
Spatial organization of biochemical cues in 3D-printed scaffolds to guide osteochondral tissue engineering†
Paula
Camacho
a,
Anne
Behre
a,
Matthew
Fainor
b,
Kelly B.
Seims
c and
Lesley W.
Chow
 *ac
*ac
aDepartment of Bioengineering, Lehigh University, Bethlehem, PA, USA
bIntegrated Degree in Engineering, Arts, and Sciences Program, Lehigh University, Bethlehem, PA, USA
cDepartment of Materials Science and Engineering, Lehigh University, Bethlehem, PA, USA. E-mail: lesley.chow@lehigh.edu
First published on 26th August 2021
Abstract
Functional repair of osteochondral (OC) tissue remains challenging because the transition from bone to cartilage presents gradients in biochemical and physical properties necessary for joint function. Osteochondral regeneration requires strategies that restore the spatial composition and organization found in the native tissue. Several biomaterial approaches have been developed to guide chondrogenic and osteogenic differentiation of human mesenchymal stem cells (hMSCs). These strategies can be combined with 3D printing, which has emerged as a useful tool to produce tunable, continuous scaffolds functionalized with bioactive cues. However, functionalization often includes one or more post-fabrication processing steps, which can lead to unwanted side effects and often produce biomaterials with homogeneously distributed chemistries. To address these challenges, surface functionalization can be achieved in a single step by solvent-cast 3D printing peptide-functionalized polymers. Peptide-poly(caprolactone) (PCL) conjugates were synthesized bearing hyaluronic acid (HA)-binding (HAbind–PCL) or mineralizing (E3–PCL) peptides, which have been shown to promote hMSC chondrogenesis or osteogenesis, respectively. This 3D printing strategy enables unprecedented control of surface peptide presentation and spatial organization within a continuous construct. Scaffolds presenting both cartilage-promoting and bone-promoting peptides had a synergistic effect that enhanced hMSC chondrogenic and osteogenic differentiation in the absence of differentiation factors compared to scaffolds without peptides or only one peptide. Furthermore, multi-peptide organization significantly influenced hMSC response. Scaffolds presenting HAbind and E3 peptides in discrete opposing zones promoted hMSC osteogenic behavior. In contrast, presenting both peptides homogeneously throughout the scaffolds drove hMSC differentiation towards a mixed population of articular and hypertrophic chondrocytes. These significant results indicated that hMSC behavior was driven by dual-peptide presentation and organization. The downstream potential of this platform is the ability to fabricate biomaterials with spatially controlled biochemical cues to guide functional tissue regeneration without the need for differentiation factors.
1. Introduction
Osteochondral (OC) tissue has been extensively studied in orthopedics due to the rising demand for improved therapies to treat diseases like osteoarthritis (OA).1,2 OA is the most common joint disease worldwide,3,4 experienced by 70% of adults over age 65.5 After damage due to injury or trauma, articular cartilage breakdown eventually results in a lesion or defect that exposes the underlying bone and predisposes a patient to develop OA.1,4,6 Unfortunately, articular cartilage has limited ability for spontaneous healing, mostly due to its avascular nature and disrupted communication with the subchondral bone following cartilage injury.7 There is no effective therapy currently available for treating cartilage degeneration. Patients ultimately require total joint replacement to treat end-stage OA.4,8 However, this approach fails to address early pathology that leads to cartilage degeneration and has significant disadvantages, especially for younger patients, due to the limited lifespan of the prosthesis.4,9To offset the need for total joint replacement, a number of surgical interventions, such as microfracture, autologous chondrocyte implantation (ACI), and autologous OC transplantation (mosaicplasty), have been developed to repair cartilage at earlier stages.8,10–12 These treatment options provide some improvement in clinical outcomes,13 but long-term data has shown significant failure rates.8,10,11,14 This is largely due to the newly formed tissue being mechanically inferior to native cartilage.4,5,8,12,15,16 Although OA was once viewed as a disease of purely mechanical cartilage degradation, it is now known to be a complex condition affecting the whole joint.4,17 Promising strategies for cartilage repair must therefore address regeneration and integration of the entire OC tissue.2,10,14,16 Functional repair of the OC unit is difficult because the transition from bone to cartilage contains biochemical, structural, and mechanical gradients necessary for normal joint function.10,14,18,19 Reproducing the unique composition and organization of this interfacial tissue has remained a persistent challenge.17,20
OC tissue consists of hyaline cartilage, a calcified cartilage layer, and subchondral bone. Each region has distinct properties that function synergistically to create a unit for load transfer. Hyaline cartilage is a dense, avascular, connective tissue that provides low-friction, load-supporting, and shock-absorbing properties to articulating joints.21,22 It is a water-rich tissue, mainly composed of chondrocytes and specific extracellular matrix (ECM) components, such as collagen type II and glycosaminoglycans (GAG).19,23 This tissue is further divided into three zones – superficial, middle, and deep. Each zone exhibits unique properties due to a specific arrangement of chondrocytes and the distinct composition, organization, and orientation of its ECM components.1,10,22–24 The calcified cartilage layer cements the non-calcified hyaline cartilage to bone22,25 and is characterized by collagen X-producing hypertrophic chondrocytes embedded in mineralized ECM.10,23 Finally, the subchondral bone is a bony lamella responsible for mechanical stability and separating cartilage from the marrow cavity.26 Development of the OC tissue gradient is guided by organization of biochemical cues that spatially regulate cell proliferation, migration, and differentiation.10,27 During OC formation, mesenchymal stem cells (MSCs) spatially differentiate into active chondrocytes, hypertrophic chondrocytes, and osteoblasts, producing an organized zoned structure with biochemical and mechanical gradients.10,28,29 Regeneration of this complex tissue requires engineering organized biomaterials that direct cells to restore native-like tissue composition and organization.20,30–35
Several biomaterial approaches, such as using tissue-derived ECM,30 incorporating mineralization,31,36,37 creating physical architecture gradients,38–40 or delivering growth factors,41,42 have been developed to guide MSC chondrogenesis and osteogenesis. Current state-of-the-art strategies for scaffold-guided OC tissue repair, including those in clinical use, involve layered scaffolds with merged components that target cartilage or bone regeneration separately.10,43 These approaches are promising but have notable disadvantages, including limited spatial control of biochemical presentation19 and risk of mechanical failure at the interface between separate layers.44 Three-dimensional (3D) printing has emerged as a useful tool to produce tunable, continuous scaffolds with spatially complex biochemical features.45,46 Scaffold materials can be functionalized with various chemical groups or bioactive cues to guide cell differentiation and encourage the secretion of tissue-specific ECM.47–50 However, surface functionalization often includes one or more post-fabrication processing steps, which can lead to unwanted side effects or undesirable changes to scaffold topography and morphology.51,52 In addition, post-fabrication functionalization often produces biomaterials with homogeneously distributed chemistries, which fail to mimic the biochemical organization found in native tissues. Achieving greater spatial biochemical control in scaffolds is of great interest33,35 to direct organized tissue formation for functional regeneration.
We developed a strategy to 3D print peptide–polymer conjugates to create scaffolds spatially functionalized with bioactive cues.53,54 Previously, this platform was used to guide local cell behavior within a single scaffold environment by spatially controlling the presentation of a well-known adhesion peptide RGDS and its negative control RGES.54 Presenting both peptides on different fibers in the same construct showed enhanced cell attachment and spreading on RGDS-functionalized fibers compared to ones presenting RGES.54 These results demonstrated that different biochemical cues can be spatially organized to guide local cell behavior within a continuous scaffold.
Leveraging our 3D printing platform, we fabricated scaffolds spatially functionalized with cartilage-promoting and bone-promoting peptides to guide organized OC tissue formation. We synthesized peptide–poly(caprolactone) (PCL) conjugates53,55 bearing hyaluronic acid (HA)-binding (HAbind–PCL) or mineralizing peptides (E3–PCL) designed to promote human MSC (hMSC) chondrogenesis56 or osteogenesis,57 respectively. HAbind–PCL includes the amino acid sequence RYPISRPRKR derived from the HA-binding region of the link protein in the aggrecan complex,58 a major component of articular cartilage, and other native HA-binding proteins.59 E3–PCL includes three glutamic acids (EEE), which have been shown to promote mineralization and induce hMSC osteogenic differentiation for bone regeneration.57 These peptide sequences were designed to bind endogenous biomolecules and promote differentiation without the addition of exogenous GAGs or differentiation factors during culture. We fabricated scaffolds with both peptides either spatially or homogeneously presented on the same scaffold (Fig. 1). Multiple printer heads were used to sequentially deposit the different peptide–polymer conjugates in discrete zones to create the dual spatial scaffolds. In contrast, we combined both peptide–polymer conjugates in a single ink to create dual mixed scaffolds. We used these scaffolds to investigate how the location of the cartilage-promoting and bone-promoting peptides within the constructs influenced hMSC differentiation and matrix formation.
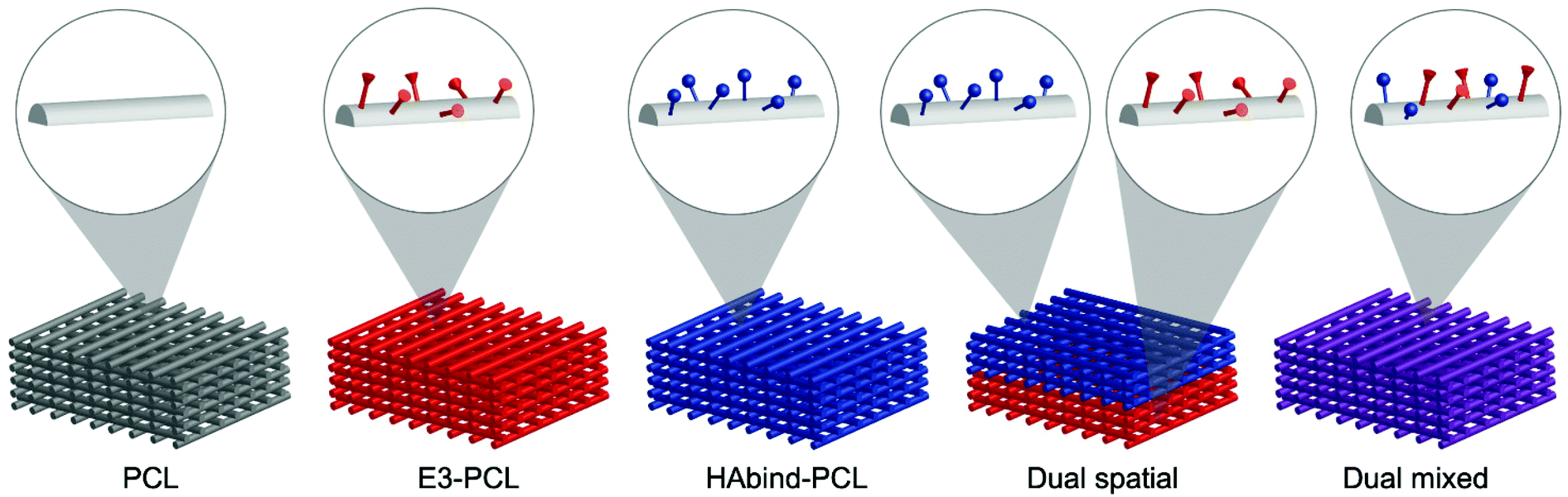 | ||
| Fig. 1 Schematic representations illustrating scaffold groups used to investigate influence of peptide presentation and location on osteochondral tissue formation. Functionalized scaffolds present cartilage-promoting (blue) or bone-promoting (red) peptides on the surface of the fibers, shown in the insets. On multi-peptide scaffolds, the peptides are presented in discrete regions (dual spatial) or homogeneously throughout the construct (dual mixed). | ||
2. Materials and methods
2.1. Materials
All details regarding materials used in this study are provided in the ESI.2.2. Synthesis of peptide–PCL conjugates
Each peptide sequence included a terminal cysteine (C) used to covalently attach the peptide to a maleimide-modified poly(caprolactone) (PCL) via Michael addition.53–55,60 The peptides were also designed with glycine (G) spacers between the C and sequence of interest to enhance presentation on the 3D-printed fiber surface. All peptides (CGGGRYPISRPRKR (HAbind), CGGGAAAEEE (E3), CGGGRYPISRPRKR(biotin) (HAbind(biotin)), and CGGGAAAEEE(azide) (E3(azide))) were synthesized, cleaved, and purified using previously described methods.54,61 Briefly, peptides were synthesized on a fluorenylmethyloxycarbonyl chloride (Fmoc)–rink–amide 4-methylbenzhydryalmine (MBHA) resin (100–200 mesh, 0.67 mmol g−1 functionalization) using Fmoc-protected amino acids. For HAbind(biotin), Fmoc–Lys(Mtt)–OH was manually coupled to the resin, and the Mtt protecting group was removed to attach biotin to the ε-amine on the lysine side chain (more details found in the ESI†). The E3(azide) peptide was synthesized with commercially available Fmoc–Lys(N3)–OH.Peptides were cleaved from the resin in 95% trifluoroacetic acid (TFA), 2.5% ultrapure water, 2.5% triisopropylsilane (TIS), and 2.5% (w/v) dithiothreitol (DTT). TFA was removed using rotary evaporation, and the peptide product was precipitated in cold diethyl ether (DEE). The precipitate was collected using centrifugation and dried overnight under vacuum. Peptides were dissolved in 5–10% acetonitrile (ACN) in ultrapure water with 0.1% TFA and purified using reversed-phase preparative high performance liquid chromatography (HPLC; Agilent 218 Prep HPLC, Agilent Technologies, Santa Clara, CA, USA) on an Agilent 5 Prep–C18 column (150 mm × 21.2 mm, 5 μm pore size, 100 Å particle size). Peptide mass and purity were verified using mass spectrometry and analytical HPLC (Fig. S1–S4†). Additional details and supporting data are provided in the ESI.†
Peptide–PCL conjugates were synthesized using methods previously described.54,61 Briefly, PCL diol (Mw 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da) was dissolved in anhydrous N-methyl pyrrolidone (NMP) under nitrogen. p-Maleimidophenyl isocyanate (PMPI) was dissolved in anhydrous NMP at 15–20 molar equivalents to PCL and added dropwise to the PCL solution while stirring under nitrogen. The reaction was continued overnight, and the resulting PCL–maleimide (PCL–mal) was precipitated in DEE. Precipitation was repeated as needed to remove excess PMPI. To conjugate the peptide, PCL–mal was reconstituted in anhydrous NMP under nitrogen. The peptides were separately dissolved in either dimethyl sulfoxide (DMSO) (for E3 and E3(azide)) or anhydrous NMP (for HAbind and HAbind(biotin)) at 8 molar equivalents and added dropwise to the PCL–mal solution while stirring under nitrogen and reacted overnight. The resulting peptide–PCL conjugates were precipitated in cold DEE, washed with ultrapure water to remove excess peptide, and dried under vacuum prior to analysis. Each synthesis step was confirmed by 1H nuclear magnetic resonance (1H NMR) (Fig. S5–S9†). Additional details and supporting data are provided in the ESI.†
000 Da) was dissolved in anhydrous N-methyl pyrrolidone (NMP) under nitrogen. p-Maleimidophenyl isocyanate (PMPI) was dissolved in anhydrous NMP at 15–20 molar equivalents to PCL and added dropwise to the PCL solution while stirring under nitrogen. The reaction was continued overnight, and the resulting PCL–maleimide (PCL–mal) was precipitated in DEE. Precipitation was repeated as needed to remove excess PMPI. To conjugate the peptide, PCL–mal was reconstituted in anhydrous NMP under nitrogen. The peptides were separately dissolved in either dimethyl sulfoxide (DMSO) (for E3 and E3(azide)) or anhydrous NMP (for HAbind and HAbind(biotin)) at 8 molar equivalents and added dropwise to the PCL–mal solution while stirring under nitrogen and reacted overnight. The resulting peptide–PCL conjugates were precipitated in cold DEE, washed with ultrapure water to remove excess peptide, and dried under vacuum prior to analysis. Each synthesis step was confirmed by 1H nuclear magnetic resonance (1H NMR) (Fig. S5–S9†). Additional details and supporting data are provided in the ESI.†
2.3. Solvent-cast 3D printing with peptide–PCL conjugates
PCL-based inks were prepared and printed using methods previously described.53,54 Peptide–PCL conjugates at desired concentrations were dissolved in 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) before adding unmodified PCL (Mn 80000 Da) at a concentration of 370 mg mL−1. All inks were 3D printed using a 3-axis EV Series automated dispensing system (Nordson EFD, Westlake, OH, USA) using a 32-gauge blunt-tip needle (100 μm inner diameter) to create 15 mm × 18 mm × ∼0.5 mm (width × length × height) cross-hatched orthogonal scaffold prints with 260 μm programmed fiber spacings (mid-point to mid-point). All scaffolds were printed with an applied pressure of 70 psi. The first layer was printed at a z-spacing of 100 μm above the printing substrate and speed of 0.4 mm s−1 while all remaining layers were printed with a print speed of 0.2 mm s−1 using 45 μm z-spacing between each subsequent layer.
