
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe molecular mechanism of P450-catalyzed amination of the pyrrolidine derivative of lidocaine: insights from multiscale simulations†
Conger Wang,
Peng Wu,
Zhanfeng Wang and
Binju Wang *
*
State Key Laboratory of Physical Chemistry of Solid Surface, Fujian Provincial Key Laboratory of Theoretical and Computational Chemistry, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, P. R. China. E-mail: wangbinju2018@xmu.edu.cn
First published on 16th August 2021
Abstract
Nitrogen heterocycles are key and prevalent motifs in drugs. Evolved variants of cytochrome P450BM3 (CYP102A1) from Bacillus megaterium employ high-valent oxo-iron(IV) species to catalyze the synthesis of imidazolidine-4-ones via an intramolecular C–H amination. Herein, we use multi-scale simulations, including classical molecular dynamics (MD) simulations, quantum mechanical/molecular mechanical (QM/MM) calculations and QM calculations, to reveal the molecular mechanism of the intramolecular C–H amination of the pyrrolidine derivative of lidocaine bearing cyclic amino moieties catalyzed by the variant RP/FV/EV of P450BM3, which bears five mutations compared to wild type. Our calculations show that overall catalysis includes both the enzymatic transformation in P450 and non-enzymatic transformation in water solution. The enzymatic transformation involves the exclusive hydroxylation of the C–H bond of the pyrrolidine derivative of lidocaine, leading to the hydroxylated intermediate, during which the substrate radical would be bypassed. The following dehydration and C–N coupling reactions are found to be much favored in aqueous situation compared to that in the non-polar protein environment. The present findings expand our understanding of the P450-catalyzed C(sp3)–H amination reaction.
1. Introdution
Nitrogen heterocycles constitute one of the most prevalent motifs in drugs.1 As such, numerous chemical approaches have been developed for the intra- and inter-molecular C–H aminations. Among these approaches, the biosynthesis of C–N bonds using enzymes represents an attractive approach concerning their benign conditions. As a versatile biocatalyst, cytochrome P450 can catalyse a wide range of reactions, such as hydroxylation, epoxidation, reductive dehalogenation, amine dealkylation and sulfoxidation.2 The coupled high-valent oxo-iron(IV) porphyrin π-radical cation species, so called compound I (Cpd I), has been well recognized as the principal oxidant of P450.2–9 Utilizing organic azides as the nitrene precursor, Fasan and co-workers found that cytochrome P450 could catalyse the intramolecular C–H amination of sulfonyl azides via the nitrene transfer reactions.10,11 In a subsequent work, Arnold and co-workers successfully engineered P450 to obtain the highly enantioselective intermolecular amination of benzylic C–H bonds.12In addition to the nitrene transfer reactions using organic azides, Wong and co-workers found the P450-mediated metabolite of the pyrrolidine derivative of lidocaine led to an unusual cyclization product,13 which is expected to be generated via a P450-Cpd I mediated intramolecular C–H amination. The following work by Wong and co-workers expand the similar reaction to a biosynthesis of a wide array of imidazolidin-4-ones using the evolved variants of cytochrome P450BM3, which may have potential bioactivity.14 Unlike the nitrene transfer reactions, P450-Cpd I mediated intramolecular C–H amination utilizes the common aminoacetamide as a substrate, which is easily available. Notably, a few P450 oxidases have been discovered to catalyse the intra- or inter-molecular C–N bond-forming reactions in recent studies.15–20 It was suggested14 that the reaction is initiated by the Cpd I-mediated H-atom abstraction (HAA) from the pyrrolidine derivative of lidocaine, leading to substrate radical and Fe(IV)–OH intermediate (Scheme 1). Next, two plausible mechanisms were proposed for P450-catalyzed amination of the pyrrolidine derivative of lidocaine. In the mechanistic route I, the one-electron oxidation of the pyrrolidine derivative of lidocaine by Cpd I may first lead to the iminium species, which may further undergo the intramolecular cyclization to form the N,N-acetal. In the alternative route II, the substrate radical may undergo a OH rebound reaction to afford the hydroxylated pyrrolidine derivative of lidocaine, which may further lose the hydroxide to form an iminium ion. Although the P450-Cpd I mediated intramolecular C–H amination is promising in the synthesis of imidazolidin-4-ones, the detailed mechanism of these processes is still unclear. For instance, how does P450 selectively oxidize the α-site C–H without oxidizing other C–H or N1–H bonds? Which mechanistic pathway is more favourable, route I or II? How is the intramolecular cyclization completed, enzymatic or non-enzymatic?
