzymatic degradation, exhibiting half-lives of 30 min and 1 h against trypsin and chymotrypsin, respectively (Fig. 4). The introduction of Cha residues at either position 7 or positions 7 and 12 resulted in improved metabolic stability against both proteases. CSP1-E1A/F7Cha exhibited a half-life of 4 h against both enzymes, whereas CSP1-F7Cha/I12Cha exhibited half-lives of 3 h and 6 h against trypsin and chymotrypsin, respectively (Fig. 4). Further analysis of the degradation products revealed that the introduction of the Cha residues did not alter the trypsin cleavage sites (R3, K6, R9, and R15), but it did slow the cleavage kinetics (Fig. S-9–S-11, ESI†). As for chymotrypsin, the analysis of the degradation products revealed that, for CSP1, chymotrypsin can cleave the peptide in all three aromatic residues (F7, F8, and F11; Fig. S-12, ESI†). Furthermore, the Cha residues in both the 7 and 12 positions could be recognized and cleaved by chymotrypsin (Fig. S-13–S-14, ESI†). Interestingly, introduction of Cha at the 7th position resulted in elimination of cleavage at F8, whereas introduction of Cha at the 12th position resulted in elimination of cleavage at F11. Overall, the introduction of Cha residues resulted in altered cleavage sites and improved peptide stability.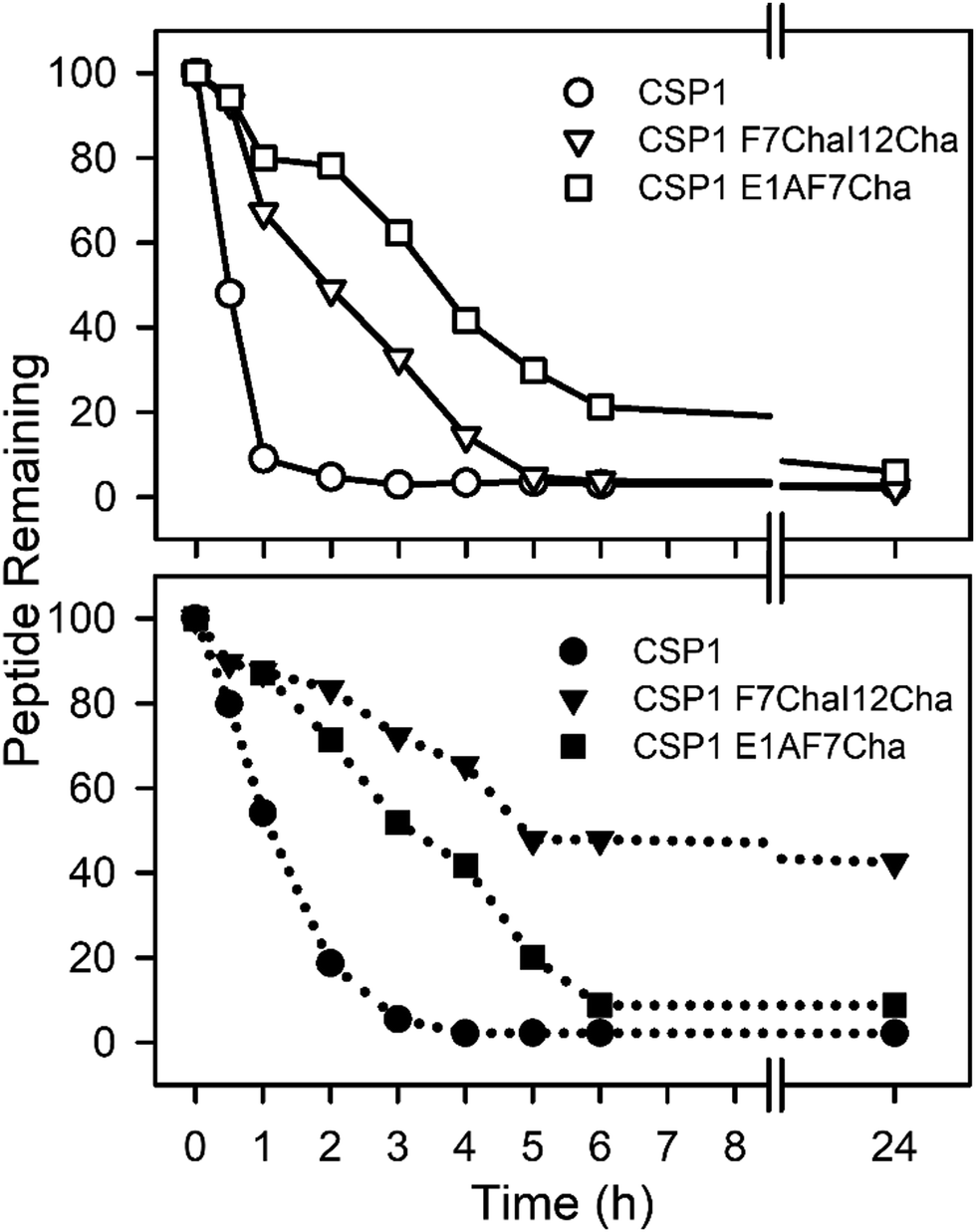 | ||
| Fig. 4 Metabolic Stability of CSP1 analogs. Peptides were incubated with trypsin (top panel) or chymotrypsin (bottom panel) and peptide degradation was monitored using analytical RP-HPLC. In the trypsin assay, CSP1 degraded first (half-life of 30 min), CSP1-F7ChaI12Cha degraded second (half-life of 3 h), and CSP1-E1AF7Cha was most stable (half-life of 4 h). In the chymotrypsin assay, CSP1 again degraded first (half-life of 1 h), however in this case CSP1-E1AF7Cha degraded second (half-life of 4 h), whereas CSP1-F7ChaI12Cha was most stable (half-life of 6 h). | ||
