
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiscovery, X-ray structure and CPP-conjugation enabled uptake of p53/MDM2 macrocyclic peptide inhibitors†
Anselm F. L.
Schneider
a,
Joerg
Kallen
b,
Johannes
Ottl
 b,
Patrick C.
Reid
c,
Sebastien
Ripoche
b,
Stephan
Ruetz
b,
Therese-Marie
Stachyra
b,
Samuel
Hintermann
b,
Christoph E.
Dumelin
b,
Christian P. R.
Hackenberger
*ad and
Andreas L.
Marzinzik
*b
b,
Patrick C.
Reid
c,
Sebastien
Ripoche
b,
Stephan
Ruetz
b,
Therese-Marie
Stachyra
b,
Samuel
Hintermann
b,
Christoph E.
Dumelin
b,
Christian P. R.
Hackenberger
*ad and
Andreas L.
Marzinzik
*b
aLeibniz-Forschungsinstitut für Molekulare Pharmakologie (FMP), Robert-Rössle-Strasse 10, Berlin 13125, Germany. E-mail: hackenbe@fmp-berlin.de
bNovartis Institutes for BioMedical Research, Novartis Campus, Basel CH-4056, Switzerland. E-mail: andreas.marzinzik@novartis.com
cPeptiDream, 3-25-23 Tonomachi, Kawasaki-Ku, Kanagawa 210-0821, Japan
dHumboldt Universität zu Berlin, Institut für Chemie, Brook-Taylor-Str. 2, Berlin 12489, Germany
First published on 26th August 2021
Abstract
Mouse double minute 2 homolog (MDM2, Hdm2) is an important negative regulator of the tumor suppressor p53. Using a mRNA based display technique to screen a library of >1012in vitro-translated cyclic peptides, we have identified a macrocyclic ligand that shows picomolar potency on MDM2. X-Ray crystallography reveals a novel binding mode utilizing a unique pharmacophore to occupy the Phe/Trp/Leu pockets on MDM2. Conjugation of a cyclic cell-penetrating peptide (cCPP) to the initially non cell-permeable ligand enables cellular uptake and a pharmacodynamic response in SJSA-1 cells. The demonstrated enhanced intracellular availability of cyclic peptides that are identified by a display technology exemplifies a process for the application of intracellular tools for drug discovery projects.
Introduction
Protein–protein interactions (PPIs) are essential networks for numerous biological processes and play a key role in all living organisms. Not surprisingly, PPIs have been very attractive for drug discovery targets during the last decades.1 Targeting PPIs is challenging due to the large, shallow, or weakly defined areas for ligand interaction, which are not well suited for small molecules (200–500 Da). Those interaction surfaces are typically highly hydrophobic with few distinct pockets making it difficult for a rule-of-five compliant small molecule to bind. Nonetheless, significant advances in novel technologies like fragment based drug discovery and high throughput screening have demonstrated that low molecular weight starting points for medicinal chemistry can be successfully developed into clinical candidates2 and become effective drugs.3 Antibodies as well as smaller fragments are well known to bind larger surfaces and have been successful in addressing targets which are difficult for small molecules;4 however, their intracellular transport is hampered and therefore often limited to extracellular PPIs.5Restoring the critical gatekeeper p53 in response to cellular damage and stress by blocking the p53/MDM2 interaction has become a very promising PPI target in cancer research. As it is one of the best characterized intracellular PPI targets with well-established biochemical and cellular assays, various highly potent and selective scaffolds disrupting the p53/MDM2 interaction have been developed. Those inhibitors show high potency in biochemical as well as cellular assays translating into efficacy models.6 One of those development candidates serves as a benchmark for our study developing peptides modulating an intracellular response.7
Linear peptides compared to wild type p53 have been discovered early on from phage displayed libraries, but those bear challenges in terms of potency, cell penetration and stability.8 In respect of superior potency and stability macrocyclic peptides are considered an appropriate modality to address the ligandability of such difficult targets, but also possess challenges concerning cell penetration. Stapled α-helical peptides emerged as a new modality to address intracellular PPIs about a decade ago and have been developed into clinical candidates for p53 dependent cancers.9,10 On the other hand, stapling peptides does not necessarily enhance affinity nor biological response by default and substantial optimization cycles from a known natural sequence are necessary to engineer highly affine molecules with a biological response in cellular studies.11,12 Alternatively, the attachment of a cyclic cell-penetrating peptide (cCPP) demonstrated an enhanced cellular delivery of a stapled peptidyl inhibitor against the MDM2/p53 interaction.13 Along those lines, very recently it was demonstrated that conjugation of cCPPs via triazole linkages to the peptide KD3, which was known to be a non-cell permeable stapled peptide inhibitor of p53/MDM2 and p53/MDMX interactions,14 enables activation of apoptosis in cancer cells.15 Alternative approaches for creating cell-permeable peptides to disrupt p53/MDMX interactions using cyclic peptide scaffolds,16 polycationic tags or medchem optimization strategies have been extensively studied.17,18
The aforementioned examples show that peptides create not only important opportunities as tool compounds for assay development and protein structure determination, but can also lead to the discovery of new binding pockets for pharmacophore mapping, even if those peptides are non-permeable. Such discoveries are fueled by the engineering of several peptide display technologies based on non-natural amino acids or in combination with chemical modification strategies, which have opened up fruitful avenues to identify ligands for difficult targets and explore their biological function.19–22 Still, a straightforward process of identifying and–if necessary–subsequently transforming non-permeable into permeable macrocyclic peptides would significantly extend the PPI repertoire, in particular for enabling intracellular target validation.
In this work we address this challenge and combine the screening of in vitro-translated macrocyclic peptides using mRNA display with a chemical modification strategy by cCPP-conjugation to identify highly potent cell-permeable macrocycles on a pharmacological target of central importance, in our case MDM2. By screening of >1012in vitro-translated macrocyclic peptides, we identified a thioether cyclic peptide that inhibits the interaction between p53 and MDM2 with picomolar potency. Subsequent X-ray crystallography revealed a novel binding mode of the peptide with the MDM2 protein. Finally, a cell-penetrating peptide (CPP)-conjugate of this cyclic peptide exhibited a mechanistic response in a cellular context thus validating the relevance of the new binding mode for drug discovery.