Four peptide-functionalized scaffold groups were created: E3–PCL; HAbind–PCL; HAbind/E3–PCL with peptides presented in opposing regions (dual spatial); HAbind–PCL and E3–PCL presented homogeneously throughout the scaffolds (dual mixed). Scaffolds for cell studies were printed with inks containing HAbind–PCL at concentrations of 1, 3, or 6 mg mL−1; E3–PCL at 6, 12, or 18 mg mL−1; or a homogeneous mixture of HAbind–PCL (3 mg mL−1) and E3–PCL (18 mg mL−1) (dual mixed). For dual spatial scaffolds, two printer heads were used to sequentially print HAbind–PCL (3 mg mL−1) and E3–PCL (18 mg mL−1) inks in discrete regions.53 Inks containing PCL only without added peptide–PCL conjugates were used to fabricate non-functionalized scaffold controls (PCL). Scaffolds from each group were imaged using scanning electron microscopy (SEM) (LEO 1550 SEM; Zeiss, Peabody, MA, USA) to confirm consistent fiber morphology and dimensions (Fig. S10†). Additional details and supporting data are provided in the ESI.†
2.4. Peptide presentation and organization in 3D-printed scaffolds
Peptides were modified with bioorthogonal groups biotin or azide to enable direct and specific labeling of peptides in the 3D-printed scaffolds.53,54 Each group (PCL, HAbind(biotin), E3(azide), dual spatial, dual mixed) was printed using sets of two inks to ensure repeatability between ink batches. Printed constructs were frozen at −20 °C for 24 hours and cut into 5 mm × 5 mm (width × length) samples using a Teflon-coated razor blade. All scaffolds were sequentially labeled with streptavidin–fluorescein isothiocyanate (streptavidin–FITC) followed by dibenzocyclooctyne–cyanine 3 (DBCO-Cy3), using methods previously described.53,54 To label biotin with streptavidin–FITC, scaffolds were blocked with 0.5% w/v bovine serum albumin (BSA) and 0.05% v/v TWEEN 20 in phosphate buffered saline (PBS) (blocking solution A) at room temperature for 20 minutes. After blocking, scaffolds were washed with PBS, incubated in 0.3 μM streptavidin–FITC in blocking solution A for 60 minutes, and washed thoroughly with PBS twice and ultrapure water for 30 minutes each. To label azide with DBCO–Cy3, scaffolds were blocked in a solution of 0.2% v/v TWEEN 20 and 0.2% v/v Triton-X in PBS (blocking solution B) for 60 minutes. After blocking, samples were washed with ultrapure water, incubated in 50 mM DBCO–Cy3 in PBS with 0.5% w/v BSA for 30 minutes, and washed for 10 minutes each in the following solutions in sequence: PBS, ultrapure water, blocking solution B, ultrapure water, 50% v/v isopropyl alcohol (IPA), 100% v/v IPA, 50% v/v IPA, and ultrapure water.Labeled scaffolds were sectioned to visualize peptide presentation throughout the constructs. Scaffolds were embedded in Optimum Cutting Temperature compound (OCT) vertically and frozen at −80 °C for 5 minutes. An OTF5000 cryostat (Bright; UK) was used to cut 30 μm cross-sections onto glass slides. Scaffold cross-sections were imaged with a Nikon C2+ laser scanning confocal fluorescence microscope (Nikon; Melville, New York, USA) using the relevant excitation wavelengths and filters for FITC and Cy3 and maintaining the same imaging settings for all samples. Labeled PCL scaffolds were used as controls. Additional details and supporting data are provided in the ESI.†
2.5. Cell seeding and culture
To prevent cell adhesion to cell culture plates, 24-well plates were coated with a two-component silicone elastomer (Sylgard 184) according to the manufacturer's instructions. Silicone-coated wells were sterilized with 70% (v/v) ethanol, rinsed with sterile water, and placed under UV light for 2 hours. Scaffolds were cut from larger prints using 3 mm diameter biopsy punches, sterilized in 70% v/v ethanol for 30 minutes, and washed with sterile water. Scaffolds were incubated in sterile 0.1% w/v BSA in water overnight, rinsed thoroughly with sterile water and left to dry overnight. Before seeding, each scaffold was immobilized in the silicon-coated plates using 0.1 mm dissection pins.Bone marrow-derived human mesenchymal stem cells (hMSC) from four donors (20–26 years old, two males (A, B) and two females (C, D); Rooster Bio) were expanded to passage 3 in cell culture flasks at a starting density of 3.3 × 103 cells per cm2. Cells were harvested at 90% confluency by treating with 0.25% trypsin–ethylenediaminetetraacetic acid (EDTA). RoosterNourish™-MSC media (Rooster Bio; USA) was used for the first passage. Growth media (Dulbecco's Modified Eagle's Medium (DMEM)-GlutaMAX™ supplemented with 10% v/v fetal bovine serum (FBS), 1% v/v antibiotic/antimycotic (anti/anti: containing amphotericin B, streptomycin, and penicillin), and 50 μg mL−1 ascorbic acid was used for the second passage and all experiments. Cells at passage 3 were harvested and prepared in a single cell suspension with a concentration of 10 × 106 cells per mL, from which 20 μL (2 × 104 cells) were placed on top of each scaffold. Plates were placed in the incubator at 37 °C and 5% CO2 for 60 minutes to allow for cell attachment. An additional 1 mL of growth media was gently added to each well before storing the seeded scaffolds back in the incubator. Samples were cultured for 7, 14, 28, or 42 days with media replaced every 2–3 days. At each timepoint for each scaffold group: five samples were placed in RNA lysis buffer for gene expression analysis; three samples were weighed and prepared for glycosaminoglycan (GAG) quantification; and three samples were transferred to 0.2% v/v Triton X-100 in water to assay alkaline phosphatase (ALP) activity. All samples were frozen and stored at −80 °C until analysis. At 28 or 42-day timepoints, three scaffolds from each group were harvested for histology and IHC. They were fixed in 4% w/v paraformaldehyde (PFA) for 12–16 hours, washed with PBS twice, and stored in fresh PBS at 4 °C.
Donor A was used to evaluate hMSC response to single-peptide scaffolds displaying different concentrations of HAbind–PCL or E3–PCL. For all other experiments, 3 mg mL−1 HAbind–PCL and/or 18 mg mL−1 E3–PCL concentrations were used. Donors A, B, and C were used to evaluate hMSC response to presenting one or both peptides on the same scaffold. Donors B, C, and D were used to evaluate hMSC response to peptide organization in dual scaffolds. Each independent experiment was performed with a single donor, so that each study measured the response across three different donors. Donor A was used in two independent experiments to ensure repeatability. Experiments using donors B and C included all five scaffold groups.
2.6. Gene expression analysis
Samples in RNA lysis buffer were thawed, vortexed, and freeze–thawed twice to improve RNA yield. RNA was extracted using Quick-RNA™ MiniPrep Kit (Zymo Research, Irvine, CA) according to the manufacturer's protocol. Concentration and purity of the extracted RNA were measured using a NanoDrop™ 2000 (Thermo Scientific; USA). Reverse transcription was performed on the RNA to synthesize cDNA using the ImProm-II™ Reverse Transcription System (Promega Corporation, Madison, WI) with oligoDT as the primer. Polymerase chain reaction (PCR) was then performed using Applied Biosystems 7300 for Real-Time PCR with a 20 μL reaction volume using SYBR© Green JumpStart™ Taq ReadyMix™ for High Throughput Quantitative PCR (Sigma-Aldrich, St Louis, MO). Primers used were obtained from previous studies and synthesized by Invitrogen (Carlsbad, CA). Expression levels of the following markers were quantified: SRY Box 9 (SOX9), runt-related transcription factor 2 (RUNX2), aggrecan (ACAN), cartilage oligomeric matrix protein (COMP), lubricin (PRG4), alkaline phosphatase (ALPL), collagen type X alpha 1 (COLXA1), osteocalcin (OC), osterix (SP7), and ribosomal 18S (housekeeping gene) (Table 1). Annealing temperatures were primer specific. Melt curves were performed after 40 cycles of PCR to ensure primer specificity. Relative gene expression was assessed using the ΔΔCT method. Results were shown as fold change normalized to the expression level of hMSCs seeded on PCL scaffolds at corresponding timepoints.| Gene | Primer | Sequence (5′ to 3′) | Annealing temperature (°C) | Ref. |
|---|---|---|---|---|
| 18S | Fwd | GTAACCCGTTGAACCCCATTC | 60 | — |
| Rev | CCATCCAATCGGTAGTAGCG | |||
| SOX9 | Fwd | AACGCCGAGCTCAGCAAG | 62 | 56 |
| Rev | ACGAACGGCCGCTTCTC | |||
| ACAN | Fwd | CACTGTTACCGCCACTTCCC | 62 | 56 |
| Rev | GACATCGTTCCACTCGCCCT | |||
| COMP | Fwd | GCAACACGGACGAGGACAAG | 64 | 62 |
| Rev | CGCCATCACTGTCCTTCTGG | |||
| PRG4 | Fwd | CTGGCCTGAATCTGTGTATTTTT | 60 | 63 |
| Rev | GTGTCGTTTCTCCATACACTGG | |||
| RUNX2 | Fwd | CCGCCTCAGTGATTTAGGGC | 61 | 64 |
| Rev | GGGTCTGTAATCTGACTCTGTCC | |||
| ALPL | Fwd | AGCACTCCCACTTCATCTGGAA | 60 | 62 |
| Rev | GAGACCCAATAGGTAGTCCACATTG | |||
| COLXA1 | Fwd | AATGCCTGTGTCTGCTTTTAC | 60 | 64 |
| Rev | ACAAGTAAAGATTCCAGTCCT | |||
| OC | Fwd | CCCAGGCGCTACCTGTATCAA | 64 | 65 |
| Rev | CTGGAGAGGAGCAGAACTGG | |||
| SP7 | Fwd | TGCTTGAGGAGGAAGTTCAC | 60 | 66 |
| Rev | AGGTCACTGCCCACAGAGTA |
2.7. Biochemical analysis
Assays to quantify of glycosaminoglycans (GAG) and DNA content and alkaline phosphatase (ALP) activity were read on a Tecan Infinite M200 Pro plate reader (Männedorf, Switzerland).To quantify GAG deposition, frozen scaffolds were lyophilized and digested using papain (0.01 M N-acetyl cysteine and 0.125 mg mL−1 papain in a phosphate buffer with 0.1 M sodium phosphate dibasic and 0.01 M ethylenediaminetetraacetic acid disodium salt dihydrate, pH 6.5) at 40 °C for 24 hours followed by 60 °C for 12 hours. Samples were analyzed for sulfated GAGs (using dimethylmethylene blue dye-binding assay, pH 3) and DNA (using Quant-iT™ PicoGreen® dsDNA Reagent). Results were represented as GAG per DNA content.