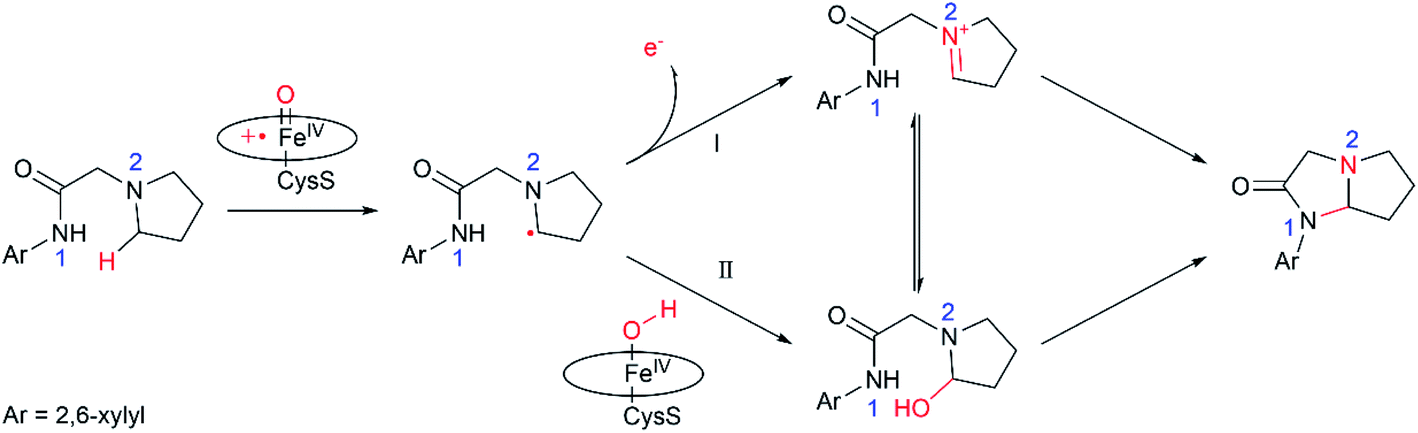 | ||
| Scheme 1 Two possible mechanisms proposed in ref. 12. | ||
To solve above issues, the combined molecular dynamics (MD) simulations, quantum mechanical/molecular mechanical (QM/MM) calculations, as well as QM calculations have been performed. QM/MM calculations can yield atomistic information on structure and mechanism of enzymatic reactions within the native environment of the protein,21–30 while the QM calculations were used to investigate the transformation of the hydroxylated intermediate in water solution. As shall demonstrated, P450-catalyzed amination of the pyrrolidine derivative of lidocaine involves both the enzymatic transformation in P450 and non-enzymatic transformation in water solution.31–34 The enzymatic transformation involves the exclusive hydroxylation of the C–H bond of the pyrrolidine derivative of lidocaine, while the following dehydration and C–N coupling reactions are found to be much favoured in aqueous situation than that in the non-polar protein environment.
2. Computational strategies and methods
2.1. Setup of the QM/MM system
Our system focuses on the RP/FV/EV variants containing R47L/Y51F/I401P/F87V/E267V mutations. The initial structure of the enzyme–substrate complex was prepared on the basis of the determined X-ray structure of P450BM3 heam domain in complex with DMSO35 (PDB code: 2J1M, with a resolution of 1.70 Å). Our setup is based on chain A, in which Zn ions are deleted since they are not necessary cofactors for P450BM3. The experimental substrate, the pyrrolidine derivative of lidocaine, was docked into the active site performed with the Rosetta ligand docking.36 According to the total energy, the top 20 docking conformations from 3000 docking results were selected for analysis. Among these 20 conformations, we found 12 of them have similar binding conformation for the substrate, in which the C3 site of the substrate and the Cpd I–O maintain a distance of 3.95–5.57 Å. Finally, we choose the conformation with the shortest distance (3.95 Å) to carry on the subsequent MD simulations. We assigned the protonation states of titratable residues (His, Glu, Asp) on the basis of pKa values from the PROPKA software37 in combination with careful visual inspection of local hydrogen-bonded networks. The histidine residues His92, His100, His138, His236, His266, His285, His361, His420 were protonated at the δ position, while His116, His171, His388, His408, His426 were protonated at the ε position. The glutamic acid residue Glu409 was protonated and all aspartic acid residues were