Results and discussion
Identification and characterization of a novel MDM2-ligand
mRNA display technologies have become one of the leading strategies to identify macrocyclic peptides against various targets including also more challenging PPIs. Highly diverse libraries of constrained peptides offer a greater chance to generate leads against difficult targets displaying novel pharmacophores with affinities comparable to those of antibodies and stability against degradation by proteolytic enzymes.23 An efficient Flexizyme enabled hit generation process based on a in vitro transcription/translation system has been developed, which enables fast screening of >1012 peptides against an immobilized target.24 Importantly, the Flexizyme based technology permits the efficient charging of different tRNAs with almost any standard or non-standard amino acid with high efficiency.25 Cyclization to thioether peptides occurs spontaneously through an incorporated N-terminal residue bearing a reactive chloro–acetyl group with the thiol from a cysteine side chain located at the C-terminus of the translated peptide. The in vitro translation system combined with the Flexizyme-mediated reprogramming of the genetic code has been previously described26 and the details of the selection conditions are provided in the ESI† (Section S4).Immobilized MDM2 was screened versus in vitro translated peptides containing a standard set of natural amino acids (Ser, Tyr, Pro, His, Arg, Thr, Asn, Val, Asp, Gly and Cys), along with the non-natural elongators N-methyl-Ser (MeS), N-methyl-Phe, N-methyl-Gly, N-methyl-Ala, and biphenylalanine (B). N-Chloroacetyl L-Phe was used in place of the initiator methionine to enable spontaneous cyclization with Cys to form thioether-cyclized peptides during the translation process (ESI,† Fig. S2). Selection and translation were performed in an iterative manner and the amino acid hit sequences were identified by next-generation sequencing. The amino acid sequence of the most enriched hit was selected for chemical synthesis and the corresponding peptide macrocycle CMR19 (Fig. 1) was generated from the linear precursor by capping the N-terminal residue with chloro–acetyl and subsequent robust ring-closure onto a Cys side chain (see ESI†). CMR19 displays a N-terminal phenylalanine–serine sequence followed by an acidic Asp residue succeeded by 3 polar Ser residues. While serine in position (4) is the only N-methylated amino acid, the serine residues in position 4 and 5 present the regular amide backbone. The next three residues presented are the hydrophobic Val(7), Pro(8), followed by polar Asn(9). Subsequently to two biphenyl at residues at position 10 and 11, Arg and Asn complete the thioether cyclic peptide formed by the Cys(14). The expendable C-terminal Gly is the point of attachment to the RNA during the display and is used as the point of conjugation for dyes or cell penetrating tags.
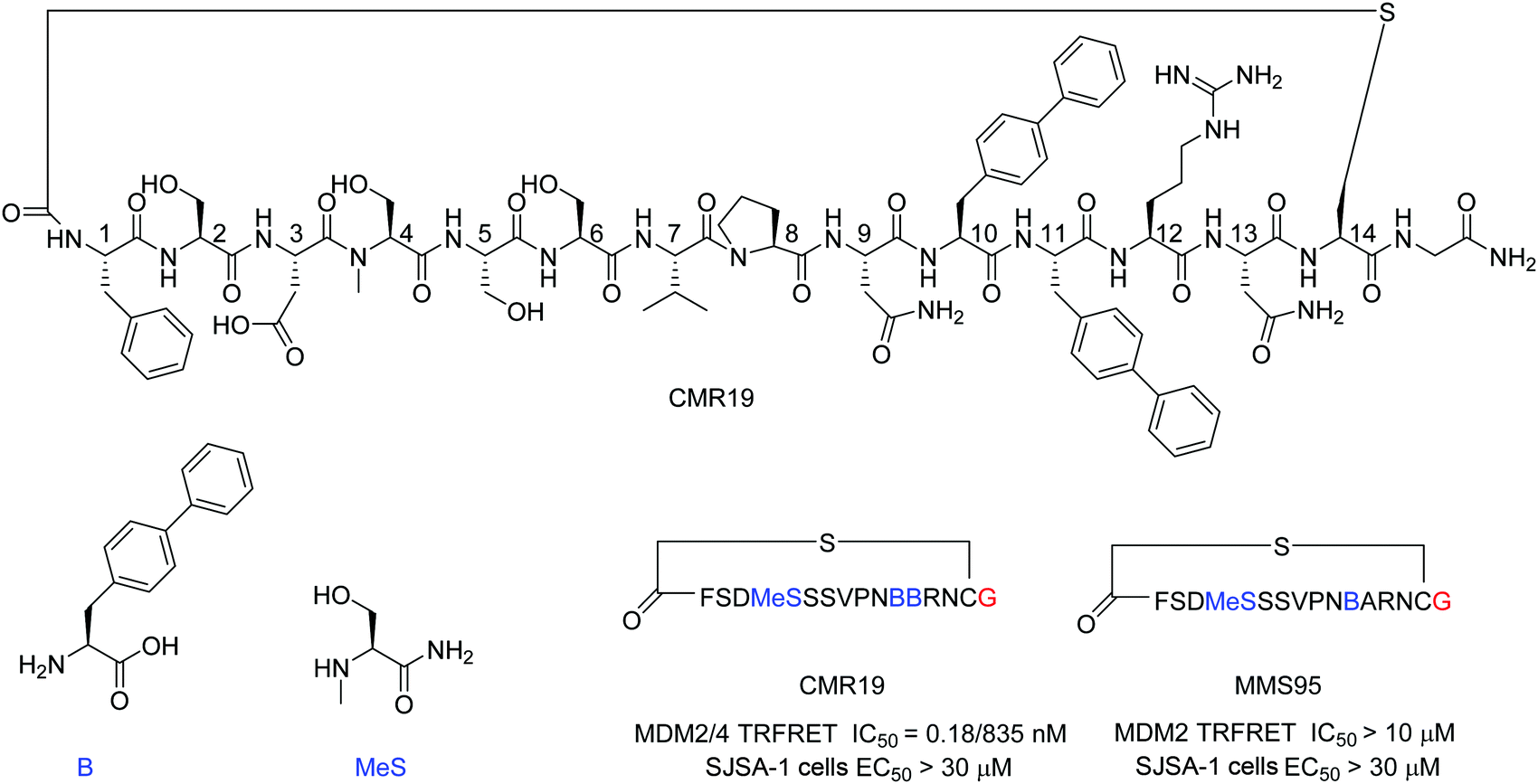 | ||
| Fig. 1 Chemical structure, MDM2/4 TR-FRET IC50 data, and SJSA EC50 cell data of macrocyclic peptide CMR19 identified from in vitro selection and of the inactive (B11A) mutant MMS95. Blue color indicates non-natural amino acids in the hit sequence and red color indicates terminal Gly used as spacer for attachment of cell penetrating moieties. | ||
Initial confirmation of the synthesized ligand was performed by assessing their ability to inhibit the interaction between MDM2 and a p53-derived peptide in a TR-FRET assay. CMR19 exhibited picomolar potency without any medicinal chemistry optimization with an IC50 value of 0.18 nM (corresponding to the calculated KI of 0.15 nM, ESI,† Section S5). In spite of this high potency in a biochemical assay, we did not observe any cytotoxic response when testing CMR19 on the MDM2 dependent SJSA-1 cells up to 30 μM. The efficiency of the ligand discovery process is shown by the fact that the highly optimized clinical small molecule inhibitor CGM097 only shows 1.7 nM potency (KI = 1.4 nM) in the same p53 displacement assay (MDM4 TR-FRET KI = 1950 nM). The macrocycle shows >4000 fold selectivity for MDM2 (KI = 0.15 nM) compared to MDM4 (KI = 740 nM). Differences in binding selectivity were also previously observed for optimized stapled KD3-peptides with picomolar affinity for MDM2, and ∼1 nM activity for MDM4.15 Due to the presence of hydrophobic pockets in the p53 binding site on MDM2, we speculated that the two biphenyl moieties may form key pharmacophores for the high binding affinity observed. Indeed, design of an inactive mutant was achieved by exchanging the biphenyl amino acid in position 11 with an Ala. The B11A mutant MMS95 showed no activity in the p53-displacement assay (>10 μM) nor on SJSA cells (>30 μM).