We next set out to evaluate the toxicity of CSP1 and the lead analogs towards mammalian cells. To this end, we performed a hemolysis assay of red blood cells (RBCs). Our results indicate that all three peptides are nontoxic, resulting in only minimal hemolysis, similar to the negative control, DMSO (Fig. 5). Overall, our results highlight the potential of introducing non-proteogenic amino acids to the CSP1 scaffold as a means to improve the pharmacological properties of the peptide without eliciting toxicity.
 | ||
| Fig. 5 Hemolytic activity of CSP-derived QS modulators on defibrinated RBCs. The CSP analogs exhibit no toxicity against RBCs. | ||
Conclusion
In this work, we set out to stabilize the CSP-ComD binding interactions through the development of single and multiple mutant CSP1 libraries consisting of bulkier, hydrophobic, and nonproteogenic substituents. Our analysis indicates that Cha and HLeu substitutions are less tolerated at positions 4 and 11 while being beneficial at positions 7, 8, and 12. These results suggest that the Cha and HLeu substitutions at 7th, 8th, and 12th positions provided a more optimal residue size and structure than the original proteogenic side chains. We hypothesize that the cyclic amino acids in the hydrophobic binding face are more open to Cha substitutions as Cha has a larger hydrophobicity index and steric value than Phe.37 These properties are not available with other natural amino acids. Additionally, Cha may be adopting conformations that Phe cannot as a planar, aromatic side chain in order to optimize hydrophobic interactions with the binding pocket of ComD1. For these reasons, our work highlights the value of using Cha in routine screens for improving peptide-protein interactions and assessing SAR in this QS system and beyond.Our rationally designed CSP1-based point and multiple mutant analogs yielded some of the most potent agonists of pneumococcal QS to date. The improved binding resulting from Cha and HLeu substitutions at positions 7, 8, and 12 indicates that there is unoccupied space in the ComD1 hydrophobic binding pocket that can be optimized. The success of some of our poly-substituted mutants demonstrate that optimized residues can be combined to further occupy this space. For example, F7Cha by itself had an EC50 value of 1.5 nM but, when combined with the I12Cha substitution, the EC50 value was reduced to 0.97 nM.
