
Nanobody-displaying porous silicon nanoparticles for the co-delivery of siRNA and doxorubicin†
Terence
Tieu
ab,
Marcin
Wojnilowicz
b,
Pie
Huda
c,
Kristofer J.
Thurecht
 c,
Helmut
Thissen
b,
Nicolas H.
Voelcker
*abde and
Anna
Cifuentes-Rius
*a
c,
Helmut
Thissen
b,
Nicolas H.
Voelcker
*abde and
Anna
Cifuentes-Rius
*a
aMonash Institute of Pharmaceutical Sciences, Monash University, Parkville Campus, Parkville, VIC 3052, Australia. E-mail: nicolas.voelcker@monash.edu; anna.cifuentesrius@monash.edu
bCommonwealth Scientific and Industrial Research Organisation (CSIRO) Manufacturing, Bayview Avenue, Clayton, VIC 3168, Australia
cCentre for Advanced Imaging, Australian Institute for Bioengineering and Nanotechnology (AIBN), ARC Centre of Excellence in Convergent Bio-Nano Science and Technology and ARC Training Centre for Innovation in Biomedical Imaging Technology, University of Queensland, Brisbane, QLD 4072, Australia
dMelbourne Centre for Nanofabrication, Victorian Node of the Australian National Fabrication Facility, Clayton, VIC 3168, Australia
eDepartment of Materials Science and Engineering, Monash University, Clayton, VIC 3168, Australia
First published on 28th October 2020
Abstract
Targeted delivery of chemotherapeutics to cancer cells has the potential to yield high drug concentrations in cancer cells while minimizing any unwanted side effects. However, the development of multidrug resistance in cancer cells may impede the accumulation of chemotherapy drugs within these, decreasing its therapeutic efficacy. Downregulation of multidrug resistance-related proteins such as MRP1 with small interfering RNA (siRNA) is a promising approach in the reversal of drug resistance. The co-delivery of doxorubicin (Dox) and siRNA against MRP1 (siMRP1) by using nanoparticles comprised of biocompatible porous silicon (pSi) presents itself as a novel opportunity to utilize the biomaterial's high loading capacity and large accessible surface area. Additionally, to increase the selectivity and retention of the delivery vehicle at the tumor site, nanobodies were incorporated onto the nanoparticle surface via a polyethylene glycol (PEG) linker directed towards either the epidermal growth factor receptor (EGFR) or the prostate specific membrane antigen (PSMA). The nanobody-displaying pSi nanoparticles (pSiNPs) demonstrated effective gene silencing, inhibiting MRP1 expression by 74 ± 6% and 74 ± 4% when incubated with EGFR-pSiNPs and PSMA-pSiNPs, respectively, in prostate cancer cells. The downregulation of MRP1 led to a further increase in cytotoxicity when both siRNA and Dox were delivered in conjunction in both cancer cell monocultures and spheroids when compared to free Dox or Dox and a scrambled sequence of siRNA. Altogether, nanobody-displaying pSiNPs are an effective carrier for the dual delivery of both siRNA and Dox for cancer treatment.
1. Introduction
In cancer, chemotherapy plays an important role as a primary form of cancer treatment in addition to traditional surgical interventions.1 However, current chemotherapy treatment is hampered by several challenges including (i) low selectivity and low tumor accumulation leading to unwanted side effects, and (ii) frequent chemotherapy dosage, having the potential of leading to the development of chemotherapy resistance.2,3 Chemotherapy resistance results from multidrug resistance mechanisms related to the expression of drug efflux pumps such as P-glycoprotein (P-gp or MDR1), multidrug resistance protein (MRP1) or breast cancer resistance protein (BRCP).4,5One of the most promising approaches for the reversal of multidrug resistance is by decreasing the expression of drug efflux proteins.6 Clinically, much of the focus in targeting multidrug resistance has been centered on the inhibition of P-gp, encoded by the MDR1 gene and commonly found in solid tumors.7,8 However, it has been shown that in some cancers such as prostate cancer, MRP1 and not MDR1 is more prevalent.9,10 MRP1 is a 190 kDa protein that is a member of the ATP-binding cassette (ABC) transporters.11 ABC transporters are active transporters, utilizing energy of adenosine triphosphate (ATP) molecules binding to transport substrates across cellular membranes. Consequently, an overexpression of ABC proteins or MRP1 is correlated with the reduced accumulation of chemotherapy drugs in cancer cells.12 Thus, by inhibiting MRP1 expression, it would be expected that the reduction in the efflux of drugs would help restore intracellular drug levels required to induce apoptosis or cytotoxicity.
RNA interference (RNAi) is a promising therapy in which strands of small interfering RNA (siRNA) have the potential to silence a chosen gene of interest.13,14 The inhibition of ABC proteins using siRNA has been studied extensively in reversing multidrug resistance in cancer.15–18 However, the delivery of siRNA to the intended cancer cells face various barriers including, (i) nonspecific distribution, (ii) rapid degradation and renal clearance, and (iii) lacking the capability of permeating cellular membranes due to the polyanionic nature and high molecular weight of siRNA molecules.13,19
Nanoparticle-based drug delivery systems allow therapeutics to be more selective and effective, resulting in enhanced treatment success and reduced side effects.20 By using nanoparticles to deliver both chemotherapeutic and siRNA, not only can the barriers associated with each therapeutic be circumvented but a synergistic effect boosting localized cytotoxicity of individual treatments can be exploited.13,20 While the co-delivery of chemotherapy and gene therapy for the improved efficacy of cancer treatment using non-viral carriers has previously been reported,15,16,21–23 pSiNPs offer many advantages such as biocompatibility and biodegradability, high loading capacity within the porous matrix and ease and tunability of its surface properties.24 We have already shown that pSiNPs can deliver chemotherapy drugs to target tissue and protect and deliver siRNA, resulting in high transfection efficiency.25,26 Moreover, we have exploited the high loading capacity of pSiNPs to co-deliver gold nanoclusters and chemotherapeutics in order to combine hyperthermia and chemotherapy. We showed that hyperthermia-inducing nanoclusters delivered via targeted pSiNPs were an effective chemosensitizer.27 Although the co-delivery of chemotherapeutic and siRNA has been reported using mesoporous silica nanoparticles,16,28,29 to date, the use of porous silicon – a highly biodegradable biomaterial as opposed to silica30 – for the co-delivery of siRNA and chemotherapy drug has not been reported. Thus, the use of siRNA technology in combination with pSiNPs to inhibit MRP1 presents itself to be a logical target in reversing the chemotherapy resistance in cancer cells.17
We have shown that pSiNPs are suitable for conjugation with moieties to recognize and target a specific cell population, which is of paramount importance to enhance therapy efficacy and reduce side effects.26,27 Among all targeting moieties, single-domain antibodies – derived from naturally occurring heavy-chain-only antibodies31 and also known as nanobodies – have been recently shown to present various advantageous properties over their conventional antibody counterparts such as their small size (∼15 kDa compared to ∼150 kDa for antibodies), high stability and a strong antigen-binding affinity.32 Here, we chose to utilize two different receptor targets for our nanobodies – the epidermal growth factor receptor (EGFR) and prostate specific membrane antigen (PSMA). EGFR overexpression has been associated with numerous cancers, including lung, breast, glioblastoma and melanoma.33,34 The amplification of EGFR generally leads to uncontrolled cancer cell division.34 PSMA on the other hand is an integral membrane protein found in prostatic tissue in which the expression of PSMA correlates with cancer aggressiveness and represents an independent indicator of poor prognosis.35
In this manuscript, we use pSiNPs modified with a fourth generation polyamidoamine (PAMAM(G4)) dendrimer and nanobody as a dual delivery platform for the co-delivery of doxorubicin (Dox) and siRNA targeting the MRP1 protein (siMRP1) (Scheme S1†). We have shown that PAMAM-G4-functionalized pSiNPs have a high siRNA loading capacity.25 The co-delivery of both Dox and siMRP1 using nanobody-displaying pSiNPs (NB-pSiNPs) represents a novel approach for the treatment of cancer with the potential to overcome drug resistance (Scheme 1). We investigate efficacy and potency of this novel nanocarrier in both 2D cancer cell culture and a 3D spheroid model. The use of spheroids – 3D cellular self-aggregates that emulate several physiological aspects of an in vivo tumor36 – provides a more rigorous and representative model of in vivo tumor characteristics when compared to cancer cell monocultures.
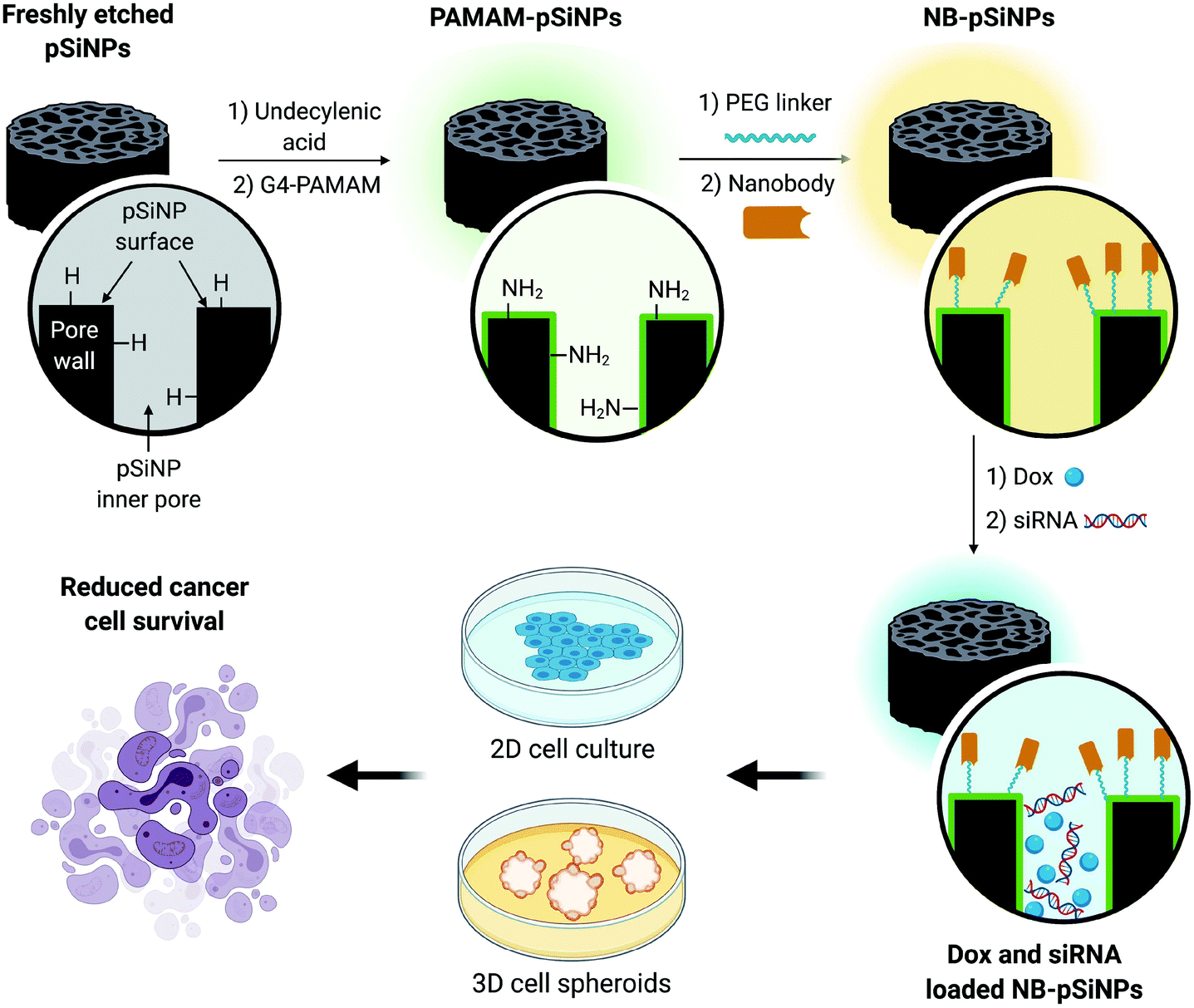 | ||
| Scheme 1 Schematic representation of the design of NB-pSiNPs and the in vitro evaluation of NB-pSiNPs loaded with doxorubicin (Dox) and siRNA against MRP1 in both 2D and 3D cell culture. Further information on the chemical functionalization steps can be found in the ESI (Scheme S1†). | ||
2. Results & discussion