deprotonated. For comparison, we also did MD simulation on Glu409-deprotonated state. Our MD simulations show that the binding conformation of the substrate are similar in both protonated and deprotonated cases, suggesting that the protonation state of Glu409 will not have significant impact on the BM3-mediated substrate oxidation (Fig. S2†). The force field for the Cpd I was parametrized using the “MCPB.py” modelling tool38,39 of AmberTools18.40 The Amber ff14SB force field41 was employed for the protein residues. The general AMBER force field (GAFF)42 was used for substrates, while the partial atomic charges were obtained from the RESP method,43 using the B3LYP/6-31G* level of theory. Sodium ions were added to the protein surface to neutralize the total charge of the systems. Finally, the resulting system was solvated in rectangular box of TIP3P waters extending up to minimum distance of 16 Å from the protein surface.2.2. Classical MD simulation
After a proper setup, the whole system was fully minimized using combined steepest descent and conjugate gradient methods. Then, the system was gently annealed from 10 to 300 K under a canonical ensemble for 50 ps. To achieve a uniform density after heating dynamics, 1 ns of density equilibration was performed under the NPT ensemble at the target temperature of 300 K and target pressure of 1.0 atm. Afterward, the system was further equilibrated for 4 ns under the NPT ensemble to get a well-settled pressure and temperature. Finally, a productive MD simulation under the NPT ensemble was conducted for 150 ns for the enzyme system. All MD simulations were performed with the GPU version of the Amber 18 package.402.3. QM/MM methodology
All QM/MM calculations were performed using ChemShell,44,45 combining Turbomole46 for the QM region and DL_POLY47 for the MM region. The electronic embedding scheme48 was used to account for the polarizing effect of the enzyme environment on the QM region. Hydrogen link atoms with the charge-shift model49 were applied to treat the QM/MM boundary. The QM subsystem is composed of the Cpd I moiety, its axial cysteine ligand Cys400, and the substrate. The whole QM region contains 116 atoms, which has the total charge of 0. The QM region was studied with the hybrid UB3LYP50–52 density functional with two levels of theory. For geometry optimization, the double-ζ basis set def2-SVP,53 labelled as B1, were used. The energies were further corrected with the larger basis set def2-TZVP53 for all atoms, labelled as B2. Dispersion corrections computed with Grimme's D3 method54–56 were included in all QM