To test for pan-group activity, our CSP1 derivatives were screened against ComD2. In general, the derivatives were much less potent against ComD2. Importantly, the two most potent pan-group activators found in our study are F7HLeu and F7Cha/I12Cha, both with EC50 values below 1 nM against ComD1 and EC50 values around 70 nM against ComD2. Even after accounting for 95% CI overlap, these two substituted analogs exhibited more than 7 to 12-fold increase in potency against both ComD receptors. However, there does not seem to be a strong correlation between the ComD1 and ComD2 activation data. For example, F8HLeu is a stronger ComD1 activator than F11Cha, yet F11Cha is a stronger ComD2 activator than F8HLeu. It is important to note that F7Cha and I12Cha by themselves were ineffective as ComD2 activators but in combination make the best ComD2 activator identified in this study. Previously, it was determined that CSP1 and CSP2 form two distinctive hydrophobic patches that are optimal for ComD1 and ComD2 binding, respectively, and that a hybrid hydrophobic patch can be achieved by a single peptide.10 Therefore, it could be that the combined substitution of Cha at F7 and I12 result in a peptide that exhibits such a hybrid hydrophobic patch and binds both receptors effectively.
All of the single and double mutants that were resynthesized with the E1A substitution displayed some degree of inhibitory activity against ComD1. E1A/F7Cha and E1A/I12HLeu proved to be the most successful combinations, displaying IC50 values 2–3 times more potent than E1A alone. Our analysis revealed that some of the peptides that showcased agonistic activity were less effective as inhibitors. For instance, E1A/I12HLeu made a better inhibitor than E1A/I12Cha and E1A/F7HLeu, even though it was less potent than both as an activator. The lack of ComD2 inhibition activity for all the E1A-based analogs further supports the previous observation completed by our lab highlighting the strict requirements for pan-group activation and inhibition, as opposed to the simple E1A modification that was found to be sufficient in converting ComD1 or ComD2 activators into competitive inhibitors.
Finally, pharmacological evaluation of the lead analogs, CSP1-F7Cha/I12Cha (lead activator) and CSP1-E1A/F7Cha (lead inhibitor), revealed that the incorporation of the Cha residues resulted in analogs that exhibit superior metabolic stability while remaining nontoxic against mammalian cells.
In conclusion, our systematic study of the hydrophobic binding surface of CSP1 revealed that larger, nonproteogenic amino acids produced improved binding relative to the proteogenic amino acids present in the native sequence. We highlighted the importance of pushing the steric limit in peptide-protein SAR and discovered the most potent ComD1 agonist and several potent S. pneumoniae QS inhibitors as a result.
Experimental section
Chemical reagents and instrumentation
All chemical reagents and solvents were purchased from Sigma-Aldrich or Chem-Impex and used without further purification. Water (18 MΩ) was purified using a Thermo Scientific Smart2Pure Pro UV/UF 16 LPH water purification system. Solid-phase resins were purchased from Chem-Impex or P3 Biosystems.Reversed-phase high-performance liquid chromatography (RP-HPLC) was performed using two Shimadzu systems each equipped with a CBM-20A communications bus module, two LC-20AT pumps, an SIL-20A auto sampler, an SPD-20A UV/VIS detector, a CTO-20A column oven, one with an FRC-10A fraction collector and one without. All RP-HPLC solvents (18 MΩ water and HPLC-grade acetonitrile (ACN)) contained 0.1% trifluoroacetic acid (TFA). Matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS) data were obtained on either a Bruker Autoflex or Bruker Microflex spectrometer equipped with a 60 Hz nitrogen laser and a reflectron. In positive ion mode, the acceleration voltage on Ion Source 1 was 19.01 kV. Exact mass (EM) data were obtained on an Agilent Technologies 6230 TOF LC/MS spectrometer. The samples were sprayed with a capillary voltage of 3500 V and the electrospray ionization (ESI) source parameters were as follows: gas temperature of 325 °C at a drying gas flow rate of 8 L min−1 at a pressure of 35 psi.
