
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBifunctional thiosquaramide catalyzed asymmetric reduction of dihydro-β-carbolines and enantioselective synthesis of (−)-coerulescine and (−)-horsfiline by oxidative rearrangement†
Manda Sathishab,
Fabiane M. Nachtigallc and
Leonardo S. Santos *a
*a
aLaboratory of Asymmetric Synthesis, Chemistry Institute of Natural Resources, Universidad de Talca, Casilla 747, 3460000 Talca, Chile. E-mail: lssantos@utalca.cl
bNúcleo Científico Multidisciplinario-DI, Universidad de Talca, Casilla 747, 3460000 Talca, Chile
cInstituto de Ciencias Químicas Aplicadas, Universidad Autónoma de Chile, Talca 3467987, Chile
First published on 21st October 2020
Abstract
Tetrahydro-β-carboline (THBC) is a tricyclic ring system that can be found in a large number of bioactive alkaloids. Herein, we report a simple and efficient method for the synthesis of enantiopure THBCs through a chiral thiosquaramide (11b) catalyzed imine reduction of dihydro-β-carbolines (17a–f). The in situ generated Pd–H employed as hydride source in the reaction of differently substituted chiral THBCs (18a–f) afforded high selectivities (R isomers, up to 96% ee) and good isolated yields (up to 88%). Moreover, the chiral thiosquaramide used also afforded exceptional catalyst activity in the syntheses of (−)-coerulescine (5) and (−)-horsfiline (6) with excellent enantioselectivities up to 98% and 93% ee, respectively, via an enantioselective oxidative rearrangement approach.
Introduction
Reduction of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond in cyclic systems is a fundamental and important reaction in the area of modern organic synthesis.1 Moreover, asymmetric reduction of the CN bond promoted by organocatalysts is a very valuable aspect in organic stereoselective synthesis.2 Generally, transition metal complexes with chiral bulky ligands are used for this type of transformation3 and a few protocols are known in biocatalysis,2c,4 as well as for chiral auxiliary assisted reduction of imines.5 The preparation of enantioselective secondary amines, important chiral drug molecules and/or asymmetric reduction of imines is an important topic in academic research as well as from an industrial point of view.6 Nowadays, widespread development has been witnessed in the asymmetric synthesis of tetrahydro-β-carboline (THBC) skeletons.7 Furthermore, there is a constant search in the development of new approaches for asymmetric synthesis using chiral organocatalysis. Because the unique tricyclic system, THBCs play key role in their bioactivities,8 for example: reserpine4a,9 is an indole alkaloid which is an alternative drug for treating hypertension. The LY23728 (1)3d,10 a simple THBC has been reported as first 5-HT2B-selective antagonist and harmicine4a,11 (2) is a rare tetracyclic pyrrolidine framework with strong antileishmania activity. Few other THBCs are competitive selective inhibitors of the monoamine oxidase type A (MAO-A) enzyme12 and also potent reuptake inhibitors of serotonin and epinephrine with greater selectivity for serotonin.12 Not only because of their bioactivity, THBCs are valuable intermediates in the production of potential drug candidates. For example, the chiral THBCs like 18c and 18f are key intermediates for the synthesis of active quinolactacin B13 (3) and the potent PDE5 inhibitor like pyrroloquinolone RWJ387273 (4),14 respectively (Fig. 1).
N bond in cyclic systems is a fundamental and important reaction in the area of modern organic synthesis.1 Moreover, asymmetric reduction of the CN bond promoted by organocatalysts is a very valuable aspect in organic stereoselective synthesis.2 Generally, transition metal complexes with chiral bulky ligands are used for this type of transformation3 and a few protocols are known in biocatalysis,2c,4 as well as for chiral auxiliary assisted reduction of imines.5 The preparation of enantioselective secondary amines, important chiral drug molecules and/or asymmetric reduction of imines is an important topic in academic research as well as from an industrial point of view.6 Nowadays, widespread development has been witnessed in the asymmetric synthesis of tetrahydro-β-carboline (THBC) skeletons.7 Furthermore, there is a constant search in the development of new approaches for asymmetric synthesis using chiral organocatalysis. Because the unique tricyclic system, THBCs play key role in their bioactivities,8 for example: reserpine4a,9 is an indole alkaloid which is an alternative drug for treating hypertension. The LY23728 (1)3d,10 a simple THBC has been reported as first 5-HT2B-selective antagonist and harmicine4a,11 (2) is a rare tetracyclic pyrrolidine framework with strong antileishmania activity. Few other THBCs are competitive selective inhibitors of the monoamine oxidase type A (MAO-A) enzyme12 and also potent reuptake inhibitors of serotonin and epinephrine with greater selectivity for serotonin.12 Not only because of their bioactivity, THBCs are valuable intermediates in the production of potential drug candidates. For example, the chiral THBCs like 18c and 18f are key intermediates for the synthesis of active quinolactacin B13 (3) and the potent PDE5 inhibitor like pyrroloquinolone RWJ387273 (4),14 respectively (Fig. 1).
