
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynergetic effect of dual co-catalysts on the activity of BiVO4 for photocatalytic carbamazepine degradation†
Beibei Wanga,
Ping Lib,
Chunlei Dub,
Yan Wangb,
Daxin Gaob,
Songtao Lib,
Liying Zhangb and
Fuyu Wen *b
*b
aEducational Technology Center, Chengde Medical University, Chengde 067000, China
bHebei Key Laboratory of Research and Development for Traditional Chinese Medicine, Chengde Medical University, Chengde 067000, China. E-mail: wenfuyu@vip.163.com
First published on 17th December 2019
Abstract
An efficient visible-light driven three components photocatalyst for carbamazepine (CBZ) degradation has been assembled by co-loading reduction cocatalyst Pt and oxidation cocatalyst Co3O4 (MnOx) on BiVO4. The apparent rate constant of the three components photocatalyst Pt/BiVO4/Co3O4 for degradation of CBZ is 54 times that of Co3O4/BiVO4 and 2.5 times that of Pt/BiVO4, which shows a synergetic effect in the photocatalytic activity. The same synergetic effect is also observed for Pt/BiVO4/MnOx. The spatial separation of the reduction and oxidation cocatalysts could reduce the recombination of the photogenerated charges, which mainly accounts for the high photocatalytic activity of the three components photocatalyst. The photocatalytic intermediates of CBZ were detected by HPLC-ESI-MS, and a deductive degradation pathway of CBZ was proposed.
1. Introduction
Pharmaceuticals and personal care products (PPCPs) are persistent organic pollutants which cannot be completely removed by conventional treatment plants.1 For instance, the removal efficiency of carbamazepine (CBZ) through wastewater treatment plants (WWTPs) is mostly below 10%.2 Drug residues in the natural environment eventually affect water quality, ecosystems and human health.3 For example CBZ is reported to be toxic even at concentrations below 100 mg L−1.4 Therefore, it is urgent to seek more efficient technologies for the degradation of CBZ-containing waste-water.Heterogeneous photocatalysis is one of the most ideal techniques because of its advantages of lower energy consumption, no secondary pollution, easy accessibility and so on.5 To date, the most widely employed semiconductor for CBZ degradation is TiO2,6–9 however, it is severely limited as a sunlight-driven photocatalyst due to large band gap and low quantum yield.10,11 Therefore substantial research efforts have aimed to develop more efficient visible-light driven photocatalyst for CBZ degradation.12–16
Many semiconductors possess suitable band gap (e.g. 2.31 eV for BiVO4 which corresponding an absorption edge at 535 nm) and sufficient band potentials for CBZ degradation but without or with low activity. The recombination of the photogenerated electrons and holes may be the main reason. To enhance the separation efficiency of the photogenerated charges, cocatalysts which serve as the active sites for the photocatalytic reactions are usually necessary.17–24 Moreover, when both the reduction and oxidation cocatalysts were co-loaded on the semiconductors, the synergistic effect on the photocatalytic activities were usually achieved.25–29 The strategy of dual cocatalysts has been successfully employed in photocatalytic water splitting,26,27,29,30 photocatalytic oxidation of some pollutants such as thiophene28,31 and some dyes.25,26,28 However, the synergistic effect of dual cocatalysts is far less investigated in the photocatalytic degradation of pharmaceuticals.
Herein we report an efficient visible-light driven three components photocatalyst for carbamazepine degradation. By co-loading reduction cocatalyst Pt and oxidation cocatalyst Co3O4 (MnOx) on BiVO4, the three components photocatalysts Pt/BiVO4/Co3O4 (MnOx) were assembled. The apparent rate constant of Pt/BiVO4/Co3O4 for degradation of carbamazepine is 54 times of Co3O4/BiVO4 and 2.5 times of Pt/BiVO4, which shows a synergetic effect in the photocatalytic activity. The present work further demonstrates the universality of the strategy of dual-cocatalysts, which would be useful to construct highly efficient photocatalyst for pharmaceuticals degradation.
2. Experimental
2.1 Catalyst preparation
All of the reagents were of analytical grade, and were used without further purification. BiVO4 was prepared by a hydrothermal process.31 The precursors NH4VO3 (12 mmol) and Bi(NO3)3·5H2O (12 mmol) were dissolved in 45 mL and 15 mL of 2.0 M nitric acid solutions, respectively. Then the above solutions were mixed to form a yellow homogeneous solution. The pH value of the solution was then adjusted to 2.0 with ammonia solution under stirring. The orange mixture was stirred for 0.5 h and aged for an additional 2 h. The orange precipitate at the bottom of the beaker was transferred to a 100 mL Teflon-lined stainless steel autoclave (70% capacity) and hydrothermal treated at 200 °C for 24 h. After the autoclave was cooled to room temperature, a vivid yellow powder was separated by filtration, washed with deionized water for more than 3 times, and then dried at 60 °C for overnight.Co3O4 (MnOx)/BiVO4 was prepared by photo-deposition method.26 Co(NO3)2 solutions (0.5 g L−1) or MnSO4 solutions (0.5 g L−1) was used as the precursor and NaIO3 was employed as the electron acceptor. Typically, 0.5 g BiVO4 powder was suspended in 100 mL NaIO3 solutions (0.01 M), then 1.0 mL Co (NO3)2 solution or MnSO4 solution was added and the suspension was then irradiated by a 300 W Xe lamp (λ > 420 nm) under continuous stirring. After 5 h photo-deposition, the suspension was filtered, washed with deionized water for more than 3 times, and finally dried at 60 °C for overnight. The preparation of Pt/BiVO4 was similar to Co3O4 (MnOx)/BiVO4 but using H2PtCl6 solution (0.374 g L−1) as the precursor and water as the hole scavenger.
Pt/BiVO4/Co3O4 (MnOx) was prepared by simultaneous photo-deposition method.27 0.5 g BiVO4 powder was suspended in 100 mL deionized water, then 6.0 mL H2PtCl6 solution and 1.0 mL Co (NO3)2 solution (MnSO4 solution) were added. After 5 h photo-deposition, the suspension was filtered, washed and finally dried at 60 °C for overnight.