Solid phase peptide synthesis
All the CSP1 analogs were synthesized using standard Fluorenyl methoxycarbonyl (Fmoc)-based solid-phase peptide synthesis (SPPS) procedures on preloaded Fmoc-L-Lys(Boc)-OH 4-benzyloxybenzyl alcohol (Wang) resin (0.59 mmol g−1) by using a Discover microwave synthesizer or Liberty1 automated peptide synthesizer (CEM Corporation). For peptides synthesized on the automated synthesizer, the resin (0.1 g) was first swelled by suspension in N,N-dimethylformamide (DMF) for 15 min at room temperature and then drained. Fmoc-protecting group removal was accomplished with treatment of the resin by 5 mL of 20% piperidine in DMF (90 s, 90 °C) followed by another 5 mL of 20% piperidine in DMF (90 s, 90 °C). The resin was then washed with DMF (3 × 5 mL) after each deprotection cycle. To couple each amino acid, Fmoc-protected amino acids (5 equiv. relative to the overall loading of the resin) were dissolved in DMF (5 mL) and mixed with 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU; 5 equiv.) and diisopropylethylamine (DIPEA; 5 equiv.). All amino acids were coupled for 5 min (30 W, 75 °C). After each coupling step, the resin was drained and washed with DMF (2 × 5 mL). This process was repeated until the desired peptide sequence was obtained. The same swelling and deprotection protocols were followed for peptides synthesized on the manual microwave synthesizer. However, 0.2 g of Wang resin was used and couplings were completed using diisopropyl carbodiimide (DIC) and Oxyma Pure with a 3.6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:3 ratio of DIC:Oxyma:AA in DMF for a final DIC concentration of 0.2 M. Each coupling was run at 75 °C for 8 min (50 W).
:3:3 ratio of DIC:Oxyma:AA in DMF for a final DIC concentration of 0.2 M. Each coupling was run at 75 °C for 8 min (50 W).
Peptide cleavage from solid support
Following coupling of the final residue, the resin was washed three times with DCM (2 mL) with manual shaking for 1 min. The resin was then washed with diethyl ether (2 mL) and dried under nitrogen stream for 3 min before transferring it into a 15 mL falcon tube. A 3 mL solution of 2.5% 18 MΩ water and 2.5% triisopropylsilane (TIPS) in 95% trifluoroacetic acid (TFA) for every 0.1 g of resin was added and the tube was shaken for 3 h at 200 rpm. Following completion of the cleavage reaction, the resin was filtered through a cotton ball, or a fritted syringe and the filtrate was transferred into a new 50 mL falcon tube. A cooled solution of diethyl ether:hexane (1:1, 45 mL, −20 °C) was added to the tube, and the tube was kept in a freezer at −20 °C for 10 min in order to precipitate the crude peptide. The pellet of the crude peptide was obtained by centrifugation of the 50 mL tube in a Beckman Coulter Allegra 6 centrifuge equipped with a GH3.8 rotor at 3000 RPM for 5 min. The supernatant was poured off and the solid peptide product was re-dissolved in 10 mL acetonitrile (ACN):water (1:1) and lyophilized for a minimum of 24 h before HPLC purification.
Peptide purification by HPLC
Crude peptides were purified using RP-HPLC. The crude peptide was dissolved in ACN:H2O (1:4; volume of ACN in water depends on the solubility of the peptide) and purified in 4 mL portions on either a Phenomenex Luna 5 μm C18 semi-preparative column (10 mm × 150 mm, 100 Å) or a Phenomenex Kinetex 5 μm C18 semi-preparative column (10 mm × 250 mm, 110 Å) with a flow rate of 5 mL min−1; mobile phase A = 18 MΩ water + 0.1% TFA and mobile phase B = ACN + 0.1% TFA. The collected fraction was lyophilized overnight and dissolved again in 5 mL ACN:H2O (1:4) for a secondary purification run. Preparative HPLC methods were used for the crude purification using a linear gradient (first prep 5% B → 45% B over 40 min and second prep 25% B → 38% B over 45 min). Fraction purity was determined through analysis on either a Phenomenex Luna 5 μm analytical C18 column (4.6 mm × 150 mm, 100 Å) or a Phenomenex Kinetex 5 μm analytical C18 column (4.6 mm × 250 mm, 110 Å) using a linear gradient (5% B → 95% B over 22 min or 27 min, respectively). Purities were determined by integration of peaks with UV detection at 220 nm. Only peptide fractions that were purified to homogeneity (>95%) were used for the biological assays. Following purification, peptides were frozen using a dry ice–acetone bath, and then lyophilized for a minimum of 24 h. Before the final masses and yields of purified peptides were determined, peptides were dissolved in 25% acetic acid in up to 1:1 ACN:water to allow removal of any residual TFA. The solution was then frozen and lyophilized for at least 24 h before the yield of the peptide was determined.