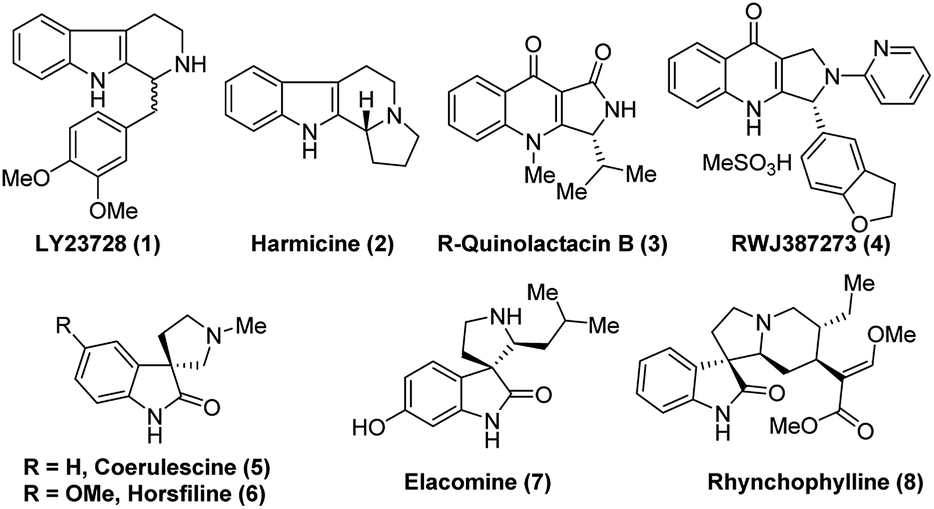 | ||
| Fig. 1 Representative examples of bioactive THBCs (1–4) and some natural spirooxindoles (5–8). | ||
Another synthon afforded by THBCs are spirooxindoles, which show unique spiro-cyclic frameworks at the C3-position of the oxindole core.15 The spirooxindole derivatives are playing important role in the recent drug discovery.16 The structurally rigid spiro-cyclic system may be a reason for the good affinity towards three-dimensional proteins to exhibit bioactivities.15,17–23 For example, the (−)-horsfiline (6) is used as an intoxicating snuff,24 spirotryprostatin A25 inhibits G2M progression of mammalian tsFT210 cells, rhynchophylline (8) shows potency against various cancer cell lines,26 corynoxine and corynoxine B show prominent activity in preventing or treating Parkinson's disease.27 These bioactivities of spirooxindole derivatives have augmented attention in organic chemists from academia as well as industry to develop various synthetic routes,28 especially in the asymmetric manner for the synthesis of chiral spirooxindoles.15,29
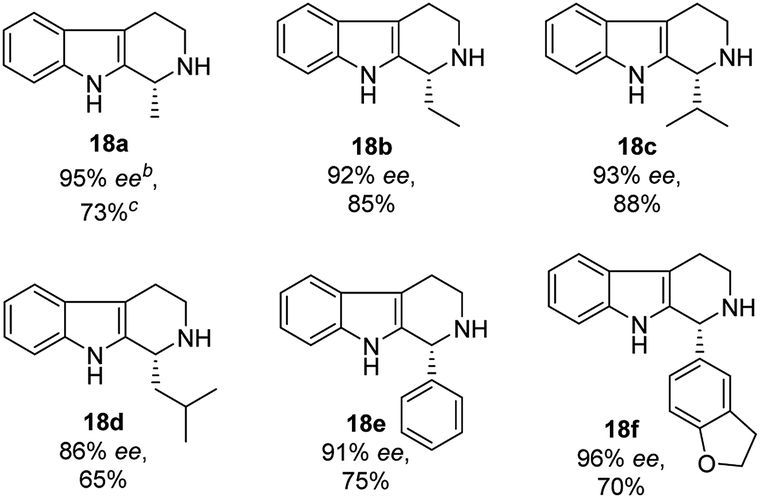
On the other hand, the chiral squaramides30 and thiosquaramides31 have proven to be excellent catalysts in the asymmetric organocatalysis. However, due to the high solubility in non-polar solvents like toluene, DCM, DCE and THF, the thiosquaramides have established as exceptional chiral organocatalysts.31 Whereas the squaramides are excellent catalysts in polar solvents.31 Rawal group developed various squaramides and thiosquaramides as chiral organocatalysts and witnessed high enantioselectivities with thiosquaramides in Michael additions of barbituric acids to nitroalkenes.31a Recently, stereoselective double Michael addition of 2-(3H)-furanone to nitroolefins,32 as well as asymmetric Michael additions of aldehydes to nitroolefins for synthesis of chiral pyrrolidines showed high enantio- and diastereoselectivities.33 Inspired by Rawal's conclusions with chiral bifunctional thiosquaramides and other consequences from recent articles, we have synthesized a known squaramide and thiosquaramide based chiral organocatalysts 11a–d (Fig. 2). In this work, the aim was to establish chiral protocols to construct bioactive natural products from DHBCs to afford enantiopure THBCs using chiral organocatalysts in asymmetric reduction reactions. The palladium hydride11 (PdH) was generated in an in situ reaction of catalytic PdCl2 with excess of Et3SiH and utilized as hydride source in the asymmetric imine reduction. The chiral THBCs 18a–f were obtained with excellent selectivities (up to 96% ee) and good yields (up to 88%). The catalytic efficiency of chiral thiosquaramide in an oxidative rearrangement was also examined to produce (−)-coerulescine (5) and (−)-horsfiline (6). Surprisingly, the products were observed with high enantioselectivities reaching 98% and 93%, respectively. Moreover, the chiral HPLC and polarimetry analyses proved the R configuration of all the products (18a–f), 5 and 6.