The electrodes of BiVO4, Pt/BiVO4/Co3O4 and Pt/BiVO4/MnOx were prepared by electrophoretic deposition of corresponding powder on FTO substrate (1 × 2 cm2),32 followed by drying in air and calcination at 573 K for 1 h.
2.2. Catalyst characterization
The prepared samples were characterized by X-ray powder diffraction (XRD) on a Rigaku D/Max-2500/PC powder diffractometer. Each sample powder was scanned using Cu Kα radiation with an operating voltage of 40 kV and an operating current of 200 mA. The scan rate of 5° min−1 was applied to record the patterns in the range of 8–80° at a step of 0.02°. Brunauer–Emmett–Teller (BET) specific surface areas of the samples were determined by nitrogen adsorption at 77 K on Micromeritics ASAP 2420 system.UV-Vis diffuse reflectance spectra (UV-Vis DRS) were recorded on a TU-1950 UV-Vis spectrophotometer (Beijing Purkinje General Instrument Co., Ltd.) equipped with an integrating sphere. The morphologies and particle sizes were examined by a Quanta 200 FEG scanning electron microscope (SEM). X-ray photoelectron spectroscopy (XPS) measurements, using a VG ESCALAB MK2 spectrometer with monochromatized Al-Kα excitation.
Electron spin resonance (ESR) signals of radicals trapped by DMPO were recorded at ambient temperature on a Brucker ESR A200 spectrometer. The samples were introduced into the home-made quartz cup inside the microwave cavity and illuminated with a 300 W Xe lamp (CERAMAX LX-300). The settings for the ESR spectrometer were as follows: sweep width, 140 G; microwave frequency, 9.82 GHz; modulation amplitude, 1 G.
Photoelectrochemical performances of the photoanodes were measured in a three-electrode setup, where Pt electrode and saturated mercury electrode were employed as counter and reference electrode, respectively. Electrolyte was 0.5 M Na2SO4 solution. A shutter was used to record both the dark and photocurrent during a single scan. A 300 W Xe lamp (Ushio-CERAMAX LX-300) and optical cutoff filter (kenko, L-42; λ > 420 nm) was used as light source. Electrochemical impedance spectroscopy (EIS) were performed on a Ivium electrochemical workstation by applying an AC voltage of 10 mV amplitude in the frequency range of 105 Hz to 10−1 Hz in 0.5 M Na2SO4.
2.3. Photocatalytic reaction
The photocatalytic degradation of carbamazepine (CBZ) was carried out using a 300 W Xe lamp and optical cut-off filter (λ > 420 nm). Normally, 100 mg photocatalyst was dispersed in 100 mL 10 mg L−1 CBZ solutions. Prior to irradiation, the suspensions were magnetically stirred in dark for 30 min to establish adsorption/desorption equilibrium between CBZ and the photocatalysts. Top irradiation was used and after different irradiation time, 3 mL suspensions were centrifuged and used for UV-Vis characterization. Calibration based on the Lambert–Beer law was used to quantify the concentration of CBZ.Total organic carbon (TOC) was measured using TOC-VCPN of Shimadzu Corporation of Japan. A combustion catalytic oxidation method of 680 °C is used. The measurement range is from 4 μg L−1 to 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 μg L−1. The degradation intermediates were detected by mass spectrometry on Agilent HPLC-6500 Q-TOF in electrospray positive ion (ESI+) mode with acetonitrile (solvent A) and ultrapure water with 0.1% V formic acid (solvent B) as mobile phase. A zorbax eclipse plus C18 column (1.8 μm, 2.1 × 50 mm) was used as separation column.