Peptide verification with mass spectrometry
During primary and secondary purification of crude peptide, peaks were verified to contain the desired peptide mass by MALDI-TOF MS. Samples were prepared using 1 μL α-cyano-4-hydroxycinammic acid (10 μg) in 1:1 H2O:ACN as a matrix and 1 μL of the desired peptide fraction. For the final verification of the peptides, a high resolution ESI-TOF MS (Tables S1 and S2, ESI†) was used to verify the exact masses of the peptides. The observed mass-to-charge (m/z) ratio of the peptide was compared to the expected m/z ratio for each peptide.
Biological reagents and strain information
All standard biological reagents were purchased from Sigma-Aldrich. Donor horse serum (defibrinated) was purchased from Sigma-Aldrich and stored at −20 °C until use in pneumococcus bacterial culture. To examine the ability of the synthesized CSP1 analogs to modulate the ComD receptors, and thus, the QS circuit in S. pneumoniae, β-galactosidase assays were performed using D39pcomX::lacZ (group I) and TIGR4pcomX::lacZ (group II) reporter strains.8Bacterial growth conditions
Freezer stocks of individual pneumococcal strains, D39pcomX::lacZ and TIGR4pcomX::lacZ, were created from 1.5 mL aliquots of overnight cultures (0.2 OD600nm) in Todd-Hewitt broth supplemented with 0.5% yeast extract (THY) and 0.5 mL glycerol, and stored at −80 °C. For the β-galactosidase experiments, bacteria from the freezer stocks were streaked into a THY agar plate containing 5% horse serum and chloramphenicol at a final concentration of 4 μg mL−1. The plate was incubated for 8–9 h in 37 °C with 5% CO2. An isolated fresh single colony was picked into sterilized cultural tube containing 5 mL of THY broth supplemented with a final concentration of 4 μg mL−1 chloramphenicol, and the culture was incubated in a CO2 incubator overnight (15 h). Overnight cultures were then diluted (1:50 for D39pcomX::lacZ; 1:10 for TIGR4pcomX::lacZ) with THY and the resulting solution was incubated in a CO2 incubator for 3–4 h, until the bacteria reached early exponential stage (OD600 values of 0.30–0.35 for D39pcomX::lacZ and 0.20–0.25 for TIGR4pcomX::lacZ) as determined by using a plate reader.
β-Galactosidase activation assays
The ability of synthetic CSP1 analogs to activate the expression of S. pneumoniae comX was determined using indicated reporter strains grown in THY (pH 7.3). An initial activation screening was performed at high concentration (10 μM) for all CSP1 analogs. Two μL of 1 mM solution of CSP1 analogs in dimethyl sulfoxide (DMSO) were added in triplicate to a clear 96-well microtiter plate. Two μL of 20 μM solution of native CSP1 (200 nM final concentration) were added in triplicate and served as the positive control for the S. pneumoniae group I strain (D39pcomX::lacZ), while two μL of 100 μM solution of CSP2 (1000 nM final concentration) were added as the positive control for the S. pneumoniae group II strain (TIGR4pcomX::lacZ). These concentrations were chosen to afford full activation of the QS circuit, as determined from the dose-dependent curves created for the native S. pneumoniae CSPs.29 Two μL of DMSO were added in triplicate and served as the negative control. Then, 198 μL of pneumococcal cultures were added to each well, the plate was incubated at 37 °C for 30 min, and the absorbance at 600 nm was measured. In order to measure the β-galactosidase activity in the pneumococcal culture, the cells were then lysed by incubating the culture for 30 min at 37 °C with 20 μL 0.1% Triton X-100 in water. Then, 100 μL of Z-buffer solution (60.2 mM Na2HPO4, 45.8 mM NaH2PO4, 10 mM KCl, and 1.0 mM MgSO4 in 18 MΩ H2O; pH was adjusted to 7.0 and sterilized before use) containing 2-nitrophenyl-β-D-galactopyranoside (ONPG) at a final concentration of 0.4 mg mL−1 was added in a new plate, followed by 100 μL of lysate, and the plate was incubated for 3 h at 37 °C. The reaction was quenched by adding 50 μL of 1 M sodium carbonate solution, and the OD420nm and OD550nm were measured using a plate reader. The results were reported as percent activation, which is the ratio between the Miller units of the analog and that of the positive control. For calculation of Miller units, please see data analysis below. Analogs that exhibited high activity (>75% activation compared to the native CSP) in the initial screening were further evaluated using a dose-dependent assay in which peptide stock solutions were diluted with DMSO in serial dilutions (either 1:2, 1:3, or 1:5) and assayed as described above. The EC50 (the concentration of a drug that gives half maximal response) value was then determined through fitting using nonlinear regression with GraphPad Prism 5.