2.1 Physicochemical characterization of pSiNPs
The fabrication of pSiNPs was reported from a previously described procedure.25 After electrochemical etching, a porous membrane was electropolished from the Si wafer and fractured into nanoparticles of broad size distribution via immersion in an ultrasonicator water bath. To obtain uniform nanoparticles, size selection via ultracentrifugation allowed for the collection of pSiNPs of ∼180 ± 5 nm in absolute ethanol (EtOH). The size of the pSiNPs was confirmed via images taken by transmission electron microscopy (TEM) (Fig. 1A) and analyzed using ImageJ. From our previous work, BET (Brunauer–Emmett–Teller) analysis was performed to evaluate the surface area and average pore size of pSiNPs. The N2 adsorption–desorption isotherm of pSiNPs showed a total surface area of 489 m2 g−1 and an average pore size of 19 nm.26 Freshly anodized pSi remains highly reactive due to the hydrogen-terminated surface. Hydrosilylation refers to the reaction of addition to the hydrogen reactive surface species with unsaturated compounds forming a solid and covalent Si–C bond. The stabilization of pSiNPs and the installment of carboxyl groups was completed when reacted with undecylenic acid via thermal hydrosilylation (UA-pSiNPs).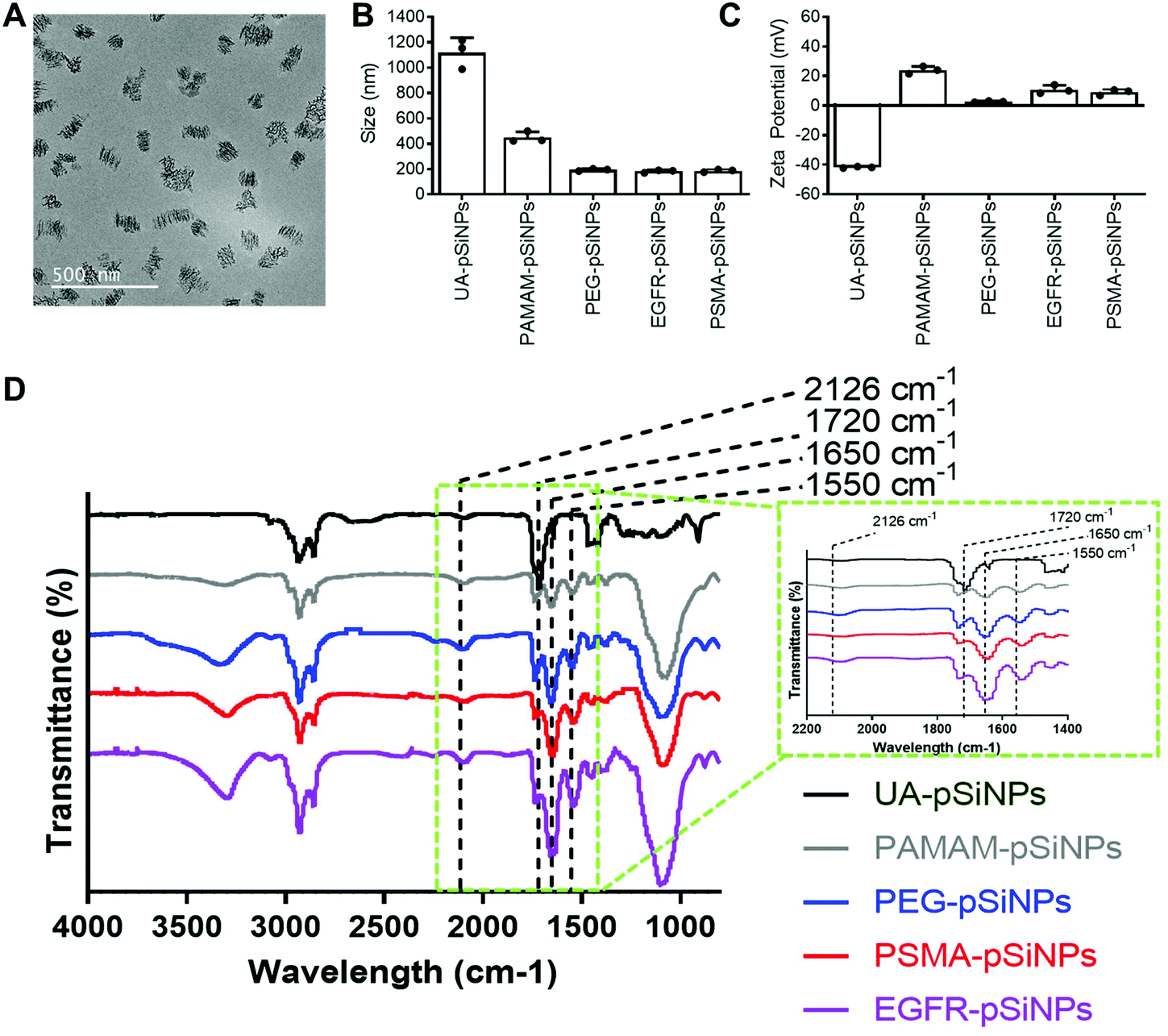 | ||
Fig. 1 Physicochemical characterization of the different functionalizations on pSiNPs by means of (A) TEM imaging of freshly etched pSiNPs (scale bar = 500 nm), (B) DLS/size measurements in PBS, (C) ζ-potential measurements. Data shown as mean ± S.D. (n = 3). (D) FTIR spectra of pSiNPs for each functionalization step. Key IR peaks seen in the spectra include the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations at 1720 cm−1 from the carboxyl groups on UA-pSiNPs, peaks corresponding to amide groups at 1650 cm−1 and 1550 cm−1, and a peak at 2126 cm−1 representing the C O stretching vibrations at 1720 cm−1 from the carboxyl groups on UA-pSiNPs, peaks corresponding to amide groups at 1650 cm−1 and 1550 cm−1, and a peak at 2126 cm−1 representing the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond from the DBCO group from the PEG linker. The inset located within the green dotted line box depicts a zoomed in region between 2200–1400 cm−1 on the spectra to highlight key IR peaks between different surface functionalizations. C bond from the DBCO group from the PEG linker. The inset located within the green dotted line box depicts a zoomed in region between 2200–1400 cm−1 on the spectra to highlight key IR peaks between different surface functionalizations. | ||
UA-pSiNPs were conjugated with a PAMAM(G4) dendrimer via EDC/NHS chemistry. In our previous work, we have shown that by functionalizing the pSiNPs surface with a PAMAM(G4) dendrimer, siRNA was loaded into the porous matrix, protecting it from degradation, and exhibited high silencing efficiency compared to other amine-rich molecules.25 The calculated siRNA loading capacity was 74%.
To prepare for the immobilization of nanobodies, PAMAM-pSiNPs were further modified with a short chain of poly(ethylene glycol) (PEG). PEG chains are not only known to increase the solubility and colloidal stability in buffer due to the hydrophilic ethylene glycol repeats,37 but also used to space the a active ligand away from the nanoparticle surface.38 The use of linkers promote greater flexibility of the active ligand and would allow for better access to cell surface receptors.38 Based on previous reports, a short chain length was chosen, as according to Yong et al. a 2-fold increase in binding was observed when a short 4 PEG unit linker was used when compared to a longer linker of 12 PEG units.39 Although the optimal PEG chain length may differ among different nanoparticles, the short PEG linker reported by Yong et al. was successfully incorporated onto the surface of PAMAM-pSiNPs. Thus, PAMAM-pSiNPs were reacted with a short PEG linker of 5 PEG units containing an N-hydroxysuccinimide (NHS) ester and dibenzocyclooctyne (DBCO) functional groups at either ends of the PEG chain (PEG-pSiNPs). The hydrodynamic diameter measured via dynamic light scattering (DLS) (Fig. 1B), ζ-potential (Fig. 1C) and Fourier Transform Infrared Spectroscopy (FTIR) spectra (Fig. 1D) were all studied for each surface modification step. The hydrodynamic diameter and ζ-potential of the modified pSiNPs were measured in phosphate buffered saline (PBS), an isotonic water-based salt-buffered solution. After the PEG linker reacted with the NHS ester with terminal amines on the dendrimer surface, PEG-pSiNPs showed a hydrodynamic diameter of 193 ± 8 nm in PBS. The clear reduction in size from 1118 ± 117 nm and 449 ± 43 nm obtained for UA-pSiNPs and PAMAM-pSiNPs, respectively, demonstrated a significant improvement in colloidal stability in PBS. The ζ-potential of PAMAM-pSiNPs was observed to be 21.5 ± 2.6 mV due to the abundance of surface amine molecules on the particle surface when compared to negatively charged UA-pSiNPs (−41.3 ± 0.4 mV) (Fig. 1C). Although the instalment of the PEG linkers was required for aforementioned reasons, an abundance of amine terminal groups from the PAMAM dendrimers was required to promote electrostatic adsorption and loading of siRNA into the pores of pSiNPs. PEG-pSiNPs retained a positive ζ-potential of 2.7 ± 0.4 mV where the optimal molar concentration of PEG towards 2 mg of PAMAM-pSiNPs was found to be 2 mM (Fig. S1†) – demonstrating that there was a balance in the ratio of PEG moieties and free amine groups for promoting electrostatic interaction with siRNA.
Successful surface modification with UA (black, Fig. 1D) was confirmed via IR analysis, where a distinct peak at 1720 cm−1 corresponded to the CO stretching vibrations from the carboxyl group. IR peaks at 1550 and 1650 cm−1 are key characteristics of the abundance of amide bonds present in PAMAM dendrimers (grey, Fig. 1D) and are representative of the N–H bending and CO stretching vibrations from these amide bonds. The appearance of a weak stretching at 2126 cm−1 in the IR spectra corresponds to the CC bond from the DBCO group at the end of the PEG linker (blue, Fig. 1D).
PEG-pSiNPs were further modified through the attachment of azide-functionalized nanobodies complementary to the EGFR and the PSMA receptors in cancer cells (NB-pSiNPs). Both nanobodies were modified with an azide positioned on a protruding loop within the protein. Due to the compact structure, the stability of nanobodies compared to other antigen-binding proteins is greatly increased.40 PEG-pSiNPs were reacted with EGFR (EGFR-pSiNPs) and PSMA (PSMA-pSiNPs) nanobodies in excess via copper-free click chemistry in order to exhaust all DBCO functional groups present on the PEG linkers. The hydrodynamic size of EGFR and PSMA-pSiNPs in PBS were observed to be 182 ± 10 nm and 186 ± 10 nm, respectively. In comparison to pSiNPs modified with conventional full antibodies, for which an increase in the hydrodynamic diameter was observed via DLS,26 no significant change in particle size was observed due to the small diameter of nanobodies. A slight increase in ζ-potential of EGFR-pSiNPs and PSMA-pSiNPs was observed at 10.9 ± 2.9 mV and 9.0 ± 1.9 mV, respectively. Furthermore, the observed increase in bending and stretching vibrations in the IR spectra (red, purple, Fig. 1D) at the amide peaks of 1550 cm−1 and 1650 cm−1, which are characteristics of protein immobilization, were attributed to the successful nanobody attachment. The IR spectra of EGFR and PSMA-pSiNPs showed a weak stretching at 2126 cm−1 attributed to residual DBCO groups, suggesting there were unreacted PEG groups after nanobody immobilization. This could be due to steric hindrance since conjugated nanobodies may hinder the accessibility to nearby adjacent DBCO-PEG groups. The supernatants after the reaction and subsequent washing steps were collected and the total amount of protein attachment was quantified via a bicinchoninic (BCA) protein assay and compared to the starting concentration of EGFR and PSMA nanobodies. The calculated amount of EGFR or PSMA nanobodies per nanoparticle was determined to be 1.22 × 104 or 1.28 × 104 molecules per nanoparticle, respectively. Further information on nanobody quantification per nanoparticle can be found in Table S1 and the ESI.†
To ensure that the NB-pSiNPs were stable, the nanoparticles were stored under simulated storage conditions (4 °C) in PBS at pH 7.4 and the hydrodynamic diameter was recorded via DLS over a 7-day period (Fig. S2A†) and 4-month period (Fig. S2B†) – where no significant change in size was measured (between 185–200 nm). Another key component in assessing the colloidal stability of NB-pSiNPs is the ability to maintain receptor specificity of the nanobodies after storage. To do so, C4–2B cells were incubated for 1 h with NB-pSiNPs stored for either 1, 3 or 7 days (Fig. S3†). No significant difference in retention at specific cell receptors was observed, underlining that the NB-pSiNPs retained their specific receptor selectivity. Taken altogether, the immobilization of both EGFR and PSMA complementary nanobodies demonstrates the versatility of the nanocarrier platform, able to react with any targeting moiety containing an azide functional group.