000 μg L−1. The degradation intermediates were detected by mass spectrometry on Agilent HPLC-6500 Q-TOF in electrospray positive ion (ESI+) mode with acetonitrile (solvent A) and ultrapure water with 0.1% V formic acid (solvent B) as mobile phase. A zorbax eclipse plus C18 column (1.8 μm, 2.1 × 50 mm) was used as separation column.
3. Results and discussion
3.1 Characterization of the photocatalysts
Fig. 1 shows the UV-Vis diffuse reflectance spectra of BiVO4, Pt/BiVO4, MnOx/BiVO4, Co3O4/BiVO4, Pt/BiVO4/MnOx and Pt/BiVO4/Co3O4. The absorption edge of the prepared BiVO4 is about 535 nm, demonstrating a strong absorption in the visible light region. No obvious shift of the absorption edge is observed when different cocatalysts were loaded on BiVO4. The XRD patterns of the as-prepared BiVO4, Pt/BiVO4/MnOx and Pt/BiVO4/Co3O4 are shown in Fig. 2. The diffraction peaks of all of the samples are identical to the peaks for the monoclinic scheelite phase of BiVO4 (JCPDS 14-0688). The crystal structures of the co-loaded BiVO4 show no changes compared with that of pure BiVO4, implying that the cocatalysts are loaded onto the surface of the BiVO4. N2 adsorption–desorption isotherms (Fig. S1†) show that the co-loading of Pt and Co3O4 (MnOx) did not change the framework of BiVO4 and the BET surface areas of the three components photocatalysts Pt/BiVO4/Co3O4 (MnOx) are comparable with BiVO4.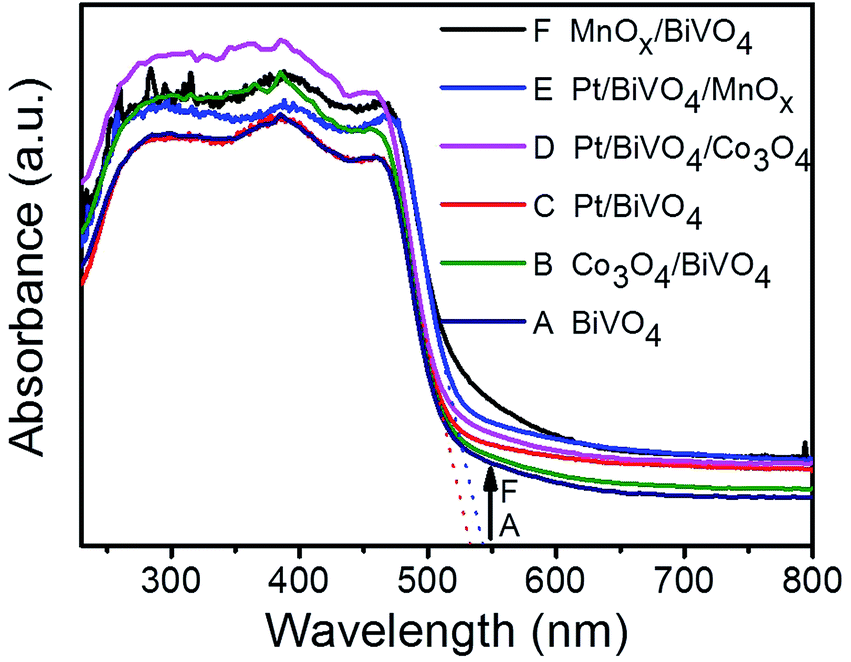 | ||
| Fig. 1 UV-Vis DRS of different photocatalysts. The contents of the deposited Pt, MnOx and Co3O4 are 0.2 wt%, 0.1 wt% and 0.1 wt%, respectively. | ||
 | ||
| Fig. 2 XRD patterns of BiVO4, Pt/BiVO4/MnOx and Pt/BiVO4/Co3O4. The contents of the deposited Pt is 0.2 wt%, Co3O4 and MnOx are all 0.1 wt%. | ||
SEM images of Pt/BiVO4/Co3O4 further demonstrate that the cocatalysts are deposited on the surface of the BiVO4 (Fig. S2†). Furthermore, as reported by Li et al.,26,27 the reduction cocatalyst Pt and oxidation cocatalyst Co3O4 may be deposited on the different facets of BiVO4, which will facilitate the charge separation of electrons and holes.
The chemical states of the loaded cocatalysts were examined by XPS characterization. The Pt 4f peak located at binding energy of 70.7 eV was observed for both three components photocatalysts, indicating the existence of the metallic Pt (0) on BiVO4 (Fig. 3). The Co 2p peaks at binding energies of 777.9 eV and 795.0 eV demonstrating that Co3O4 was deposited on BiVO4 (Fig. 4a).26 The Mn 2p peaks are located at binding energies of 653.5 eV and 641.9 eV, which are between those of Mn2O3 and MnO2 (Fig. 4b). Based on this, the deposited Mn species can be ascribed to MnOx, where x is between 1.5 and 2.0.27
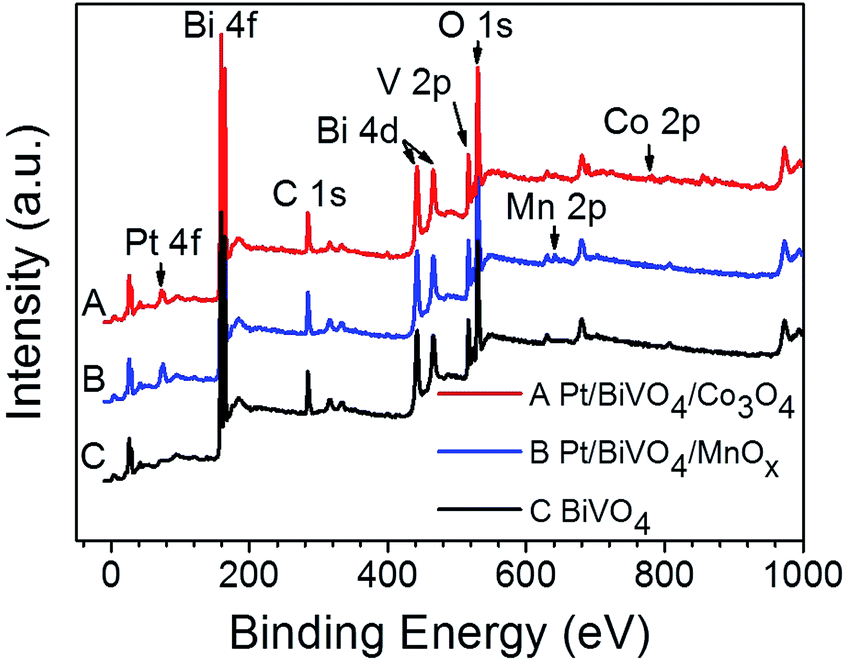 | ||
| Fig. 3 XPS spectra of BiVO4, Pt/BiVO4/Co3O4 and Pt/BiVO4/MnOx. The contents of the deposited Pt is 0.2 wt%, Co3O4 and MnOx are all 0.1 wt%. | ||
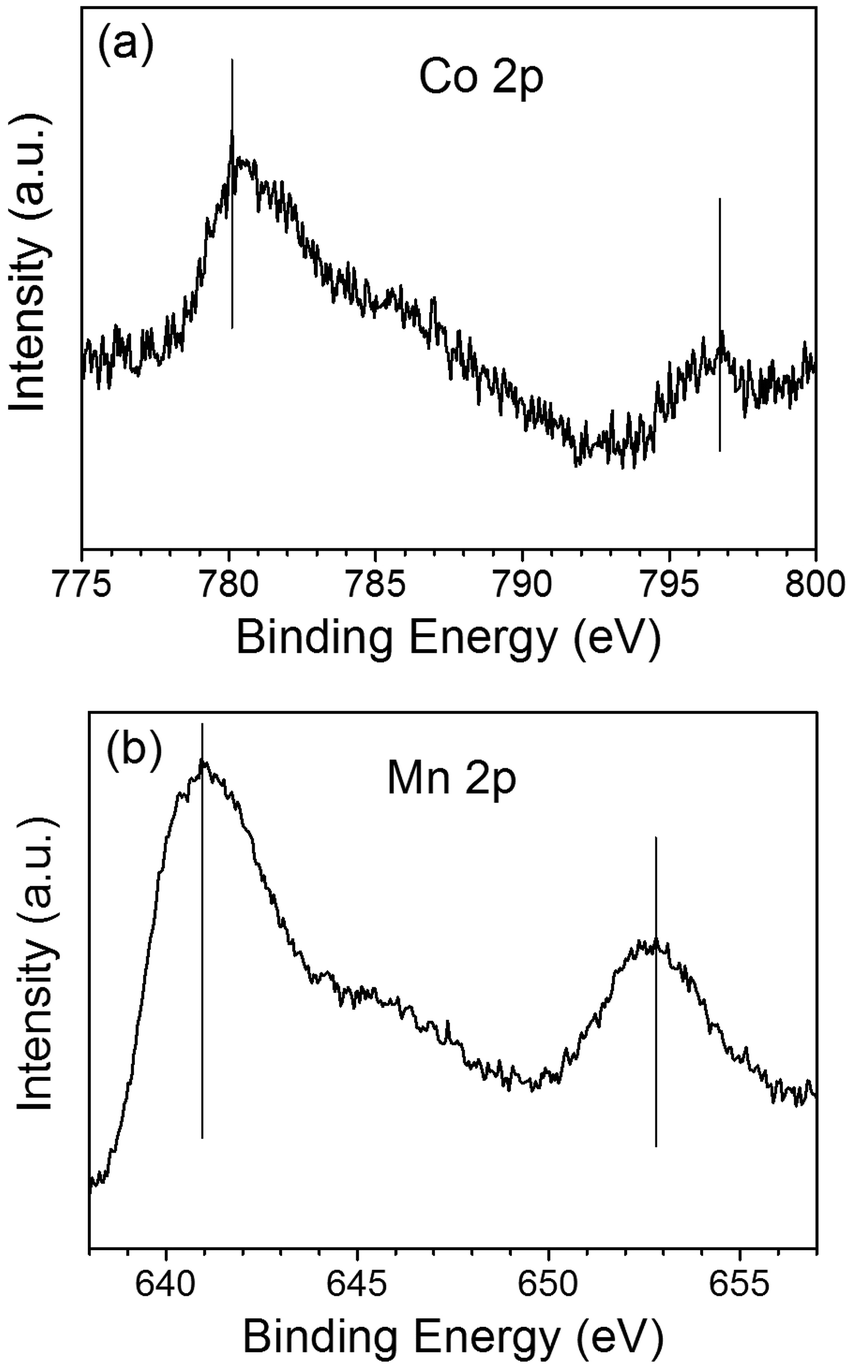 | ||