calculations. All the transition states (TSs) were located by relaxed potential energy surface (PES) scans followed by full TS optimizations using the dimer optimizer implemented in the DL-FIND code.57 For comparison, we also scanned the energy barrier of H-abstraction at the larger basis set def2-TZVP directly. Our calculations show that B3LYP/def2-TZVP yields the comparable barrier for H-abstraction (ΔE‡ = 17.9 kcal without ZPE, see Fig. S4†) as B3LYP/def2-SVP//def2-TZVP (ΔE‡ = 18.7 kcal without ZPE). All these indicate our selected basis sets are quite reliable.B3LYP has already been proven to be a successful25,26,29,31,58–68 functional for studying iron-based metalloenzymes. The benchmark study of H-abstraction by an iron-oxo species found UB3LYP was the best functional for such systems.69 All QM/MM calculations focus on the doublet state, since both the doublet and quartet states are almost degenerate and show similar reactivities in HAA reactions.
2.4. The QM calculations
All QM calculations were performed with the Gaussian 16 software package.70 The geometries of all the TSs, reactants and intermediates involved in the reaction were fully optimized using the SMD continuum solvation model71 at the B3LYP/6-31G(d) level of theory. Harmonic frequency calculations were performed using the equilibrium geometries to confirm the existence of first-order saddle points and local minima on the potential energy surfaces and to estimate the zero-point energies, as well as the thermal and entropic corrections. The connections between the stable structures and the transition states were ascertained by analysing the corresponding imaginary frequency modes, as well as by limited intrinsic reaction coordinate (IRC) calculations. The energies of the B3LYP/6-31G(d)-optimized structures were further refined by single-point calculations at the BMK/6-311++G(d,p) level.3. Results and discussion
3.1. Substrate binding in the active site of the P450 variant
The substrate was first docked into the active site of variant P450BM3 R47L/Y51F/I401P/F87V/E267V, which was found to have the highest activity.14 Fig. 1a shows the MD equilibrated active structure of the Cpd I of P450BM3 variants RP/FV/EV in complex with substrate, while Fig. 1b presents the distance fluctuations between Cpd I–O and the H1 atom, the H2 atoms of the substrate throughout the MD trajectory. It is seen that the substrate maintains a relatively stable conformation in the active site, with its H1 and H2 atoms being both close to the O atom of Cpd I (Fig. 1b and S1†). In addition, we found that Lys69 forms stable