β-Galactosidase inhibition assays
Analogs that exhibited low competence activation in the initial screening were evaluated for competitive inhibition. The ability of synthesized CSP1 analogs to inhibit the expression of comX by outcompeting native pneumococcal CSPs (CSP1 or CSP2) for the receptor binding site was evaluated using the same assay conditions as described above, except that in this case native CSP was added to every well at a set concentration (2 μL, 50 nM final concentration of CSP1 for group I; 250 nM final concentration of CSP2 for group II) that was chosen to afford full activation of the QS circuit, as determined from the dose-dependent curves created for the native pneumococcal CSPs. Two μL of native CSP (5 μM solution of CSP1 for group I; 25 μM solution of CSP2 for group II) and 2 μL of DMSO were added to the same well in triplicate and served as the positive control. Four μL of DMSO were added in triplicate and served as the negative control. Then, 196 μL of bacterial cultures were added to each well and the plate was incubated at 37 °C for 30 min. The procedure for lysis, incubation with ONPG and all the measurements were as described in the β-galactosidase activation assay. Analogs that exhibited significant competitive inhibition in the initial screening were further evaluated using a dose-dependent assay where a series of dilutions of the CSP1 analogs was prepared similarly to those made for the EC50 assay, and the IC50 (the concentration of an inhibitor where the response or binding is reduced by half) was then determined by using GraphPad Prism 5.Analysis of activation/inhibition data
Miller units were calculated using the following formula:Abs420 is the absorbance of o-nitrophenol (ONP). Abs550 is the scatter from cell debris, which, when multiplied by 1.75 approximates the scatter observed at 420 nm. t is the duration of incubation with ONPG in minutes, v is volume of lysate in milliliters, and Abs600 reflects cell density.