Co-crystal structure of CMR19 with MDM2
The first crystal structure of MDM2 bound to a 15-residue transactivation domain peptide of p53 had shown that MDM2 has a deep hydrophobic cleft on which the p53 peptide binds as an amphipathic α-helix.27 Three key amino acids covering the triad of p53 amino acids Phe19, Trp23, and Leu26 insert deeply into the MDM2 clefts by hydrophobic interactions.The co-crystal structure of ligand CMR19 in complex with MDM2 (Fig. 2) revealed that the non-canonical biphenyl side chains in positions 10 and 11 occupy the Leu- and Trp-pockets of MDM2. Interestingly, the Phe-pocket of MDM2 is less occupied by Val(7) of CMR19 than the Phe of p53 in MDM2.28 Both biphenyls are penetrating deeply into the MDM2 protein. The almost parallel arrangement of the adjacent biphenyls is stabilized by an edge-to-face interaction between the opposite aromatic rings. All amides of the CMR19 backbone are in trans configuration including the N-methylated Ser(4), except Pro(8) is cis. In comparison with the X-ray of the clinically evaluated CGM097 (white structure in Fig. 2B) the biphenyl group (10) of CMR19 (blue structure) penetrates more deeply into Leu-pocket than corresponding group of CGM097.
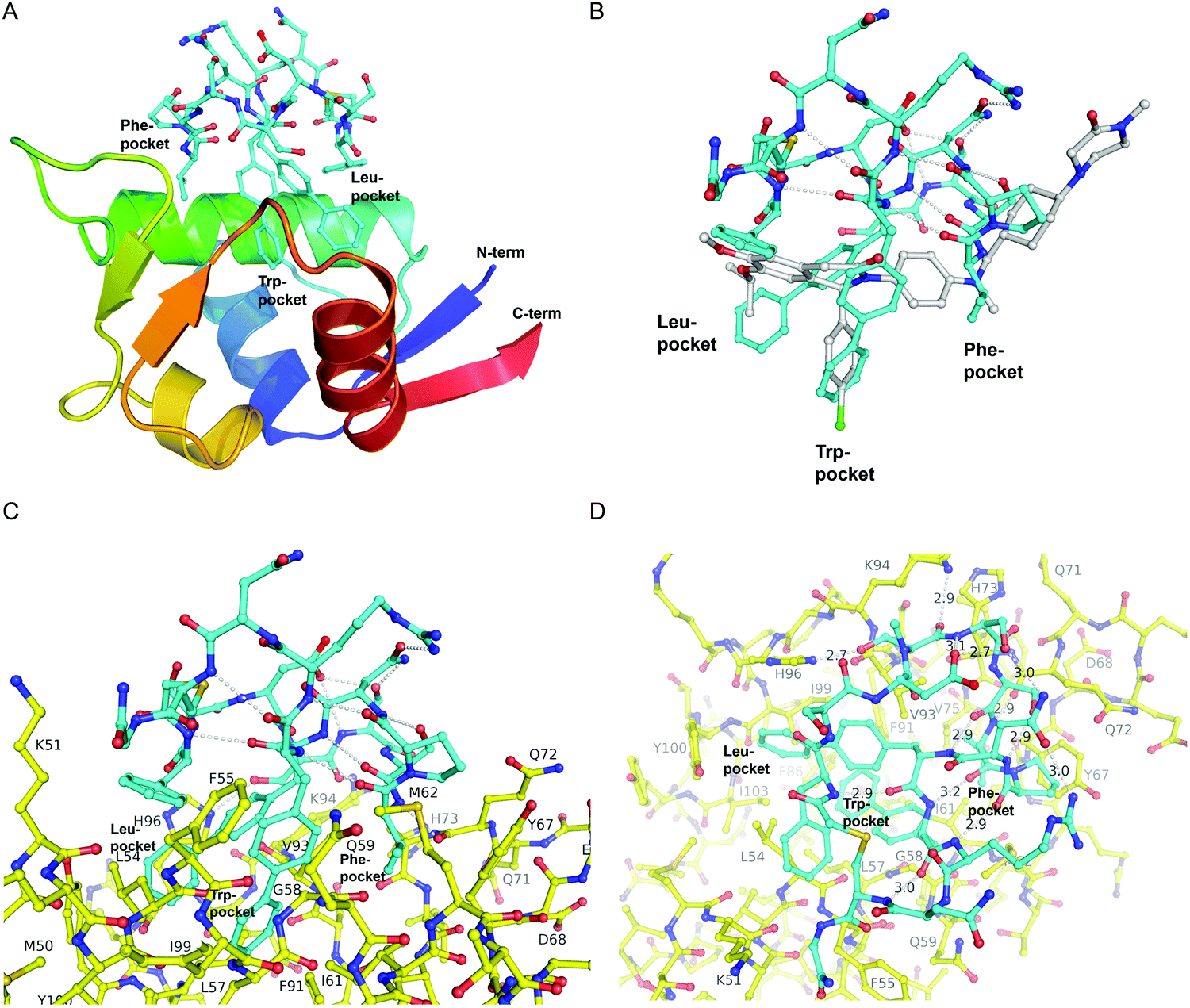 | ||
| Fig. 2 X-Ray structure of CMR19 in complex with MDM2. (A) Overview showing the N-terminal domain of MDM2 as a ribbon-model (color ramped from N-terminus in blue to C-terminus in red) and CMR19 as a stick-model (carbons in cyan, nitrogens in blue, oxygens in red, sulfurs in brown). The Leu- and Trp-pockets of MDM2 are deeply occupied by biphenyl groups, the Phe-pocket by Val-Pro. (B) Superposition of CMR19 (carbons in cyan) and the clinical trial compound CGM097 (carbons in white) bound to MDM2. The biphenyl group of CMR19 penetrates more deeply into the Leu-pocket than the corresponding group of CGM097. On the other hand, for CGM097 the Cl-phenyl makes slightly deeper contacts than the corresponding biphenyl group in the Trp-pocket. The orientation of (B) and (C) is rotated by 180 deg around a vertical axis, relative to (A). (C) CMR19 (carbons in cyan) forms an intricate intra-molecular hydrogen bond network which stabilizes the MDM2-bound conformation. The “upper half” of CMR19 does not interact with MDM2 (carbons in yellow) but stabilizes the conformation of the ligand. The non-canonical biphenyl side chains of CMR19 in positions 10 and 11 occupy the Leu- and Trp-pockets of MDM2, while the side chain of Val(7) occupies the Phe-pocket. (D) View rotated by 90 deg towards viewer around a horizontal axis, relative to (C). Selected direct intra- and inter-molecular hydrogen bond interactions are depicted in white (water molecules not shown). CMR19 makes direct inter-molecular hydrogen bonds between the backbone NH of Val(7) and the backbone CO–Gln(72) (distance 2.9 Å), between the side chain OH of MeS(4) and the side chain of His(96) (distance 2.7 Å), and the backbone CO of MeS(4) and the side chain of K(94) (distance 2.9 Å). The coordinates for MDM2/CMR19 have been deposited in the PDB databank (PDB access code = 7NUS). | ||
Interestingly, a substantial part of CMR19 is not involved in direct binding interaction with MDM2 (Fig. 2C). There is a complex intramolecular hydrogen bonding network in place that stabilizes the distinct conformation of the peptide and directs the three main pharmacophoric substituents. Solvent exposed polar amino acids Ser(2) and Asn(13) are not part of that network whereas hydrogen bonds between the polar side chains of Asp(3), Ser(5), Ser(6), Asn(9), and Arg(12) and the backbone of the macrocycle are. In addition, several intramolecular hydrogen bonds between backbone CO and backbone NH are stabilizing the conformation, e.g. NH of Arg(12) with CO of Pro(8), NH of Cys(14) with CO of B(11), and NH of Phe(1) with CO of B(10).