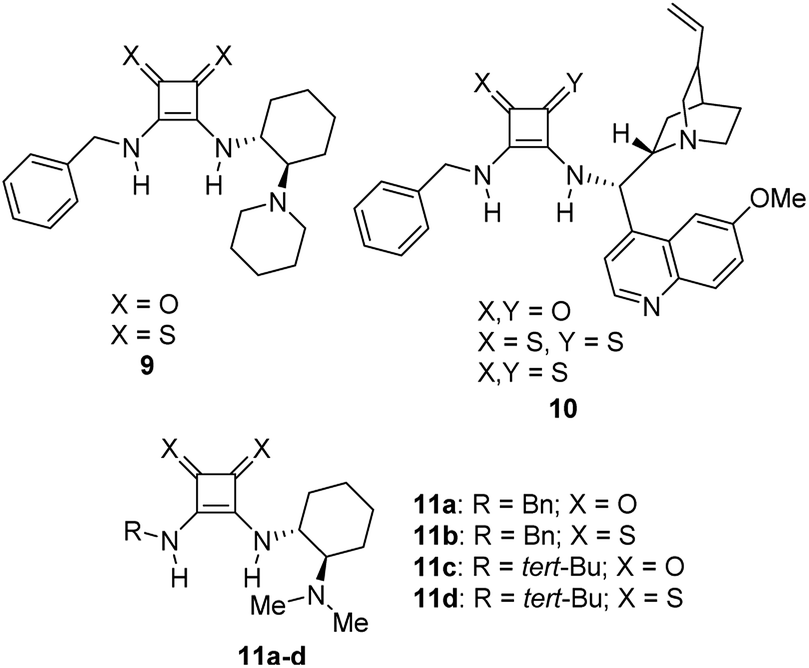 | ||
| Fig. 2 Few known chiral squaramides and thiosquaramides (9–11) and the organocatalysts employed in this work (11a–d). | ||
Results and discussion
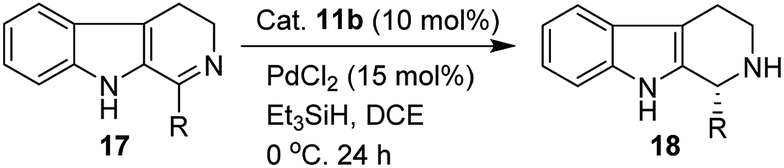
We have synthesized the chiral squaramides 11a and 11c and thiosquaramides 11b and 11d employing literature protocols31a (Scheme 1), 17a–f and 20a–b were prepared as displayed in Scheme 2. From our previous experience11 and other literature reports,34 we choose the PdH as hydride source that showed good enantioselectivity in similar approaches, which can be generated in situ by treatment of catalytic PdCl2 (15 mol%) and excess of Et3SiH. Initially, the asymmetric reduction of imine 17b employing 11a–d (10 mol%) was performed in 1,2-dichloroethane (DCE) and THF for 2 h at 0 °C. The reaction mixtures were analyzed by chiral HPLC, which shown the chiral THBC 18b in 53%, 88%, 40%, and 62% ee in case of DCE, and 50%, 70%, 38% and 60% ee in THF with 11a, 11b, 11c and 11d respectively in moderate yields (entries 1–8, Table 1). It is observed that the thiosquaramide 11b catalyzed well compared with 11a, 11c and 11d. Thiosquaramides seem to be good catalysts for this reaction, it may be due to the poor solubility of squaramides that afforded low catalysts activity. Further, 5 mol% of 11b was utilized to test the catalytic efficiency and achieved only 85% ee with low yield (45%, entry 9, Table 1). However, the selectivity and yield improved dramatically when the reaction was performed up to 24 h at 0 °C (entry 10, Table 1) with catalyst 11b (10 mol%). Further increment in catalyst load (15 mol%) was ineffective on selectivity as well as yield (entry 11, Table 1). These observations indicate just 10 mol% of catalyst load is sufficient for the asymmetric imine reduction. Furthermore, the reaction was also examined in other solvents such as MeOH, DCM and water/DCM systems (entries 13–15, Table 1). Moderate selectivities (73% ee and 80% ee) and good yields (80% and 82%) were observed in aprotic solvents THF and DCM (entries 12 and 14, Table 1). Whereas only traces of products were observed in the protic solvents like MeOH as well as water/DCM systems (entries 13 and 15, Table 1). Based on the above methodology outcomes, the asymmetric imine reduction tool was fixed as 10 mol% of thiosquaramide catalyst (11b) in DCE as solvent for 24 h at 0 °C.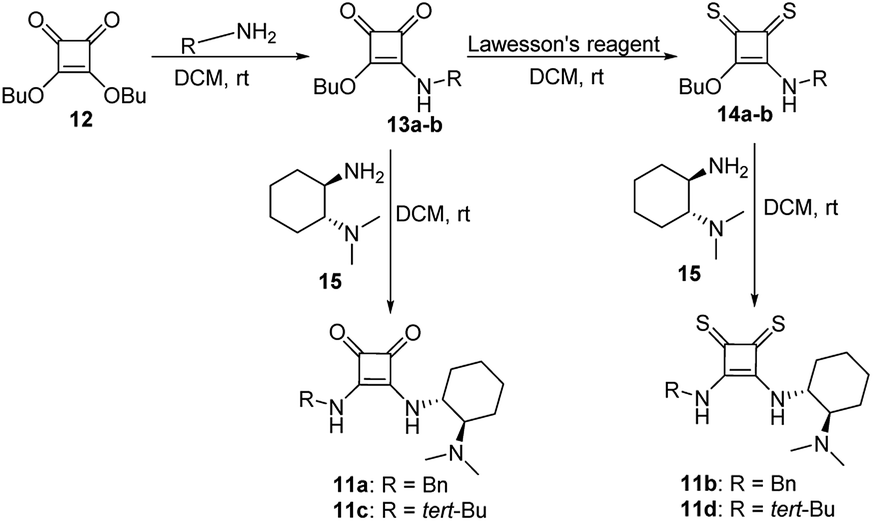 | ||
| Scheme 1 Synthesis of chiral thiosquaramide (11a–d). | ||
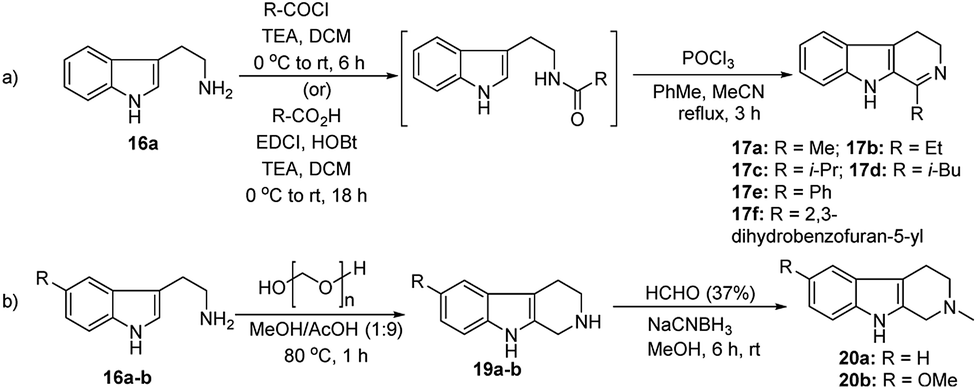 | ||
| Scheme 2 Synthesis of (a) DHBCs 17a–f and (b) N-methyl THBCs 20a–b. | ||
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Mol% | Solvent | Time (h) | Yieldb (%) | eec (%) |
| a Reactions were performed using 17b (1 mmol), 11 (mol%), PdCl2 (15 mol%) Et3SiH (4 mmol) solvent, for the given time.b Isolated yield.c The enantiomeric excess (ee) was determined by chiral HPLC.d Imine 17b was recovered and the yield calculated by isolated product amount. | ||||||
| 1 | 11a | 10 | DCE | 2 | 30d | 53 |
| 2 | 11b | 10 | DCE | 2 | 60d | 88 |