2.2 Loading capacity and release kinetics of pSiNPs
The high loading capacity of pSiNPs is a key characteristic that makes pSi an attractive biomaterial for drug delivery.24 NB-pSiNPs were evaluated for their capacity to load both Dox and siRNA molecules. pSiNPs were first immersed in a 1 mg mL−1 solution of Dox in RNAse-free water overnight at 4 °C, washed twice in RNAse-free water to remove loosely-bound Dox molecules, followed by incubating with siRNA molecules for 1 h at 4 °C. A loading capacity of 10.9 ± 0.6%, 12.3 ± 0.5%, 12.1 ± 0.6%, and 12.3 ± 0.5% w/w of Dox was calculated for PAMAM-pSiNPs, PEG-pSiNPs, EGFR-pSiNPs and PSMA-pSiNPs, respectively (Fig. 2A and Table S2†). The fact that the loading did not decrease after nanobody immobilization suggests that the nanobodies, despite their small molecular size (approximately 4 nm long and 2.5 nm wide32 as compared to the average pore size, which was measured to be 13 ± 3 nm), are predominantly localized on the outer NP surface and do not block the pores. When the Dox-loaded pSiNPs were incubated with the loading solution with siRNA targeting the MRP1 protein, a siRNA loading capacity of 20.9 ± 1.3%, 14.9 ± 0.2%, 13.5 ± 0.7%, and 13.8 ± 0.4% w/w was calculated for PAMAM-pSiNPs, PEG-pSiNPs, EGFR-pSiNPs and PSMA-pSiNPs, respectively (Fig. 2A and Table S2†). A decrease in siRNA loading was observed between the PAMAM-pSiNPs and the pSiNPs with the addition of the PEG linkers. This was to be expected as the availability of surface amines had been reduced through the conjugation of the PEG linker, reducing possible amine sites to promote electrostatic interactions with the phosphate backbone in siRNA molecules. Additionally, PEG has been known to reduce biomolecule surface interactions which would contribute to the decrease in siRNA loading.41 There was no significant difference in the loading of Dox or siRNA when nanobodies were conjugated to the PEG linkers.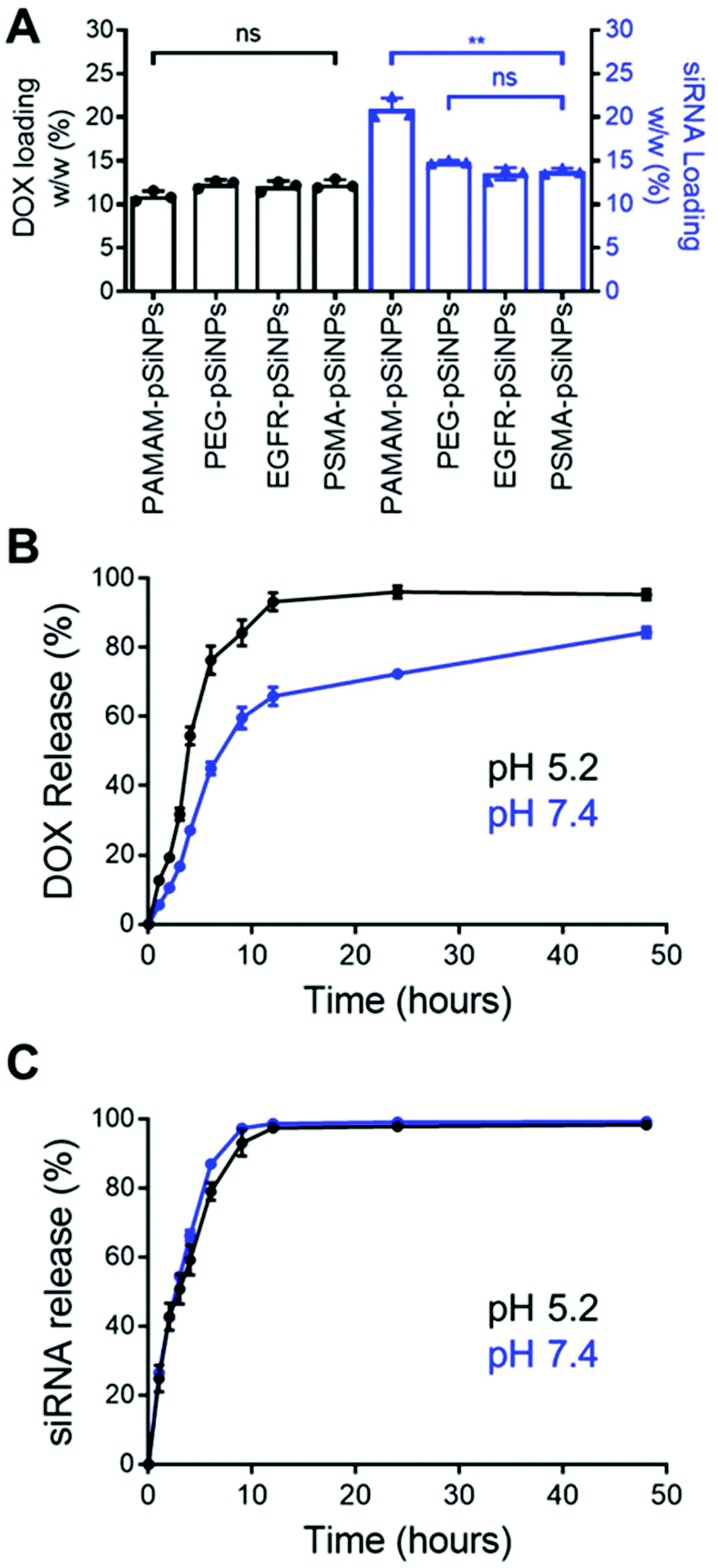 | ||
| Fig. 2 (A) Doxorubicin and siRNA loading (w/w) percentage in NB-pSiNPs. Data shown as the mean ± S.D. (n = 3, **p ≤ 0.01). Release profile of (B) Dox and (C) siRNA from PSMA-pSiNPs over 48 h in PBS at pH 5.2 (black) and pH 7.4 (blue) under agitation at 37 °C. Data shown as the mean ± S.D. (n = 4). | ||
In vitro release profiles were established for the NB-pSiNPs by incubating the particles in PBS at 37 °C and at both acidic pH (5.2), which is a characteristic of the acidic tumor microenvironment, and at neutral pH (7.4). Release kinetics of PSMA-pSiNPs were recorded over 48 h for both Dox and siMRP1 (Fig. 2B and C). At physiological pH, 45.1 ± 3.7% of Dox was released after 6 h while a faster release was seen over the same time at pH 5.2 (Fig. 2B, blue), reaching 76.3 ± 4.1% of released Dox (Fig. 2B, black). This pH-dependent release kinetics is attributed to the protonation of the primary amine group on Dox molecules at acidic pH, exhibiting better aqueous solubility and weaker bonding towards positively-charged pSiNPs.42,43 Since it is known that pSi remains stable under acidic conditions, we can confirm that the faster release profile of Dox at a pH of 5.2 was not due to nanoparticle degradation.44 There was no difference in siRNA release kinetics from PSMA-pSiNPs at acidic and physiological pH, with 97.5 ± 0.8% and 98.7 ± 0.4% of siRNA released after 12 h, respectively (Fig. 2C). The release of siRNA differs from our previous reports where after 12 h, only 13.2% of the siRNA had been released.25 We hypothesize that the reduction in amine terminals due to the attachment of PEG linkers led to a reduction in electrostatic interaction between the nanoparticle surface and the phosphate backbone of siRNA molecules – which is supported by the lower positive zeta potential observed in both PEG-pSiNPs and NB-pSiNPs. Additionally, there was no significant difference in release kinetics between PSMA-pSiNPs and EGFR-pSiNPs or PEG-pSiNPs, confirming that the presence of nanobodies or nanobody type did not affect the release behavior of the pSiNPs (Fig. 2 and Fig. S4†). The pH-responsive release pattern for Dox observed in NB-pSiNPs, with a lower extent of release at physiological pH, would enable more Dox molecules to be retained within the nanoparticles upon reaching the tumor site. Furthermore, the faster release of siRNA in comparison to Dox could be used to our advantage to favor a faster siRNA complexation with the RNA-induced silencing complex inside the cytoplasm, leading to an earlier onset for gene silencing. Additionally, the release of siMRP1 to inhibit MRP1 expression has been shown to improve therapeutic outcomes by sensitizing cancer cells to Dox.17,45 Therefore, the release kinetics for both Dox and siRNA observed are suitable for systemic administration and in line with previous pSiNP formulations encapsulating small drug molecules.46–48 For example, increased accumulation of pSiNPs has been observed in a glioblastoma model after 2 h post-intravenous injection, limiting any off-target toxicity from premature release of held cargo.46 Thus, for NB-pSiNPs, <10% of Dox was released under physiological conditions after 2 h, which would greatly limit the amount of Dox released prior to accumulation at the tumor site.