hydrogen-bonding networks with the two N atoms of substrate, suggesting Lys69 is key for controlling the stability and the binding conformation of the substrate. In addition to hydrogen-bonding interactions from Lys69, the substrate is also stabilized by hydrophobic interactions with Val87, Thr327, Val267, Thr268, Leu437 and Thr438. Notably, the site mutation of F87A is found to reduce steric hindrance, enabling the substrate to approach Cpd I, while E267V is expected to increase the hydrophobic interaction with the substrate, thus enhancing the binding affinity of the substrate.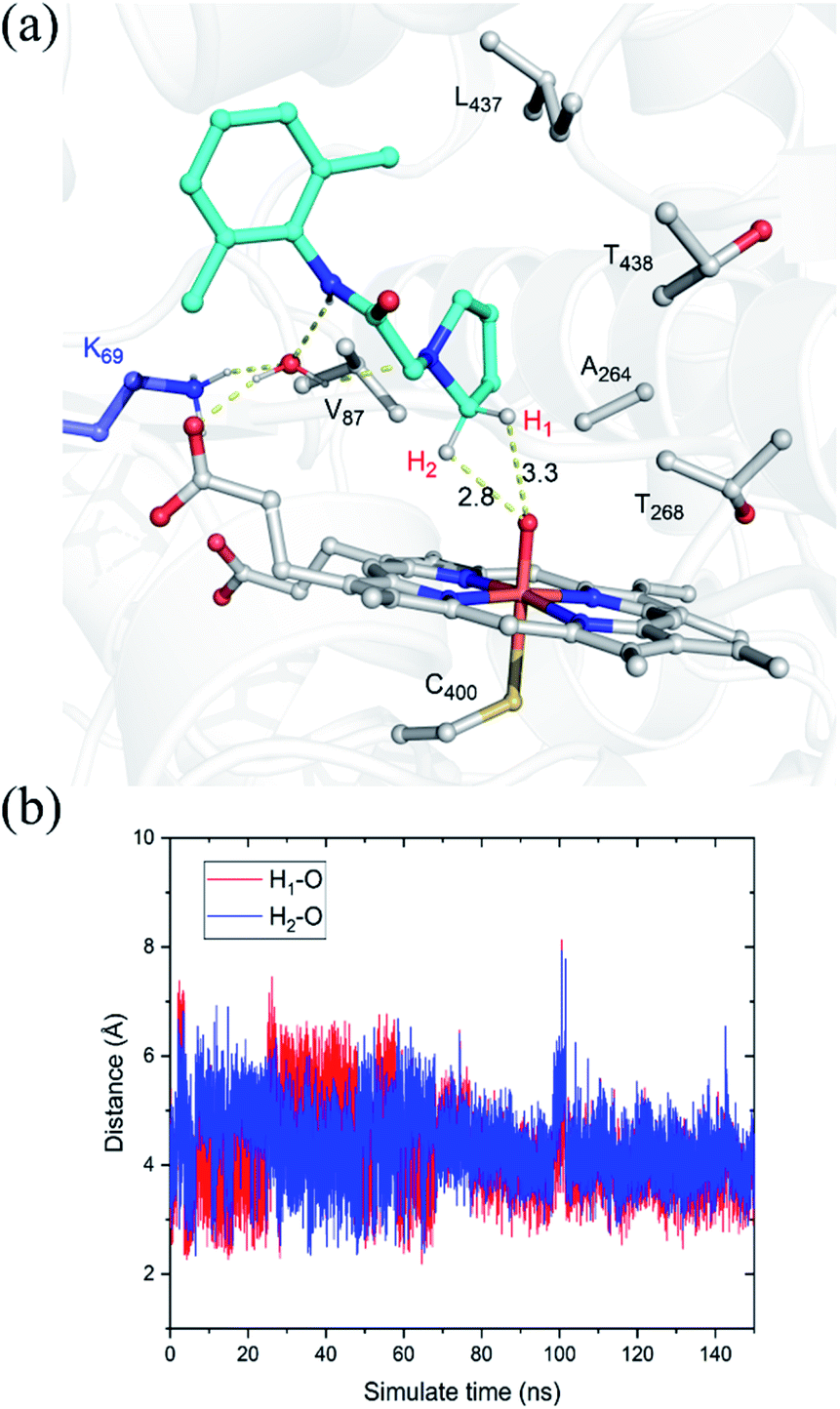 | ||
| Fig. 1 (a) QM(UB3LYP/B1)/MM optimized structure of the Cpd I with the bound substrate, pyrrolidine derivative of lidocaine, taken from a representative snapshot in the equilibrated MD trajectory. Key distances are given in angstroms. Most hydrogen atoms of C–H bonds have been omitted for clarity. (b) Distance fluctuations between Cpd I–O and the H1 atom (red), the H2 atom (blue) of the substrate throughout the MD trajectory. | ||
3.2. QM/MM and QM studies on the amination of the pyrrolidine derivative of lidocaine
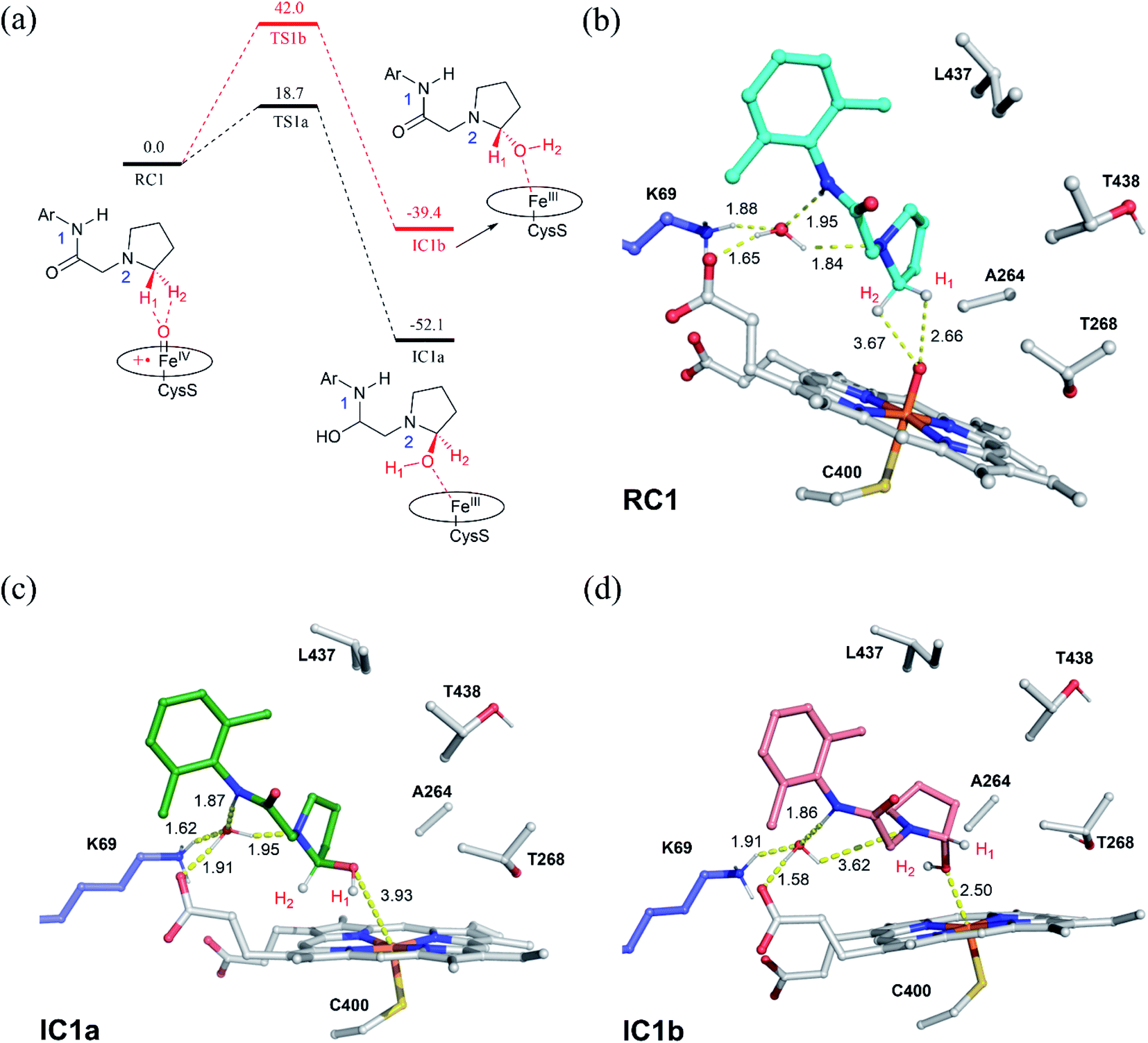 | ||
| Fig. 2 (a) QM(UB3LYP/B2)/MM relative energies (kcal mol−1) for the hydroxylation of the pyrrolidine derivative of lidocaine by the P450 variant. Key intermediates along the reaction energy profile are schematically drawn. (b) QM/MM optimized structures for the reactant complexes of Cpd I/substrate (RC1). (c) The R-chirality hydroxylated intermediate (IC1a) and (d) the S-chirality hydroxylated. | ||
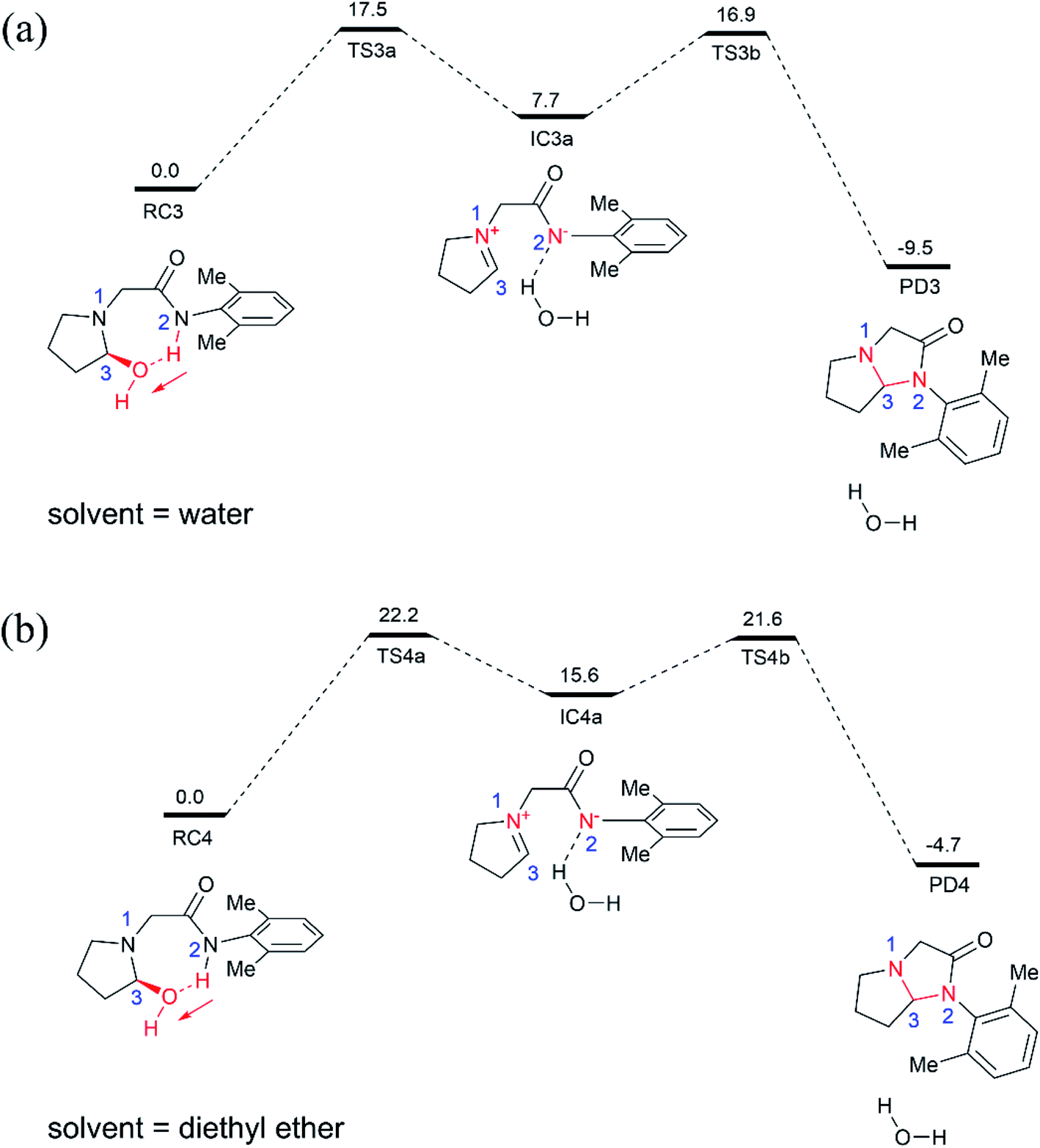 | ||
| Fig. 3 BMK/6-311++G(d,p) relative Gibbs energies (in kcal mol−1) for the cyclization of the hydroxylated intermediate in water (a) and diethyl ether (b) solution, shown along with schematic drawings of key intermediates along the reaction pathway. The red arrows highlight the direction of proton transfer. | ||
For comparison, we also examined the cyclization of the hydroxylated intermediate in diethyl ether solution (Fig. 3b), which has a dielectric constant of 4.2 and thus can mimic the hydrophobic nonpolar environment of active site of enzyme pocket. As shown in Fig. 3b, the overall cyclization reaction requires a high free energy barrier of 22.2 kcal mol−1. Notably, the zwitterion intermediate (IC4a) generated in diethyl ether solution is quite unstable, 15.6 kcal mol−1 higher than the corresponding reactant species (RC4), suggesting that highly polar water solvent can stabilize the intermediate and speed-up the cyclization reaction.