CMR19 makes additional critical contacts with MDM2 (Fig. 2D), including hydrogen bonds between the backbone NH of Val(7) and the backbone CO–Gln(72) (distance 2.9 Å), between the side chain OH of MeS(4) and the side chain of His(96) (distance 2.7 Å), and the backbone CO of MeS(4) and the side chain of K(94) (distance 2.9 Å). Additional figures illustrating the interactions (and electron density) are shown in the ESI† and a pymol-session file is included in the ESI.† His(96) of MDM2 is replaced in MDM4 by a proline residue, so the CMR19 side chains of MeS(4) and the bi-phenyl(10) cannot form the key interactions, which correlates with the more than thousand fold loss in affinity.
During the screening process the C-terminal Gly is the point of attachment to the oligonucleotide linking the genotype to the phenotype and any attachment to the Gly usually does not interfere with the binding to the protein. This is evident from the crystal structure showing that the C-terminal amide is pointing vertically away from the protein (left part of ligand above Phe(1) in Fig. 2C). In general, C-terminal modifications of thioether macrocyclic peptides have been described as strategies for transitioning peptides to cell permeable leads.29
Cellular uptake of CPP-conjugated peptide inhibitors
The generated MDM2-inhibitor and the negative control were functionalized at the C-terminus to confer cell-permeability. Cell-penetrating peptides (CPPs) are useful tools that have been used to transport a variety of cargoes into cells.30–32 Alternatively, reactive disulfides have also been shown to enable the cellular delivery of peptide cargoes.33 Both options were evaluated in the context of this work. For this, derivatives of the CMR19 and MMS95 peptides were synthesized either with a cysteine, as a handle for the attachment of a cell-penetrating peptide via a disulfide bond, or with asparagusic acid (AspA), a reactive disulfide.33 As a cell-penetrating peptide, a cyclic deca-arginine (cR10) was used.34,35 Additionally, to evaluate the cellular uptake of the peptides, fluorescent versions of CMR19 bearing the cysteine or AspA handles were synthesized (Fig. 3).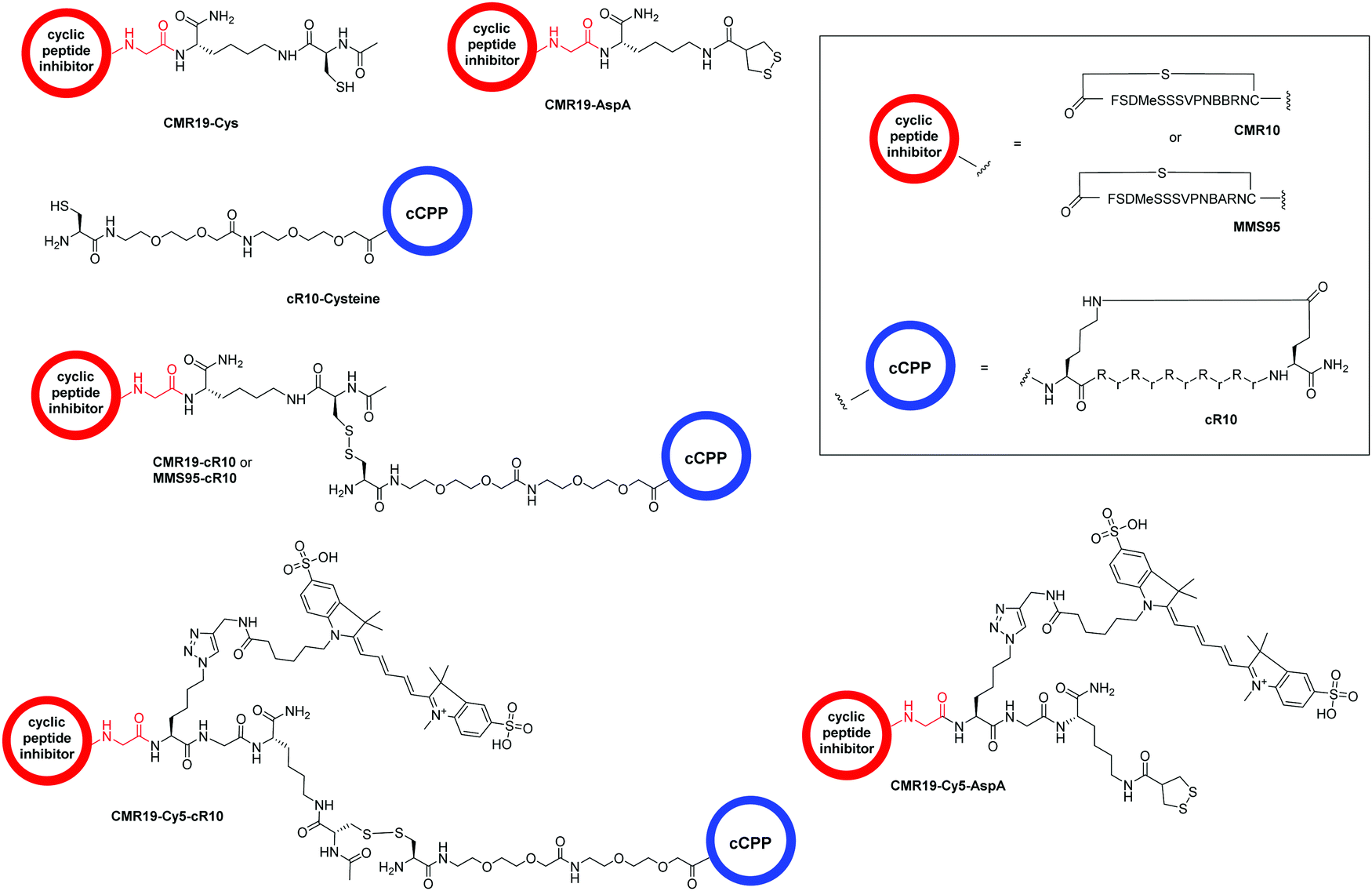 | ||
| Fig. 3 Structure of the conjugated moieties to CMR19 and MMS95. Gly shown in red indicates the C-terminal Gly spacer next to Cys(14) shown in Fig. 1. | ||
For the generation of disulfide-linked conjugates of the inhibitor peptides with the cR10-CPP, the inhibitors were first activated with Ellman's reagent (5,5′-dithiobis-(2-nitrobenzoic acid)). The activated species was purified, followed by addition of the cysteine-functionalized cR10 peptide and another purification by HPLC. Procedures, UV chromatograms and mass spectra showing the purity and identity of the peptides are provided in the ESI.†
The cellular uptake of the fluorescent peptides was evaluated by fluorescence microscopy. The peptides were incubated with SJSA-1 cells at concentrations ranging from 500 nM to 5 μM for 2 hours at 37 °C. The cells were then washed and imaged live using confocal laser scanning microscopy (full dataset in ESI†). Even at the highest concentration, the AspA and unmodified cysteine peptide show barely any intracellular fluorescence, suggesting ineffective cellular uptake (Fig. 4, upper row, and bottom right). The conjugate of the peptide with the cR10-CPP showed intracellular Cy5 fluorescence, which correlated with increasing externally applied concentration (Fig. 4, lower row). At the 500 nM and 1 μM concentrations the distribution of the fluorescence within the cell has a punctate pattern, suggesting predominantly endosomal entrapment. At the 5 μM concentration the fluorescent pattern observed indicates a broader cytosolic distribution of the peptide. This is in line with previous observations that suggest a concentration dependent mode of uptake for cell-penetrating peptides.36 Based on these results, the CPP-conjugated peptides were chosen as the most promising candidates for further studies.