ALP activity was quantified in samples stored in 0.2% v/v Triton X-100, using an Alkaline Phosphatase Diethanolamine Activity Kit (Sigma-Aldrich, St Louis, MO). Samples were incubated at 37 °C until there was a visible color change (∼60 minutes). Results were shown as ALP activity per minutes of incubation per DNA content (determined using Quant-iT™ PicoGreen ® dsDNA Reagent).
2.8. Tissue sectioning and staining
Fixed samples stored in PBS were used for histology and immunohistochemistry (IHC). Orientation of dual spatial scaffolds was tracked through culture and harvesting to evaluate spatial tissue formation.Scaffolds for Alcian blue, Alizarin Red, and collagen staining were cut in half and infiltrated with sucrose the day prior to sectioning. Samples were incubated in a series of solutions for 60 minutes each: 10% w/v sucrose in PBS, 30% w/v sucrose in PBS, 50/50 mixture of 30% w/v sucrose in PBS, and OCT and then stored in fresh OCT overnight at 4 °C. Scaffolds were embedded in OCT with the cut side facing up and frozen at −80 °C for 5 minutes. An OTF5000 cryostat (Bright; UK) was used to cut 20 μm cross-sections onto glass slides. To visualize sulfated GAG deposition, samples were stained with 1% w/v Alcian blue dye in 3% acetic acid (pH 2.5) and counterstained with 0.1% w/v Nuclear Fast Red solution. To label calcium deposition, samples were stained with 2% w/v Alizarin Red solution (pH 4.1–4.3). Histological samples were imaged on a Nikon Eclipse Ts2R microscope (Nikon; Melville, New York, USA).
Immunohistochemistry with antibodies for collagen type I (Abcam, ab34710), type II (Abcam, ab34712), and type X (GeneTex, GTX37732) was used to quantify collagen deposition. Sections were permeabilized with Proteinase K for 4 minutes, washed with PBS, and blocked in 5% v/v goat serum in PBS for 90 minutes. Samples were incubated with primary antibodies (1:150 dilution in 5% v/v goat serum) overnight at 4 °C, washed with 0.1% v/v TWEEN 20 in PBS (three times for 5 minutes per wash), and incubated in Alexa Fluor 488 secondary antibody (Abcam, ab150077, 1:200 dilution in 5% v/v goat serum) for 2 hours at room temperature. Samples were washed with 0.1% v/v TWEEN 20 in PBS three times for 5 minutes per wash and stained with Hoechst solution (1:1000 dilution in 5% v/v goat serum) for 10 minutes. Finally, sections were washed with 0.1% v/v TWEEN 20 in PBS three times for 5 minutes per wash and running water for 2 minutes. All samples were imaged with a Nikon C2+ laser scanning confocal fluorescence microscope (Nikon; Melville, New York, USA) using the relevant excitation wavelengths and filters for FITC and DAPI and maintaining the same imaging settings for all samples. Sections incubated with 5% v/v goat serum instead of the primary antibody were used as negative controls.
2.9. Statistical analysis
Results were obtained from independent experiments using a single donor, with minimum three scaffolds per donor of each type for every timepoint. Welch ANOVA with Dunnett T3 post-hoc was performed for experiments with only one donor. Mixed ANOVA with Sidak post-hoc was performed for pooled experiments with three donors, considering scaffold type as the within-group variable and donor as the between-group variable. This technique was used to determine if any differences seen resulted from an interaction between scaffold group and donor or only one factor. This ensured all donors contributed to statistical significance and any effects observed were not driven by a single donor. Differences between groups were considered significant for p-values <0.1. Data were represented as mean ± standard deviation. Statistical analyses were performed using SPSS (version 25, IBM software, USA).3. Results and discussion
3.1. Conjugate synthesis and 3D printing of peptide-functionalized scaffolds
Synthesis and purification steps for all peptides and peptide–PCL conjugates were confirmed using mass spectrometry, analytical HPLC, and 1H NMR (see ESI†). Inks containing PCL with peptide–PCL conjugates were successfully solvent-cast 3D-printed into peptide-functionalized scaffolds. Scanning electron microscopy (SEM) of all scaffold groups showed that the overall fiber morphology and surface topography were unaffected by the addition of the conjugates (Fig. S10†).Scaffolds were labeled with streptavidin–FITC and DBCO–Cy3 to confirm peptide presentation and localization. Fluorescent microscopy images of scaffold cross-sections confirmed HAbind(biotin) and E3(azide) peptides were present on the surface of peptide-functionalized scaffolds. The biotin and azide functional groups on the peptides selectively react with their complementary pair, streptavidin67 and DBCO,68 respectively. Fluorophores conjugated to these bioorthogonal groups therefore allowed for highly specific labeling of each peptide on the scaffold surface. As expected, the non-functionalized PCL scaffolds showed no fluorescence (Fig. 2A). On single-peptide scaffolds, Cy3 fluorescence was only present on scaffolds printed with E3(azide)–PCL (Fig. 2B) while FITC fluorescence was only seen on HAbind(biotin)–PCL scaffolds (Fig. 2C). In dual spatial scaffolds, labeling confirmed zonal organization of peptides with FITC-tagged HAbind(biotin) peptides in the top region and Cy3-tagged E3(azide) peptides in the bottom region (Fig. 2D). Comparing fluorescence to corresponding bright-field images demonstrated peptides were present throughout the entire construct.
 | ||
| Fig. 2 Fluorescent (left) and bright-field (right) images of 3D-printed scaffold cross-sections to visualize peptide presentation and organization. Cross-sections of scaffolds 3D printed with (A) PCL, (B) E3(azide)–PCL, (C) HAbind(biotin)–PCL, and (D) E3(azide)–PCL and HAbind(biotin)–PCL in opposing zones (dual spatial) labeled with both streptavidin–FITC and DBCO–Cy3 showed HAbind(biotin) (green) and E3(azide) (red) localization on peptide-functionalized scaffolds. Bright field images showed peptides were presented throughout the entire construct. | ||
Scaffolds were cultured in growth media for up to 60 days and labeled with fluorophores to visualize peptide presentation (details in ESI†). Fluorescence quantification demonstrated that the peptides were still available on the surface of peptide-functionalized scaffolds compared to PCL controls (Fig. S11†).
3.2. Cell response to 3D-printed scaffolds with single-peptide or dual-peptide presentation
We evaluated hMSC response to single-peptide and spatially organized dual-peptide scaffolds cultured under growth conditions. Interestingly, we discovered that having both peptides present in the same scaffold led to a synergistic enhancement of both chondrogenic and osteogenic responses compared to single-peptide scaffolds (Fig. 3). Chondrogenesis is driven by expression of the SOX9 transcription factor, which promotes increased glycosaminoglycan (GAG) deposition in the matrix.29,69,70 Although we did not see a significant upregulation of SOX9 at the timepoints investigated, dual spatial scaffolds developed significantly higher GAG accumulation at day 28 compared to HAbind–PCL and PCL scaffolds (p = 0.002 and p = 0.015, respectively) (Fig. 3A). These data suggested that dual spatial scaffolds promoted hMSC chondrogenesis with cells producing a GAG-rich extracellular matrix (ECM).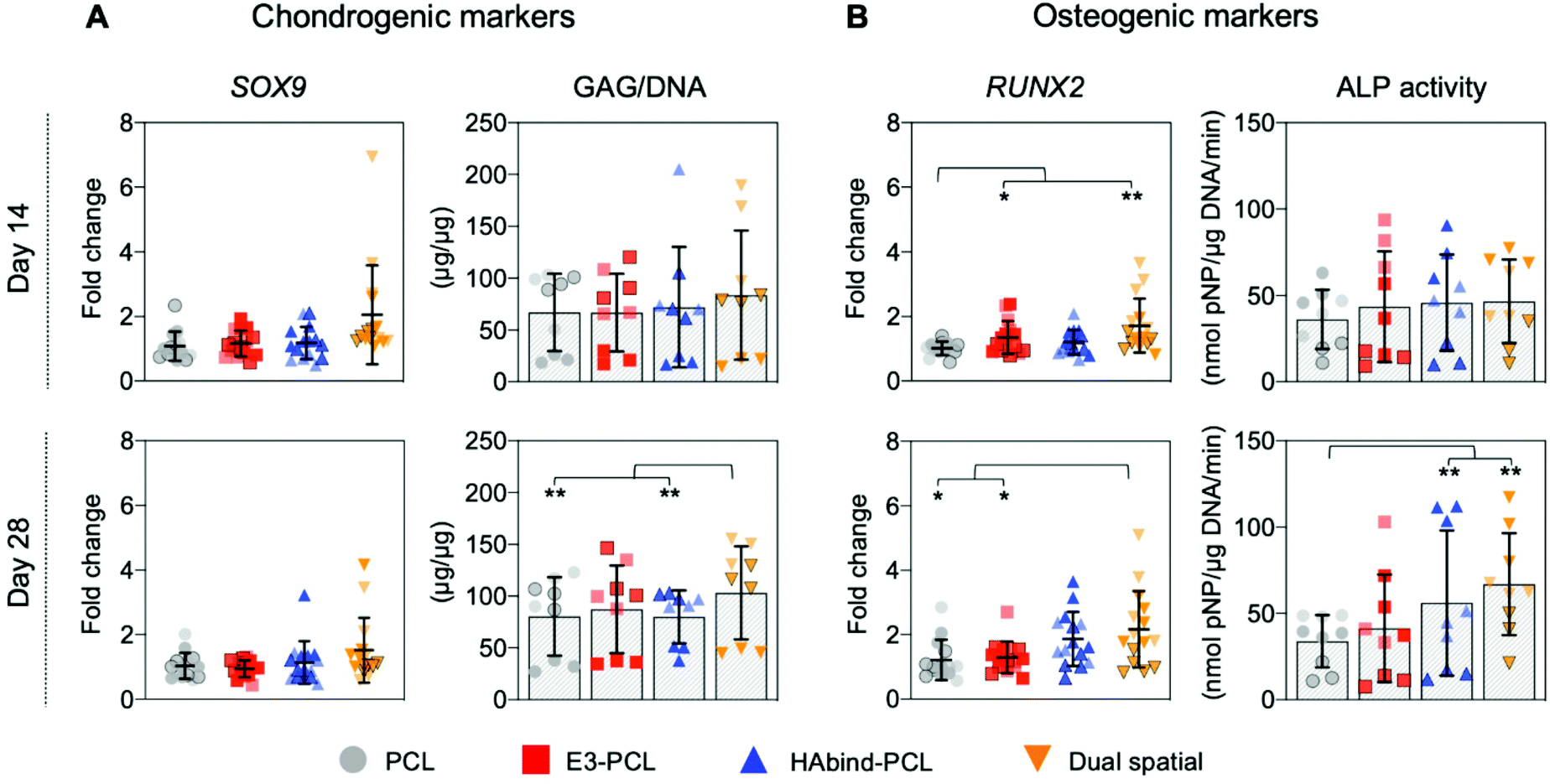 | ||
| Fig. 3 Evaluation of hMSC gene expression and biochemical analysis in response to single-peptide and dual-peptide 3D-printed scaffolds. (A) Chondrogenic and (B) osteogenic markers measured in constructs cultured for up to 28 days without added differentiation factors. Data from three independent experiments with different donors represented in different shades (light = donor A; medium with black border = donor B; dark = donor C). SOX9 and RUNX2 gene expression was measured by RT-qPCR, normalized to 18S, and reported as fold-difference relative to PCL at each time point (4–5 scaffolds per donor in each scaffold group; n = 14–15 scaffolds total per group). Biochemical analysis of GAG content was normalized to DNA, and ALP activity was normalized to DNA and time of reaction (3 scaffolds per donor in each scaffold group; n = 9 scaffold total per group). Data is presented as mean ± SD (significance *p < 0.1, **p < 0.05). | ||
Dual spatial scaffolds showed a more significant osteogenic response, with upregulation of RUNX2 at day 14 compared to PCL (p = 0.021) and at day 28 compared to both PCL and E3–PCL (p = 0.054 and p = 0.076, respectively) (Fig. 3B). Increased expression of the RUNX2 transcription factor characterizes hMSC terminal differentiation to hypertrophic chondrocytes and posterior osteogenesis, which leads to higher alkaline phosphatase (ALP) activity.70–73 Gene expression results correlated with higher ALP activity at day 28 in dual spatial scaffolds compared to PCL (p = 0.002) (Fig. 3B). No significant differences in DNA content were observed between scaffold groups at each timepoint investigated (Fig. S13†). Together, these data indicated that dual spatial scaffolds stimulated hMSC differentiation towards hypertrophy or osteogenesis. The observed synergistic effect suggests that each peptide plays a role in both chondrogenesis and osteogenesis, and their combination amplifies hMSC differentiation.