2.3 Cellular association of pSiNPs
The expression of EGFR and PSMA were assessed via Western blotting in three separate cell lines: (i) C4–2B – a clinically relevant cell derivative subline of the human prostate cancer cell line LNCaP,49 (ii) C32 – a human melanoma cell line, and (iii) HEK293-WT – a human embryonic kidney cell line (Fig. 3A and Fig. S5†). Overexpression of EGFR was observed in C4–2B and C32 cells when compared to HEK293-WT cells. Conversely, PSMA was only detected in C4–2B cells.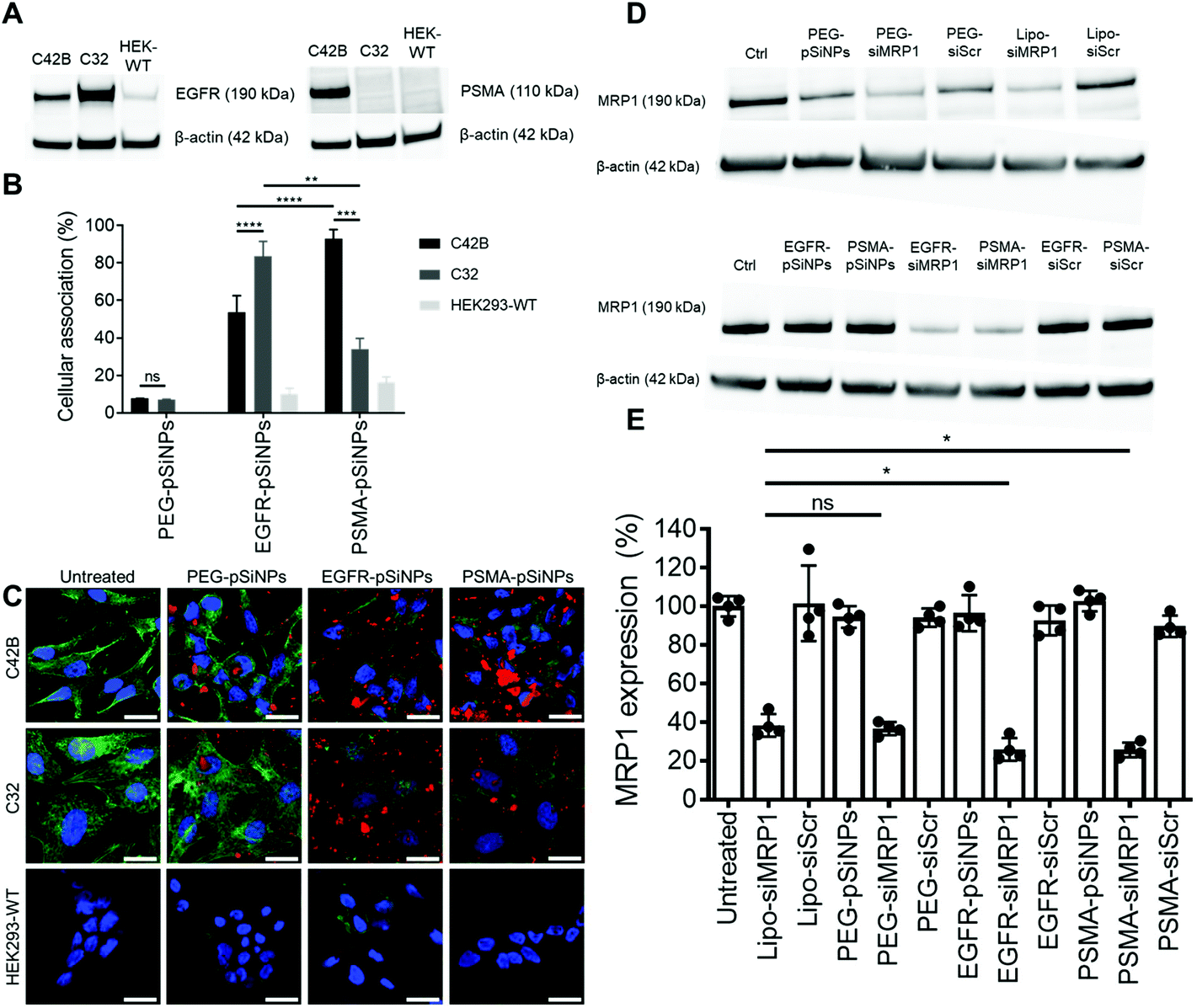 | ||
| Fig. 3 (A) EGFR and PSMA expression via Western blotting on C4–2B, C32 and HEK293-WT cell lines. (B) Cellular association studies of NB-pSiNPs analyzed via flow cytometry where cellular association was compared to a negative control of untreated cells. Data shown as a mean ± S.D; (n = 4, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001). (C) Confocal microscopy images of C4–2B, C32 and HEK293-WT cells treated for 1 h with NB-pSiNPs. The nuclei were stained with Hoechst 33342 (blue, the cytoskeleton stained with phalloidin-TRITC (green), and NB-pSiNPs labeled with Cy5 (red) (scale bar = 40 μm). (D and E) MRP1 expression in C4–2B cells as measured by (D) Western blotting and (E) quantified when treated either with 50 μg mL−1 of pSiNPs or positive and negative controls including Lipofectamine (Lipo). Data shown as a mean ± S.D. (n = 4, *p ≤ 0.05). | ||
EGFR-pSiNPs and PSMA-pSiNPs (and PEG-pSiNPs) were labeled with Cyanine-5 (Cy5) to assess their retention capabilities towards their respective cell surface receptors. Their cellular association was evaluated against the three cell lines via flow cytometry and confocal microscopy. Cellular association was measured as a percentage of positive cells when compared to an untreated control. There was an increase in cellular association when cells were treated with NB-pSiNPs (Fig. 3B and Fig. S6†). For all three cell lines, PEG-pSiNPs had associated with <8% of cells. Conversely, for C32 and C4–2B cells, 84 ± 8% and 54 ± 9% of cells had EGFR-pSiNPs associated with the cells, respectively. Furthermore, for C32 and C4–2B cells, 34 ± 6% and 93 ± 5% of cells had PSMA-pSiNPs associated with them after 1 h incubation. The cellular association data demonstrated that there was preferential accumulation of EGFR-pSiNPs with C32 cells, understandably as the melanoma cells showed a high level of EGFR expression (Fig. 3A and Fig. S5†). Similarly, for C4–2B cells, PSMA-pSiNPs showed more association with the prostate carcinoma cells in which a high level of PSMA expression was evident as seen via Western blotting (Fig. 3A and Fig. S5†). Thus, the >90% cellular association in C4–2B compared to the 34% association in C32 cells, was most likely due to the expression of PSMA – demonstrating that PSMA-pSiNPs were better retained at the complementary surface receptor. Furthermore, the NB-pSiNPs were investigated with two breast cancer cell lines (MDA-MB-231BO and AT3 cells) to further verify the receptor selectivity of the nanoparticles (Fig. S7†). MDA-MB-231BO are a bone metastasis variant model of triple negative breast cancer that has shown high levels of EGFR expression,26 while AT3 cells are a breast cancer cell line of mouse origin.50 Similar trends as those observed in the C32 cells were observed in the MDA-MB-231 cells where EGFR-pSiNPs (57 ± 6%) showed higher cellular association compared to PEG-pSiNPs (13 ± 6%) and PSMA-pSiNPs (32 ± 5%). AT3 cells acted as a further negative control, where cellular association for all three nanoparticle variants was <11%. As the AT3 cells were of mice origin, the NB-pSiNPs did not selectively interact with said cells most probably due to a lower affinity of the human nanobodies towards mice EGFR or PSMA receptors as a result of interspecies differences. Interestingly, for C32 and MDA-MB-231BO cells, there was significant association of PSMA-pSiNPs as compared to those of HEK293-WT and AT3 cells. We hypothesize that this could be due to the expression of folate receptor-α (FOLR1) as it has been reported that there is a weak correlation between FOLR1 and PSMA (also known as folate hydrolase 1, FOLH1).51 As FOLR1 expression is prevalent in MDA-MB-231 cells52 and melanoma cells,53 the close relation to FOLH1 led to significant cellular association (34 ± 6% for C32 and 32 ± 5% for MDA-MB-231BO cells) when compared to HEK293-WT and AT3 cells due to the quasi-specificity of PSMA-pSiNPs. To further corroborate the flow cytometry data, cellular association was investigated via confocal microscopy. Confocal microscopy images displayed a similar trend as the flow cytometry data, where increased particle accumulation can be seen when C4–2B and C32 cells were treated with EGFR-pSiNPs and PSMA-pSiNPs as compared to PEG-pSiNPs (Fig. 3C). Far less particle accumulation was detected in HEK293-WT images – supporting the lower cellular association percentages as determined via flow cytometry. Therefore, from the cellular association data generated via flow cytometry and confocal microscopy, it was observed that PSMA-pSiNPs showed a greater extent of association compared to EGFR-pSiNPs, suggesting that PSMA may be a better receptor target, especially in the case of prostate cancer.
2.4 Knockdown of MRP1 in C4–2B cells
NB-pSiNPs were assessed in the delivery of siMRP1 towards C4–2B cells to study the downregulation of MRP1 via Western blotting. Lipofectamine RNAiMAX, a commercial transfection reagent, was used as a point of comparison and positive control. After 72 h, MRP1 expression was inhibited by 64 ± 3% when incubated with PEG-pSiNPs for 4 h compared to a reduction in expression of 62 ± 6% for Lipofectamine at the same concentration of siRNA loaded in the pSiNPs (52.6 nM) (Fig. 3D and E). In comparison, there was a significant decrease in MRP1 expression of 74 ± 6% and 74 ± 4% when incubated with EGFR-pSiNPs and PSMA-pSiNPs, respectively. No significant gene silencing of MRP1 was observed when the pSiNPs were loaded with a scrambled sequence of siRNA, validating that the downregulation of MRP1 was sequence specific and through the effective delivery of siMRP1 by pSiNPs.2.5 Cytotoxicity of Dox/siRNA loaded pSiNPs
Cellular viability of the dual delivery of Dox and siRNA via pSiNPs was evaluated using an ATP activity-based luminescent cell viability assay (Fig. 4). C4–2B, C32 and HEK293-WT cells were incubated with PEG-pSiNPs, EGFR-pSiNPs or PSMA-pSiNPs alone or loaded with either Dox, siRNA (either siMRP1 or scrambled siRNA) or both therapeutics (Dox and siMRP1) at a particle concentration of 50 μg mL−1 for 1 h before the cells were copiously washed to remove any free and unbound pSiNPs. A particle concentration of 50 μg mL−1 (∼2.4 μM of Dox and ∼5 μM of siMRP1) displayed similar cytotoxicity towards C4–2B cells when compared to a concentration of 100 μg mL−1 – thus, 50 μg mL−1 was the chosen particle concentration for all experiments (Fig. S8†). No cytotoxicity was observed when treated with the functionalized pSiNPs only or when loaded with siMRP1 or a scrambled sequence (Fig. S9†). After 72 h, 32 ± 3% of C4–2B cells reminded alive when treated with PSMA-pSiNPs loaded with Dox. In comparison, when loaded with both Dox and siMRP1, 23 ± 1% of cells remained viable (Fig. 4A). When assessed after 96 h, C4–2B viability was further decreased to 14 ± 2% after treatment with PSMA-pSiNPs loaded with both therapeutics (Fig. 4B). No difference in cytotoxicity was observed between the positive controls of free Dox, free Dox and siMRP1 or a scrambled sequence was observed after incubating with C4–2B cells after 96 h (Fig. S10†). The decrease in cell viability between 72 and 96 h was attributed to the effective downregulation of MRP1, sensitizing the C4–2B cells to Dox, indicating that the downregulation of MRP1 presents itself to be a viable approach in reducing chemoresistance. Thus, the cell viability for C32 and HEK293-WT cells was only evaluated after 96 h. | ||
| Fig. 4 Cell viability as evaluated using an ATP-based luminescent cell viability assay. NB-pSiNPs (50 μg mL−1) loaded with Dox and/or MRP1 (and appropriate controls) were incubated with cells for 1 h and then washed away with copious amounts of PBS. (A and B) Cell viability results for C4–2B prostate cancer cells after (A) 72 h and (B) 96 h. (C) Cell viability results for C32 melanoma cells after 96 h. (D) Cell viability results for HEK293-WT cells after 96 h. Cell viability was compared with untreated cells and cells treated with 10% DMSO which represented the negative and positive control, respectively. A matching concentration of free Dox (2.4 μM) to the amount released in 1 h by NB-pSiNPs determined in the in vitro release profile was used as a control. Data shown as a mean ± S.D. (n = 4, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001). | ||
C32 cells treated with EGFR-pSiNPs loaded with Dox and siMRP1 displayed a cell viability of 29 ± 6% (Fig. 4C). When loaded with both therapeutics, PSMA-pSiNPs exhibited a cell viability of 38 ± 5%. The difference in cell viability in both cells, C4–2B and C32, for EGFR-pSiNPs and PSMA-pSiNPs correlate with the differences seen in particle accumulation and the receptor expression pattern. This observation was further supported by the HEK293-WT cell viability, where cell viability towards PEG-pSiNPs (48 ± 3%) was similar to EGFR-pSiNPs (40 ± 2%) and PSMA-pSiNPs (45 ± 5%). In all cases, the nanoparticles outperformed free Dox at a matching concentration (2.4 μM) – demonstrating that the pSiNP delivery system was effective in increasing cellular cytotoxicity.
2.6 Cellular association and cytotoxicity of pSiNPs in C4–2B spheroids
Further investigation into the efficacy and potency of the NB-pSiNPs, the cellular association and cytotoxicity were evaluated in a C4–2B spheroid model. Spheroids are 3D cellular architectures that recapitulate in vivo tumor characteristics with respect to growth kinetics, cell signaling pathway activity and gene expression, as well as hypoxic and proliferative gradients.54 Therefore, spheroids provide a cost-effective, high-throughput pseudo-preclinical model with greater clinical relevance than conventional monolayer cell assays.C4–2B spheroids were treated with PEG-pSiNPs, EGFR-pSiNPs or PSMA-pSiNPs for 1 h before numerous washes with PBS to remove any free or loosely bound pSiNPs from the spheroid culture. After 24 h, when the spheroids were disassociated, only 9 ± 2% of cells display fluorescence signal when treated with PEG-pSiNPs and analyzed via flow cytometry (Fig. 5A). Conversely, when treated with EGFR-pSiNPs or PSMA-pSiNPs, the percentage of cellular association was observed to be 73 ± 8% and 88 ± 5%, respectively. Confocal images further verified that an increase in fluorescence representative of pSiNPs was witnessed when the spheroids were treated with EGFR-pSiNPs or PSMA-pSiNPs when compared to PEG-pSiNPs (Fig. 5B), where more fluorescence was found in the core of the spheroids. This was more evident when the spheroids were treated and fixed after 1 h, where there was a lack of fluorescence in the core of the spheroids when treated with PEG-pSiNPs (Fig. S11†).