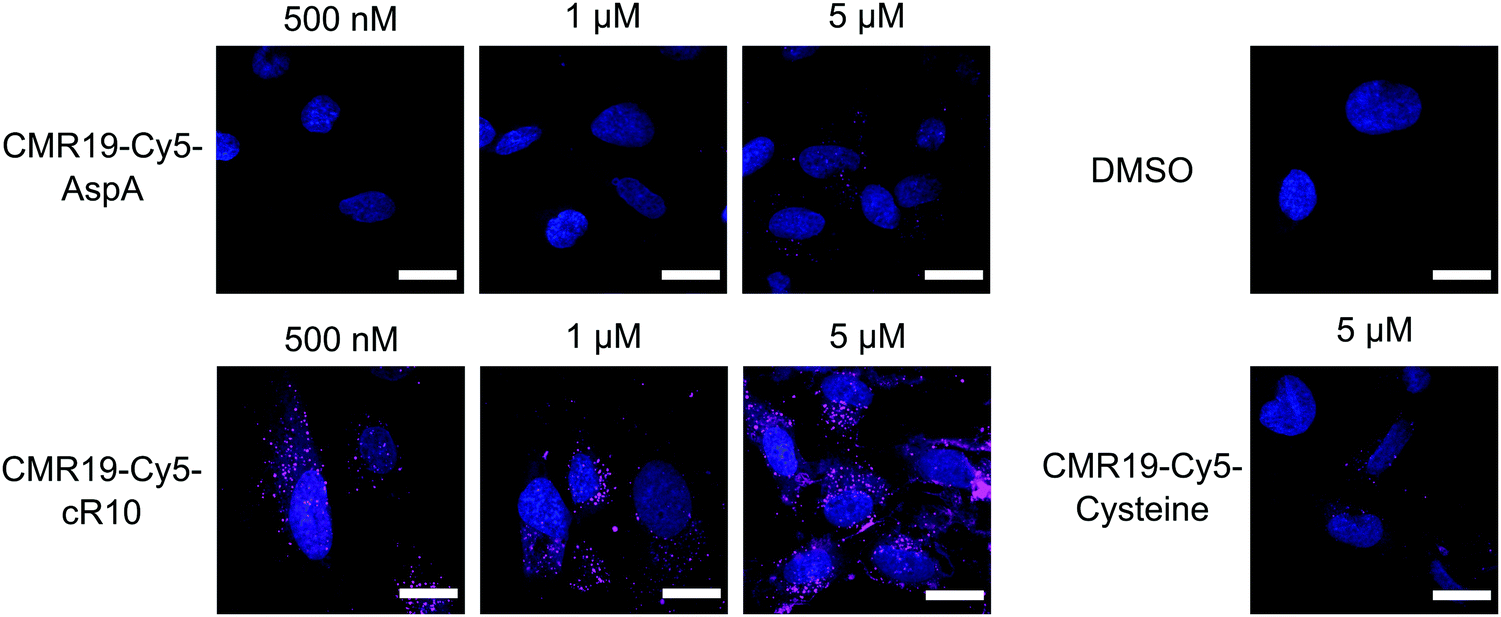 | ||
| Fig. 4 Cellular uptake of Cy5-modified CMR19 peptides. SJSA-1 cells were treated with the peptides at the indicated concentrations, then counterstained with Hoechst 33342. Shown are merge images of the Hoechst and Cy5 channels. Scale bars 20 μm. | ||
p53-Mediated cytotoxicity of cell-permeable MDM2-inhibitors
Since the CMR19-peptide is proposed to block the MDM2/p53 protein–protein interaction, it should be cytotoxic in the MDM2-dependent SJSA-1 cell line. We probed the cytotoxicity of the synthesized peptide-inhibitors in cell proliferation assay on SJSA-1 cells using the WST-1 assay (Fig. 5 and full graphs with all tested conditions in ESI,† Fig. S7). As a control, we also tested the inhibitors in the p53-mutant cell line SW-480, which should not show a cytotoxic effect of the inhibitors.37 The cells were incubated for 24 hours in presence of varying concentrations of the peptide inhibitors. Additionally, NVP-CGM097 was used as a positive control.7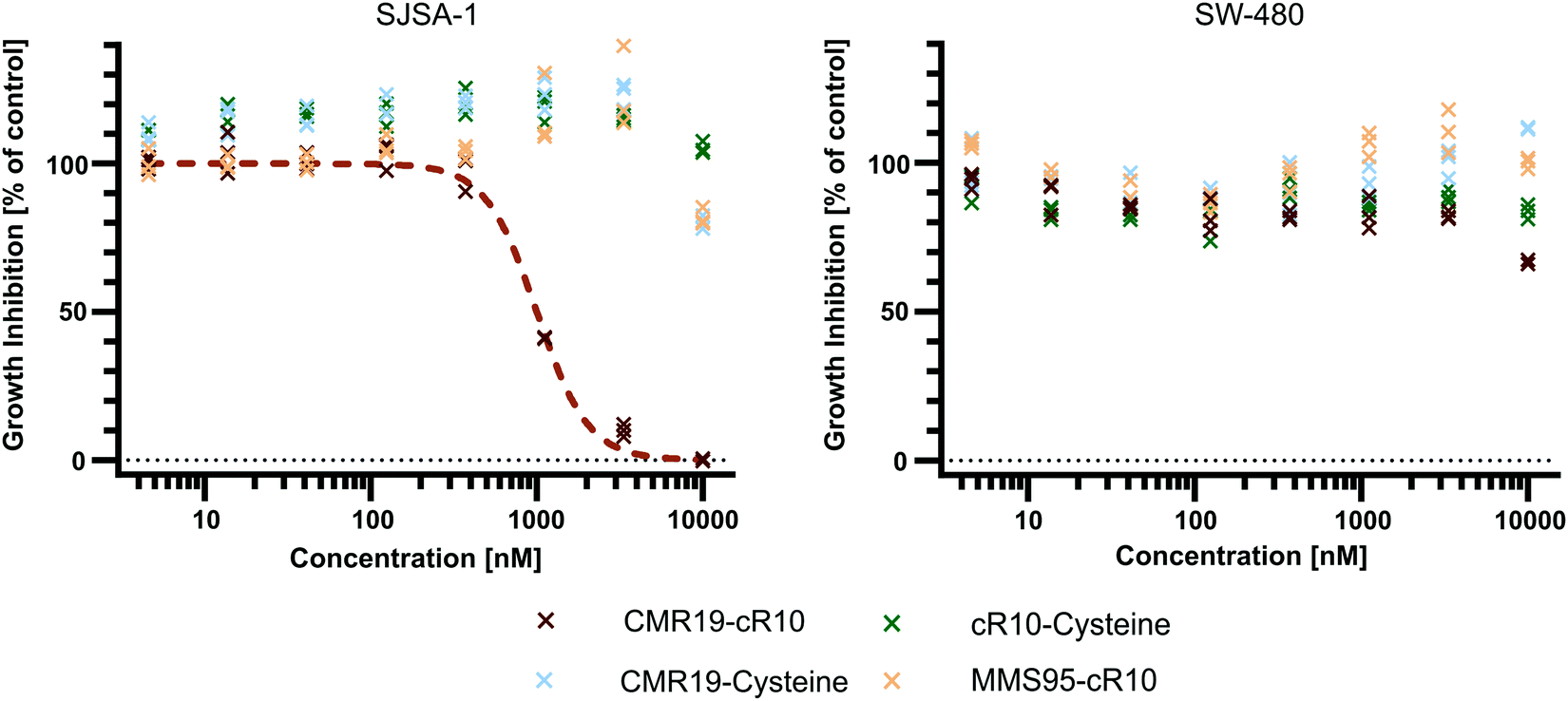 | ||
| Fig. 5 Cell proliferation assay with SJSA-1 and SW-480 cells treated with peptide inhibitors of MDM2 and the NVP-CGM097 small molecule. | ||
Only the CMR19-cR10 conjugate and the positive control showed efficient growth inhibition in SJSA-1 cells, with similar GI50 values around 1 μM. The peptide conjugate also showed no growth inhibition in the SW-480 cell line. The cR10-derivative of the inactive control MMS95, which previously showed no binding to MDM2 in the TR-FRET assay, had no growth inhibitory effect in this concentration range on either cell line, and neither did the unmodified AspA-modified inhibitor-peptides, or the cell-penetrating peptide alone (Fig. 5 and ESI,† Fig. S7).
Next, we wanted to confirm that the cytotoxicity occurs by inhibiting MDM2. When the p53/MDM2 interaction is inhibited, the MDM2 protein accumulates in the nucleus promoting p53 translation.38 We treated cells with the cR10-CPP fusions of CMR19 and MMS95, as well as with the positive control NVP-CGM097 for 4 hours at 37 °C. The cells were then fixed and nuclear MDM2 was detected using immunofluorescence. As expected based on our previous findings, the small molecule inhibitor NVP-CGM097 and the cCPP-conjugated CMR19 peptide led to an increased enrichment of MDM2 in the nuclei compared to the DMSO control (Fig. 6a, top 2 rows). In contrast, the inactive mutant MMS95 did not result in increased nuclear staining (Fig. 6a, lower 2 rows) Additionally, we did not observe unspecific membrane disruption caused by the peptides after 4 hours in SJSA-1 cells.
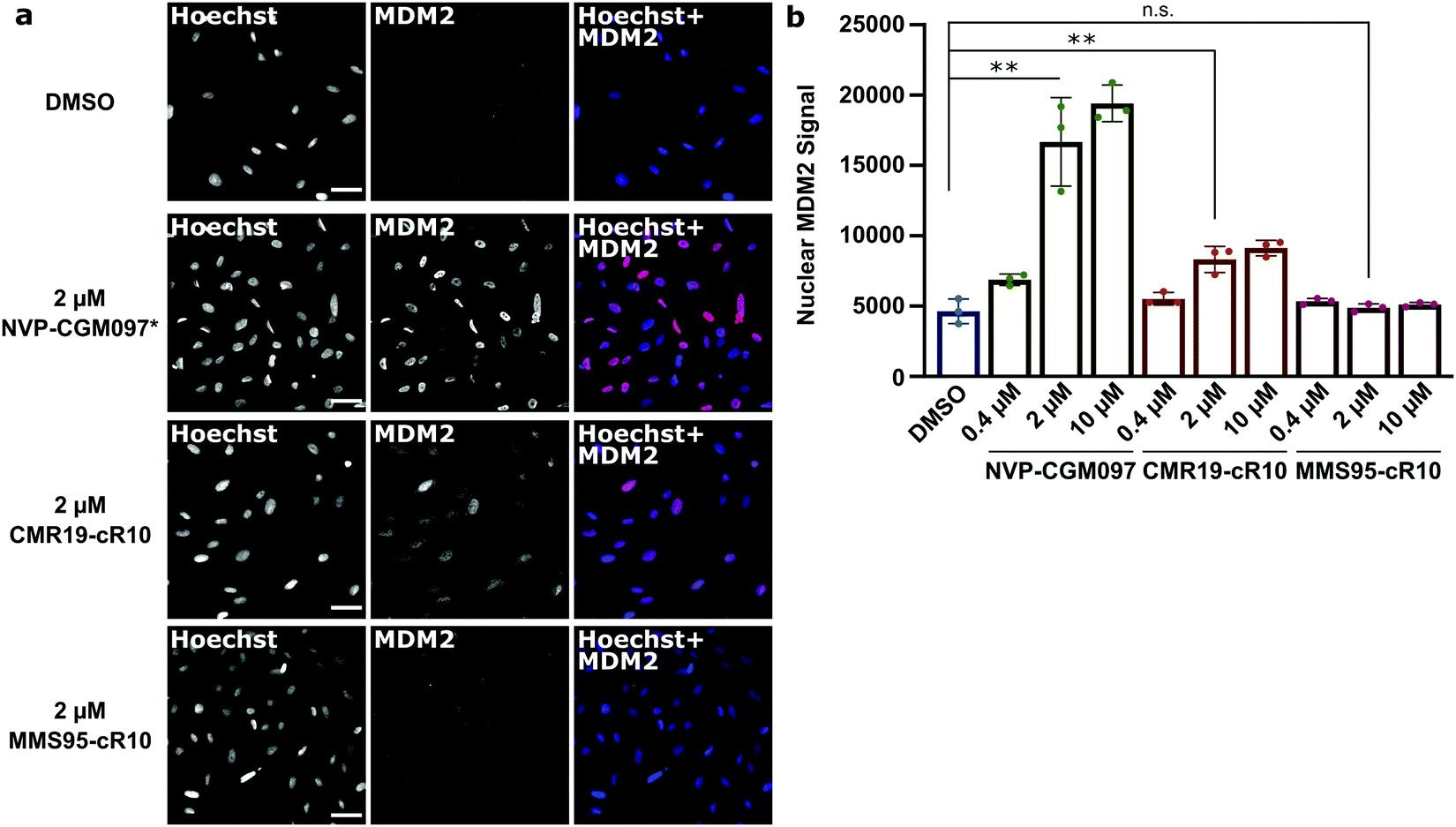 | ||