The single-peptide scaffolds were designed with peptides that stimulated hMSC chondrogenesis or osteogenesis in prior literature.56,57,74,75 Hyaluronic acid-binding peptides have been shown to promote MSC chondrogenesis in hydrogel systems,56,74 while peptide sequences with negatively charged glutamic acid residues were found to promote MSC osteogenesis by inducing nucleation and growth of apatite-like calcium phosphate (CaP) crystals on polymeric nanofibers.57,75 However, these studies used chondrogenic56,74 or osteogenic57,75 differentiation media, which may confound peptide activity. Here, single-peptide scaffolds had the opposite effect to what was expected. After 28 days in culture, HAbind–PCL scaffolds showed lower GAG deposition compared to dual spatial (p = 0.002) and higher ALP activity compared to PCL (p < 0.001) (Fig. 3). This data suggested HAbind–PCL scaffolds promoted terminal differentiation and osteogenesis. This conflicting result suggested that peptide charge may be playing a significant role in our system. HAbind is a net positively charged peptide, and positively charged surfaces have been shown to induce osteogenic differentiation of MSCs.76 In contrast, E3–PCL scaffolds demonstrated higher expression of RUNX2 at day 14 compared to PCL scaffolds (p = 0.087) (Fig. 3B), which indicated potential for the E3 peptide to stimulate hypertrophic differentiation of hMSCs. However, this upregulation was not seen at day 28 and did not lead to higher ALP activity, implying that E3–PCL scaffolds do not drive osteogenic behavior. We therefore investigated if peptide concentration influenced hMSC behavior by comparing scaffolds printed with different concentrations of HAbind–PCL (1, 3, 6 mg mL−1) or E3–PCL (6, 12, 18 mg mL−1) to PCL only controls (Fig. S12†). No overall trend was found with any specific concentration, suggesting that peptide surface concentration does not significantly influence differentiation behavior.
These results further confirmed that having both peptides in the scaffold is necessary to stimulate osteogenic and chondrogenic responses in cultured hMSCs in the absence of differentiation factors. Previous studies evaluated these peptides independently and under differentiation media conditions,56,57,74,75 making it difficult to compare directly to our studies. Chondrogenic and osteogenic factors added to differentiation media, such as dexamethasone77 or bone morphogenetic protein (BMP-2),78 also play a role in both differentiation pathways. In the absence of these cues, the observed effect of our multi-peptide scaffolds suggests that each peptide plays a role in both chondrogenesis and osteogenesis. Therefore, the combination of peptides was necessary to stimulate hMSC differentiation.
Matrix formation was evaluated after 28 days in culture by histological staining with Alcian blue for GAG accumulation and Alizarin Red for calcium deposition and immunohistochemistry (IHC) against collagen type I, II, and X. Histology showed complete cellular infiltration and matrix deposition throughout all constructs (Fig. S14†). Alcian blue staining showed higher GAG accumulation in dual spatial scaffolds compared to PCL and HAbind–PCL scaffolds (Fig. S14†). This correlated with the GAG content quantified using biochemical assays (Fig. 3A). All scaffolds presented similar Alizarin Red staining, suggesting no differences in calcium deposition, or mineralization, between groups. Mineralization is considered a functional in vitro endpoint reflecting advanced cell differentiation in osteogenic cultures.79 Increasing mineral deposition has been correlated with an increase followed by a decrease in ALP activity.79,80 After 28 days of culture, dual spatial scaffolds showed higher ALP activity than PCL (Fig. 3B), suggesting that longer culture times may be necessary to observe measurable differences in calcium deposition. However, scaffolds in this study were not cultured in osteogenic differentiation media, which limits propensity for mineralization.80
Immunohistochemical staining showed homogeneous deposition of collagen I in all constructs, indicating immature matrix formation15,28 (Fig. 4). Monolayer expansion of hMSCs typically leads to expression of collagen I, which can be maintained even after differentiation is initiated.15 Even though collagen I expression is characteristic of fibrocartilage,15,81 the deposition seen in the constructs may be an artefact due to the hMSC expansion in tissue culture plastic previous to seeding.15 As chondrogenesis progresses, production of collagen type II continuously increases.15,28 Positive staining for collagen II was observed in all scaffold groups, with the highest intensity found in dual spatial samples. Collagen II constitutes 90–95% of native cartilage ECM.19,23 The IHC data indicated the dual spatial scaffolds enhanced cartilage-specific collagen formation (Fig. 4). Collagen X deposition was only observed in dual spatial scaffolds. Collagen X is a ubiquitous product of hypertrophic chondrocytes found in the calcified cartilage layer.19 These IHC results strongly suggested that presenting both peptides provided sufficient signals to direct hMSC behavior in the absence of added differentiation factors.
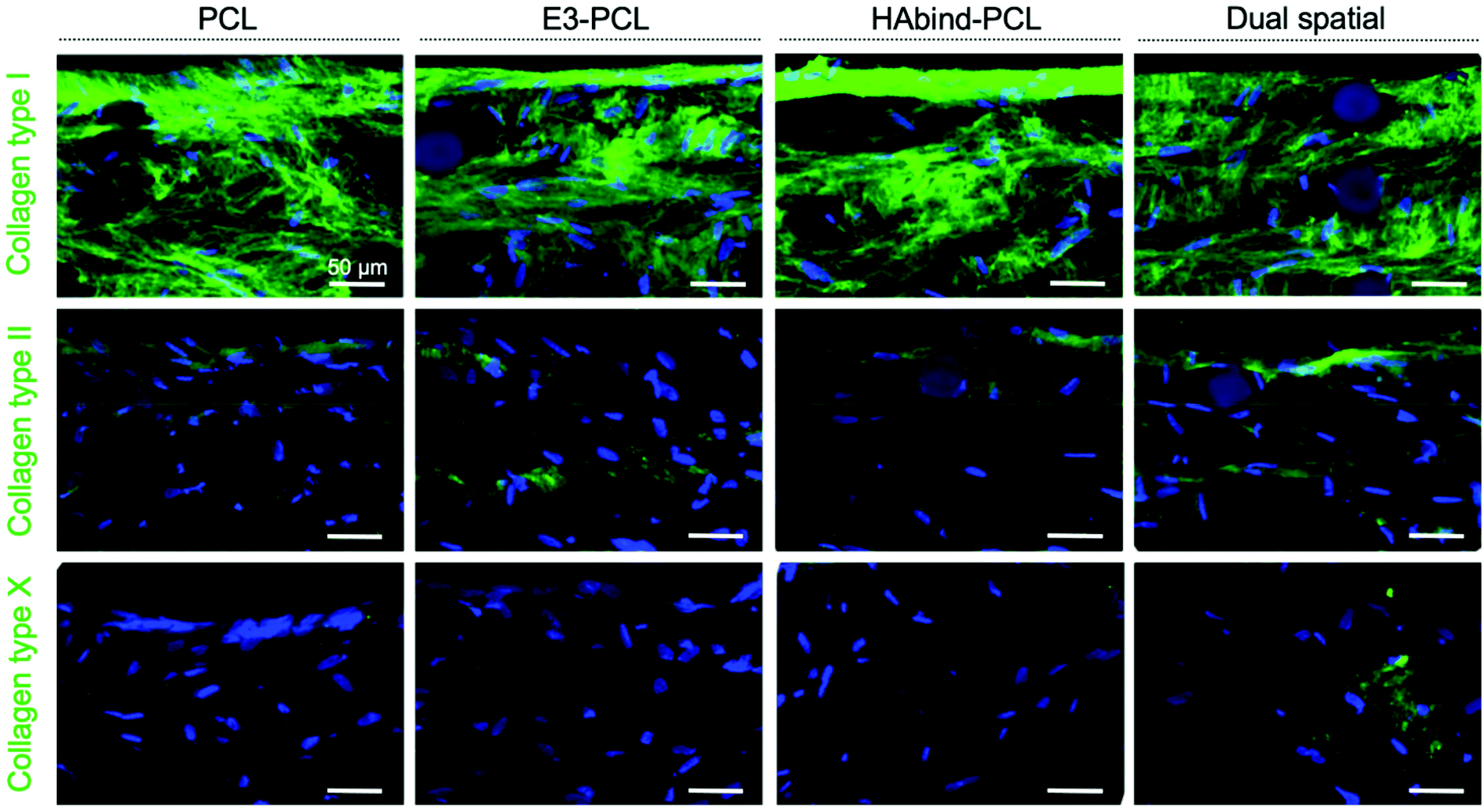 | ||
| Fig. 4 Collagen (type I, II, X) deposition in single-peptide and dual-peptide 3D-printed scaffolds after 28 days of culture. Representative confocal microscopy images of histological cross-sections showing immunohistochemical staining for collagen type I, II and X (top to bottom; Alexa Fluor 488, green) and cell nuclei (Hoescht Dye, blue) in PCL, E3–PCL, HAbind–PCL, and dual spatial scaffolds (L to R). All scaffold groups presented immature tissue formation, seen by positive staining for collagen I. Collagen II deposition was also found in all groups. Dual spatial scaffolds presented more intense collagen II staining and early production of collagen X in some areas of the construct. (Scale bar = 50 μm.) | ||
Dual-peptide presentation significantly enhanced hMSC differentiation across the three different donors used. However, some differences between donors were observed. A mixed model ANOVA was utilized to analyze overall scaffold effects, while accounting for possible differences in donor response. For example, donor C showed lower overall GAG deposition at both timepoints compared to donors A and B (Fig. 3A), while donor B presented overall lower ALP activity compared to donors A and C (Fig. 3B). These variations can be explained by differences in chondrogenic or osteogenic potential between donors82,83 and the inherent heterogeneity of bone marrow MSC populations.70,84
3.3. Cell response to multi-peptide spatial organization in 3D-printed scaffolds
The ability to spatially localize bioactive cues is critical to direct local cellular activities and promote organized OC tissue formation.10,27 Our 3D printing approach introduced the ability to control local cell behavior by tailoring the spatial presentation of multiple peptides within a single construct.54 The synergistic effect of presenting both cartilage-promoting and bone-promoting peptides in one scaffold motivated us to investigate how dual-peptide organization influenced hMSC differentiation and matrix deposition. We compared three scaffold groups: PCL, dual spatial, and dual mixed. Dual spatial scaffolds presented HAbind–PCL and E3–PCL in opposing zones of the same construct (Fig. 2D). Dual mixed scaffolds were 3D printed using one ink containing both HAbind–PCL and E3–PCL. Fluorescence imaging of dual mixed scaffolds labeled with streptavidin–FITC and DBCO–Cy3 confirmed presentation of both peptides throughout the entire printed construct (Fig. 5). | ||
| Fig. 5 Fluorescent and corresponding bright-field images of the cross-section of a dual mixed 3D-printed scaffold. The scaffold labeled with both streptavidin–FITC (green) and DBCO–Cy3 (red) was sectioned to show the homogeneous distribution and colocalization (yellow) of HAbind(biotin) and E3(azide) peptides throughout the construct. Bright field images showed both peptides were homogeneously presented throughout the entire construct. | ||
The synergistic effect seen in dual-peptide scaffolds in Fig. 3 was supported in a separate experiment with a different group of donors. No significant differences in DNA content were observed between scaffold groups at each timepoint investigated (Fig. S16†). At day 14, dual spatial and dual mixed scaffolds upregulated SOX9 expression compared to unmodified PCL scaffolds (p = 0.006 and p = 0.024, respectively) (Fig. 6A and Fig. S15†). Dual spatial and dual mixed scaffolds also showed increased expression of RUNX2 compared to PCL scaffolds after 14 days in culture (p = 0.004 and p = 0.015, respectively) (Fig. 6A and Fig. S15†). SOX9 specifies commitment and differentiation of MSCs toward the chondrogenic lineage29,85,86 while RUNX2 is expressed through chondrocyte hypertrophy and osteogenic differentiation.73,87 These results indicated that dual-peptide scaffolds promoted differentiation of a mixed chondrogenic and osteogenic cell population.