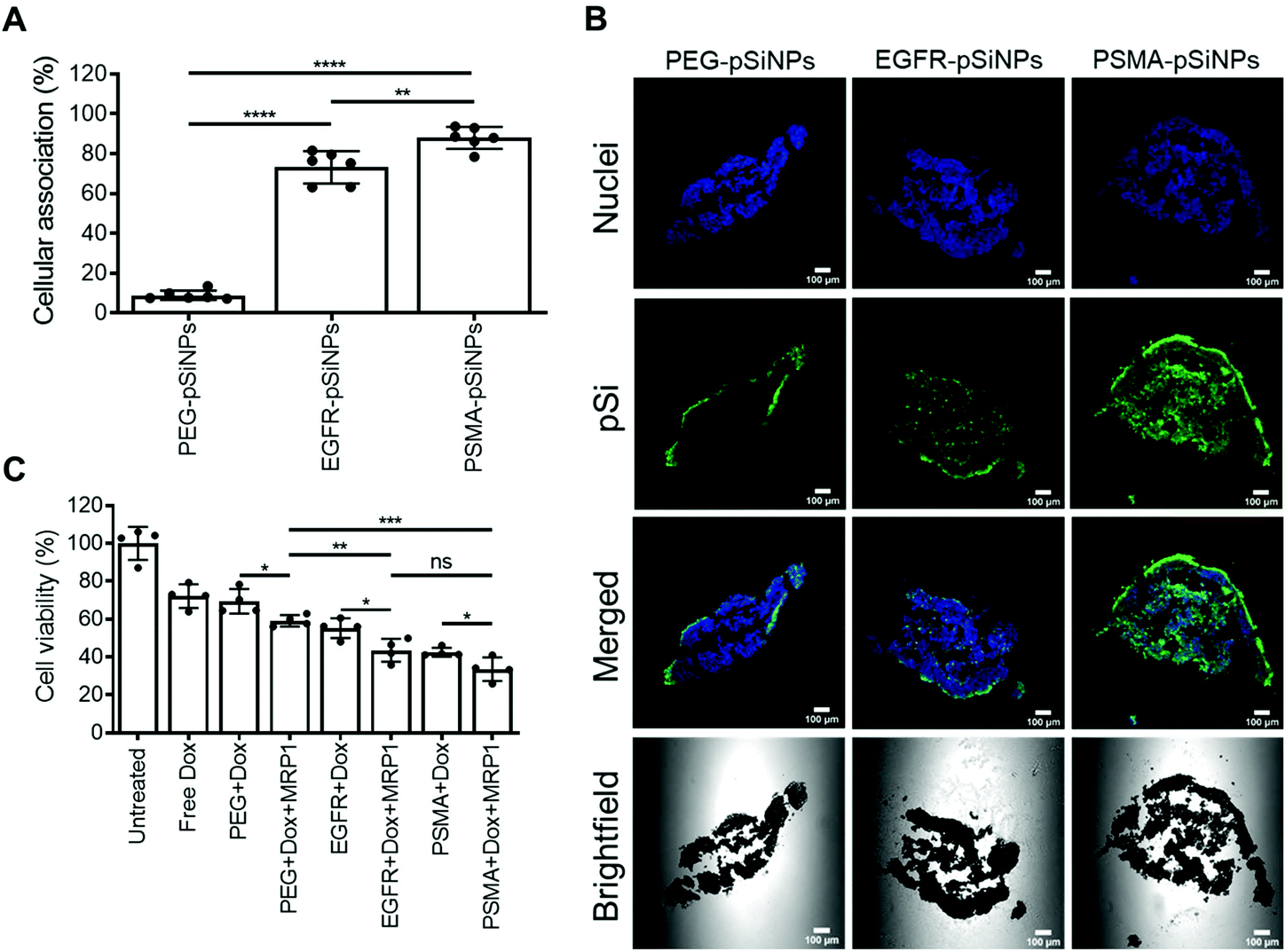 | ||
| Fig. 5 (A) Cellular association studies of nanobody-displaying pSiNPs (NB-pSiNPs) analyzed by flow cytometry. Cellular association was compared to a negative control of untreated C4–2B spheroids. Data shown as a mean ± S.D. (n = 6, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001). (B) Confocal microscopy images of the middle slice of C4–2B spheroids treated for 1 h with NB-pSiNPs, washed and then fixed after 24 h, where the nuclei were stained with Hoechst 33342 (blue), and pSiNPs labeled with Cy5 (green) (scale bar = 100 μm). (C) Cell viability results C4–2B spheroids treated with NB-pSiNPs (50 μg mL−1) loaded with Dox and/or MRP1 for 1 h and further incubated for a total of 96 h. Cell viability was compared with untreated cells and cells treated with 10% DMSO which represented the negative and positive control, respectively. A matching concentration of free Dox (2.4 μM) to the amount released in 1 h by NB-pSiNPs determined in the in vitro release profile was used as a control. Data shown as a mean ± S.D. (n = 4, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001). | ||
The cytotoxicity of the dual therapeutic delivery system was also studied in the C4–2B spheroids. The spheroids were incubated with pSiNPs loaded with either Dox, siRNA, or a combination of therapeutics for 1 h and cell viability was assessed after 96 h (Fig. 5C and Fig. S12†). A similar trend was observed for the 2D culture and spheroids. PEG-pSiNPs, EGFR-pSiNPs and PSMA-pSiNPs loaded with Dox showed that 69 ± 7%, 55 ± 5% and 43 ± 2% of cells remained viable, respectively. The downregulation of MRP1 when delivered with Dox using PEG-pSiNPs led to a further decrease in cell viability (59 ± 3% viable), supporting the results obtained in the 2D monocultures. An increase in cytotoxicity was observed when the two therapeutics were delivered using EGFR-pSiNPs (44 ± 6% viable cells) and PSMA-pSiNPs (34 ± 6% viable cells). Therefore, the enhanced cellular association in spheroids resulted to the higher cytotoxicity induced by EGFR-pSiNPs and PSMA-pSiNPs compared to spheroids treated with PEG-pSiNPs. Collectively, these results show that the enhanced retention of NB-pSiNPs at cell receptor sites allowed for increased cellular uptake to effectively deliver both doxorubicin and siMRP1.
In the present proof-of-principle study, we aimed to demonstrate that NB-pSiNPs are an effective nanoformulation, having shown the different nanobodies were preferentially retained on different cancer cell types depending on receptor expression. Colloidally stable nanoparticles are critical for clinical translation. NB-pSiNPs were shown to be stable over a four-month period, retaining their selectivity to their complementary receptor. Furthermore, we also show that NB-pSiNPs successfully penetrate C4–2B spheroids and co-deliver siMRP1 and Dox for improved cytotoxicity compared to relevant controls. However, the current spheroid model is limited to a singular cell type, where the human tumor microenvironment consists of many different cell types as well as dense stroma surrounding tumor cells.55 In the future, the specificity of NB-pSiNPs should be studied further, especially in an in vivo setting, where the biodistribution of NB-pSiNPs is important in assessing targeting capabilities.
3. Conclusion
In conclusion, we fabricated NB-pSiNPs with PAMAM and PEG coatings that display preferential cellular association due to increased retention at EGFR or PSMA receptors by the nanobody moieties. The nanoparticles were demonstrated to deliver both Dox and siMRP1 in both cancer cell monocultures and spheroids. Through the dual delivery of both a chemotherapeutic and siRNA, an increase in cytotoxicity was observed when MRP1 was downregulated by siMRP1. PSMA-pSiNPs decreased MRP1 expression by 74 ± 4%. When comparing PSMA-pSiNPs loaded with only Dox or with both Dox and siRNA, cellular viability in prostatic cancer cells decreased from 29 ± 5% to 14 ± 2%, respectively, displaying a 2-fold decrease when the drug efflux was reduced through downregulation of MRP1. A similar phenomenon was observed in a 3D prostatic cancer spheroid model, where cellular viability decreased from 43 ± 2% to 34 ± 6%, respectively. Altogether NB-pSiNPs appear to be an effective dual drug delivery system for the simultaneous delivery of siRNA and chemotherapeutics, and are a promising addition to cancer therapy tools due to their enhanced cancer cell selectivity and retention capabilities.4. Experimental
4.1 Materials
Single-crystalline silicon wafers were purchased from Siltronix (Archamps, France). Hydrofluoric acid (HF; 49%) was purchased from J. T. Baker (Center Valley, PA, USA). Absolute ethanol (EtOH), dimethylformamide (DMF), dichloromethane (DCM) and triethylamine (TEA) were purchased from Merck (Australia). Doxorubicin hydrochloride (Dox, 15007) was purchased from Sapphire Bioscience. DBCO-PEG5-NHS ester (A102P) was purchased from Click Chemistry Tools (Scottsdale, AZ, USA). 7-Aminoactinmycin D (7-AAD) was purchased from BD Sciences (Franklin Lakes, NJ, USA). RPMI-1640 (11875119), DMEM (11966025), Opti-MEM (31985088), GlutaMAX (35050061), Ultrapure RNAse-free water (10977015), Lipofectamine RNAiMAX (13778075) were purchased from Life Technologies (Mulgrave, VIC, Australia). siMRP1 (labeled with Cy5) (5′ GAGGCUUUGAUCGUCAAGUTT 3′) and a control sequence (5′ UUCUCCGAACGUGUCACGUTT 3′) were purchased from GenePharma Co. Ltd (Shanghai, China). Undecylenic acid (124672), 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide hydrochloride (EDC HCl, 03459), N-hydroxysuccinimide (NHS, 130672) and all other chemicals were purchased from Sigma Aldrich unless stated otherwise.4.2 Porous silicon nanoparticle preparation
pSiNPs were prepared by anodic electrochemical etching from a p-type silicon wafer (0.00055–0.001 Ω cm, Siltronix, France) in an electrolyte composed of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 HF(49%):EtOH as previously described.25 The perforated etching procedure alternated between current densities of 5 mA cm−2 for 20 s and 139 mA cm−2 for 0.2 s for 1000 cycles. The perforated layer was then removed from the wafer by etching at a constant current density of 139 mA cm−2 for 60 s in a 1:1 HF(49%):EtOH solution. The freestanding pSi film was sonicated in an ultrasonicator water bath in absolute EtOH for 24 h into different sized nanoparticles. pSiNPs were collected by centrifugation. First, the nanoparticles were centrifuged at 2000 RCF for 6 min, and the supernatant was collected. Afterward, the supernatant was centrifuged at 22000 RCF for 10 min. The two-step process was repeated an additional time and the final pellet consisted of ∼180 nm nanoparticles.
:1 HF(49%):EtOH as previously described.25 The perforated etching procedure alternated between current densities of 5 mA cm−2 for 20 s and 139 mA cm−2 for 0.2 s for 1000 cycles. The perforated layer was then removed from the wafer by etching at a constant current density of 139 mA cm−2 for 60 s in a 1:1 HF(49%):EtOH solution. The freestanding pSi film was sonicated in an ultrasonicator water bath in absolute EtOH for 24 h into different sized nanoparticles. pSiNPs were collected by centrifugation. First, the nanoparticles were centrifuged at 2000 RCF for 6 min, and the supernatant was collected. Afterward, the supernatant was centrifuged at 22000 RCF for 10 min. The two-step process was repeated an additional time and the final pellet consisted of ∼180 nm nanoparticles.