4. Conclusion
In summary, this work reports multi-scale calculations on the mechanistic nature of the P450-mediated amination of the pyrrolidine derivative of lidocaine. The QM/MM calculations show that the Cpd I active species of P450 selectively hydroxylate the C–H bond of the pyrrolidine derivatives of lidocaine and subsequent barrierless rebound of OH group leads to the hydroxylated intermediate, during which the substrate radical would be bypassed. Further QM calculations show that the hydroxylated intermediate is readily dehydrated to afford a zwitterion intermediate. Finally, a facile C–N cyclization reaction in the zwitterion intermediate generates the product of the imidazolidine-4-ones. In particular, our calculations show that the cyclization reaction is more favoured in water solution than that in the nonpolar solvent due to the stabilization effect of the polar environment on the zwitterion intermediate, suggesting that the efficient amination of the pyrrolidine derivative of lidocaine may require both the enzymatic transformation in P450 and non-enzymatic transformation in water solution. Thus, our calculations demonstrate a feasible mechanism for the 450-mediated amination of the pyrrolidine derivative of lidocaine, which could be different from other P450-catalyzed amination reactions (Fig. 4).10,11,17,72–74 | ||
| Fig. 4 Proposed catalytic mechanism for P450-catalyzed amination of the pyrrolidine derivative of lidocaine. | ||
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Key Research and Developement Program of China (2019YFA0906400), NSFC (No. 22073077, 21933009 and 21907082).References
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- S. Shaik, S. Cohen, Y. Wang, H. Chen, D. Kumar and W. Thiel, Chem. Rev., 2010, 110, 949–1017 CrossRef CAS PubMed.
- D. Kumar, H. Hirao, L. Que Jr and S. Shaik, J. Am. Chem. Soc., 2005, 127, 8026–8027 CrossRef CAS PubMed.
- S. Shaik, H. Hirao and D. Kumar, Acc. Chem. Res., 2007, 40, 532–542 CrossRef CAS PubMed.
- Y. Wei, E. L. Ang and H. Zhao, Curr. Opin. Chem. Biol., 2018, 43, 1–7 CrossRef CAS PubMed.
- F. P. Guengerich, ACS Catal., 2018, 8, 10964–10976 CrossRef CAS PubMed.
- J. B. Wang, Q. Huang, W. Peng, P. Wu, D. Yu, B. Chen, B. Wang and M. T. Reetz, J. Am. Chem. Soc., 2020, 142, 2068–2073 CrossRef CAS PubMed.
- Z. Li, Y. Jiang, F. P. Guengerich, L. Ma, S. Li and W. Zhang, J. Biol. Chem., 2020, 295, 833–849 CrossRef.
- T. Coleman, A. M. Kirk, R. R. Chao, M. N. Podgorski, J. S. Harbort, L. R. Churchman, J. B. Bruning, P. V. Bernhardt, J. R. Harmer, E. H. Krenske, J. J. De Voss and S. G. Bell, ACS Catal., 2021, 11, 1995–2010 CrossRef CAS.
- R. Singh, M. Bordeaux and R. Fasan, ACS Catal., 2014, 4, 546–552 CrossRef CAS PubMed.
- R. Singh, J. N. Kolev, P. A. Sutera and R. Fasan, ACS Catal., 2015, 5, 1685–1691 CrossRef CAS PubMed.
- C. K. Prier, R. K. Zhang, A. R. Buller, S. Brinkmann-Chen and F. H. Arnold, Nat. Chem., 2017, 9, 629–634 CrossRef CAS PubMed.
- X. Ren, J. A. Yorke, E. Taylor, T. Zhang, W. Zhou and L. L. Wong, Chemistry, 2015, 21, 15039–15047 CrossRef CAS PubMed.