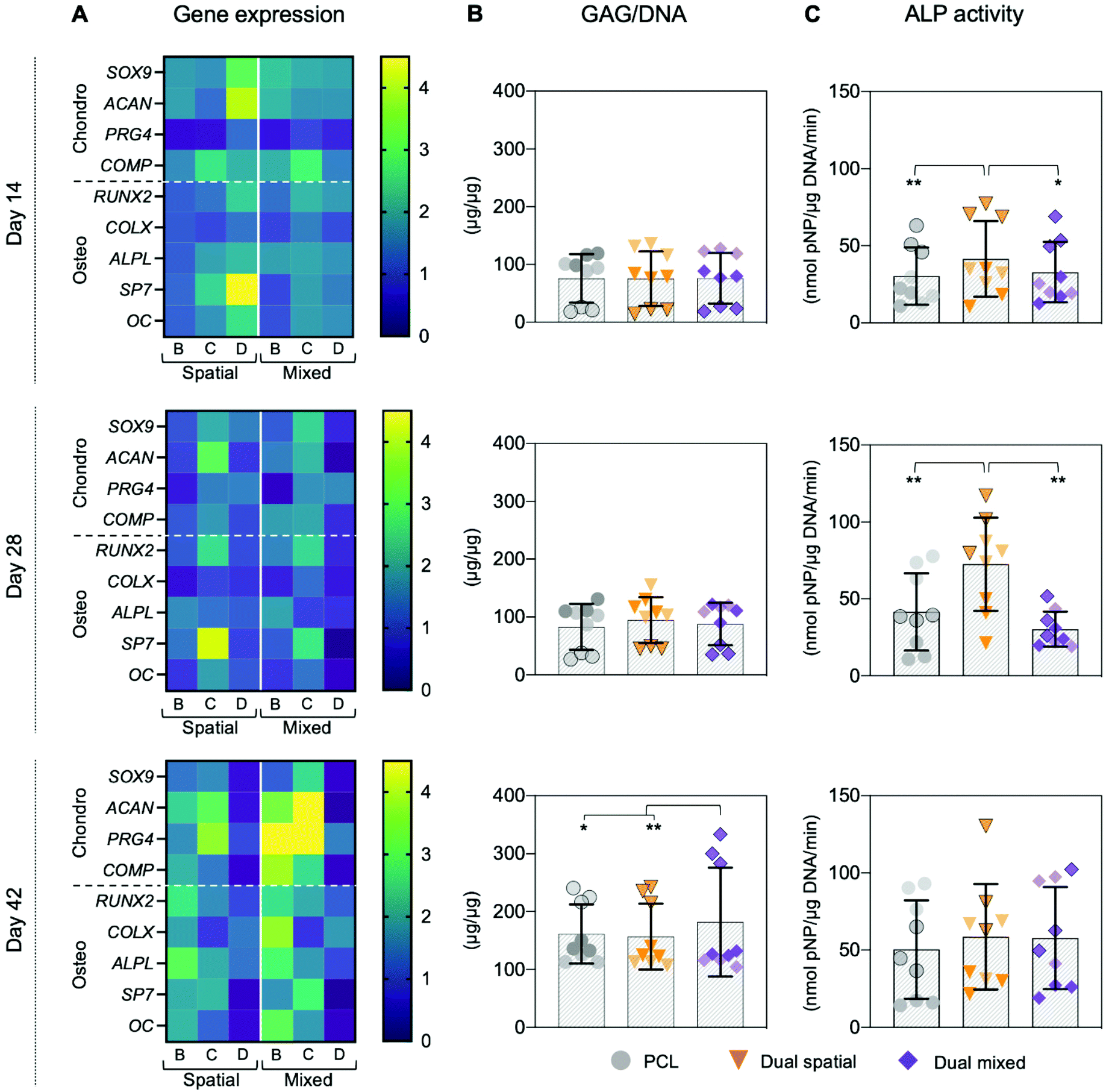 | ||
| Fig. 6 Evaluation of hMSC gene expression and biochemical analysis in response to dual-peptide 3D-printed scaffolds. Constructs were cultured for 14, 28, and 42 days without added differentiation factors in three independent experiments. (A) Gene expression was measured by RT-qPCR, normalized to 18S, and reported as fold-difference relative to PCL at each time point (4–5 scaffolds per donor in each scaffold group; n = 14–15 scaffolds total per group). Data from three different donors, each represented in different columns of the heatmap. Biochemical analysis of (B) GAG content was normalized to DNA, while (C) ALP activity was normalized to DNA and time of reaction (3 scaffolds per donor in each scaffold group; n = 9 scaffolds total per group). Data from three different donors represented as different shades (light = donor B; medium with black border = donor C; dark = donor D). Data is presented as mean ± SD (significance *p < 0.1, **p < 0.05). | ||
Interestingly, dual spatial and dual mixed scaffolds showed significant differences from each other when additional tissue-specific markers were investigated, illustrating how multi-peptide organization affected cell response. Specifically, distributing HAbind and E3 peptides homogeneously throughout the dual mixed scaffold drove hMSCs towards a stable chondrogenic phenotype while presenting both peptides in opposing zones of the dual spatial scaffold promoted osteogenesis. When prechondrocytes differentiate into fully committed and active chondrogenic cells, they express high levels of cartilage ECM components like aggrecan (ACAN), link protein (Cartl1), and cartilage oligomeric matrix protein (COMP).29 At day 14, both dual spatial and dual mixed scaffolds stimulated early upregulation of ACAN compared to PCL (p = 0.041 and p = 0.005, respectively). However, later timepoints showed that dual mixed scaffolds promoted more chondro-like behavior compared to dual spatial scaffolds. Significant upregulation of ACAN was also observed at day 42 in dual mixed scaffolds compared to both dual spatial (p = 0.028) and PCL (p = 0.003) scaffolds (Fig. 6A and Fig. S15†). Aggrecan is the major proteoglycan in articular cartilage ECM88 and is present at high concentrations in the deep zone of the OC tissue.23 Additionally, dual mixed scaffolds upregulated COMP gene expression compared to PCL at days 28 (p = 0.065) and 42 (p = 0.007). Cartilage oligomeric matrix protein is a component of adult articular cartilage produced by fully committed chondrocytes.89,90 Dual mixed scaffolds also showed higher expression of PRG4 compared to dual spatial (p = 0.011) and PCL scaffolds (p < 0.001) at day 42. This gene encodes for proteoglycan 4, also known as the superficial zone protein (SZP) or lubricin, found only in the superficial zone of articular cartilage.23,91,92 High expression of PRG4 is characteristic of terminally differentiated articular chondrocytes.29 This increased expression of multiple chondrogenic ECM genes correlated with higher GAG deposition in dual mixed scaffolds after 42 days of culture compared to dual spatial (p = 0.001) and PCL scaffolds (p = 0.068) (Fig. 6B). In contrast, dual spatial scaffolds did not present significant upregulation in chondrogenic ECM gene expression compared to PCL, suggesting this effect was driven by homogeneous multi-peptide presentation.
Dual mixed scaffolds also promoted higher expression of hypertrophic gene markers, including collagen X (COLX), alkaline phosphatase protein (ALPL), and osteocalcin (OC). Hypertrophic chondrocytes are found in the OC tidemark where they express RUNX2, COLX, ALPL, OC, and induce mineralization of the cartilage matrix.29,92 Dual mixed scaffolds showed upregulation of ALPL compared to PCL at day 14 (p = 0.043) and at day 42 (p = 0.007). Additionally, dual mixed scaffolds showed higher expression of RUNX2 (p = 0.011), COLX (p = 0.002), and OC (p = 0.075) compared to PCL at day 42 (Fig. 6A and Fig. S15†). Together, these results suggested dual mixed scaffolds drive hMSC differentiation towards a mixed population of articular and hypertrophic chondrocytes. Both of these phenotypes are present in native OC tissue and are responsible for producing the organized ECM structure.10,28,29 Dual mixed scaffolds therefore have potential to guide formation of articular and calcified cartilage layers in organized OC tissue.
Dual spatial scaffolds showed upregulation of osteogenic genes compared to PCL and dual mixed scaffolds. Osteogenic differentiation of hMSCs is characterized by expression of markers that include RUNX2 and osterix (SP7) as well as bone matrix genes ALPL, collagen type I, bone sialoprotein, and OC.71,80,93 During hMSC differentiation, RUNX2 is crucial for osteoblast lineage commitment but is later downregulated for further bone maturation.93 Dual spatial scaffolds showed upregulation of RUNX2 compared to PCL scaffolds after 14 days in culture (p = 0.004) (Fig. 6A and Fig. S15†). However, there was no significant difference in RUNX2 expression between dual spatial and PCL scaffolds at days 28 and 42. At day 14, dual spatial scaffolds also showed increased expression of SP7 compared to PCL (p = 0.006) and dual mixed scaffolds (p = 0.036). SP7 is a downstream target of RUNX2, which promotes terminal chondrocyte differentiation94 and commitment to osteogenesis.71 Additionally, dual spatial scaffolds upregulated ALPL compared to PCL at days 14 (p = 0.011) and day 42 (p = 0.002), as well as higher OC expression at day 14 compared to PCL (p = 0.076). Upregulation of osteogenic genes corresponded to higher ALP activity in dual spatial scaffolds compared to PCL and dual mixed scaffolds at day 14 (p = 0.017 and p = 0.067, respectively) and day 28 (p = 0.002 and p = 0.001, respectively) (Fig. 6C).
Since chondrocyte hypertrophy leads to terminal differentiation and osteogenesis, some markers including RUNX2, ALPL, and OC overlap in these two populations.29,71 This overlap made it difficult to distinguish between the hypertrophic and osteogenic cell behavior seen in dual mixed and dual spatial scaffolds. However, there were key differences between the two dual-peptide scaffold groups. Dual mixed scaffolds showed higher COLX expression at day 42 compared to PCL while dual spatial showed no significant difference to PCL. Collagen type X is produced exclusively by pre-hypertrophic and hypertrophic chondrocytes.29,93 Additionally, dual mixed scaffolds did not upregulate SP7 at any of the timepoints, showing limited osteogenic behavior. In contrast, this marker of commitment to the osteogenic lineage71 was expressed at higher levels in dual spatial scaffolds at day 7 compared to PCL. Together, these results suggested that scaffolds presenting HAbind and E3 peptides in discrete zones drive hMSC differentiation towards osteogenesis. Therefore, dual spatial scaffolds have potential to promote hMSC terminal differentiation and formation of subchondral bone.
The data collectively showed that the spatial organization of cartilage-promoting and bone-promoting peptides influenced hMSC differentiation and tissue formation. Specifically, dual spatial scaffolds directed hMSCs to differentiate into an osteogenic phenotype while dual mixed scaffolds promoted more articular and hypertrophic chondrogenic behavior. Histological evaluation of constructs after 42 days in culture showed tissue maturation in all scaffolds. Staining with Alcian blue showed similar GAG accumulation in all scaffolds. The longer culture time also allowed for calcium deposition, especially in dual spatial scaffolds compared to PCL and dual mixed scaffolds (Fig. S17†). The presence of calcium suggested higher matrix mineralization in dual spatial scaffolds,79 consistent with increased osteogenic behavior found in our gene expression and biochemical data (Fig. 6). These results point to local and global cell–cell communication that influences differentiation and matrix formation. Mesenchymal stem cells are known to secrete a spectrum of paracrine factors, known as the secretome, that influences several biological processes, including cell differentiation.95,96 The specific composition of the secretome is affected by environmental stimuli.97 In dual spatial and dual mixed scaffolds, cells experienced different local microenvironments and thus are expected to produce different secretome compositions that drive different differentiation responses.
Higher collagen deposition, especially collagen type II and X, was observed in all scaffold groups at day 42 (Fig. 7) compared to previous scaffolds evaluated after only 28 days in culture (Fig. 4). These IHC results suggested an improved collagen II:collagen I ratio, characteristic of articular cartilage,15,81 and increased collagen X production as cells undergo hypertrophy.28,29 We did not observe measurable differences in collagen deposition between scaffold groups, although gene expression for differentiation markers was affected. Traditional chondrogenic and osteogenic induction of hMSCs requires weeks of culture in differentiation media with growth factors to see significant histological changes in matrix formation.98 However, our scaffolds showed deposition of collagen II and X, key markers of OC tissue, despite being cultured in growth media without added differentiation factors. These IHC results indicated that the responses observed were driven by multi-peptide presentation and organization.