Metabolic stability
Enzymatic stability studies of CSP1 analogs was carried out in aqueous PBS solution (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 1.8 mM KH2PO4; pH 7.4). Peptide stocks (1 mM) were made in 18 mΩ H2O and final working concentration was 0.33 mM in PBS. Protease (trypsin or chymotrypsin) stock solution (25 μg mL−1; diluted from a 2.5 mg mL−1 solution) was made in PBS solution. Protease solution was added to the peptide solution to afford a final concentration of 0.05 μg mL−1 and then the solution was incubated at 37 °C with shaking (200 rpm) for 24 h. Aliquots (100 μL) were taken at 0, 0.5, 1, 2, 3, 4, 5, 6, and 24 h time points and mixed with 20 μL acetonitrile to stop the enzymatic activity. Then, 100 μL were injected via an autosampler and analyzed immediately for peptide degradation by analytical RP-HPLC. During the analytical RP-HPLC runs, the degradation products were manually collected and analyzed by MALDI-TOF MS. The digested peptide fractions at the end of the experiment (24 h time point) were also analyzed on a high-resolution ESI-TOF LC-MS to elucidate the peptide degradation pattern and confirm the identity of the peptide fragments.Hemolysis assay
The toxicity of synthetic CSP analogs was evaluated through hemolysis of red blood cells (RBCs). The hemolysis assay was performed as previously described with minor modifications.38 Briefly, 2 μL of a 1 mM CSP analog stock solution in DMSO were plated in triplicate in a clear bottom 96-well microtiter plate. Then, 198 μL of fresh THY media was added to each well. A positive control was prepared by adding 2 μL of a 1% Triton X solution to 198 μL THY media, and a negative control was prepared by adding 2 μL DMSO to 198 μL fresh THY media. One mL of defibrinated rabbit RBCs were centrifuged down, then the supernatants containing plasma and pre-lysed RBCs were pipetted out and the pelleted RBCs were resuspended in approximately 1 mL PBS. The process was repeated until the pre-lysed RBCs were completely removed (two to three washes, until the supernatant was clear). The washed RBCs were resuspended in PBS to a total volume of 1 mL, then 15 μL aliquots were added to each sample in the 96-well plate, and the plate was incubated for 30 min at 37 °C. Following incubation, the 96-well plate was centrifuged at 2000 rpm for 5 min at 4 °C and 25 μL of supernatants were transferred to a new plate containing 175 μL of 18 mΩ H2O, and the absorbance at 541 nm was recorded. Three independent trials were performed, and data is presented as the percent hemolysis relative to the 0.01% Triton X positive control.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by a grant from the National Science Foundation (CHE-1808370). The S. pneumoniae D39pcomX::lacZ and TIGR4pcomX::lacZ reporter strains were a generous gift from G. W. Lau (University of Illinois at Urbana−Champaign).References
- C. M. Waters and B. L. Bassler, Annu. Rev. Cell Dev. Biol., 2005, 21, 319–346 CrossRef CAS PubMed.
- D. N. McBrayer, C. D. Cameron and Y. Tal-Gan, Org. Biomol. Chem., 2020, 18, 7273–7290 RSC.
- B. L. Bassler and R. Losick, Cell, 2006, 125, 237–246 CrossRef CAS PubMed.
- M. R. Parsek and E. Greenberg, Trends Microbiol., 2005, 13, 27–33 CrossRef CAS PubMed.
- L. S. Håvarstein, G. Coomaraswamy and D. A. Morrison, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 11140–11144 CrossRef PubMed.
- D. L. Hava and A. Camilli, Mol. Microbiol., 2002, 45, 1389–1406 CAS.
- E. A. Meighen, Microbiol. Rev., 1991, 55, 123–142 CrossRef CAS PubMed.
- L. Zhu and G. W. Lau, PLoS Pathog., 2011, 7, e1002241 CrossRef CAS PubMed.
- B. Koirala, J. Lin, G. W. Lau and Y. Tal-Gan, ChemBioChem, 2018, 19, 2380–2386 CrossRef CAS PubMed.
- Y. Yang, J. Lin, A. Harrington, G. Cornilescu, G. W. Lau and Y. Tal-Gan, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 1689–1699 CrossRef CAS PubMed.
- C. Duan, L. Zhu, Y. Xu and G. W. Lau, PLoS One, 2012, 7, e44710 CrossRef CAS PubMed.
- M. Lella and Y. Tal-Gan, Pept. Sci., 2021, e24222 CAS.
- T. A. Milly and Y. Tal-Gan, RSC Chem. Biol., 2020, 1, 60–67 RSC.
- J. R. Brannon and M. Hadjifrangiskou, Drug Des., Dev. Ther., 2016, 10, 1795 CAS.
- B. LaSarre and M. J. Federle, Microbiol. Mol. Biol. Rev., 2013, 77, 73–111 CrossRef CAS PubMed.
- S. Mehr and N. Wood, Paediatr. Respir. Rev., 2012, 13, 258–264 CrossRef PubMed.
- K. L. O'brien, L. J. Wolfson, J. P. Watt, E. Henkle, M. Deloria-Knoll, N. McCall, E. Lee, K. Mulholland, O. S. Levine and T. Cherian, Lancet, 2009, 374, 893–902 CrossRef.