| Fig. 6 Quantification of MDM2 activation in response to inhibition of p53:MDM2 interactions by immunofluorescence. (a), Confocal microscopy images of MDM2-immunofluorescence in SJSA-1 cells treated with inhibitor-peptides or NVP-CGM097 at the indicated concentrations. * = the fluorescent signal for the NVP-CGM097 positive control was brighter than for the other inhibitors and so the contrast was set two-fold lower. Scale bars 20 μm. (b), Quantification of nuclear MDM2 in the immunofluorescence experiment at different inhibitor concentrations. Shown are single values and mean ± SD, n = 3, ** = P < 0.005 in unpaired t-test. | ||
Furthermore, by using the Hoechst stain as mask for the nuclei, we could use an automated script to quantify nuclear MDM2 fluorescence for the different inhibitors at different concentrations (Fig. 6b). We observed that both NVP-CGM097 and the CMR19 inhibitors show concentration-dependent enrichment of MDM2, while MMS95 does not.
Conclusions
In summary, we suggest that our mRNA display based method can be used to screen for highly potent and selective macrocycles against MDM2. Expressing peptides containing non-natural amino acids was key to discover a new binding site. To the best of our knowledge, we describe for the first time that two biphenyls and a valine residue mimic the helical arrangement of Phe/Trp/Leu of p53. These new pharmacophore represent novel opportunities for the design of small molecule inhibitors of the p53/MDM2 interaction. For the effective translation of a new binding site or new binding mode into drug discovery efforts, it is important to validate the mechanism of action in a cellular setting. The subsequent chemical conjugation of the cyclic peptide presenting a Cys at the C-terminus with cR10 by disulfide formation is well established and is transferable to essentially all ligands identified by mRNA display. By cellular quantification we have established a pharmacodynamic response of our new inhibitor in cells confirming the relevance of the new pharmacophore in a cellular context. Our work has been complemented by many other discoveries of bioactive macrocycles by mRNA display methods and applications of our method to other intracellular targets are under investigation.23 Our results demonstrate that the discovery process works well for MDM2 indicating that a similar approach is feasible for other intracellular PPI targets. Thus, this discovery process could significantly broaden the application of cyclic peptides in evaluating new biological targets, imaging approaches and drug discovery in general.Author contributions