| 3 | 11c | 10 | DCE | 2 | 34d | 40 |
| 4 | 11d | 10 | DCE | 2 | 55d | 62 |
| 5 | 11a | 10 | THF | 2 | 27d | 50 |
| 6 | 11b | 10 | THF | 2 | 55d | 70 |
| 7 | 11c | 10 | THF | 2 | 30d | 38 |
| 8 | 11d | 10 | THF | 2 | 53d | 60 |
| 9 | 11b | 5 | DCE | 2 | 45d | 85 |
| 10 | 11b | 10 | DCE | 24 | 85 | 92 |
| 11 | 11b | 15 | DCE | 24 | 85 | 92 |
| 12 | 11b | 10 | THF | 24 | 80 | 73 |
| 13 | 11b | 10 | MeOH | 24 | Trace | — |
| 14 | 11b | 10 | DCM | 24 | 82 | 80 |
| 15 | 11b | 10 | Water/DCM | 24 | Trace | — |
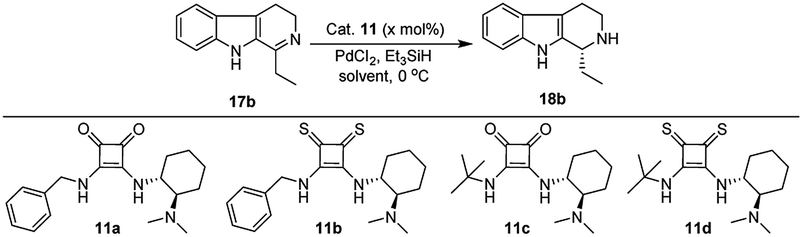
With the proven reaction conditions in hand, a series of DHBCs 17a–f with different substituents were explored (Table 2). Thus, DHBCs evenly undergone asymmetric reduction in presence of catalytic chiral thiosquaramide (11b) and PdCl2/Et3SiH, and we observed the chiral THBCs in good yields and enantioselectivities (18a–f, Table 2). Typically, substituents at C1 position of the DHBCs slightly altered the enantioselectivities. For example, the imine (17a) with methyl group gave the chiral THBC (18a) with good selectivity (95% ee) and moderate yield (73%). Moreover, chiral THBCs (18b and 18c) with ethyl and isopropyl groups also shown significant selectivities (92% ee and 93% ee, respectively) and high yields (85% and 88%), as depicted in Table 2. However, the 1-isobutyl THBC (18d) was obtained with slightly low selectivity (86% ee, Table 2), which may be explained due to an unfavorable transition state of isobutyl DHBC (17d) with chiral catalyst as proposed in Fig. 3. Despite, the chiral 1-phenyl THBC (18e) that displayed moderate selectivity (91% ee, Table 2), gratifyingly the 2,3-dihydrobenzofuranyl THBC (18f) demonstrated high enantioselectivity (96% ee, Table 2).
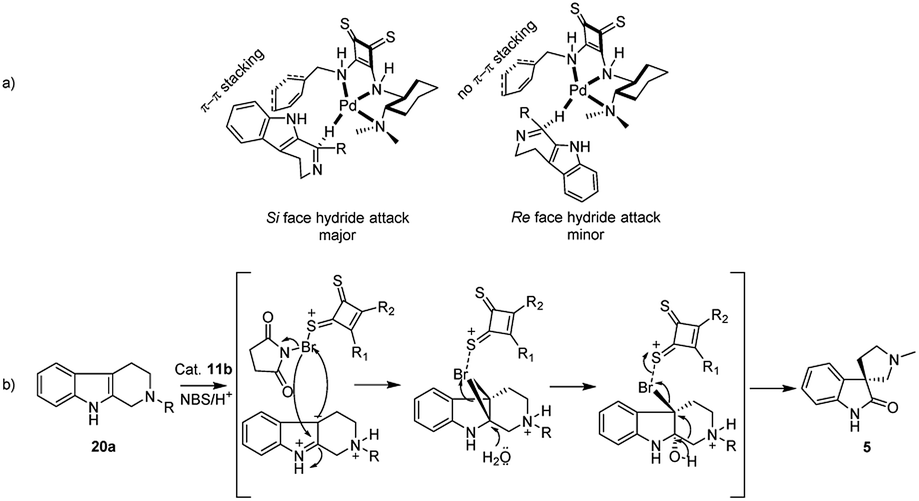 | ||
| Fig. 3 Plausible mechanism for the approach (major Si face and minor Re face) of hydride to the DHBC 17 in the presence of chiral thiosquaramide catalyst (11b), and the catalyzed oxidative rearrangement. | ||
Furthermore, we also explored an alternative enantiomeric oxidative rearrangement of THBC. Several approaches for the syntheses of chiral quinolactacins13 and RWJ387273![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 14 were described. Quinolactacins shows activity against tumor necrosis factor production, and RWJ387273 exhibited promising male erectile dysfunction (MED) PDE5 inhibition. However, the key step in both the syntheses approaches was the asymmetric reduction of imine to corresponding amines. Generally, most of the reports applied Noyori asymmetric transfer hydrogenation of cyclic imine (17c and 17f) to the total synthesis of chiral quinolactacin B (3) and potential PDE5 inhibitor RWJ387273 (4). Although the Noyori hydrogenation35 is an excellent methodology, which includes Ru catalyst and extremely flammable hydrogen gas, considering the cost effect, mildness of protocol and environmental point of view, it is proposed a new formal synthetic route for the R-quinolactacin B (3) and potential PDE5 inhibitor RWJ387273 (4), as depicted in Scheme 3.