4.3 Preparation of PAMAM-pSiNPs
PAMAM-pSiNPs were prepared following a previously described method.25 After pSiNPs collection, the stabilization of the pSi surface and ready access to a carboxyl group was employed. Briefly, a 10 mL aliquot of undecylenic acid was liquefied under N2 to remove all trace water and oxygen from the system for 20 min. Approximately 10 mg of pSiNPs was added to the solution and allowed to reach for 24 h at 120 °C under a gentle N2 stream. Afterward, the undecylenic acid functionalized pSiNPs were washed 1× in DCM and 3× in absolute EtOH by centrifugation.The undecylenic acid functionalized pSiNPs were washed 2× in DMF to remove trace EtOH. A 40 mM solution of 40 mM solution of EDC HCl with 0.2 equiv. of TEA and a 20 mM solution of NHS were prepared separately in DMF. 5 mg of pSiNPs were resuspended in a 1:1 ratio of EDC:NHS. Afterward, a solution of generation 4 polyamidoamine (PAMAM) dendrimers was added to the reaction mixture for a final concentration of 10 mM. The reaction was left under agitation for 3 h at RT. The newly functionalized PAMAM-pSiNPs were washed 2× in ice cold MilliQ water, 2× in 70% EtOH and 2× in absolute EtOH. The PAMAM-pSiNPs were stored in EtOH until further use.
4.4 Expression and purification of azide functional nanobodies
DNA coding sequences for 9G856 and JVZ-00757 nanobodies, containing C-terminal 6xHis tags, were synthesized by GeneArt (Thermo Fisher) and ligated into pET26b (Novagen) after the PelB coding sequence. Amber codons were inserted by site directed mutagenesis, substituting amino acid position G42 for 9G8 and G44 for JVZ-007. Mutations were carried out by PCR using PrimeSTAR Max DNA polymerase (TaKaRa) and overlapping primers containing substituting base pairs. PCR products were confirmed on a 1% agarose gel and product treated with DpnI (NEB), following transformation into DH5α cells, grown over night at 37 °C on LB agar with kanamycin (50 μg mL−1). Single colonies were grown over night at 37 °C 200 RPM in LB with kanamycin and plasmids purified using NucleoSpin Plasmid EasyPure kit (Macherey-Nagel). Mutations were confirmed by sequencing.Mutated plasmids were co-transformed with pEVOL-pAzF (gifted by Peter Schultz (Addgene plasmid # 31186))58 into competent BL21(DE3) cells and grown over night at 37 °C on LB agar with kanamycin (30 μg mL−1) and chloramphenicol (25 μg mL−1). Single colonies were grown over night at 37 °C and 200 RPM in LB with kanamycin and chloramphenicol. 500 mL of TB containing kanamycin and chloramphenicol was inoculated with 5 mL overnight culture and grown to OD600 ∼ 1, then induced with 0.5 mM IPTG and 0.05% arabinose and media supplemented with 1 mM 4-azido-L-phenylalanine (Iris Biotech GMBH). Induced culture incubated over night at 20 °C and 200 rpm. Cells were harvested by centrifugation and periplasmic extracts prepared by means of chloroform extraction as previously described.59
Cleared periplasmic extract was purified by affinity chromatography following recommended protocol for HisTrap HP (GE). Imidazole was removed from eluate by dialysis.
4.5 Functionalization of EGFR-PEG-PAMAM(G4)-pSiNPs and PSMA-PEG-PAMAM-pSiNPs
PAMAM-pSiNPs were washed 2× in MilliQ water to remove any trace EtOH. PAMAM-pSiNPs were then resuspended in 900 μL of 0.1 M NaHCO3 buffer at pH 8.3. 100 μL of 20 mM of DBCO-PEG5-NHS Ester for a final concentration of 2 mM. PAMAM-pSiNPs were left under agitation with DBCO-PEG5-NHS ester for 24 h at RT. The PEG-PAMAM-pSiNPs were then washed 2× in absolute EtOH and 2× in MilliQ water to remove unbound PEG from the nanoparticles.PEG-PAMAM-pSiNPs (PEG-pSiNPs) were incubated with azide-functionalized anti-EGFR or anti-PSMA nanobodies in PBS (pH 7.4) for 24 h at 4 °C. 1 mg of PEG-pSiNPs was reacted with a 0.4 mg mL−1 solution of anti-EGFR or anti-PSMA nanobody in 1 mL of PBS for 24 h at 4 °C. Afterward, the EGFR-PEG-PAMAM-pSiNPs (EGFR-pSiNPs) and PSMA-PEG-PAMAM-pSiNPs (PSMA-pSiNPs) were washed 3× with PBS and stored until further use.
For fluorescence labeling of PEG-pSiNPs, EGFR-pSiNPs, and PSMA-pSiNPs, prior to the nanobody attachment, 5 mg of PEG-pSiNPs were washed 2× in MilliQ water and resuspended in 980 μL of 0.1 M NaHCO3. 20 μL of 20 mM Cyanine-5-NHS Ester (Cy5, Lumiprobe) was added dropwise and left under agitation for 3 h at RT. The Cy5-labeled PEG-pSiNPs were washed 3× in MilliQ water to separate the free Cy5-NHS Ester from the nanoparticles.
4.6 Loading of doxorubicin and siRNA in pSiNPs
200 μg of PEG-pSiNPs, EGFR-pSiNPs, and PSMA-pSiNPs were washed 2× in RNAse-free water. pSiNPs were then dispersed in a 200 μL of Dox (1 mg mL−1) loading solution and left under agitation for 24 h at 4 °C. After drug loading, the particles were pelleted via ultracentrifugation, and the supernatant was collected for quantification of the amount of Dox loaded. pSiNPs were washed 2× in RNAse-free water to remove any loosely bound Dox molecules. Subsequently, Dox-loaded pSiNPs were dispersed in 200 μL of Cy5-tagged siRNA for 1 h at 4 °C. Dox/siRNA-loaded pSiNPs were then pelleted and the supernatant collected for further quantification of the amount of siRNA loaded. Dox/siRNA-loaded pSiNPs were washed 2× in RNAse-free water to remove any loosely bound siRNA molecules. The loading amount of Dox and siRNA in the pSiNPs were determined by subtracting the amount of Dox/siRNA in the corresponding supernatants and washing steps from the initial amount of Dox/siRNA added to the pSiNPs. The supernatants were read in a black 96-well plate (100 μL per well) in duplicates for Dox (λex = 470 nm, λem = 595 nm) and Cy5 (λex = 649 nm, λem = 666 nm) on a PerkinElmer EnSpire multimode plate reader. The siRNA loading supernatant was also measured via UV-VIS (Nanodrop 2000, Thermo Fisher) to cross validate the siRNA loading amount. All loading samples were done in triplicates (n = 3).4.7 Release kinetics
100 μg of PEG-pSiNPs, EGFR-pSiNPs, and PSMA-pSiNPs were resuspended in 200 μL of PBS (either pH 5.0 or 7.4) and left under gentle agitation in a 37 °C shaking incubator. At time points of 0, 1, 2, 3, 4, 6, 9, 12, 24 and 48 h, the pSiNPs were pelleted via ultracentriguation at 25000 RCF for 5 min. 100 μL of the supernatant was removed and replaced with 100 μL of prewarmed PBS, briefly sonicated (∼2 s) and placed back into the shaking incubator. The supernatant were then transferred into a black 384-well plate (25 μL per well) and read on a fluorescence plate reader for Dox and Cy5 fluorescence, where an average of the four wells was used as the released amount at the set time point. The total release percentage was calculated as a percentage from the total loaded amount for Dox and Cy5-siRNA as established previously. The release supernatant was also measured via UV-Vis to corroborate the in vitro siRNA release profile. All experiments were done in quadruplicates (n = 4).
4.8 Transmission electron microscopy
pSiNPs were imaged by transmission electron microscopy (JEOL JEM-2100F) equipped with a field emission gun. The pSiNPs samples were deposited on Formvar film-coated copper grids (ProSciTech, Australia). Images were acquired at 200 kV accelerating voltage.4.9 Dynamic light scattering and ζ potential measurements
The mean hydrodynamic diameter of pSiNPs, size distribution, polydispersity index (PDI), and ζ-potential of pSiNPs were analysed by dynamic light scattering using a Zetasizer Nano ZX (Malvern, UK). A scattering angle of 173° and a temperature of 25 °C were used with pSiNPs dispersed in 0.1 M PBS.4.10 Fourier transform infrared spectroscopy
pSiNPs that were stored in PBS were washed 2× in MilliQ water to remove any free salt in the solution and resuspended in absolute EtOH. A 2 μL aliquot of pSiNPs (1 mg mL−1) in EtOH was spotted on a flat high resistivity (3–6 Ω cm) p-type silicon wafer and air-dried. Analysis was conducted on a Hyperion 1000 Fourier transform infrared spectrophotometer microscope coupled to a Vertex 70 IR source (Bruker, Germany) and a liquid-N2-cooled MCT detector. Spectra were acquired between 650 and 4000 cm−1 at a resolution of 4 cm−1 for 64 scans.4.11 ICP-OES analysis to determine pSi concentration
A 0.5 mL aliquot of sample was warmed to dryness before being digested in 1 mL of 69% HNO3 and 2–3 drops of 48% HF before being made up to a suitable volume. Once the reaction had ceased, a calculated quantity of triethanolamine was added to neutralize the sample to pH 7. The sample was then diluted as required and analyzed by a Varian 730-ES axial ICP-OES. Certified multi-element solutions were used to check the accuracy of the calibration standards and the method used.4.12 Cell culture
C4–2B were maintained in RPMI 1640 medium supplemented with 10% FBS. C32 human melanoma cells were maintained in RPMI 1640 medium supplemented with 10% FBS and 1% GlutaMAX. HEK 293-WT (ATCC CRL-1573) human embryonic kidney cells were maintained in DMEM supplemented with 5% FBS. MDA-MB-231BO and AT3 (ATCC CRL-2375) cells were maintained in DMEM supplemented with 10% FBS and 1% GlutaMAX. Prof. Lisa Butler (Prostate Cancer Research Group at the South Australian Health and Medical Research Institute) kindly provided C4–2B human prostate carcinoma cell line. Prof. Claudine Bonder (Vascular Biology Laboratory at the Centre for Cancer Biology) kindly provided the C32 human melanoma cell line. Dr Jacqui McGovern (Institute of Health and Biomedical Innovation) kindly provided the MDA-MB-231BO cell line. Cells were cultured at 37 °C and 5% CO2 and experiments were conducted on cells that were passaged at least twice after thawing from the frozen stock. Cells were discarded after 10 subsequent passages, with a new frozen stock thawed afterward. Cells were routinely tested negative for mycoplasma contamination by using PlasmoTest™ Mycoplasma Detection Kit (rep-pt1, Invivogen, San Diego, CA, USA).4.13 Colloidal stability of NB-pSiNPs determined via DLS and fluorescence plate reader
The mean hydrodynamic diameter was measured as per section 4.9. 1 mg of PEG-pSiNPs, EGFR-pSiNPs or PSMA-pSiNPs was suspended in 1 mL of PBS and stored at 4 °C, protected from light. At day 1, 3, and 7, 100 μL was removed from the stock solution and 900 μL of PBS was added to the sample. The particles were then transferred to a polystyrene cuvette for hydrodynamic diameter measurements via DLS.To measure cell receptor specificity of NB-pSiNPs, 500 μg of Cy5-tagged PEG pSiNPs, EGFR-pSiNPs or PSMA-pSiNPs was suspended in 500 μL of PNBS and stored at 4 °C, protected from light. At day 0, 2, and 6 (the day before) C4–2B cells were seeded in a black 96-well plate at a cell density of 1 × 104 and left to attach overnight in growth medium. At day 1, 3 and 7, 50 μL was removed from the pSiNP stock solution and pelleted at 20000 RCF for 5 min. The nanoparticles were then resuspended in 1 mL of RPMI 1640 supplemented with 10% FBS (for a final particle concentration of 50 μg mL−1). C4–2B cells were treated with 100 μL of each pSiNP solution (50 μg mL−1) for 1 h and then washed copiously (5×) with PBS to remove any free or loosely adhered particles from the cell surface. Afterward, the wells were replenished with 100 μL of PBS and the fluorescence was read on a PerkinElmer EnSpire multimode plate reader for Cy5 fluorescence (λex = 649 nm, λem = 666 nm).