A. F. L. S., C. P. R. H. and A. L. M. conceived experiments and wrote the manuscript. A. F. L. S. synthesized and characterized final disulfide peptide conjugates, performed uptake, cell viability and microscopy experiments. P. R., J. O., A. L. M. planned display experiments, analyzed NGS data, and deconvoluted hits that led to the identification of CMR19. J. K. performed the X-ray experiments, solved the structure, analysed the data, and co-wrote the manuscript. S. R. developed and performed the SJSA cellular assay. T. M. S. performed TRFRET experiments, analysed the data and commented the manuscript. S. R., S. H., A. L. M. designed peptides, planned the peptide synthesis, and summarized experimental data in the ESI.† S. R. performed the syntheses and the chemical characterization. C. E. D. contributed conceptually to the cell experiments and commented on the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We acknowledge Aude Izaac, Rene Hemmig, Matteo Fischer, and K. Kemnitz-Hassanin for their excellent technical assistance. The crystallographic experiments were performed on the X10SA beamline at the Swiss Light Source, Paul Scherrer Institute, Villigen, Switzerland. This work was supported by grants from the Deutsche Forschungsgemeinschaft (SPP 1623 and RTG 2473, Projektnummer: 392923329) to C. P. R. H. (HA 4468/9-1, 9-2), the GIF, the German–Israeli Foundation for Scientific Research and Development, and the Fonds der Chemischen Industrie (FCI) to C. P. R. H. and A. F. L. S. (Chemiefonds fellowship).References
- D. E. Scott, A. R. Bayly, C. Abell and J. Skidmore, Nat. Rev. Drug Discovery, 2016, 15, 533–550 CrossRef CAS PubMed.
- H. Lu, Q. Zhou, J. He, Z. Jiang, C. Peng, R. Tong and J. Shi, Signal Transduction Targeted Ther., 2020, 5, 213 CrossRef PubMed.
- N. Papadantonakis and H. P. Erba, Adv. Cell Gene Ther., 2020, 3 Search PubMed.
- D. Schumacher, J. Helma, A. F. L. Schneider, H. Leonhardt and C. P. R. Hackenberger, Angew. Chem., Int. Ed., 2018, 57, 2314–2333 CrossRef CAS PubMed.
- S. Du, S. S. Liew, L. Li and S. Q. Yao, J. Am. Chem. Soc., 2018, 140, 15986–15996 CrossRef CAS PubMed.
- K. R. Patel and H. D. Patel, Curr. Med. Chem., 2020, 27, 3706–3734 CrossRef CAS PubMed.
- P. Holzer, K. Masuya, P. Furet, J. Kallen, T. Valat-Stachyra, S. Ferretti, J. Berghausen, M. Bouisset-Leonard, N. Buschmann, C. Pissot-Soldermann, C. Rynn, S. Ruetz, S. Stutz, P. Chene, S. Jeay and F. Gessier, J. Med. Chem., 2015, 58, 6348–6358 CrossRef CAS PubMed.
- M. Pazgier, M. Liu, G. Zou, W. Yuan, C. Li, C. Li, J. Li, J. Monbo, D. Zella, S. G. Tarasov and W. Lu, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4665–4670 CrossRef CAS PubMed.
- Y. S. Chang, B. Graves, V. Guerlavais, C. Tovar, K. Packman, K. H. To, K. A. Olson, K. Kesavan, P. Gangurde, A. Mukherjee, T. Baker, K. Darlak, C. Elkin, Z. Filipovic, F. Z. Qureshi, H. L. Cai, P. Berry, E. Feyfant, X. G. E. Shi, J. Horstick, D. A. Annis, A. M. Manning, N. Fotouhi, H. Nash, L. T. Vassilev and T. K. Sawyer, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E3445–E3454 CrossRef CAS PubMed.