14 were described. Quinolactacins shows activity against tumor necrosis factor production, and RWJ387273 exhibited promising male erectile dysfunction (MED) PDE5 inhibition. However, the key step in both the syntheses approaches was the asymmetric reduction of imine to corresponding amines. Generally, most of the reports applied Noyori asymmetric transfer hydrogenation of cyclic imine (17c and 17f) to the total synthesis of chiral quinolactacin B (3) and potential PDE5 inhibitor RWJ387273 (4). Although the Noyori hydrogenation35 is an excellent methodology, which includes Ru catalyst and extremely flammable hydrogen gas, considering the cost effect, mildness of protocol and environmental point of view, it is proposed a new formal synthetic route for the R-quinolactacin B (3) and potential PDE5 inhibitor RWJ387273 (4), as depicted in Scheme 3.
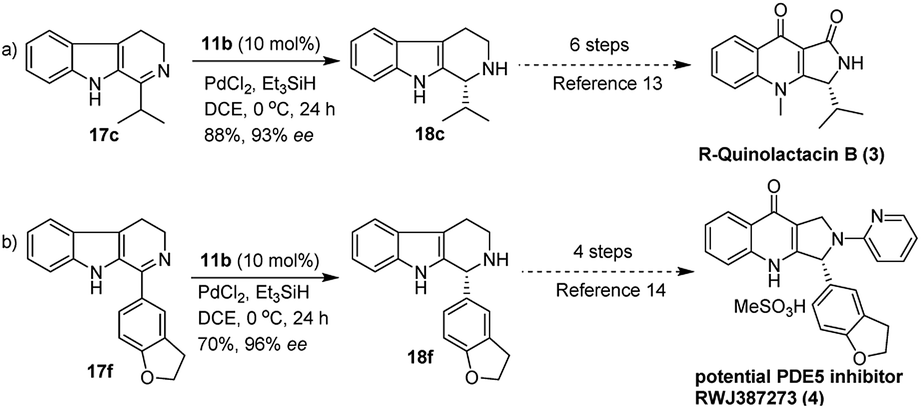 | ||
| Scheme 3 Formal synthesis of (R)-quinolactacin (3) and potential PDE5 inhibitor RWJ387273 (4) through 11b catalyzed asymmetric reduction of 17c and 17f. | ||
Next following our interest in complex structures, it was explored the conversion of N-methyl tetrahydro-β-carbolines to spirooxindoles through a bifunctional activity of organocatalyst 11a–d. Thus, the conversion from N-methyl tetrahydro-β-carboline 20a to spirooxindole 5 have been carried out using N-bromosuccinimide (NBS, 1 equiv., Scheme 2) and isolated products with satisfactory yields (58–85%) after 20 min reaction time, as depicted in Table 3. In the catalytic activity optimization, initially we tested 10 mol% of thiosquaramides 11a–d for the asymmetric synthesis of spirooxindole 5 in THF/water/AcOH (1:1:1). The reaction mixtures were analysed by chiral HPLC and evidenced that all the catalysts 11a–d catalysed the reaction considerably (78%, 98%, 69% and 85% ee respectively, entries 1–4, Table 3). However, reaction with 11b gave excellent selectivity and good yield (98% ee and 85%, entry 2, Table 3). We considered 11b as suitable catalyst for this reaction, further tested with 5 mol% and we noticed only 68% ee (entry 9, Table 3). This reaction was found to be optimal with 10 mol% of 11b. However, the asymmetric reactions without AcOH afforded only racemic mixtures even after 24 h (entries 5–8, Table 3). This may be due to the low solubility of 20a in the absence of AcOH, which may be needed to protonate the basic nitrogen to soluble in the solvent mixture. It might indicate that the asymmetric oxidative rearrangement may require protonation to the nitrogen atom at β-position of carboline to form the stable transition state with thiosquaramide catalyst leading to yield the product in an enantioselective manner. Product 5 was not observed without the use of aqueous solvent mixtures (entries 12 and 13, Table 3). Hence, we examined solvent mixtures such as THF/water (1:1), DCE/water (1:1), THF/water/AcOH (1:1:1), DCE/water/AcOH (1:1:1) and identified that the chiral reaction was slightly efficient in THF/water/AcOH (98% ee, entry 2, Table 3) than in DCE/water/AcOH (87% ee, entry 11, Table 3). The reaction temperature also affected drastically the enantioselectivities and witnessed good selectivity at 0 °C, while poor selectivities were observed at room temperature (entry 10, Table 3). Reactions carried out at temperatures below −10 °C (−20 to −78 °C) did not give the desired product.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Mol% | Solvents | Temp. (°C) | Yield (%) | eeb (%) |
| a All the reactions were performed using tetrahydro-β-carboline 20a (1 equiv.), NBS (1 equiv.) for 20 min.b The enantiomeric excess (ee) was determined by chiral HPLC.c The reaction was stirred for 24 h. | ||||||
| 1 | 11a | 10 | THF/water/AcOH (1:1:1) |
0 | 83 | 78 |
| 2 | 11b | 10 | THF/water/AcOH (1:1:1) |
0 | 85 | 98 |
| 3 | 11c | 10 | THF/water/AcOH (1:1:1) |
0 | 78 | 69 |
| 4 | 11d | 10 | THF/water/AcOH (1:1:1) |
0 | 80 | 85 |
| 5c | 11b | 10 | THF/water (1:1) |
rt | 65 | Racemic |
| 6c | 11b | 10 | DCE/water (1:1) |
rt | 60 | Racemic |
| 7c | 11b | 10 | THF/water (1:1) |
0 | 63 | Racemic |
| 8c | 11b | 10 | DCE/water (1:1) |
0 | 58 | Racemic |
| 9 | 11b | 5 | THF/water/AcOH (1:1:1) |
0 | 80 | 68 |
| 10 | 11b | 10 | THF/water/AcOH (1:1:1) |
rt | 83 | 78 |
| 11 | 11b | 10 | DCE/water/AcOH (1:1:1) |
0 | 84 | 87 |
| 12 | 11b | 10 | THF | 0 | — | — |
| 13 | 11b | 10 | DCE | 0 | — | — |
Based on above results, it was fixed the protocol as 1 equivalent of NBS, 10 mol% of thiosquaramide catalyst 11b, THF/water/AcOH (1:1:1) as solvent system, and reaction temperature at 0 °C for 20 min to give the key spirooxindole for the asymmetric synthesis of (−)-coerulescine (5) in 98% ee and 85% yield. Furthermore, after reaching the optimal reaction conditions, we also explored a short synthesis of (−)-horsfiline (6) with 93% ee and 90% yield, as shown in Scheme 4.