4.14 Expression of EGFR and PSMA in C4–2B, C32 and HEK293-WT cells
C4–2B, C32 and HEK293-WT cells were lysed in ice cold RIPA buffer supplemented with a protease cocktail inhibitor (P8340, Sigma Aldrich). Protein concentration was determined using a BCA assay kit (Thermo Fisher Scientific). 20 μg of protein was mixed with NuPAGE sample reducing agent (Thermo Fisher Scientific) and LDS sample buffer (Thermo Fisher Scientific) and heated at 70 °C for 10 min. Afterward, the denatured proteins were electrophoresed in a 4–12% bis–tris gel (Thermo Fisher Scientific) and transferred onto nitrocellulose membranes. The membranes were blocked with 3% filtered BSA in TBS-T (0.1% Tween 20 in TBS) for 1 h, followed by incubation with either anti-EGFR (SAB5600138, Sigma Aldrich (1:1000)) or anti-PSMA primary antibody (SAB4300352, Sigma Aldrich (1:1000)) in 3% BSA in TBS-T overnight under gentle agitation at 4 °C. The following day, membranes were washed copiously with TBS-T and incubated with a secondary antibody (goat anti-rabbit HRP, ab6721, Abcam (1:10000)) in PBS-T (0.1% Tween 20 in PBS) for 1 h at RT. After washing, membranes were developed with SuperSignal West Pico PLUS Chemiluminescent substrate (34577, Thermo Fisher Scientific) and imaged using a ChemiDoc imaging system (Biorad). β-Actin was used as a housekeeping control (β-actin primary antibody (ab6276, Abcam (1:1500)) and goat anti-mouse secondary antibody (1:10000)). MRP1 expression was measured via densitometry analysis of the bands using ImageJ – normalized to the integrated density of the β-actin housekeeping protein band. The experiment was done in triplicates, at different cell passages to ensure consistent protein expression throughout the study.
4.15 Preparation of C4–2B spheroids
C4–2B spheroids were adapted from a previously reported method.60 C4–2B cells were seeded into ultra-low attachment spheroid 96-well plates (Corning 4515, USA) at a seeding density of 1 × 104 cells per well in growth media and centrifuged at 300 RCF for 5 min. Growth and formation of spheroids were monitored under a Brightfield microscope and media was replenished every third day until compact spheroids were formed (typically 7–9 days after seeding).4.16 Cellular association via confocal microscopy
C4–2B, C32 and HEK293-WT cells were seeded in 8-well glass chamber slides (Thermo Fisher) at a cell density of 1 × 104 and left to attach overnight in their respective growth medium. Cells were washed 2× in PBS and PEG-pSiNPs, EGFR-pSiNPs or PSMA-pSiNPs were added at a concentration of 50 μg mL−1 in 100 μL of Opti-MEM and allowed to incubate for 1 h. Afterward, the media was removed and the cells were washed 3× in PBS to remove free or loosely bound pSiNPs from the cell surface and replaced with respective growth media. After either 1 or 24 h, cells were washed 2× in PBS and fixed in 4% paraformaldehyde for 20 min at RT. The wells were washed 2× in PBS and permeabilized with 0.1% Triton X-100 in PBS for 10 min at RT. The wells were washed 2× in PBS and incubated with Hoechst 33342 (1:5000, Thermo Fisher) and phalloidin-TRITC (1:300, Sigma Aldrich) for 30 min at RT. Afterward, the glass slide was washed 2× in PBS, the well dividers removed, and mounted with Prolong Diamond Antifade Mountant (Thermo Fisher). Images were taken on a confocal fluorescence microscope (Leica TCS SP8, Leica Microsystems).
C4–2B spheroids were formed as previously described in section 4.15. Spheroids were then incubated with either PEG-pSiNPs, EGFR-pSiNPs or PSMA-pSiNPs labeled with Cy5 at a concentration of 50 μg mL−1 for 1 h. Afterward, the spheroids were washed 3× in PBS to remove any free or loosely bound nanoparticles and replaced with growth media. After 24 h, the spheroids were washed 2× in PBS and then fixed in 4% PFA in PBS overnight at 4 °C. The fixed spheroids were washed 2× in PBS and stored in a solution of 20% sucrose in PBS until sectioning. After dehydration in 20% sucrose, the spheroids were embedded in optimal cutting temperature compound (OCT) and 20 μm sections were obtained using a Cryostat Leica CM1950 (Leica Biosystems, Australia). The sections were washed 2× in PBS to remove OCT from the glass slides and stained with Hoechst 33342 (0.2 mg mL−1) for 1 h. The sections were imaged and analyzed using a Nikon Eclipse Ti laser-scanning confocal microscope using a 20 × 0.75 NA objective and processed using Fiji (ImageJ).
4.17 Cellular association via flow cytometry
C4–2B, C32 and HEK293-WT cells were seeded in 12-well plates at a cell density of 1 × 105 and left to attach overnight in their respective growth medium. Cells were washed 2× in PBS and PEG-pSiNPs, EGFR-pSiNPs or PSMA-pSiNPs were added at a concentration of 50 μg mL−1 in 1 mL of Opti-MEM and allowed to incubate for 1 h. The Opti-MEM media was removed and cells washed 3× in PBS to remove free or loosely bound pSiNPs from the cell surface and replaced with their respective growth media. After 24 h, cells were washed 2× in PBS and then detached using 200 μL of trypsin–EDTA (0.25%) per well. The trypsin was inactivated with 800 μL of growth media and the cells collected into centrifuge tubes. Afterward, cells were pelleted via centrifugation at 200 RCF for 5 min and the pellet was washed 1× in PBS. The cells were then stained with 7-AAD to assess cell viability for 5 min on ice, and then washed 2× in PBS. Finally, cells were resuspended in FACS buffer (1× PBS containing 10% FBS, 2 mM EDTA, and 0.1% NaN3) and kept on ice until analysis.C4–2B spheroids were formed as previously described in section 4.15. For the spheroid samples, 50 μg mL−1 of PEG-pSiNPs, EGFR-pSiNPs or PSMA-pSiNPs were incubated with spheroids for 1 h. The spheroids were then washed 3× in PBS to remove any free pSiNPs from the wells and replaced with 100 μL of growth media. After 24 h, the spheroids were washed 2× in PBS and then six spheroids from each group were collected into an Eppendorf tube and centrifuged at 300 RCF for 5 min. The spheroid pellet was washed 1× in PBS and the spheroids were disassociated with 100 μL of Accutase (Sigma Aldrich) for 20 min at RT. The disassociated cells were centrifuged at 300 RCF for 5 min and washed 1× in PBS. The cells were resuspended in FACS buffer and kept on ice until analysis.
Samples were analyzed by flow cytometry (BD FACS Canto II) for Cy5 fluorescence. Cellular association percentage was calculated as the number of cells that displayed fluorescence when compared to untreated cells/spheroids. All experiments were completed in quadruplicates.
4.18 Cell viability
C4–2B, C32 and HEK293-WT cells were seeded in a 96-well plate at a cell density of 1 × 104 and left for overnight attachment in their respective growth medium. Cells were washed 1× in PBS and PEG-pSiNPs, EGFR-pSiNPs or PSMA-pSiNPs loaded with either Dox, siMRP1, siScr or a combination of two therapeutics were added at a concentration of 50 μg mL−1 in 100 μL of Opti-MEM and allowed to incubate for 1 h. An equivalent amount of free Dox equivalent to the concentration of Dox released from pSiNPs after 1 h at pH 5.0 (1.32 μg mL−1) as determined via the in vitro release profile was used as a comparison. Afterward, the cells were washed 2× in PBS, replaced with fresh growth medium and incubated for a further 72 or 96 h. After incubation, cell viability was determined using an ATP-based luminescence cell viability assay (CellTiter-Glo, Promega, Madison, WI, USA) according to the manufacturer's protocol. Each experiment was performed in quadruplicate and compared to a negative (untreated cells) and positive control (10% DMSO). Luminescence was measured on a PerkinElmer EnSpire multimode plate reader.C4–2B spheroids were formed as previously described in section 4.15. Once formed, spheroids were treated with either PEG-pSiNPs, EGFR-pSiNPs or PSMA-pSiNPs loaded with either Dox, siMRP1, siScr or a combination of two therapeutics were added at a concentration of 50 μg mL−1 in 100 μL of Opti-MEM and allowed to incubate for 1 h. An equivalent amount of free Dox equivalent to the concentration of Dox released from pSiNPs after 1 h (2.4 μM) was used as a comparison. After the 1 h incubation, the spheroids were washed 2× in PBS and fresh growth media was added and further incubated for 96 h. After incubation, cellular viability was evaluated using an ATP-based luminescent cell viability assay according to the manufacturer's protocol. Each experiment was performed in quadruplicate and compared to a negative (untreated cells) and positive control (10% DMSO in growth media). Luminescence was measured on a PerkinElmer EnSpire multimode plate reader.
4.19 Knockdown of MRP1 via Western blotting
C4–2B cells were seeded in 6-well plates at a cell density of 2 × 105 and left to attach overnight. The cells were then washed 2× in PBS and then incubated with PEG-pSiNPs, EGFR-pSiNPs or PSMA-pSiNPs loaded with either siMRP1 or siScr and relevant controls (unloaded particles and commercial Lipofectamine RNAiMAX prepared lipoplexes as comparisons) at a concentration of 50 μg mL−1 in 1 mL of OptiMEM for 4 h. Afterward, the cells were washed 2× in PBS to remove unbound pSiNPs and replaced with growth medium and allowed to incubate for 72 h.To study the expression of MRP1, protein lysates were analyzed via Western blotting. After 72 h, cells were washed 2× in PBS and directly lysed with 150 μL of ice cold RIPA lysis buffer supplemented with a protease inhibitor cocktail. Protein concentration was quantified using a BCA assay kit. 20 μg of protein was mixed with NuPAGE sample reducing agent and LDS sample buffer and heated at 70 °C for 10 min. The proteins were electrophoresed in 4–12% bis–tris gels and transferred onto nitrocellulose membranes. Membranes were blocked with 3% filtered BSA in TBS-T for 1 h, followed by incubation with anti-MRP1 primary antibody (ab24102, Abcam (1:50)) in 3% BSA in TBS-T overnight under gentle agitation at 4 °C. Membranes were washed thoroughly with TBS-T and incubated with a secondary antibody (goat anti-mouse HRP, 1705047, BioRad (1:10000)) in PBS-T for 1 h at RT. After washing, membranes were developed with SuperSignal West Pico PLUS Chemiluminescent substrate and imaged using a ChemiDoc imaging system (Biorad). β-Actin was used as a housekeeping control (β-actin primary antibody (ab6276, Abcam (1:1500)) and goat antimouse secondary antibody (1:10000)). MRP1 expression was measured via densitometry analysis of the bands using ImageJ, normalized to the integrated density of the β-actin housekeeping protein band.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was performed in part at the Melbourne Centre for Nanofabrication (MCN) in the Victorian Node of the Australian National Fabrication Facility (ANFF). We acknowledge use of facilities within the Monash Centre for Electron Microscopy (MCEM). We thank Winston Liew, Chris Sheedy, and the Biophysics group from CSIRO Manufacturing for performing ICP-OES analysis. We thank BioRender.com for the creation of the schemes. T. T. and N. H. V. acknowledge support from the Office of the Chief Executive and CSIRO Manufacturing. T. T. acknowledges support of an Australian Government RTP scholarship. P. H. acknowledges support from the Carlsberg Foundation. K. J. T. acknowledges fellowship funding from the NHMRC (Career Development Fellowship; APP1148582). P. H. and K. J. T. acknowledge support from the ARC Centre of Excellence in Convergent Bio-Nano Science & Technology (CBNS). A. C.-R. acknowledges the National Health & Medical Research Council (NHMRC) of Australia (Grant GNT1112432).References
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS
.