| Fig. 4 The Co 2p XPS spectra of Co3O4 (a) and Mn 2p XPS spectra of MnOx (b) deposited on BiVO4. The contents of the deposited Co3O4 and MnOx are all 0.1 wt%. | ||
3.2 The effect of cocatalyst on photocatalytic degradation of carbamazepine
Fig. 5 shows the kinetic data of the photocatalytic degradation of CBZ on different photocatalysts. BiVO4 without loaded cocatalyst exhibited a low removal (ca. 2%) in 120 min, and the removal was slightly enhanced when the oxidation cocatalyst MnOx or Co3O4 was loaded on BiVO4. When reduction cocatalyst Pt was deposited on BiVO4, the removal of CBZ was enhanced to about 28% in 120 min. However, when Pt and Co3O4 were co-loaded on BiVO4, the removal was significantly improved to ca. 56%, much higher than those of Pt/BiVO4 and Co3O4/BiVO4. Same trend was also observed when Pt and MnOx were co-loaded on BiVO4, yielding a removal of ca. 53% in 120 min, which is much higher than those of Pt/BiVO4 and MnOx/BiVO4. | ||
| Fig. 5 Photocatalytic degradation of CBZ on different photocatalysts. Reaction conditions: 100 mL CBZ solution with initial concentration of 10 mg L−1, catalyst dosage 1 g L−1, the contents of the deposited Pt is 0.2 wt%, Co3O4 and MnOx are all 0.1 wt% and 300 W Xe lamp (λ > 420 nm), top irradiation. | ||
TOC analysis was carried out to evaluate the mineralization extent of CBZ on BiVO4 and Pt/BiVO4/Co3O4 photocatalysts (Fig. S3†). As shown in Fig. S3,† the TOC removal of CBZ in the presence of Pt/BiVO4/Co3O4 is also much higher than that of BiVO4. Long-term circulation measurement was carried out to check the stability of the photocatalyst Pt/BiVO4/Co3O4. As shown in Fig. S4,† the photocatalytic activity of Pt/BiVO4/Co3O4 can be well maintained even after three runs, only less than 5% decreasing, indicating the excellent stability and durability of the three components photocatalyst.
The plots of ln(C0/C) versus the reaction time (t) is presented in Fig. 6. All of the fitted curves show high corresponding apparent coefficients, revealing that the photocatalytic degradation of CBZ over different photocatalysts is in line with pseudo-first-order kinetic. The values of the rate constant k for different photocatalysts are listed in Table 1. The single loading of either oxidation cocatalyst (entry 2 and 3) or reduction cocatalyst (entry 4) can somewhat enhance the activity of BiVO4, demonstrating the roles of cocatalysts played in the facilitating of charge transfer of photogenerated holes or electrons; however, the enhancement is limited. The promotion caused by the reduction cocatalyst loading is more obvious than the oxidation cocatalyst loading, indicating that the reduction half-reaction may be the rate-determining step.26 When reduction and oxidation cocatalysts were co-loaded on BiVO4, the rate constants (entry 5 and 6) were significantly improved. The k for Pt/BiVO4/MnOx is 23 times of MnOx/BiVO4 and 2.3 times of Pt/BiVO4. Similarly, the k for Pt/BiVO4/Co3O4 is 54 times of Co3O4/BiVO4 and 2.5 times of Pt/BiVO4. Both of the three components photocatalysts show obvious synergetic effect of the dual cocatalysts (k5 > k3 + k4, k6 > k2 + k4). This demonstrates that only when the reduction and oxidation reactions were accelerated simultaneously we can get the much higher reaction rate than any other one.