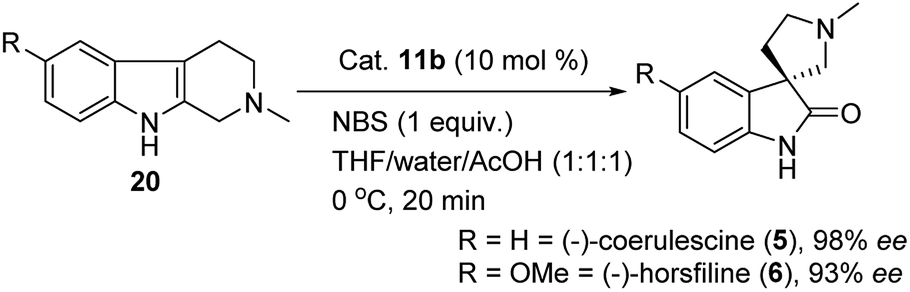 | ||
| Scheme 4 Thiosquaramide 11b catalyzed asymmetric synthesis of (−)-coerulescine (5) and (−)-horsfiline (6). | ||
Conclusions
In conclusion, we have synthesized four known squaramides and thiosquaramide based organocatalysts (11a–d) and 11b was identified as excellent organocatalyst for the asymmetric imine reduction. Under the optimized conditions, considerable yields (65–88%) and good enantioselectivities (88–96%) were observed for the synthesis of chiral tetrahydro-β-carbolines (18a–f). All THBCs were obtained with R configuration that could be explained by Si face approach of hydride in the transition state, as rationalized in Fig. 3. Moreover, formal syntheses of R-quinolactacin (3) and potential PDE5 inhibitor RWJ387273 (4) via asymmetric reduction of 17c and 17f, which can be avoided through metal catalyst and hazardous reagents. Additionally, we perform a chiral thiosquaramide 11b catalyzed enantioselective synthesis of natural spirooxindoles (−)-coerulescine (5) and (−)-horsfiline (6) through enantioselective rearrangement in a short approach, which afforded good yield and high enantioselectivities (up to 98% ee) in the direct oxidative rearrangement.Conflicts of interest
There are no conflicts to declare.Acknowledgements
L. L. S. and F. M. N. thank FONDECYT (Project #1180084) and M. S. is grateful to Dirección de Investigación Vicerrectoría Académica-Universidad de Talca, Talca, Chile for financial support for this work.Notes and references
- (a) Z. Nadine, M. Christian, M. H. Stefan and G. Harald, Nat. Commun., 2018, 9, 1949 CrossRef; (b) I. Jakub and W. Elżbieta, Org. Biomol. Chem., 2018, 16, 7296 RSC; (c) F. F. Douglas, B. Matthew, Mc. J. Cooper and D. R. Karl, Tetrahedron Lett., 1996, 37, 6227 CrossRef.
- (a) Z. Yang, Z. Ding, F. Chen, Y. M. He, N. Yang and Q. H. Fan, Eur. J. Org. Chem., 2017, 1973 CrossRef CAS; (b) K. A. Nolin, R. W. Ahn, Y. Kobayashi, J. J. Kennedy-Smith and F. D. Toste, Chemistry, 2010, 16, 9555 CrossRef CAS; (c) S. Hussain, F. Leipold, H. Man, E. Wells, S. P. France, K. R. Mulholland, G. Grogan and N. J. Turner, ChemCatChem, 2015, 7, 579 CrossRef CAS.
- (a) J. Václavík, P. Šot, J. Pecháček, B. Vilhanová, O. Matuška, M. Kuzma and P. Kačer, Molecules, 2014, 19, 6987 CrossRef; (b) P. A. Dub and J. C. Gordon, Nat. Rev. Chem., 2018, 2, 396 CrossRef CAS; (c) N. Uematsu, A. Fujii, S. Hashiguchi, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1996, 118, 4916 CrossRef CAS; (d) C. Li and J. Xiao, J. Am. Chem. Soc., 2008, 130, 13208 CrossRef CAS; (e) V. S. Shende, S. H. Deshpande, S. K. Shingote, A. Joseph and A. A. Kelkar, Org. Lett., 2015, 17, 2878 CrossRef CAS.
- (a) D. Ghislieri, D. Houghton, A. P. Green, S. C. Willies and N. J. Turner, ACS Catal., 2013, 3, 2869 CrossRef CAS; (b) Y. Mirabal-Gallardo, M. P. Soriano and L. S. Santos, Tetrahedron: Asymmetry, 2013, 24, 440 CrossRef CAS; (c) F. Leipold, S. Hussain, D. Ghislieri and N. J. Turner, ChemCatChem, 2013, 5, 3505 CrossRef CAS; (d) M. Espinoza-Moraga, T. Petta, M. Vasquez-Vasquez, V. F. Laurie, L. A. Moraes and L. S. Santos, Tetrahedron: Asymmetry, 2010, 21, 1988 CrossRef CAS.