- X. K. Ren, J. A. O'Hanlon, M. Morris, J. Robertson and L. L. Wong, ACS Catal., 2016, 6, 6833–6837 CrossRef CAS.
- F. He, T. Mori, I. Morita, H. Nakamura, M. Alblova, S. Hoshino, T. Awakawa and I. Abe, Nat. Chem. Biol., 2019, 15, 1206–1213 CrossRef CAS PubMed.
- I. Morita, T. Mori, T. Mitsuhashi, S. Hoshino, Y. Taniguchi, T. Kikuchi, K. Nagae, N. Nasu, M. Fujita, T. Ohwada and I. Abe, Angew. Chem., Int. Ed., 2020, 59, 3988–3993 CrossRef CAS PubMed.
- V. Steck, J. N. Kolev, X. Ren and R. Fasan, J. Am. Chem. Soc., 2020, 142, 10343–10357 CrossRef CAS PubMed.
- V. V. Shende, Y. Khatri, S. A. Newmister, J. N. Sanders, P. Lindovska, F. Yu, T. J. Doyon, J. Kim, K. N. Houk, M. Movassaghi and D. H. Sherman, J. Am. Chem. Soc., 2020, 142, 17413–17424 CrossRef CAS PubMed.
- D. H. Scharf, P. Chankhamjon, K. Scherlach, J. Dworschak, T. Heinekamp, M. Roth, A. A. Brakhage and C. Hertweck, Chembiochem, 2021, 22, 336–339 CrossRef CAS PubMed.
- I. Morita, T. Mori and I. Abe, Chemistry, 2021, 27, 2963–2972 CrossRef CAS PubMed.
- A. Warshel and M. Levitt, J. Mol. Biol., 1976, 103, 227–249 CrossRef CAS PubMed.
- H. Lin and D. G. Truhlar, Theor. Chem. Acc., 2006, 117, 185–199 Search PubMed.
- H. M. Senn and W. Thiel, Angew. Chem., Int. Ed., 2009, 48, 1198–1229 CrossRef CAS PubMed.
- M. W. van der Kamp and A. J. Mulholland, Biochemistry, 2013, 52, 2708–2728 CrossRef CAS PubMed.
- D. Kumar, W. Thiel and S. P. de Visser, J. Am. Chem. Soc., 2011, 133, 3869–3882 CrossRef CAS PubMed.
- P. Schyman, W. Lai, H. Chen, Y. Wang and S. Shaik, J. Am. Chem. Soc., 2011, 133, 7977–7984 CrossRef CAS PubMed.
- A. Warshel, Angew. Chem., Int. Ed., 2014, 53, 10020–10031 CrossRef CAS PubMed.
- B. Wang, C. Li, K. D. Dubey and S. Shaik, J. Am. Chem. Soc., 2015, 137, 7379–7390 CrossRef CAS PubMed.
- K. D. Dubey, B. Wang and S. Shaik, J. Am. Chem. Soc., 2016, 138, 837–845 CrossRef CAS PubMed.
- K. D. Dubey, B. Wang, M. Vajpai and S. Shaik, Chem. Sci., 2017, 8, 5335–5344 RSC.
- B. J. Wang, Z. C. Cao, D. A. Sharon and S. Shaik, ACS Catal., 2015, 5, 7077–7090 CrossRef CAS.
- B. Wang, E. M. Johnston, P. Li, S. Shaik, G. J. Davies, P. H. Walton and C. Rovira, ACS Catal., 2018, 8, 1346–1351 CrossRef CAS.
- H. Zhou, B. Wang, F. Wang, X. Yu, L. Ma, A. Li and M. T. Reetz, Angew. Chem., Int. Ed., 2019, 58, 764–768 CrossRef CAS PubMed.
- L. Wang, W. Song, B. Wang, Y. Zhang, X. Xu, J. Wu, C. Gao, J. Liu, X. Chen, J. Chen and L. Liu, ACS Catal., 2021, 11, 2808–2818 CrossRef CAS.
- J. Kuper, T. S. Wong, D. Roccatano, M. Wilmanns and U. Schwaneberg, J. Am. Chem. Soc., 2007, 129, 5786–5787 CrossRef CAS PubMed.
- G. Lemmon and J. Meiler, Methods Mol. Biol., 2012, 819, 143–155 CrossRef CAS PubMed.
- C. R. Sondergaard, M. H. Olsson, M. Rostkowski and J. H. Jensen, J. Chem. Theory Comput., 2011, 7, 2284–2295 CrossRef CAS PubMed.
- P. Li and K. M. Merz Jr, J. Chem. Inf. Model., 2016, 56, 599–604 CrossRef CAS PubMed.
- P. Li and K. M. Merz Jr, Chem. Rev., 2017, 117, 1564–1686 CrossRef CAS PubMed.