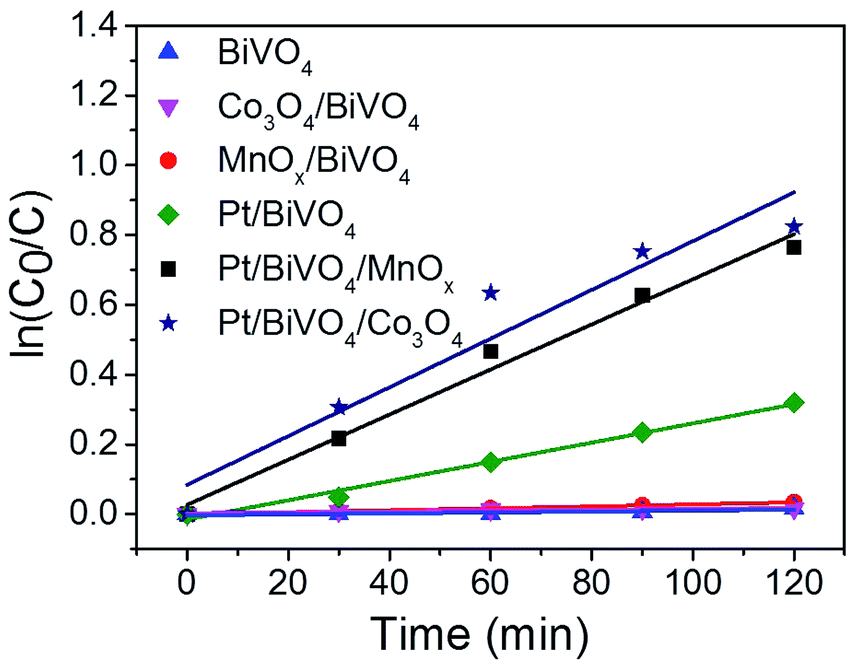 | ||
| Fig. 6 Kinetic fitting of the CBZ degradation on different photocatalysts. Reaction conditions: 100 mL CBZ solution with initial concentration of 10 mg L−1, catalyst dosage 1 g L−1, the contents of the deposited Pt is 0.2 wt%, Co3O4 and MnOx are all 0.1 wt% and 300 W Xe lamp (λ > 420 nm), top irradiation. | ||
| Entry | Photocatalysts | ka (×10−3 min−1) |
|---|---|---|
| a The apparent first-order rate constant k is calculated from the equation: ln(C0/C) = kt, where C0 is the initial concentration and C is the concentration at certain time t. Reaction conditions: 100 mL CBZ solution with initial concentration of 10 mg L−1, catalyst dosage 1 g L−1, the contents of the deposited Pt is 0.2 wt%, Co3O4 and MnOx are all 0.1 wt% and 300 W Xe lamp (λ > 420 nm), top irradiation. | ||
| 1 | BiVO4 | 0.12 |
| 2 | Co3O4/BiVO4 | 0.13 |
| 3 | MnOx/BiVO4 | 0.28 |
| 4 | Pt/BiVO4 | 2.75 |
| 5 | Pt/BiVO4/MnOx | 6.46 |
| 6 | Pt/BiVO4/Co3O4 | 6.98 |
The roles of cocatalysts played in facilitating photo-generated charges transfer can be further demonstrated by the photoelectrochemical measurements. As shown in Fig. S5,† the photocurrent density of the three components photocatalysts Pt/BiVO4/Co3O4 and Pt/BiVO4/MnOx are higher than that of bare BiVO4. This demonstrating that due to the co-loading of cocatalysts, the photo-generated charges can be more efficiently trapped, which makes more photo-generated holes survived on the three components photocatalysts. Moreover, the photocurrent of Pt/BiVO4/Co3O4 is higher than Pt/BiVO4/MnOx, demonstrating that the photogenerated holes (h+) would transfer easier from BiVO4 to Co3O4 than to MnOx, which cause the superior performance of Pt/BiVO4/Co3O4 than Pt/BiVO4/MnOx on the photocatalytic CBZ degradation. The electrochemical impedance spectra (EIS) of BiVO4 and Pt/BiVO4/Co3O4 further demonstrate the role of cocatalysts played in facilitating photo-generated charges transfer. As shown in Fig. S6,† the arc radius on the EIS plot of Pt/BiVO4/Co3O4 is smaller than that of BiVO4, implying that the co-loading of Pt and Co3O4 make photo-generated charges transfer easier.
3.3 Identification of main reactive species
To identify the main reactive species in the photocatalytic degradation process, trapping experiments were conducted on Pt/BiVO4/Co3O4 photocatalyst and the results are shown in Fig. 7. The addition of ammonium oxalate (AO, scavengers of h+ radical) has significant inhibition on CBZ degradation, indicating that h+ is crucial active species. The addition of ascorbic acid (VC, scavengers of ·O2− radical) and isopropyl alcohol (IPA, scavengers of ·OH radical) have feeble inhibition, which reveals that ·O2− and ·OH serve as synergistic active species but not as the most crucial active species. Similar results were observed for Pt/BiVO4/MnOx photocatalyst (Fig. S7†). | ||
| Fig. 7 Effect of scavengers on photocatalytic degradation of CBZ over Pt/BiVO4/Co3O4 photocatalyst. Reaction conditions: 100 mL CBZ solution with initial concentration of 10 mg L−1, scavenger concentration 50 mmol L−1, catalyst dosage 1 g L−1, the contents of the deposited Pt and Co3O4 are 0.2 wt% and 0.1 wt% respectively and 300 W Xe lamp (λ > 420 nm), top irradiation. | ||
In order to gain direct evidence for the reactive radicals involved in the photocatalytic process, ESR analysis was employed using DMPO as trapping agent. As shown in Fig. 8, the sextet ESR signal centered at g = 2.0065 can be assigned to DMPO-O2˙−. These results provide evidence of ·O2− formed in the presence of the photocatalyst BiVO4 and Pt/BiVO4/Co3O4. Moreover, the signal of ·O2− generated after 10 min of illumination on Pt/BiVO4/Co3O4 is more obvious than those for BiVO4, demonstrating the roles of cocatalysts played in facilitating charge separation and transfer.