- (a) M. Espinoza-Moraga, A. G. Caceres and L. S. Santos, Tetrahedron Lett., 2009, 50, 7059 CrossRef CAS; (b) O. Forero-Doria, L. S. Santos, F. M. Nachtigall and N. Shankaraiah, Comb. Chem. High Throughput Screening, 2017, 20, 696 CrossRef CAS.
- (a) A. E. Laine, C. Lood and A. M. Koskinen, Molecules, 2014, 19, 1544 CrossRef; (b) A. Spindler, K. Stefan and M. Wiese, J. Med. Chem., 2016, 59, 6121 CrossRef CAS; (c) S. Xiao, X. X. Shi, J. Xing, J. J. Yan, S. L. Liu and W. D. Lu, Tetrahedron: Asymmetry, 2009, 20, 2090 CrossRef CAS; (d) Y. Zhang, Q. He, H. Ding, X. Wu and Y. Xie, Org. Prep. Proced. Int., 2005, 37, 99 CrossRef CAS.
- (a) Q. Yin, S. G. Wang and S. L. You, Org. Lett., 2013, 15, 2688 CrossRef CAS; (b) M. Bandini, A. Melloni, F. Piccinelli, R. Sinisi, S. Tommasi and A. Umani-Ronchi, J. Am. Chem. Soc., 2006, 128, 1424 CrossRef CAS; (c) E. Xie, A. Rahmana and X. Lin, Org. Chem. Front., 2017, 4, 1407 RSC.
- (a) Y. Liu, H. Song, Y. Huang, J. Li, S. Zhao, Y. Song, P. Yang, Z. Xiao, Y. Liu, Y. Li, H. Shang and Q. Wang, J. Agric. Food Chem., 2014, 62, 9987 CrossRef CAS; (b) J. Galisteo and T. Herraiz, J. Agric. Food Chem., 2003, 51, 7156 CrossRef.
- (a) E. McQueen, A. Doyle and F. Smirk, Nature, 1954, 174, 1015 CrossRef CAS; (b) C. Tsioufis and C. Thomopoulos, Pharmacol. Res., 2017, 125B, 266 CrossRef; (c) B. Wang, N. K. Choudhry, J. J. Gagne, J. Landon and A. S. Kesselheim, Am. Heart J., 2015, 169, 379 CrossRef.
- J. E. Audia, D. A. Evrard, G. R. Murdoch, J. J. Droste, J. S. Nissen, K. W. Schenck, P. Fludzinski, V. L. Lucaites, D. L. Nelson and M. L. Cohen, J. Med. Chem., 1996, 39, 2773 CrossRef CAS.
- W. A. da Silva, M. T. Rodrigues Jr, N. Shankaraiah, R. B. Ferreira, C. K. Z. Andrade, R. A. Pilli and L. S. Santos, Org. Lett., 2009, 11, 3238 CrossRef CAS.
- H. Rommelspacher, H. Kauffmann, C. H. Cohnitz and H. Coper, Naunyn-Schmiedeberg's Arch. Pharmacol., 1977, 298, 83 CrossRef CAS.
- (a) N. Shankaraiah, W. A. da Silva, C. K. Z. Andrade and L. S. Santos, Tetrahedron Lett., 2008, 49, 4289 CrossRef CAS; (b) X. Zhang, W. Jiang and Z. Sui, J. Org. Chem., 2003, 68, 4523 CrossRef CAS.
- (a) N. Shankaraiah and L. S. Santos, Tetrahedron Lett., 2009, 50, 520 CrossRef CAS; (b) B. Willemsens, I. Vervest, D. Ormerod, W. Aelterman, C. Fannes, N. Mertens, I. E. Marko and S. Lemaire, Org. Process Res. Dev., 2006, 10, 1275 CAS; (c) S. Lemaire, B. Willemsens and I. E. Markó, Synlett, 2007, 5, 709 Search PubMed.
- (a) Y. Na, C. Haiying, A. W. Eric, S. Pei-Yong and Z. Jia, ACS Infect. Dis., 2016, 2, 382 CrossRef; (b) Z. Bing, Y. Yaxi, S. Jingjing, L. Zhi and L. Yuanchao, J. Org. Chem., 2013, 78, 2897 CrossRef; (c) C. Li, C. Chan, A. C. Heimann and S. J. Danishefsky, Angew. Chem., Int. Ed., 2007, 46, 1444 CrossRef CAS; (d) J. D. White, Y. Li and D. C. Ihle, J. Org. Chem., 2010, 75, 3569 CrossRef CAS.
- (a) B. Assem, S. Mohammad, M. G. Hussien, M. A. M. Abdullah, F. E. S. Fardous, A. B. Farid, A. M. M. E. Yaseen and A. G. G. Hazem, RSC Adv., 2018, 8, 14335 RSC; (b) C. Linwei, X. Jialin, S. Hongjian, L. Yuxiu, G. Yucheng, W. Lizhong and W. Qingmin, J. Agric. Food Chem., 2016, 64, 6508 CrossRef; (c) Y. Bin, Z. Yi-Chao, S. Xiao-Jing, Q. Ping-Ping and L. Hong-Min, Anti-Cancer Agents Med. Chem., 2016, 16, 1315 CrossRef.
- (a) G. J. Mei and F. Shi, Chem. Commun., 2018, 54, 6607 RSC; (b) M. Pelay-Gimeno, G. Adrian, K. Oliver and N. G. Tom, Angew. Chem., Int. Ed., 2015, 54, 8896 CAS.
- (a) Y. Yan-Tao, Z. Jun-Fang, L. Guochao, X. Hai-Jiang and Y. Bin, Curr. Med. Chem., 2018, 25, 2233 Search PubMed; (b) G. Bhaskar, Y. Arun, C. Balachandran, C. Saikumar and P. T. Perumal, Eur. J. Med. Chem., 2012, 51, 79 CrossRef CAS.