- L. S. Jabr-Milane, L. E. van Vlerken, S. Yadav and M. M. Amiji, Cancer Treat. Rev., 2008, 34, 592–602 CrossRef CAS
- M. M. Gottesman, Annu. Rev. Med., 2002, 53, 615–627 CrossRef CAS
- J. I. Fletcher, R. T. Williams, M. J. Henderson, M. D. Norris and M. Haber, Drug Resist. Updates, 2016, 26, 1–9 CrossRef
- R. W. Robey, K. M. Pluchino, M. D. Hall, A. T. Fojo, S. E. Bates and M. M. Gottesman, Nat. Rev. Cancer, 2018, 18, 452–464 CrossRef CAS
- R. Krishna and L. D. Mayer, Eur. J. Pharm. Sci., 2000, 11, 265–283 CrossRef CAS
- T. M. Grogan, C. M. Spier, S. E. Salmon, M. Matzner, J. Rybski, R. S. Weinstein, R. J. Scheper and W. S. Dalton, Blood, 1993, 81, 490–495 CrossRef CAS
- H. Burger, J. A. Foekens, M. P. Look, M. E. Meijer-van Gelder, J. G. M. Klijn, E. A. C. Wiemer, G. Stoter and K. Nooter, Clin. Cancer Res., 2003, 9, 827–836 CAS
- J. Zalcberg, X. F. Hu, A. Slater, J. Parisot, S. El-Osta, P. Kantharidis, S. T. Chou and J. D. Parkin, Prostate Cancer Prostatic Dis., 2000, 3, 66–75 CrossRef CAS
- C. Sánchez, P. Mendoza, H. R. Contreras, J. Vergara, J. A. McCubrey, C. Huidobro and E. A. Castellón, Prostate, 2009, 69, 1448–1459 CrossRef
- S. P. C. Cole, J. Biol. Chem., 2014, 289, 30880–30888 CrossRef CAS
- J. Pan, S. A. Attia, M. A. Subhan, N. Filipczak, L. P. Mendes, X. Li, S. S. K. Yalamarty and V. P. Torchilin, Mol. Pharmaceutics, 2020, 17, 1638–1647 CrossRef CAS
- K. A. Whitehead, R. Langer and D. G. Anderson, Nat. Rev. Drug Discovery, 2009, 8, 129–138 CrossRef CAS
- S. Rietwyk and D. Peer, ACS Nano, 2017, 11, 7572–7586 CrossRef CAS
- H. Meng, M. Liong, T. Xia, Z. Li, Z. Ji, J. I. Zink and A. E. Nel, ACS Nano, 2010, 4, 4539–4550 CrossRef CAS
- H. Meng, W. X. Mai, H. Zhang, M. Xue, T. Xia, S. Lin, X. Wang, Y. Zhao, Z. Ji, J. I. Zink and A. E. Nel, ACS Nano, 2013, 7, 994–1005 CrossRef CAS
- W. Y. Tong, M. Alnakhli, R. Bhardwaj, S. Apostolou, S. Sinha, C. Fraser, T. Kuchel, B. Kuss and N. H. Voelcker, J. Nanobiotechnol., 2018, 16, 38 CrossRef
- H. M. Aliabadi, B. Landry, P. Mahdipoor, C. Y. M. Hsu and H. Uludağ, Eur. J. Pharm. Biopharm., 2012, 81, 33–42 CrossRef CAS
- D. Bumcrot, M. Manoharan, V. Koteliansky and D. W. Y. Sah, Nat. Chem. Biol., 2006, 2, 711–719 CrossRef CAS
- D. Rosenblum, N. Joshi, W. Tao, J. M. Karp and D. Peer, Nat. Commun., 2018, 9, 1410 CrossRef
- A. M. Chen, M. Zhang, D. Wei, D. Stueber, O. Taratula, T. Minko and H. He, Small, 2009, 5, 2673–2677 CrossRef CAS
- M. Wu, J. Li, X. Lin, Z. Wei, D. Zhang, B. Zhao, X. Liu and J. Liu, Biomater. Sci., 2018, 6, 1457–1468 RSC
- W. Sun, X. Chen, C. Xie, Y. Wang, L. Lin, K. Zhu and X. Shuai, Biomacromolecules, 2018, 19, 2248–2256 CrossRef CAS
- T. Tieu, M. Alba, R. Elnathan, A. Cifuentes-Rius and N. H. Voelcker, Adv. Ther., 2019, 2, 1800095 CrossRef
- T. Tieu, S. Dhawan, V. Haridas, L. M. Butler, H. Thissen, A. Cifuentes-Rius and N. H. Voelcker, ACS Appl. Mater. Interfaces, 2019, 11, 22993–23005 CrossRef CAS
- M. Landgraf, C. A. Lahr, I. Kaur, A. Shafiee, A. Sanchez-Herrero, P. W. Janowicz, A. Ravichandran, C. B. Howard, A. Cifuentes-Rius, J. A. McGovern, N. H. Voelcker and D. W. Hutmacher, Biomaterials, 2020, 240, 119791 CrossRef CAS
- A. Cifuentes-Rius, A. Ivask, E. Sporleder, I. Kaur, Y. Assan, S. Rao, D. Warther, C. A. Prestidge, J.-O. Durand and N. H. Voelcker, Small, 2017, 13, 1701201 CrossRef
- T. Xia, M. Kovochich, M. Liong, H. Meng, S. Kabehie, S. George, J. I. Zink and A. E. Nel, ACS Nano, 2009, 3, 3273–3286 CrossRef CAS
- S. Wang, X. Liu, S. Chen, Z. Liu, X. Zhang, X.-J. Liang and L. Li, ACS Nano, 2019, 13, 274–283 CrossRef CAS
- M. H. Kafshgari, M. Alnakhli, B. Delalat, S. Apostolou, F. J. Harding, E. Mäkilä, J. J. Salonen, B. J. Kuss and N. H. Voelcker, Biomater. Sci., 2015, 3, 1555–1565 RSC
- S. Muyldermans, Annu. Rev. Biochem., 2013, 82, 775–797 CrossRef CAS
- I. Van Audenhove and J. Gettemans, EBioMedicine, 2016, 8, 40–48 CrossRef
- B. Boone, K. Jacobs, L. Ferdinande, J. Taildeman, J. Lambert, M. Peeters, M. Bracke, P. Pauwels and L. Brochez, J. Cutaneous Pathol., 2011, 38, 492–502 CrossRef
- S. Sigismund, D. Avanzato and L. Lanzetti, Mol. Oncol., 2018, 12, 3–20 CrossRef
- U. Haberkorn, M. Eder, K. Kopka, J. W. Babich and M. Eisenhut, Clin. Cancer Res., 2016, 22, 9–15 CrossRef CAS
- G. Lazzari, P. Couvreur and S. Mura, Polym. Chem., 2017, 8, 4947–4969 RSC
- J. V. Jokerst, T. Lobovkina, R. N. Zare and S. S. Gambhir, Nanomedicine, 2011, 6, 715–728 CrossRef CAS
- J. F. Stefanick, J. D. Ashley, T. Kiziltepe and B. Bilgicer, ACS Nano, 2013, 7, 2935–2947 CrossRef CAS
- K. W. Yong, D. Yuen, M. Z. Chen and A. P. R. Johnston, ACS Appl. Mater. Interfaces, 2020, 12, 5593–5600 CrossRef CAS
- D. Schumacher, J. Helma, A. F. L. Schneider, H. Leonhardt and C. P. R. Hackenberger, Angew. Chem., Int. Ed., 2018, 57, 2314–2333 CrossRef CAS
- A. L. Hook, N. H. Voelcker and H. Thissen, Acta Biomater., 2009, 5, 2350–2370 CrossRef CAS
- A. Zhang, L. Yao and M. An, Chem. Commun., 2017, 53, 12826–12829 RSC
- A. Janoniene, Z. Liu, L. Baranauskiene, E. Mäkilä, M. Ma, J. Salonen, J. Hirvonen, H. Zhang, V. Petrikaite and H. A. Santos, ACS Appl. Mater. Interfaces, 2017, 9, 13976–13987 CrossRef CAS
-
M. J. Sailor, in Handbook of Porous Silicon, ed. L. Canham, Springer International Publishing, Cham, 2021, pp. 1–24, DOI:10.1007/978-3-319-04508-5_37-1
- M. Gao, L. Miao, M. Liu, C. Li, C. Yu, H. Yan, Y. Yin, Y. Wang, X. Qi and J. Ren, Oncotarget, 2016, 7, 59714 CrossRef
- R. H. Kang, J.-E. Jang, E. Huh, S. J. Kang, D.-R. Ahn, J. S. Kang, M. J. Sailor, S. G. Yeo, M. S. Oh, D. Kim and H. Y. Kim, Nanoscale Horiz., 2020, 5, 1213–1225 RSC
- S. Hussain, J. Joo, J. Kang, B. Kim, G. B. Braun, Z.-G. She, D. Kim, A. P. Mann, T. Mölder, T. Teesalu, S. Carnazza, S. Guglielmino, M. J. Sailor and E. Ruoslahti, Nat. Biomed. Eng., 2018, 2, 95–103 CrossRef CAS
- T. Yong, X. Zhang, N. Bie, H. Zhang, X. Zhang, F. Li, A. Hakeem, J. Hu, L. Gan, H. A. Santos and X. Yang, Nat. Commun., 2019, 10, 3838 CrossRef
- J. L. Gillis, L. A. Selth, M. M. Centenera, S. L. Townley, S. Sun, S. R. Plymate, W. D. Tilley and L. M. Butler, Oncotarget, 2013, 4, 691 CrossRef
- S. Loi, S. Pommey, B. Haibe-Kains, P. A. Beavis, P. K. Darcy, M. J. Smyth and J. Stagg, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 11091–11096 CrossRef CAS
- A. Cheung, J. Opzoomer, K. M. Ilieva, P. Gazinska, R. M. Hoffmann, H. Mirza, R. Marlow, E. Francesch-Domenech, M. Fittall, D. D. Rodriguez, A. Clifford, L. Badder, N. Patel, S. Mele, G. Pellizzari, H. J. Bax, S. Crescioli, G. Petranyi, D. Larcombe-Young, D. H. Josephs, S. Canevari, M. Figini, S. Pinder, F. O. Nestle, C. Gillett, J. F. Spicer, A. Grigoriadis, A. N. J. Tutt and S. N. Karagiannis, Clin. Cancer Res., 2018, 24, 5098–5111 CrossRef CAS
- B. M. Necela, J. A. Crozier, C. A. Andorfer, L. Lewis-Tuffin, J. M. Kachergus, X. J. Geiger, K. R. Kalari, D. J. Serie, Z. Sun, A. M. Aspita, D. J. O'Shannessy, J. D. Maltzman, A. E. McCullough, B. A. Pockaj, H. E. Cunliffe, K. V. Ballman, E. A. Thompson and E. A. Perez, PLoS One, 2015, 10, e0122209 CrossRef
- L. Sánchez-del-Campo, M. F. Montenegro, J. Cabezas-Herrera and J. N. Rodríguez-López, Pigm. Cell Melanoma Res., 2009, 22, 588–600 CrossRef
- S. Nath and G. R. Devi, Pharmacol. Ther., 2016, 163, 94–108 CrossRef CAS
- M. Moro, G. Bertolini, R. Caserini, C. Borzi, M. Boeri, A. Fabbri, G. Leone, P. Gasparini, C. Galeone, G. Pelosi, L. Roz, G. Sozzi and U. Pastorino, Sci. Rep., 2017, 7, 6689 CrossRef
- R. C. Roovers, M. J. W. D. Vosjan, T. Laeremans, R. el Khoulati, R. C. G. de Bruin, K. M. Ferguson, A. J. Verkleij, G. A. M. S. van Dongen and P. M. P. van Bergen en Henegouwen, Int. J. Cancer, 2011, 129, 2013–2024 CrossRef CAS
- K. L. S. Chatalic, J. Veldhoven-Zweistra, M. Bolkestein, S. Hoeben, G. A. Koning, O. C. Boerman, M. de Jong and W. M. van Weerden, J. Nucl. Med., 2015, 56, 1094–1099 CrossRef CAS
- J. W. Chin, S. W. Santoro, A. B. Martin, D. S. King, L. Wang and P. G. Schultz, J. Am. Chem. Soc., 2002, 124, 9026–9027 CrossRef CAS
- G. F. Ames, C. Prody and S. Kustu, J. Bacteriol., 1984, 160, 1181–1183 CrossRef CAS
- S. Balaban, Z. D. Nassar, A. Y. Zhang, E. Hosseini-Beheshti, M. M. Centenera, M. Schreuder, H.-M. Lin, A. Aishah, B. Varney, F. Liu-Fu, L. S. Lee, S. R. Nagarajan, R. F. Shearer, R.-A. Hardie, N. L. Raftopulos, M. S. Kakani, D. N. Saunders, J. Holst, L. G. Horvath, L. M. Butler and A. J. Hoy, Mol. Cancer Res., 2019, 17, 949–962 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0bm01335h |
| This journal is © The Royal Society of Chemistry 2021 |