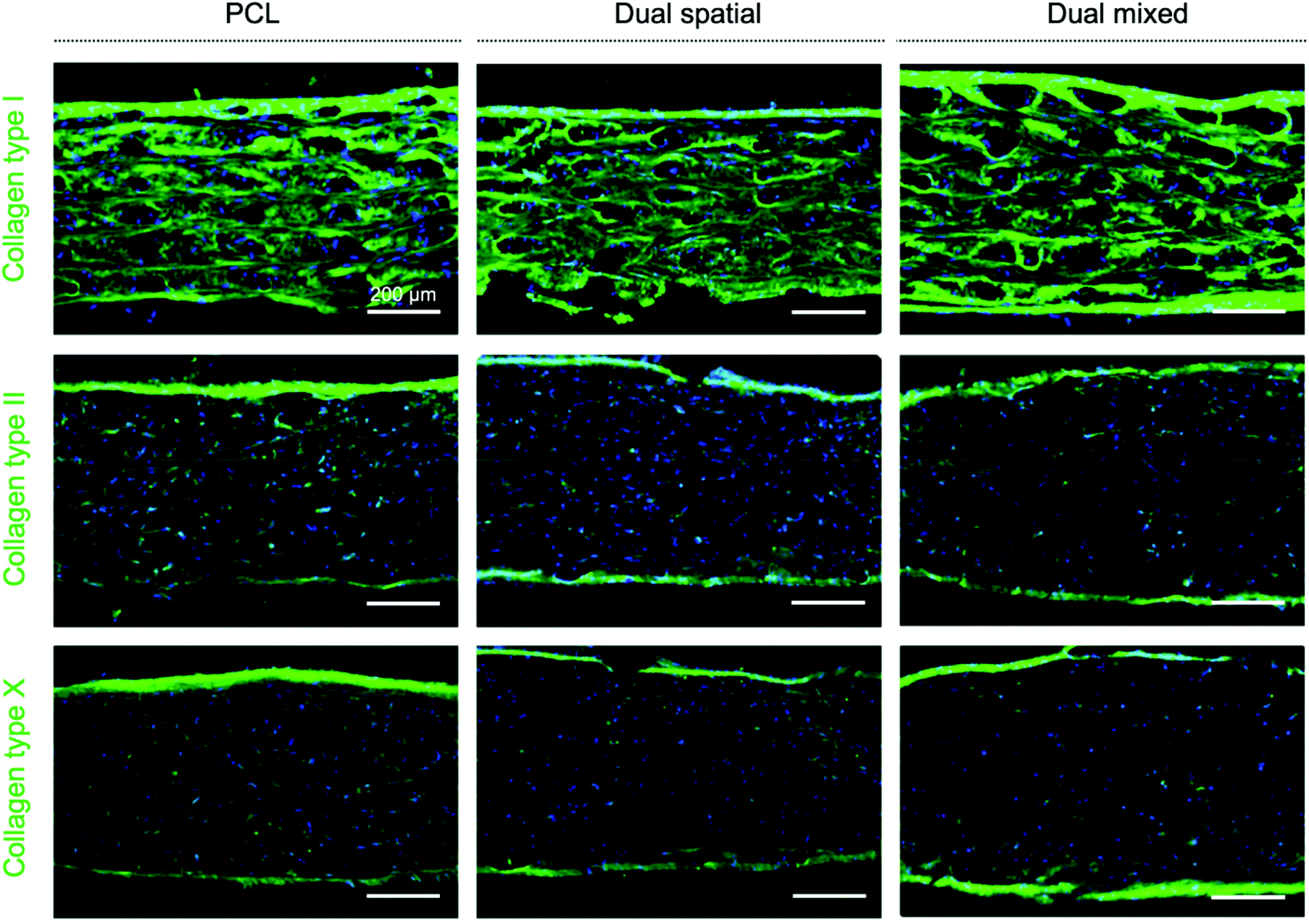 | ||
| Fig. 7 Collagen (type I, II, X) deposition in dual-peptide 3D-printed scaffolds after 42 days of culture. Representative confocal microscopy images of histological cross-sections showing immunohistochemical staining for collagen type I, II, and X (top to bottom) (Alexa Fluor 488 – green) and cell nuclei (Hoescht Dye – blue) in PCL, dual spatial, and dual mixed scaffolds (L to R). All scaffold groups presented immature tissue formation, seen by positive staining for collagen I. Collagen II deposition was also found in all groups. Dual mixed scaffolds presented more intense collagen II staining while dual spatial and PCL showed more collagen X deposition in the constructs. (Scale bar = 200 μm.) | ||
Differences in hMSC response between donors were also observed in dual-peptide studies due to variations in chondrogenic and osteogenic potential across donors. Donor C showed lower overall GAG deposition at days 14 and 28 compared to donors A and B (Fig. 6B). These results correlated with responses shown in Fig. 3A. However, this effect was reversed at day 42 with donor C showing the highest GAG deposition in all scaffolds. Donor C also showed consistently higher ALP activity in dual spatial scaffolds compared to PCL and dual mixed scaffolds at all timepoints (Fig. 6C). These results indicated that donor C may have higher chondrogenic and osteogenic potential than donors B and D overall. Notably, donor C was the youngest of all the donors used, and age has been shown to influence hMSC differentiation capacity.99,100 Donor D showed a large upregulation of chondrogenic and osteogenic gene expression in dual spatial scaffolds at day 14 compared to PCL. However, this effect was not seen at days 28 and 42 (Fig. 6A), suggesting donor D had the lowest chondrogenic and osteogenic potential. Also, donor D had similar overall GAG deposition in all scaffolds at all timepoints. This donor was not used in previous experiments comparing single-peptide and dual-peptide scaffolds (Fig. 3), which may explain the differences in PCL and dual spatial scaffolds between the multiple experiments. Despite the variability in donor differentiation capacity, we observed significant differences overall between scaffold groups, supporting our findings that peptide location influences hMSC behavior. Furthermore, the osteogenic response in dual spatial scaffolds detected in the previous experiments (Fig. 3) was also observed with an expanded set of donors (Fig. 6).
These studies illustrated how controlling the spatial presentation of biochemical cues is crucial to influencing hMSC behavior and organized tissue formation. This work exploited 3D printing with peptide–polymer conjugates to fabricate scaffolds presenting cartilage-promoting and bone-promoting peptides either homogeneously throughout the construct or in opposing zones. We found that the spatial organization of these peptides significantly influenced hMSC differentiation and ECM deposition. Dual mixed scaffolds promoted hMSC chondrogenic differentiation, introducing the potential to guide the formation of articular and calcified cartilage layers. In contrast, dual spatial scaffolds promoted hMSC terminal differentiation, which can be used to enhance subchondral bone regeneration and integration. This versatile strategy therefore enables highly controlled fabrication of complex scaffolds to direct regeneration of functional OC tissue.
4. Conclusions
This work demonstrated a single-step functionalization strategy using solvent-cast 3D printing with peptide–polymer conjugates to fabricate scaffolds to guide tissue regeneration. The specific peptides used in this study were selected to promote hMSC chondrogenesis and osteogenesis to drive functional osteochondral tissue formation. The presence of both peptides in the same scaffold resulted in a synergistic enhancement of both osteogenic and chondrogenic responses compared to unfunctionalized and single-peptide scaffolds. Furthermore, spatial organization of both peptides in the same construct influenced hMSC differentiation and tissue formation. Specifically, dual mixed scaffolds supported articular and hypertrophic chondrogenic behavior while dual spatial scaffolds guided hMSCs to differentiate into an osteogenic phenotype. Notably, these significant results occurred without the addition of exogenous GAGs or differentiation factors during culture, indicating that dual-peptide presentation and organization were sufficient to guide hMSC behavior.This modular strategy can be used to explore the combined influence of biochemical cues, organization, and physical architecture on tissue formation. For example, future studies with scaffolds presenting different combinations and arrangements of single-peptide and dual mixed layers can be conducted to identify optimal scaffold designs to guide OC tissue formation. In addition, printing parameters and patterns can be modified to determine complementary architectures that enhance hMSC response. The downstream potential of this approach is the ability to generate biomaterials with spatially controlled cues to guide in situ functional tissue regeneration without the need for added differentiation factors.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors would like to thank Dr Hannah L. Dailey for help with experimental design, the late Dr William G. De Long, Jr for help with funding acquisition, Hafiz Busari for help with peptide and conjugate synthesis, Dr Peter Schwarzenberg for help with optimizing 3D printing parameters, Dr E. Thomas Pashuck and Samuel Rozans for help with LC-MS, and Dr Rhiannon Grant for help with cell culture training and tissue characterization protocols. The authors also acknowledge Lehigh's Electron Microscopy and Nanofabrication Facility, Health Research Hub (HRH), and Department of Chemistry Shared Instrumentation. This work made use of the Lehigh University NMR Facility. The Bruker Neo 500 MHz NMR was acquired through NSF-MRI-1725883, with additional support from Lehigh University.This work was generously supported by a Faculty Early Career Development (CAREER) Award (DMR 1944914 to LWC) from the NSF, a grant from the Pennsylvania Department of Health through the Commonwealth University Research Enhancement (CURE; #4100077064) Program (LWC), a grant from the Foundation for Orthopedic Trauma (FOT #4018-18), Lehigh's Collaborative Research Opportunity (CORE) Grant, a grant with Polysciences, Inc. from the Commonwealth of Pennsylvania, Department of Community and Economic Development, through the Pennsylvania Infrastructure Technology Alliance (PITA) as well as start-up funds awarded to LWC from Lehigh University. Additional support from Lehigh University was provided as a Clare Boothe Luce research scholarship awarded to AB, a Grant for Experiential Learning in Health (GELH) awarded to MF, and a President's Scholarship awarded to KBS.
References
- P. J. Yang and J. S. Temenoff, Engineering Orthopedic Tissue Interfaces, Tissue Eng., Part B, 2009, 15(2), 127–141, DOI:10.1089/ten.teb.2008.0371.
- W. L. Grayson, P. G. Chao, D. Marolt, D. L. Kaplan and G. Vunjak-Novakovic, Engineering Custom-Designed Osteochondral Tissue Grafts, Trends Biotechnol., 2008, 26(4), 181–189, DOI:10.1016/j.tibtech.2007.12.009.
- M. Falah, G. Nierenberg, M. Soudry, M. Hayden and G. Volpin, Treatment of Articular Cartilage Lesions of the Knee, Int. Orthop., 2010, 34(5), 621–630, DOI:10.1007/s00264-010-0959-y.
- S. Glyn-Jones, A. J. R. Palmer, R. Agricola, A. J. Price, T. L. Vincent, H. Weinans and A. J. Carr, Osteoarthritis, Lancet, 2015, 386(9991), 376–387, DOI:10.1016/S0140-6736(14)60802-3.
- L. Kock, C. C. Van Donkelaar and K. Ito, Tissue Engineering of Functional Articular Cartilage: The Current Status, Cell Tissue Res., 2012, 347(3), 613–627, DOI:10.1007/s00441-011-1243-1.
- C. N. van Dijk, M. L. Reilingh, M. Zengerink and C. J. A. van Bergen, Osteochondral Defects in the Ankle: Why Painful?, Knee Surg., Sports Traumatol. Arthrosc., 2010, 18(5), 570–580, DOI:10.1007/s00167-010-1064-x.
- I. Gadjanski and G. Vunjak-Novakovic, Challenges in Engineering Osteochondral Tissue Grafts with Hierarchical Structures, Expert Opin. Biol. Ther., 2015, 15(11), 1583–1599, DOI:10.1517/14712598.2015.1070825.
- E. A. Makris, A. H. Gomoll, K. N. Malizos, J. C. Hu and K. A. Athanasiou, Repair and Tissue Engineering Techniques for Articular Cartilage, Nat. Rev. Rheumatol., 2015, 11(1), 21–34, DOI:10.1038/nrrheum.2014.157.
- B. W. Schreurs and G. Hannink, Total Joint Arthroplasty in Younger Patients: Heading for Trouble?, Lancet, 2017, 389(10077), 1374–1375, DOI:10.1016/S0140-6736(17)30190-3.
- A. Di Luca, C. Van Blitterswijk and L. Moroni, The Osteochondral Interface as a Gradient Tissue: From Development to the Fabrication of Gradient Scaffolds for Regenerative Medicine, Birth Defects Res., Part C, 2015, 105(1), 34–52, DOI:10.1002/bdrc.21092.
- H. Kwon, W. E. Brown, C. A. Lee, D. Wang, N. Paschos, J. C. Hu and K. A. Athanasiou, Surgical and Tissue Engineering Strategies for Articular Cartilage and Meniscus Repair, Nat. Rev. Rheumatol., 2019, 15(9), 550–570, DOI:10.1038/s41584-019-0255-1.
- B. J. Huang, J. C. Hu and K. A. Athanasiou, Cell-Based Tissue Engineering Strategies Used in the Clinical Repair of Articular Cartilage, Biomaterials, 2016, 98, 1–22, DOI:10.1016/j.biomaterials.2016.04.018.
- B. M. Devitt, S. W. Bell, K. E. Webster, J. A. Feller and T. S. Whitehead, Surgical Treatments of Cartilage Defects of the Knee: Systematic Review of Randomised Controlled Trials, Knee, 2017, 24(3), 508–517, DOI:10.1016/j.knee.2016.12.002.
- M. Tamaddon, L. Wang, Z. Liu and C. Liu, Osteochondral Tissue Repair in Osteoarthritic Joints: Clinical Challenges and Opportunities in Tissue Engineering, Bio-Des. Manuf., 2018, 1(2), 101–114, DOI:10.1007/s42242-018-0015-0.
- A. R. Armiento, M. Alini and M. J. Stoddart, Articular Fibrocartilage - Why Does Hyaline Cartilage Fail to Repair?, Adv. Drug Delivery Rev., 2019, 146, 289–305, DOI:10.1016/j.addr.2018.12.015.
- L. Zhou, V. O. Gjvm, J. Malda, M. J. Stoddart, Y. Lai, R. G. Richards, K. Ki-wai Ho and L. Qin, Innovative Tissue-Engineered Strategies for Osteochondral Defect Repair and Regeneration: Current Progress and Challenges, Adv. Healthcare Mater., 2020, 2001008, 1–22, DOI:10.1002/adhm.202001008.
- B. Johnstone, M. Alini, M. Cucchiarini, G. Dodge, D. Eglin, F. Guilak, H. Madry, A. Mata, R. Mauck and C. Semino, et al., Tissue Engineering for Articular Cartilage Repair – the State of the Art, Eur. Cells Mater., 2013, 25(May), 248–267, DOI:10.22203/eCM.v025a18.
- S. P. Nukavarapu and D. L. Dorcemus, Osteochondral Tissue Engineering: Current Strategies and Challenges, Biotechnol. Adv., 2013, 31(5), 706–721, DOI:10.1016/j.biotechadv.2012.11.004.