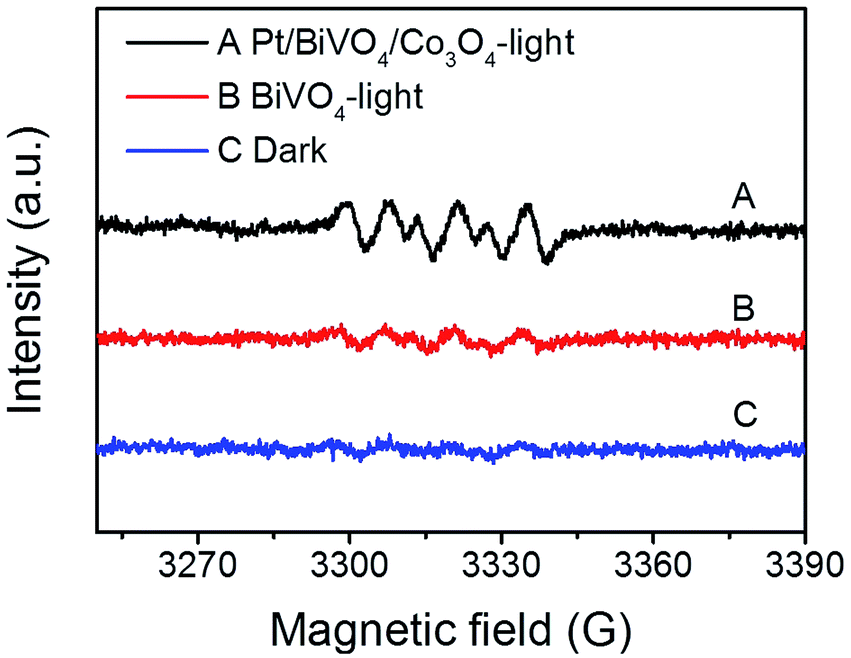 | ||
| Fig. 8 In situ ESR spectra of DMPO-O2˙− generated by BiVO4 and Pt/BiVO4/Co3O4 after 10 min of visible-light irradiation (λ > 420 nm). The signal obtained in the presence of photocatalyst but without light irradiation is denoted as “Dark”. | ||
3.4 Degradation pathway of CBZ photocatalyzed by Pt/BiVO4/Co3O4
Based on the LC-ESI-MS results (Table S1 and Fig. S8†), the possible degradation pathway of CBZ photocatalyzed by Pt/BiVO4/Co3O4 was proposed in Fig. 9. First of all, alkene double bond of CBZ was attack by ·OH radical and transferred into intermediate P6 and P9.33–35 Intermediate P6 was further hydroxylated to generate P2 which might be further oxidized to generate P8.35 As for intermediate P9, it might be further oxidized by ·O2− and photo-generated h+ to convert into P4 and P7.34 After aldehyde oxidation reaction in P4, P5 was generated and further transferred to P8.35 Product P7 might undergo several successive steps including deamination and decarboxylation to produce P1 and P3.7,34,36 Further oxidized by ·OH, h+ and ·O2−, P3 and P8 could be finally mineralized into CO2 and H2O via ring–rupturing reaction.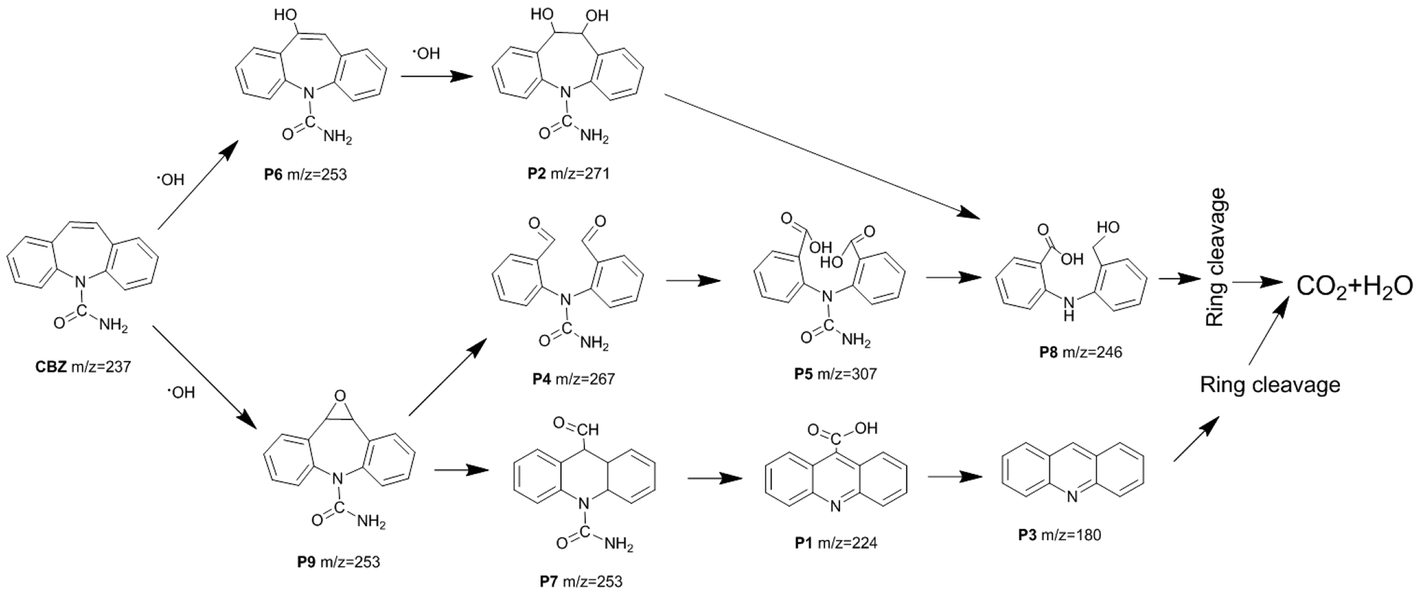 | ||
| Fig. 9 Proposed degradation pathway of CBZ by Pt/BiVO4/Co3O4 photocatalyst. | ||
The proposed mechanism of photocatalytic degradation of CBZ on Pt/BiVO4/Co3O4 (MnOx) is shown in Fig. 10. Under visible light irradiation, photo-generated electrons would transfer from the conduction band (CB) of BiVO4 to the reduction cocatalyst Pt, on which the adsorbed O2 is reduced to ·O2− radical. Meanwhile, the holes would migrate from the valence band (VB) of BiVO4 to the oxidation cocatalyst Co3O4 (MnOx), where the adsorbed CBZ or H2O are oxidized to CO2 and ·OH respectively. The produced ·O2− and ·OH also participate in the degradation of CBZ, which is consistent with the trapping experiment results. The co-loading of both the reduction cocatalyst Pt and oxidation cocatalyst Co3O4 (MnOx) is beneficial for the efficient separation and transfer of the photo-generated electrons and holes, accounts for the high photocatalytic activity of CBZ degradation.