- (a) R. R. Kumar, S. Perumal, P. Senthilkumar, P. Yogeeswari and D. Sriram, J. Med. Chem., 2008, 51, 5731 CrossRef CAS; (b) S. M. Rajesh, S. Perumal, C. J. Menéndez, Y. Perumal and D. Sriram, Med. Chem. Commun., 2011, 2, 626 RSC.
- (a) B. Yu, D. Q. Yu and H. M. Liu, Eur. J. Med. Chem., 2015, 97, 673 CrossRef CAS; (b) Y. Arun, K. Saranraj, C. Balachandran and P. T. Perumal, Eur. J. Med. Chem., 2014, 74, 50 CrossRef CAS; (c) W. Dao-Cai, F. Chen, X. Yong-Mei, Y. Shun and S. Hang, Arabian J. Chem., 2019, 12, 1918 CrossRef.
- W. Yong-Chao, W. Jun-Liang, S. B. Kevin, Z. Jiang-Wei, Z. Qiu-Mei, P. Ya-Dan, Y. Li-Jun and C. Xue-Bing, RSC Adv., 2018, 8, 5702 RSC.
- H. Saoussen, B. Sarra, N. A. Tarunkumar, P. R. Jignesh, P. François, S. Armand, A. Moheddine, K. Michael, R. Yoann, M. K. Marek and R. A. Dhanji, New J. Chem., 2015, 39, 520 RSC.
- (a) Y. Kia, H. Osman, R. S. Kumar, A. Basiri and V. Murugaiyah, Bioorg. Med. Chem., 2014, 22, 1318 CrossRef CAS; (b) I. A. Abdulrahman, S. K. Raju, A. Natarajan, B. Alireza, K. Yalda, A. A. Mohamed, F. Mehvish and M. A. Vikneswaran, Molecules, 2015, 20, 2296 CrossRef.
- G. K. Mukund, P. D. Attrimuni, W. C. Sanjay, S. B. Ajit, B. S. Yunnus, R. B. Deekshaputra, P. D. Mayur and R. D. Nagorao, Beilstein J. Org. Chem., 2010, 6, 876 CrossRef.
- R. S. Paul, O. Hiroyuki, U. Takeo and M. W. Robert, Tetrahedron, 2002, 58, 6311 CrossRef.
- L. Hanwool, H. B. Seung, H. L. Jong, K. Chulwon, K. Jeong-Hyeon, L. Seok-Geun, C. Arunachalam, A. A. Sulaiman, M. Y. Woong, U. Jae-Young, S. Gautam and S. A. Kwang, Int. J. Mol. Sci., 2017, 18, 1095 CrossRef.
- L. L. Chen, J. X. Song, J. H. Lu, Z. W. Yuan, L. F. Liu, S. S. Durairajan and M. Li, J. Neuroimmune Pharmacol., 2014, 9, 380 CrossRef.
- (a) H. Chen and D. Shi, J. Comb. Chem., 2010, 12, 571 CrossRef CAS; (b) C. She-Jie, L. Yi-Feng, X. Jiang-Cheng, Z. Hui, Z. Qing and Z. Peng-Fei, Adv. Synth. Catal., 2011, 353, 371 CrossRef.
- (a) Q. Liu, S. Li, X. Y. Chen, K. Rissanen and D. Enders, Org. Lett., 2018, 20, 3622 CrossRef CAS; (b) Y. Bin, X. Hui, Y. De-Quan and L. Hong-Min, Beilstein J. Org. Chem., 2016, 12, 1000 CrossRef; (c) D. C. Prakash, H. Bor-Cherng, W. Chao-Lin and L. Gene-Hsiang, ACS Omega, 2019, 4, 655 CrossRef; (d) T. Mukaiyama, K. Ogata, I. Sato and Y. Hayashi, Chem.–Eur. J., 2014, 20, 13583 CrossRef CAS; (e) G. Lakshmaiah, T. Kawabata, M. Shang and K. Fuji, J. Org. Chem., 1999, 64, 1699 CrossRef CAS.
- (a) J. P. Malerich, K. Hagihara and V. H. Rawal, J. Am. Chem. Soc., 2008, 130, 14416 CrossRef CAS; (b) W. Yang and D. M. Du, Chem. Commun., 2011, 47, 12706 RSC; (c) L. Bing-Yu and D. Da-Ming, Adv. Synth. Catal., 2018, 360, 3164 CrossRef.
- (a) M. Rombola, C. S. Sumaria, T. D. Montgomery and V. H. Rawal, J. Am. Chem. Soc., 2017, 139, 5297 CrossRef CAS; (b) N. Sándor, D. Gyula, K. Péter, F. Zsuzsanna, S. András, B. Júlia, H. Tibor, M. Béla, K. Levente, D. László, H. Péter and K. József, New J. Chem., 2019, 43, 5948 RSC; (c) P. Rodríguez-Ferrer, N. Daniel, M. Alicia, M. A. José and P. Rafael, Eur. J. Org. Chem., 2019, 2019, 6539 CrossRef.
- M. Yang, C. Chen, X. Yi, Y. Li, X. Wu, Q. Li and S. Ban, Org. Biomol. Chem., 2019, 17, 2883 RSC.
- O. Kristína, B. Stanislav, M. Mária and S. Radovan, ChemistrySelect, 2019, 4, 8870 CrossRef.
- M. Mirza-Aghayan, R. Boukherroub, M. Bolourtchian and M. Rahimifard, J. Organomet. Chem., 2007, 692, 5113 CrossRef CAS.
- (a) R. Noyori and S. Hashiguchi, Acc. Chem. Res., 1997, 30, 97 CrossRef CAS; (b) C. V. Sandoval, T. Ohkuma, K. Muñiz and R. Noyori, J. Am. Chem. Soc., 2003, 125, 13490 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra07705d |
| This journal is © The Royal Society of Chemistry 2020 |