- S. Ansari, S. Khorshidi and A. Karkhaneh, Engineering of Gradient Osteochondral Tissue: From Nature to Lab, Acta Biomater., 2019, 87, 41–54, DOI:10.1016/j.actbio.2019.01.071.
- M. A. Barajaa, L. S. Nair and C. T. Laurencin, Bioinspired Scaffold Designs for Regenerating Musculoskeletal Tissue Interfaces, Regener. Eng. Transl. Med., 2020, 6(4), 451–483, DOI:10.1007/s40883-019-00132-3.
- S. Camarero-Espinosa, B. Rothen-Rutishauser, E. J. Foster and C. Weder, Articular Cartilage: From Formation to Tissue Engineering, Biomater. Sci., 2016, 4(5), 734–767, 10.1039/C6BM00068A.
- P. Orth, C. Peifer, L. Goebel, M. Cucchiarini and H. Madry, Comprehensive Analysis of Translational Osteochondral Repair: Focus on the Histological Assessment, Prog. Histochem. Cytochem., 2015, 50(3), 19–36, DOI:10.1016/j.proghi.2015.10.001.
- A. R. Poole, T. Kojima, T. Yasuda, F. Mwale, M. Kobayashi and S. Laverty, Composition and Structure of Articular Cartilage, Clin. Orthop. Relat. Res., 2001, 391(391 SUPPL.), S26–S33, DOI:10.1097/00003086-200110001-00004.
- V. C. Mow, A. Ratcliffe and A. Robin Poole, Cartilage and, Diarthrodial Joints as Paradigms for Hierarchical Materials and Structures, Biomaterials, 1992, 13(2), 67–97, DOI:10.1016/0142-9612(92)90001-5.
- M. Wong and D. Carter, Articular Cartilage Functional Histomorphology and Mechanobiology: A Research Perspective, Bone, 2003, 33(1), 1–13, DOI:10.1016/S8756-3282(03)00083-8.
- H. Madry, C. N. van Dijk and M. Mueller-Gerbl, The Basic Science of the Subchondral Bone, Knee Surg., Sports Traumatol. Arthrosc., 2010, 18(4), 419–433, DOI:10.1007/s00167-010-1054-z.
- H. M. Kronenberg, Developmental Regulation of the Growth Plate, Nature, 2003, 423(6937), 332–336, DOI:10.1038/nature01657.
- I. Gadjanski, K. Spiller and G. Vunjak-Novakovic, Time-Dependent Processes in Stem Cell-Based Tissue Engineering of Articular Cartilage, Stem Cell Rev. Rep., 2012, 8(3), 863–881, DOI:10.1007/s12015-011-9328-5.
- V. Lefebvre and P. Smits, Transcriptional Control of Chondrocyte Fate and Differentiation, Birth Defects Res., Part C, 2005, 75(3), 200–212, DOI:10.1002/bdrc.20048.
- G. M. Cunniffe, P. J. Díaz-Payno, E. J. Sheehy, S. E. Critchley, H. V. Almeida, P. Pitacco, S. F. Carroll, O. R. Mahon, A. Dunne and T. J. Levingstone, et al., Tissue-Specific Extracellular Matrix Scaffolds for the Regeneration of Spatially Complex Musculoskeletal Tissues, Biomaterials, 2019, 188(July 2018), 63–73, DOI:10.1016/j.biomaterials.2018.09.044.
- B. A. Harley, A. K. Lynn, Z. Wissner-Gross, W. Bonfield, I. V. Yannas and L. J. Gibson, Design of a Multiphase Osteochondral Scaffold III: Fabrication of Layered Scaffolds with Continuous Interfaces, J. Biomed. Mater. Res., Part A, 2009, 92(3), 1078–1093, DOI:10.1002/jbm.a.32387.
- N. H. Dormer, M. Singh, L. Wang, C. J. Berkland and M. S. Detamore, Osteochondral Interface Tissue Engineering Using Macroscopic Gradients of Bioactive Signals, Ann. Biomed. Eng., 2010, 38(6), 2167–2182, DOI:10.1007/s10439-010-0028-0.
- M. T. Frassica and M. A. Grunlan, Perspectives on Synthetic Materials to Guide Tissue Regeneration for Osteochondral Defect Repair, ACS Biomater. Sci. Eng., 2020, 6(8), 4324–4336, DOI:10.1021/acsbiomaterials.0c00753.
- C. Li, J. P. Armstrong, I. J. Pence, W. Kit-Anan, J. L. Puetzer, S. Correia Carreira, A. C. Moore and M. M. Stevens, Glycosylated Superparamagnetic Nanoparticle Gradients for Osteochondral Tissue Engineering, Biomaterials, 2018, 176, 24–33, DOI:10.1016/j.biomaterials.2018.05.029.
- C. Loebel and J. A. Burdick, Engineering Stem and Stromal Cell Therapies for Musculoskeletal Tissue Repair, Cell Stem Cell, 2018, 22(3), 325–339, DOI:10.1016/j.stem.2018.01.014.
- H. Kang, Y. Zeng and S. Varghese, Functionally Graded Multilayer Scaffolds for in Vivo Osteochondral Tissue Engineering, Acta Biomater., 2018, 78, 365–377, DOI:10.1016/j.actbio.2018.07.039.
- G. Filardo, F. Perdisa, M. Gelinsky, F. Despang, M. Fini, M. Marcacci, A. P. Parrilli, A. Roffi, F. Salamanna and M. Sartori, et al., Novel Alginate Biphasic Scaffold for Osteochondral Regeneration: An in Vivo Evaluation in Rabbit and Sheep Models, J. Mater. Sci. Mater. Med., 2018, 29(6), 74, DOI:10.1007/s10856-018-6074-0.
- D. L. Dorcemus, E. O. George, C. N. Dealy and S. P. Nukavarapu, Harnessing External Cues: Development and Evaluation of an In Vitro Culture System for Osteochondral Tissue Engineering, Tissue Eng., Part A, 2017, 23(15–16), 719–737, DOI:10.1089/ten.tea.2016.0439.
- A. Di Luca, I. Lorenzo-Moldero, C. Mota, A. Lepedda, D. Auhl, C. Van Blitterswijk and L. Moroni, Tuning Cell Differentiation into a 3D Scaffold Presenting a Pore Shape Gradient for Osteochondral Regeneration, Adv. Healthcare Mater., 2016, 5(14), 1753–1763, DOI:10.1002/adhm.201600083.
- M. Castilho, V. Mouser, M. Chen, J. Malda and K. Ito, Bi-Layered Micro-Fibre Reinforced Hydrogels for Articular Cartilage Regeneration, Acta Biomater., 2019, 95, 297–306, DOI:10.1016/j.actbio.2019.06.030.
- A. Di Luca, M. Klein-Gunnewiek, J. G. Vancso, C. A. van Blitterswijk, E. M. Benetti and L. Moroni, Covalent Binding of Bone Morphogenetic Protein-2 and Transforming Growth Factor-B3 to 3D Plotted Scaffolds for Osteochondral Tissue Regeneration, Biotechnol. J., 2017, 12(12), 1–9, DOI:10.1002/biot.201700072.
- S. Lu, J. Lam, J. E. Trachtenberg, E. J. Lee, H. Seyednejad, J. J. J. P. van den Beucken, Y. Tabata, M. E. Wong, J. A. Jansen and A. G. Mikos, et al., Dual Growth Factor Delivery from Bilayered, Biodegradable Hydrogel Composites for Spatially-Guided Osteochondral Tissue Repair, Biomaterials, 2014, 35(31), 8829–8839, DOI:10.1016/j.biomaterials.2014.07.006.
- L.-P. Yan, J. M. Oliveira, A. L. Oliveira and R. L. Reis, Current Concepts and Challenges in Osteochondral Tissue Engineering and Regenerative Medicine, ACS Biomater. Sci. Eng., 2015, 1(4), 183–200, DOI:10.1021/ab500038y.
- A. Seidi, M. Ramalingam, I. Elloumi-Hannachi, S. Ostrovidov and A. Khademhosseini, Gradient Biomaterials for Soft-to-Hard Interface Tissue Engineering, Acta Biomater., 2011, 7(4), 1441–1451, DOI:10.1016/j.actbio.2011.01.011.
- A.-V. Do, B. Khorsand, S. M. Geary and A. K. Salem, 3D Printing of Scaffolds for Tissue Regeneration Applications, Adv. Healthcare Mater., 2015, 4(12), 1742–1762, DOI:10.1002/adhm.201500168.
- C. Li, L. Ouyang, J. P. K. Armstrong and M. M. Stevens, Advances in the Fabrication of Biomaterials for Gradient Tissue Engineering, Trends Biotechnol., 2021, 39(2), 150–164, DOI:10.1016/j.tibtech.2020.06.005.
- H. Rashidi, J. Yang and K. M. Shakesheff, Surface Engineering of Synthetic Polymer Materials for Tissue Engineering and Regenerative Medicine Applications, Biomater. Sci., 2014, 2(10), 1318–1331, 10.1039/C3BM60330J.
- A. R. Armiento, M. J. Stoddart, M. Alini and D. Eglin, Biomaterials for Articular Cartilage Tissue Engineering: Learning from Biology, Acta Biomater., 2018, 65, 1–20, DOI:10.1016/j.actbio.2017.11.021.
- M. M. Stevens, Exploring and Engineering the Cell Surface Interface, Science, 2005, 310(5751), 1135–1138, DOI:10.1126/science.1106587.
- F. R. Maia, S. J. Bidarra, P. L. Granja and C. C. Barrias, Functionalization of Biomaterials with Small Osteoinductive Moieties, Acta Biomater., 2013, 9(11), 8773–8789, DOI:10.1016/j.actbio.2013.08.004.
- X.-Y. Sun, R. Shankar, H. G. Börner, T. K. Ghosh and R. J. Spontak, Field-Driven Biofunctionalization of Polymer Fiber Surfaces during Electrospinning, Adv. Mater., 2007, 19(1), 87–91, DOI:10.1002/adma.200601345.
- R. Gentsch, F. Pippig, S. Schmidt, P. Cernoch, J. Polleux and H. G. Börner, Single-Step Electrospinning to Bioactive Polymer Nanofibers, Macromolecules, 2011, 44(3), 453–461, DOI:10.1021/ma102847a.
- P. Camacho, M. Fainor, K. B. Seims, J. W. Tolbert and L. W. Chow, Fabricating Spatially Functionalized 3D-Printed Scaffolds for Osteochondral Tissue Engineering, J. Biol. Methods, 2021, 8(1), e146, DOI:10.14440/jbm.2021.353.
- P. Camacho, H. Busari, K. B. Seims, P. Schwarzenberg, H. L. Dailey and L. W. Chow, 3D Printing with Peptide–Polymer Conjugates for Single-Step Fabrication of Spatially Functionalized Scaffolds, Biomater. Sci., 2019, 7(10), 4237–4247, 10.1039/C9BM00887J.
- L. W. Chow, A. Armgarth, J.-P. St-Pierre, S. Bertazzo, C. Gentilini, C. Aurisicchio, S. D. McCullen, J. A. M. Steele and M. M. Stevens, Peptide-Directed Spatial Organization of Biomolecules in Dynamic Gradient Scaffolds, Adv. Healthcare Mater., 2014, 3(9), 1381–1386, DOI:10.1002/adhm.201400032.
- P. A. Parmar, J.-P. St-Pierre, L. W. Chow, J. L. Puetzer, V. Stoichevska, Y. Y. Peng, J. A. Werkmeister, J. A. M. Ramshaw and M. M. Stevens, Harnessing the Versatility of Bacterial Collagen to Improve the Chondrogenic Potential of Porous Collagen Scaffolds, Adv. Healthcare Mater., 2016, 5(13), 1656–1666, DOI:10.1002/adhm.201600136.
- O. Karaman, A. Kumar, S. Moeinzadeh, X. He, T. Cui and E. Jabbari, Effect of Surface Modification of Nanofibres with Glutamic Acid Peptide on Calcium Phosphate Nucleation and Osteogenic Differentiation of Marrow Stromal Cells, J. Tissue Eng. Regener. Med., 2016, 10(2), E132–E146, DOI:10.1002/term.1775.
- P. F. Goetinck, N. S. Stirpe, P. A. Tsonis and D. Carlone, The Tandemly Repeated Sequences of Cartilage Link Protein Contain the Sites for Interaction with Hyaluronic Acid, J. Cell Biol., 1987, 105(5), 2403–2408, DOI:10.1083/jcb.105.5.2403.
- B. Yang, B. L. Yang, R. C. Savani and E. A. Turley, Identification of a Common Hyaluronan Binding Motif in the Hyaluronan Binding Proteins RHAMM, CD44 and Link Protein, EMBO J., 1994, 13(2), 286–296, DOI:10.1002/j.1460-2075.1994.tb06261.x.
- D. P. Nair, M. Podgórski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, The Thiol-Michael Addition Click Reaction: A Powerful and Widely Used Tool in Materials Chemistry, Chem. Mater., 2014, 26(1), 724–744, DOI:10.1021/cm402180t.
- L. W. Chow, Electrospinning Functionalized Polymers for Use as Tissue Engineering Scaffolds, in Biomaterials for Tissue Engineering. Methods in Molecular Biology, ed. K. Chawla, Humana Press, New York, NY, 2018, vol. 1758, pp. 27–39. DOI:10.1007/978-1-4939-7741-3_3.