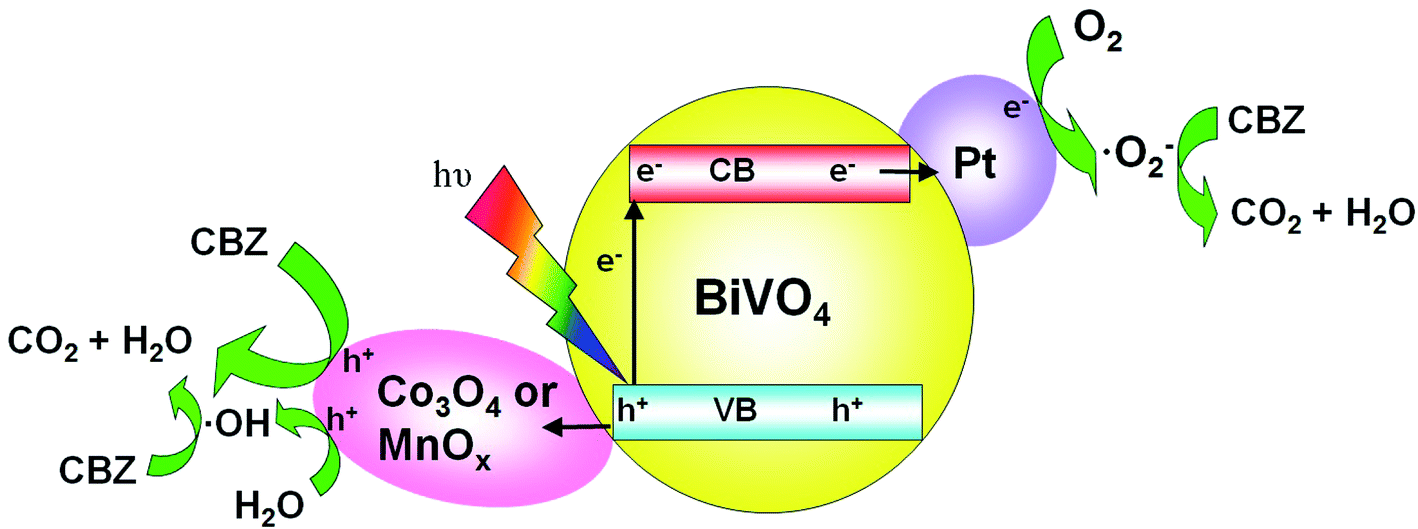 | ||
| Fig. 10 Schematic description of the mechanism for the photocatalytic degradation of CBZ on Pt/BiVO4/Co3O4 (MnOx) photocatalyst under visible light irradiation. | ||
4. Conclusion
A synergetic effect in photocatalytic degradation of CBZ was achieved when reduction cocatalyst Pt and oxidation cocatalyst Co3O4 (MnOx) were co-loaded on BiVO4. The co-loading of both the reduction cocatalyst and oxidation cocatalyst can facilitate the separation and transfer of the photo-generated electrons and holes, which is responsible for the high photocatalytic activity of the three components photocatalysts. This work further demonstrates the universality of the strategy of dual-cocatalysts, which would be useful to construct highly efficient photocatalyst for pharmaceuticals degradation.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was financially supported by Scientific Research Fund of the Hebei Education Department (No. BJ2014002), Natural Science Foundation of Hebei Province, China (No. B2016406012 and H2017406022), National Natural Science Foundation of China (No. 81473101 and 81001401) and Undergraduate Innovation and Entrepreneurship Training Program of Chengde Medical University (No. 201424 and 201603). The authors thank Prof. Can Li, Prof. Xu Zong, Dr Jingfeng Han, Dr Sheng Ye and Xianwen Zhang for the help on characterizations of the photocatalysts.References
- J. Rivera-Utrilla, M. Sanchez-Polo, M. A. Ferro-Garcia, G. Prados-Joya and R. Ocampo-Perez, Chemosphere, 2013, 93, 1268–1287 CrossRef CAS PubMed
.