- D. A. Case, I. Y. Ben-Shalom, S. R. Brozell, D. S. Cerutti, T. E. Cheatham III, V. W. D. Cruzeiro, T. A. Darden, R. E. Duke, D. Ghoreishi, M. K. Gilson, H. Gohlke, A. W. Goetz, D. Greene, R. Harris, N. Homeyer, Y. Huang, S. Izadi, A. Kovalenko, T. Kurtzman, T. S. Lee, S. LeGrand, P. Li, C. Lin, J. Liu, T. Luchko, R. Luo, D. J. Mermelstein, K. M. Merz, Y. Miao, G. Monard, C. Nguyen, H. Nguyen, I. Omelyan, A. Onufriev, F. Pan, R. Qi, D. R. Roe, A. Roitberg, C. Sagui, S. Schott-Verdugo, J. Shen, C. L. Simmerling, J. Smith, R. SalomonFerrer, J. Swails, R. C. Walker, J. Wang, H. Wei, R. M. Wolf, X. Wu, L. Xiao, D. M. York and P. A. Kollman, AMBER 2018, University of California, San Francisco, 2018 Search PubMed.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- C. I. Bayly, P. Cieplak, W. D. Cornell and P. A. Kollman, J. Phys. Chem., 1993, 97, 10269–10280 CrossRef CAS.
- P. Sherwood, A. H. de Vries, M. F. Guest, G. Schreckenbach, C. R. A. Catlow, S. A. French, A. A. Sokol, S. T. Bromley, W. Thiel, A. J. Turner, S. Billeter, F. Terstegen, S. Thiel, J. Kendrick, S. C. Rogers, J. Casci, M. Watson, F. King, E. Karlsen, M. Sjovoll, A. Fahmi, A. Schafer and C. Lennartz, J. Mol. Struct.: THEOCHEM, 2003, 632, 1–28 CrossRef CAS.
- S. Metz, J. Kastner, A. A. Sokol, T. W. Keal and P. Sherwood, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 101–110 CAS.
- R. Ahlrichs, M. Bar, M. Haser, H. Horn and C. Kolmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- W. Smith and T. R. Forester, J. Mol. Graphics, 1996, 14, 136–141 CrossRef CAS PubMed.
- D. Bakowies and W. Thiel, J. Phys. Chem., 1996, 100, 10580–10594 CrossRef CAS.
- A. H. de Vries, P. Sherwood, S. J. Collins, A. M. Rigby, M. Rigutto and G. J. Kramer, J. Phys. Chem. B, 1999, 103, 6133–6141 CrossRef CAS.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1992, 97, 9173–9177 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- J. Kastner, J. M. Carr, T. W. Keal, W. Thiel, A. Wander and P. Sherwood, J. Phys. Chem. A, 2009, 113, 11856–11865 CrossRef CAS PubMed.
- H. Liu, J. Llano and J. W. Gauld, J. Phys. Chem. B, 2009, 113, 4887–4898 CrossRef CAS PubMed.
- M. G. Quesne, R. Latifi, L. E. Gonzalez-Ovalle, D. Kumar and S. P. de Visser, Chemistry, 2014, 20, 435–446 CrossRef CAS PubMed.
- B. Wang, D. Usharani, C. Li and S. Shaik, J. Am. Chem. Soc., 2014, 136, 13895–13901 CrossRef CAS PubMed.
- H. M. Senn, J. Kastner, J. Breidung and W. Thiel, Can. J. Chem., 2009, 87, 1322–1337 CrossRef CAS.
- K. Sen and J. C. Hackett, J. Am. Chem. Soc., 2010, 132, 10293–10305 CrossRef CAS PubMed.
- R. Lonsdale, K. T. Houghton, J. Zurek, C. M. Bathelt, N. Foloppe, M. J. de Groot, J. N. Harvey and A. J. Mulholland, J. Am. Chem. Soc., 2013, 135, 8001–8015 CrossRef CAS PubMed.
- E. Salanouve, G. Bouzemame, S. Blanchard, E. Derat, M. Desage-El Murr and L. Fensterbank, Chemistry, 2014, 20, 4754–4761 CrossRef CAS PubMed.
- X. Song, J. Lu and W. Lai, Phys. Chem. Chem. Phys., 2017, 19, 20188–20197 RSC.
- K. Bian, S. A. P. Lenz, Q. Tang, F. Chen, R. Qi, M. Jost, C. L. Drennan, J. M. Essigmann, S. D. Wetmore and D. Li, Nucleic Acids Res., 2019, 47, 5522–5529 CrossRef CAS PubMed.
- C. Hui, W. Singh, D. Quinn, C. Li, T. S. Moody and M. Huang, Phys. Chem. Chem. Phys., 2020, 22, 21696–21706 RSC.
- S. A. Lenz, D. Li and S. D. Wetmore, DNA Repair, 2020, 96, 102944 CrossRef CAS PubMed.
- R. Ramanan, K. D. Dubey, B. Wang, D. Mandal and S. Shaik, J. Am. Chem. Soc., 2016, 138, 6786–6797 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision B.01, Wallingford CT, 2016 Search PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- Z. Li, D. J. Burnell and R. J. Boyd, J. Phys. Chem. B, 2017, 121, 10859–10868 CrossRef CAS PubMed.
- Z. J. Jia, S. Gao and F. H. Arnold, J. Am. Chem. Soc., 2020, 142, 10279–10283 CrossRef CAS PubMed.
- Z. Fu, L. Yang, D. Sun, Z. Qu, Y. Zhao, J. Gao and Y. Wang, Dalton Trans., 2020, 49, 11099–11107 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra04564d |
| This journal is © The Royal Society of Chemistry 2021 |