- W. K. Grier, R. A. Sun Han Chang, M. D. Ramsey and B. A. C. Harley, The Influence of Cyclic Tensile Strain on Multi-Compartment Collagen-GAG Scaffolds for Tendon-Bone Junction Repair, Connect. Tissue Res., 2019, 60(6), 530–543, DOI:10.1080/03008207.2019.1601183.
- A. J. Roelofs, J. Zupan, A. H. K. Riemen, K. Kania, S. Ansboro, N. White, S. M. Clark and C. De Bari, Joint Morphogenetic Cells in the Adult Mammalian Synovium, Nat. Commun., 2017, 8(1), 15040, DOI:10.1038/ncomms15040.
- P. A. Parmar, S. C. Skaalure, L. W. Chow, J. P. St-Pierre, V. Stoichevska, Y. Y. Peng, J. A. Werkmeister, J. A. M. Ramshaw and M. M. Stevens, Temporally Degradable Collagen-Mimetic Hydrogels Tuned to Chondrogenesis of Human Mesenchymal Stem Cells, Biomaterials, 2016, 99, 56–71, DOI:10.1016/j.biomaterials.2016.05.011.
- F. Klatte-Schulz, S. Pauly, M. Scheibel, S. Greiner, C. Gerhardt, J. Hartwig, G. Schmidmaier and B. Wildemann, Characteristics and Stimulation Potential with BMP-2 and BMP-7 of Tenocyte-Like Cells Isolated from the Rotator Cuff of Female Donors, PLoS One, 2013, 8(6), e67209, DOI:10.1371/journal.pone.0067209.
- F. Zhu, M. S. Friedman, W. Luo, P. Woolf and K. D. Hankenson, The Transcription Factor Osterix(SP7) Regulates BMP6-Induced Human Osteoblast Differentiation, J. Cell. Physiol., 2012, 227(6), 2677–2685, DOI:10.1002/jcp.23010.
- E. P. Diamandis and T. K. Christopoulos, The Biotin-(Strept)Avidin System: Principles and Applications in Biotechnology, Clin. Chem., 1991, 37(5), 625–636, DOI:10.1093/clinchem/37.5.625.
- M. R. Karver, R. Weissleder and S. A. Hilderbrand, Bioorthogonal Reaction Pairs Enable Simultaneous, Selective, Multi-Target Imaging, Angew. Chem., 2012, 124(4), 944–946, DOI:10.1002/ange.201104389.
- H. Tsuchiya, H. Kitoh, F. Sugiura and N. Ishiguro, Chondrogenesis Enhanced by Overexpression of Sox9 Gene in Mouse Bone Marrow-Derived Mesenchymal Stem Cells, Biochem. Biophys. Res. Commun., 2003, 301(2), 338–343, DOI:10.1016/S0006-291X(02)03026-7.
- R. A. Somoza, J. F. Welter, D. Correa and A. I. Caplan, Chondrogenic Differentiation of Mesenchymal Stem Cells: Challenges and Unfulfilled Expectations, Tissue Eng., Part B, 2014, 20(6), 596–608, DOI:10.1089/ten.teb.2013.0771.
- G. S. Stein, J. B. Lian, A. J. Van Wijnen, J. L. Stein, M. Montecino, A. Javed, S. K. Zaidi, D. W. Young, J. Y. Choi and S. M. Pockwinse, Runx2 Control of Organization, Assembly and Activity of the Regulatory Machinery for Skeletal Gene Expression, Oncogene, 2004, 23(24), 4315–4329, DOI:10.1038/sj.onc.1207676.
- B. Johnstone, T. M. Hering, A. I. Caplan, V. M. Goldberg and J. U. Yoo, In Vitro Chondrogenesis of Bone Marrow-Derived Mesenchymal Progenitor Cells, Exp. Cell Res., 1998, 238(1), 265–272, DOI:10.1006/excr.1997.3858.
- M. M. J. Caron, P. J. Emans, A. Cremers, D. A. M. Surtel, M. M. E. Coolsen, L. W. van Rhijn and T. J. M. Welting, Hypertrophic Differentiation during Chondrogenic Differentiation of Progenitor Cells Is Stimulated by BMP-2 but Suppressed by BMP-7, Osteoarthrotic Cartilage, 2013, 21(4), 604–613, DOI:10.1016/j.joca.2013.01.009.
- S. A. Unterman, M. Gibson, J. H. Lee, J. Crist, T. Chansakul, E. C. Yang and J. H. Elisseeff, Hyaluronic Acid-Binding Scaffold for Articular Cartilage Repair, Tissue Eng., Part A, 2012, 18(23–24), 2497–2506, DOI:10.1089/ten.tea.2011.0711.
- D. Barati, J. D. Walters, S. R. Pajoum Shariati, S. Moeinzadeh and E. Jabbari, Effect of Organic Acids on Calcium Phosphate Nucleation and Osteogenic Differentiation of Human Mesenchymal Stem Cells on Peptide Functionalized Nanofibers, Langmuir, 2015, 31(18), 5130–5140, DOI:10.1021/acs.langmuir.5b00615.
- W. Zhang, N. Liu, H. Shi, J. Liu, L. Shi, B. Zhang, H. Wang, J. Ji and P. K. Chu, Upregulation of BMSCs Osteogenesis by Positively-Charged Tertiary Amines on Polymeric Implants via Charge/INOS Signaling Pathway, Sci. Rep., 2015, 5, 1–10, DOI:10.1038/srep09369.
- H. Oshina, S. Sotome, T. Yoshii, I. Torigoe, Y. Sugata, H. Maehara, E. Marukawa, K. Omura and K. Shinomiya, Effects of Continuous Dexamethasone Treatment on Differentiation Capabilities of Bone Marrow-Derived Mesenchymal Cells, Bone, 2007, 41(4), 575–583, DOI:10.1016/j.bone.2007.06.022.
- N. Zhou, Q. Li, X. Lin, N. Hu, J. Y. Liao, L. B. Lin, C. Zhao, Z. M. Hu, X. Liang and W. Xu, et al., BMP2 Induces Chondrogenic Differentiation, Osteogenic Differentiation and Endochondral Ossification in Stem Cells, Cell Tissue Res., 2016, 366(1), 101–111, DOI:10.1007/s00441-016-2403-0.
- C. D. Hoemann, H. El-Gabalawy and M. D. McKee, In Vitro Osteogenesis Assays: Influence of the Primary Cell Source on Alkaline Phosphatase Activity and Mineralization, Pathol. Biol., 2009, 57(4), 318–323, DOI:10.1016/j.patbio.2008.06.004.
- N. Jaiswal, S. E. Haynesworth, A. I. Caplan and S. P. Bruder, Osteogenic Differentiation of Purified, Culture-Expanded Human Mesenchymal Stem Cells in Vitro, J. Cell. Biochem., 1997, 64(2), 295–312, DOI:10.1002/(SICI)1097-4644(199702)64:2<295::AID-JCB12>3.0.CO;2-I.
- A. J. Freemont and J. Hoyland, Lineage Plasticity and Cell Biology of Fibrocartilage and Hyaline Cartilage: Its Significance in Cartilage Repair and Replacement, Eur. J. Radiol., 2006, 57(1), 32–36, DOI:10.1016/j.ejrad.2005.08.008.
- L. D. Solorio, C. D. Dhami, P. N. Dang, E. L. Vieregge and E. Alsberg, Spatiotemporal Regulation of Chondrogenic Differentiation with Controlled Delivery of Transforming Growth Factor-B1 from Gelatin Microspheres in Mesenchymal Stem Cell Aggregates, Stem Cells Transl. Med., 2012, 1(8), 632–639, DOI:10.5966/sctm.2012-0039.
- C. Loebel, E. M. Czekanska, M. Bruderer, G. Salzmann, M. Alini and M. J. Stoddart, In Vitro Osteogenic Potential of Human Mesenchymal Stem Cells Is Predicted by Runx2/Sox9 Ratio, Tissue Eng., Part A, 2015, 21(1–2), 115–123, DOI:10.1089/ten.tea.2014.0096.
- D. G. Phinney, G. Kopen, W. Righter, S. Webster, N. Tremain and D. J. Prockop, Donor Variation in the Growth Properties and Osteogenic Potential of Human Marrow Stromal Cells, J. Cell. Biochem., 1999, 75(3), 424–436, DOI:10.1002/(SICI)1097-4644(19991201)75:3<424::AID-JCB8>3.0.CO;2-8.
- H. Yasuda and C. Oh, Regulation of SOX9 Activity During Chondrogenesis, in Encyclopedia of Bone Biology, Elsevier, 2020, pp. 548–559. DOI:10.1016/B978-0-12-801238-3.62212-2.
- G. Zhou, Q. Zheng, F. Engin, E. Munivez, Y. Chen, E. Sebald, D. Krakow and B. Lee, Dominance of SOX9 Function over RUNX2 during Skeletogenesis, Proc. Natl. Acad. Sci. U. S. A., 2006, 103(50), 19004–19009, DOI:10.1073/pnas.0605170103.
- P. Dy, W. Wang, P. Bhattaram, Q. Wang, L. Wang, R. T. Ballock and V. Lefebvre, Sox9 Directs Hypertrophic Maturation and Blocks Osteoblast Differentiation of Growth Plate Chondrocytes, Dev. Cell, 2012, 22(3), 597–609, DOI:10.1016/j.devcel.2011.12.024.
- C. Kiani, L. Chen, Y. J. Wu, A. J. Yee and B. B. Yang, Structure and Function of Aggrecan, Cell Res., 2002, 12(1), 19–32, DOI:10.1038/sj.cr.7290106.
- M. B. Goldring, K. Tsuchimochi and K. Ijiri, The Control of Chondrogenesis, J. Cell. Biochem., 2006, 97(1), 33–44, DOI:10.1002/jcb.20652.
- S. Koelling, T. S. Clauditz, M. Kaste and N. Miosge, Cartilage Oligomeric Matrix Protein Is Involved in Human Limb Development and in the Pathogenesis of Osteoarthritis, Arthritis Res. Ther., 2006, 8(3), 1–10, DOI:10.1186/ar1922.
- E. E. Coates and J. P. Fisher, Engineering Superficial Zone Chondrocytes from Mesenchymal Stem Cells, Tissue Eng., Part C, 2014, 20(8), 630–640, DOI:10.1089/ten.tec.2013.0224.
- S. P. Grogan, S. F. Duffy, C. Pauli, J. A. Koziol, A. I. Su, D. D. D'Lima and M. K. Lotz, Zone-Specific Gene Expression Patterns in Articular Cartilage, Arthritis Rheum., 2013, 65(2), 418–428, DOI:10.1002/art.37760.
- M. Bruderer, R. Richards, M. Alini and M. Stoddart, Role and Regulation of RUNX2 in Osteogenesis, Eur. Cells Mater., 2014, 28, 269–286, DOI:10.22203/eCM.v028a19.
- C. F. Liu, W. E. Samsa, G. Zhou and V. Lefebvre, Transcriptional Control of Chondrocyte Specification and Differentiation, Semin. Cell Dev. Biol., 2017, 62, 34–49, DOI:10.1016/j.semcdb.2016.10.004.
- W. Y. Lee and B. Wang, Cartilage Repair by Mesenchymal Stem Cells: Clinical Trial Update and Perspectives, J. Orthop. Transl., 2017, 9, 76–88, DOI:10.1016/j.jot.2017.03.005.
- M. Brennan, P. Layrolle and D. J. Mooney, Biomaterials Functionalized with MSC Secreted Extracellular Vesicles and Soluble Factors for Tissue Regeneration, Adv. Funct. Mater., 2020, 1909125, 1–21, DOI:10.1002/adfm.201909125.
- T. H. Qazi, D. J. Mooney, G. N. Duda and S. Geissler, Biomaterials That Promote Cell-Cell Interactions Enhance the Paracrine Function of MSCs, Biomaterials, 2017, 140, 103–114, DOI:10.1016/j.biomaterials.2017.06.019.
- R. Olivares-Navarrete, E. M. Lee, K. Smith, S. L. Hyzy, M. Doroudi, J. K. Williams, K. Gall, B. D. Boyan and Z. Schwartz, Substrate Stiffness Controls Osteoblastic and Chondrocytic Differentiation of Mesenchymal Stem Cells without Exogenous Stimuli, PLoS One, 2017, 12(1), e0170312, DOI:10.1371/journal.pone.0170312.
- S. Zhou, J. S. Greenberger, M. W. Epperly, J. P. Goff, C. Adler, M. S. Leboff and J. Glowacki, Age-Related Intrinsic Changes in Human Bone-Marrow-Derived Mesenchymal Stem Cells and Their Differentiation to Osteoblasts, Aging Cell, 2008, 7(3), 335–343, DOI:10.1111/j.1474-9726.2008.00377.x.
- P. Ganguly, J. J. El-Jawhari, P. V. Giannoudis, A. N. Burska, F. Ponchel and E. A. Jones, Age-Related Changes in Bone Marrow Mesenchymal Stromal Cells: A Potential Impact on Osteoporosis and Osteoarthritis Development, Cell Transplant., 2017, 26(9), 1520–1529, DOI:10.1177/0963689717721201.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1bm00859e |
| This journal is © The Royal Society of Chemistry 2021 |