- M. N. Saleh, M. R. Patel, T. M. Bauer, S. Goel, G. S. Falchook, G. I. Shapiro, K. Y. Chung, J. R. Infante, R. M. Conry, G. Rabinowits, D. S. Hong, J. S. Wang, U. Steidl, G. Naik, V. Guerlavais, V. Vukovic, D. A. Annis, M. Aivado and F. Meric-Bernstam, Clin. Cancer Res., 2021 DOI:10.1158/1078-0432.CCR-21-0715.
- T. Okamoto, K. Zobel, A. Fedorova, C. Quan, H. Yang, W. J. Fairbrother, D. C. Huang, B. J. Smith, K. Deshayes and P. E. Czabotar, ACS Chem. Biol., 2013, 8, 297–302 CrossRef CAS PubMed.
- Q. Chu, R. E. Moellering, G. J. Hilinski, Y. W. Kim, T. N. Grossmann, J. T. H. Yeh and G. L. Verdine, MedChemComm, 2015, 6, 111–119 RSC.
- P. G. Dougherty, J. Wen, X. Pan, A. Koley, J. G. Ren, A. Sahni, R. Basu, H. Salim, G. Appiah Kubi, Z. Qian and D. Pei, J. Med. Chem., 2019, 62, 10098–10107 CrossRef CAS PubMed.
- G. Philippe, Y. H. Huang, O. Cheneval, N. Lawrence, Z. Zhang, D. P. Fairlie, D. J. Craik, A. D. de Araujo and S. T. Henriques, Biopolymers, 2016, 106, 853–863 CrossRef CAS PubMed.
- G. J. Philippe, A. Mittermeier, N. Lawrence, Y. H. Huang, N. D. Condon, A. Loewer, D. J. Craik and S. T. Henriques, ACS Chem. Biol., 2021, 16, 414–428 CrossRef CAS PubMed.
- N. Lawrence, G. J.-B. Philippe, P. J. Harvey, N. D. Condon, A. H. Benfield, O. Cheneval, D. J. Craik and S. Troeira Henriques, RSC Chem. Biol., 2020, 1, 405–420 RSC.
- L. K. Buckton, M. N. Rahimi and S. R. McAlpine, Chemistry, 2021, 27(5), 1487–1513 CrossRef CAS PubMed.
- M. R. Naylor, A. T. Bockus, M. J. Blanco and R. S. Lokey, Curr. Opin. Chem. Biol., 2017, 38, 141–147 CrossRef CAS PubMed.
- Z. Zhang, R. Gao, Q. Hu, H. Peacock, D. M. Peacock, S. Dai, K. M. Shokat and H. Suga, ACS Cent. Sci., 2020, 6, 1753–1761 CrossRef CAS PubMed.
- J. E. Townend and A. Tavassoli, ACS Chem. Biol., 2016, 11, 1624–1630 CrossRef CAS PubMed.
- S. Kalhor-Monfared, M. R. Jafari, J. T. Patterson, P. I. Kitov, J. J. Dwyer, J. M. Nuss and R. Derda, Chem. Sci., 2016, 7, 3785–3790 RSC.
- K. Maola, J. Wilbs, J. Touati, M. Sabisz, X. D. Kong, A. Baumann, K. Deyle and C. Heinis, Angew. Chem., Int. Ed., 2019, 58, 11801–11805 CrossRef CAS PubMed.
- Y. Huang, M. M. Wiedmann and H. Suga, Chem. Rev., 2019, 119, 10360–10391 CrossRef CAS PubMed.
- K. Kashiwagi and P. C. Reid, US Pat., US2012/208720, 2012 Search PubMed.
- Y. Goto, T. Katoh and H. Suga, Nat. Protoc., 2011, 6, 779–790 CrossRef CAS PubMed.
- T. Ishizawa, T. Kawakami, P. C. Reid and H. Murakami, J. Am. Chem. Soc., 2013, 135, 5433–5440 CrossRef CAS PubMed.
- P. H. Kussie, S. Gorina, V. Marechal, B. Elenbaas, J. Moreau, A. J. Levine and N. P. Pavletich, Science, 1996, 274, 948–953 CrossRef CAS PubMed.
- J. Kallen, A. Izaac, S. Chau, E. Wirth, J. Schoepfer, R. Mah, A. Schlapbach, S. Stutz, A. Vaupel, V. Guagnano, K. Masuya, T. M. Stachyra, B. Salem, P. Chene, F. Gessier, P. Holzer and P. Furet, ChemMedChem, 2019, 14, 1305–1314 CrossRef CAS PubMed.
- L. J. Walport, R. Obexer and H. Suga, Curr. Opin. Biotechnol, 2017, 48, 242–250 CrossRef CAS PubMed.
- H. Derakhshankhah and S. Jafari, Biomed. Pharmacother., 2018, 108, 1090–1096 CrossRef CAS PubMed.
- A. Falanga, L. Lombardi, E. Galdiero, V. D. Genio and S. Galdiero, Future Med. Chem., 2020, 12, 1431–1446 CrossRef CAS PubMed.
- N. Nischan, H. D. Herce, F. Natale, N. Bohlke, N. Budisa, M. C. Cardoso and C. P. Hackenberger, Angew. Chem., Int. Ed., 2015, 54, 1950–1953 CrossRef CAS PubMed.
- G. Gasparini, G. Sargsyan, E. K. Bang, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2015, 54, 7328–7331 CrossRef CAS PubMed.
- H. D. Herce, D. Schumacher, A. F. L. Schneider, A. K. Ludwig, F. A. Mann, M. Fillies, M. A. Kasper, S. Reinke, E. Krause, H. Leonhardt, M. C. Cardoso and C. P. R. Hackenberger, Nat. Chem., 2017, 9, 762–771 CrossRef CAS PubMed.
- A. F. L. Schneider, A. L. D. Wallabregue, L. Franz and C. P. R. Hackenberger, Bioconjugate Chem., 2019, 30, 400–404 CrossRef CAS PubMed.
- F. Duchardt, M. Fotin-Mleczek, H. Schwarz, R. Fischer and R. Brock, Traffic, 2007, 8, 848–866 CrossRef CAS PubMed.
- L. T. Vassilev, B. T. Vu, B. Graves, D. Carvajal, F. Podlaski, Z. Filipovic, N. Kong, U. Kammlott, C. Lukacs, C. Klein, N. Fotouhi and E. A. Liu, Science, 2004, 303, 844–848 CrossRef CAS PubMed.
- M. Gajjar, M. M. Candeias, L. Malbert-Colas, A. Mazars, J. Fujita, V. Olivares-Illana and R. Fahraeus, Cancer Cell, 2012, 21, 25–35 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cb00056j |
| This journal is © The Royal Society of Chemistry 2021 |