- Y. J. Zhang, S. U. Geissen and C. Gal, Chemosphere, 2008, 73, 1151–1161 CrossRef CAS
- T. Heberer, Toxicol. Lett., 2002, 131, 5–17 CrossRef CAS PubMed
- K. Fent, A. A. Weston and D. Caminada, Aquat. Toxicol., 2006, 76, 122–159 CrossRef CAS PubMed
- P. A. K. Reddy, P. V. L. Reddy, E. Kwon, K. H. Kim, T. Akter and S. Kalagara, Environ. Int., 2016, 91, 94–103 CrossRef CAS PubMed
- S. Murgolo, V. Yargeau, R. Gerbasi, F. Visentin, N. El Habra, G. Ricco, I. Lacchetti, M. Carere, M. L. Curri and G. Mascolo, Chem. Eng. J., 2017, 318, 103–111 CrossRef CAS
- M. Nawaz, W. Miran, J. Jang and D. S. Lee, Appl. Catal., B, 2017, 203, 85–95 CrossRef CAS
- A. Carabin, P. Drogui and D. Robert, J. Taiwan Inst. Chem. Eng., 2015, 54, 109–117 CrossRef CAS
- A. Y. C. Tong, R. Braund, D. S. Warren and B. M. Peake, Cent. Eur. J. Chem., 2012, 10, 989–1027 CAS
- A. Cincinelli, T. Martellini, E. Coppini, D. Fibbi and A. Katsoyiannis, J. Nanosci. Nanotechnol., 2015, 15, 3333–3347 CrossRef CAS PubMed
- D. Kanakaraju, B. D. Glass and M. Oelgemoller, Environ. Chem. Lett., 2014, 12, 27–47 CrossRef CAS
- Y. X. Gao, G. Yu, K. Liu, S. B. Deng, B. Wang, J. Huang and Y. J. Wang, Chem. Eng. J., 2017, 330, 157–165 CrossRef CAS
- X. Chen, W. Y. Lu, T. F. Xu, N. Li, Z. X. Zhu, G. Q. Wang and W. X. Chen, Chem. Eng. J., 2017, 328, 853–861 CrossRef CAS
- L. Lin, H. Y. Wang and P. Xu, Chem. Eng. J., 2017, 310, 389–398 CrossRef CAS
- L. Lin, H. Y. Wang, W. B. Jiang, A. R. Mkaouar and P. Xu, J. Hazard. Mater., 2017, 333, 162–168 CrossRef CAS PubMed
- Y. B. Ding, G. L. Zhang, X. R. Wang, L. H. Zhu and H. Q. Tang, Appl. Catal., B, 2017, 202, 528–538 CrossRef CAS
- J. H. Yang, D. G. Wang, H. X. Han and C. Li, Acc. Chem. Res., 2013, 46, 1900–1909 CrossRef CAS PubMed
- F. Y. Wen and C. Li, Acc. Chem. Res., 2013, 46, 2355–2364 CrossRef CAS PubMed
- K. Wenderich and G. Mul, Chem. Rev., 2016, 116, 14587–14619 CrossRef CAS PubMed
- T. D. Nguyen, Q. T. P. Bui, T. B. Le, T. M. Altahtamouni, K. B. Vu, D. V. N. Vo, N. T. H. Le, T. D. Luu, S. S. Hong and K. T. Lim, RSC Adv., 2019, 9, 23526–23534 RSC
- D. Gao, W. Liu, Y. Xu, P. Wang, J. Fan and H. Yu, Appl. Catal., B, 2020, 260, 118190 CrossRef CAS
- S. C. Sun, Y. C. Zhang, G. Q. Shen, Y. T. Wang, X. L. Liu, Z. W. Duan, L. Pan, X. W. Zhang and J. J. Zou, Appl. Catal., B, 2019, 243, 253–261 CrossRef CAS
- H. G. Yu, R. R. Yuan, D. D. Gao, Y. Xu and J. G. Yu, Chem. Eng. J., 2019, 375, 121934 CrossRef CAS
- M. H. Ai, J. W. Zhang, R. J. Gao, L. Pan, X. W. Zhang and J. J. Zou, Appl. Catal., B, 2019, 256, 117805 CrossRef CAS
- R. G. Li, X. P. Tao, R. T. Chen, F. T. Fan and C. Li, Chem.–Eur. J., 2015, 21, 14337–14341 CrossRef CAS PubMed
- R. G. Li, H. X. Han, F. X. Zhang, D. G. Wang and C. Li, Energy Environ. Sci., 2014, 7, 1369–1376 RSC
- R. G. Li, F. X. Zhang, D. G. Wang, J. X. Yang, M. R. Li, J. Zhu, X. Zhou, H. X. Han and C. Li, Nat. Commun., 2013, 4, 7 Search PubMed
- F. Lin, Y. N. Zhang, L. Wang, Y. L. Zhang, D. G. Wang, M. Yang, J. H. Yang, B. Y. Zhang, Z. X. Jiang and C. Li, Appl. Catal., B, 2012, 127, 363–370 CrossRef CAS
- B. J. Ma, F. Y. Wen, H. F. Jiang, J. H. Yang, P. L. Ying and C. Li, Catal. Lett., 2010, 134, 78–86 CrossRef CAS
- L. C. Mu, Y. Zhao, A. L. Li, S. Y. Wang, Z. L. Wang, J. X. Yang, Y. Wang, T. F. Liu, R. T. Chen, J. Zhu, F. T. Fan, R. G. Li and C. Li, Energy Environ. Sci., 2016, 9, 2463–2469 RSC
- F. Lin, D. Wang, Z. Jiang, Y. Ma, J. Li, R. Li and C. Li, Energy Environ. Sci., 2012, 5, 6400–6406 RSC
- D. E. Wang, R. G. Li, J. Zhu, J. Y. Shi, J. F. Han, X. Zong and C. Li, J. Phys. Chem. C, 2012, 116, 5082–5089 CrossRef CAS
- X. Y. Gao, X. C. Zhang, Y. W. Wang, S. Q. Peng, B. Yue and C. M. Fan, Chem. Eng. J., 2015, 273, 156–165 CrossRef CAS
- Y. Guo, P. F. Wang, J. Qian, Y. H. Ao, C. Wang and J. Hou, Appl. Catal., B, 2018, 234, 90–99 CrossRef CAS
- J. Cao, W. S. Nie, L. Huang, Y. B. Ding, K. L. Lv and H. Q. Tang, Appl. Catal., B, 2019, 241, 18–27 CrossRef CAS
- J. Xu, L. Li, C. S. Guo, Y. Zhang and W. Meng, Appl. Catal., B, 2013, 130, 285–292 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra07152k |
| This journal is © The Royal Society of Chemistry 2019 |