- N. J. Croucher, S. R. Harris, C. Fraser, M. A. Quail, J. Burton, M. Van Der Linden, L. McGee, A. Von Gottberg, J. H. Song and K. S. Ko, Science, 2011, 331, 430–434 CrossRef CAS PubMed.
- J. Cornick and S. Bentley, Microbes Infect., 2012, 14, 573–583 CrossRef CAS PubMed.
- W. P. Hanage, C. Fraser, J. Tang, T. R. Connor and J. Corander, Science, 2009, 324, 1454–1457 CrossRef CAS PubMed.
- L. S. Håvarstein, R. Hakenbeck and P. Gaustad, J. Bacteriol., 1997, 179, 6589–6594 CrossRef PubMed.
- S. Shekhar, R. Khan, D. M. Ferreira, E. Mitsi, E. German, G. H. Rørvik, D. Berild, K. Schenck, K. Kwon and F. Petersen, Front. Immunol., 2018, 9, 747 CrossRef PubMed.
- E. Pestova, L. Håvarstein and D. Morrison, Mol. Microbiol., 1996, 21, 853–862 CrossRef CAS PubMed.
- F. M. Hui, L. Zhou and D. A. Morrison, Gene, 1995, 153, 25–31 CrossRef CAS PubMed.
- O. Ween, P. Gaustad and L. S. Håvarstein, Mol. Microbiol., 1999, 33, 817–827 CrossRef CAS PubMed.
- M. S. Lee and D. A. Morrison, J. Bacteriol., 1999, 181, 5004–5016 CrossRef CAS PubMed.
- A. Dagkessamanskaia, M. Moscoso, V. Hénard, S. Guiral, K. Overweg, M. Reuter, B. Martin, J. Wells and J. P. Claverys, Mol. Microbiol., 2004, 51, 1071–1086 CrossRef CAS PubMed.
- A. M. Whatmore, V. A. Barcus and C. G. Dowson, J. Bacteriol., 1999, 181, 3144–3154 CrossRef CAS PubMed.
- Y. Yang, B. Koirala, L. A. Sanchez, N. R. Phillips, S. R. Hamry and Y. Tal-Gan, ACS Chem. Biol., 2017, 12, 1141–1151 CrossRef CAS PubMed.
- Y. Yang, G. Cornilescu and Y. Tal-Gan, Biochemistry, 2018, 57, 5359–5369 CrossRef CAS PubMed.
- O. Johnsborg, P. E. Kristiansen, T. Blomqvist and L. S. Håvarstein, J. Bacteriol., 2006, 188, 1744–1749 CrossRef CAS PubMed.
- B. Koirala, R. A. Hillman, E. K. Tiwold, M. A. Bertucci and Y. Tal-Gan, Beilstein J. Org. Chem., 2018, 14, 1769–1777 CrossRef CAS PubMed.
- T. A. Milly, E. R. Engler, K. S. Chichura, A. R. Buttner, B. Koirala, Y. Tal-Gan and M. A. Bertucci, ChemBioChem, 2021, 22, 1940–1947 CrossRef CAS PubMed.
- D. Rooklin, A. E. Modell, H. Li, V. Berdan, P. S. Arora and Y. Zhang, J. Am. Chem. Soc., 2017, 139, 15560–15563 CrossRef CAS PubMed.
- W. Chan and P. White, Fmoc solid phase peptide synthesis: a practical approach, OUP Oxford, 1999 Search PubMed.
- B. Koirala and Y. Tal-Gan, ChemBioChem, 2020, 21, 340–345 CrossRef CAS PubMed.
- J. L. Fauchere, M. Charton, L. B. Kier, A. Verloop and V. Pliska, Int. J. Pept. Protein Res., 1988, 32, 269–278 CrossRef CAS PubMed.
- R. W. Mull and Y. Tal-Gan, ACS Chem. Biol., 2021, 16, 2834–2844 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: HPLC traces and MS data, initial screening results, dose response curves for CSP1 analogs, and additional metabolic stability figures. See DOI: 10.1039/d1cb00224d |
| This journal is © The Royal Society of Chemistry 2022 |