
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Characterisation of hydration and nanophase separation during the temperature response in hydrophobic/hydrophilic elastin-like polypeptide (ELP) diblock copolymers†
Katharina
Widder
a,
Sarah R.
MacEwan‡
b,
Elisabeth
Garanger
c,
Vanesa
Núñez
a,
Sébastien
Lecommandoux
c,
Ashutosh
Chilkoti
b and
Dariush
Hinderberger
 *a
*a
aInstitut für Chemie, Martin-Luther-Universität Halle-Wittenberg, Von-Danckelmann-Platz 4, 06120 Halle (Saale), Germany. E-mail: dariush.hinderberger@chemie.uni-halle.de
bDepartment of Biomedical Engineering, Duke University, 136 Hudson Hall, Campus Box 90281, Durham, North Carolina 27708, USA
cLaboratoire de Chimie des Polymères Organiques, CNRS UMR 5629, Université de Bordeaux, Bordeaux-INP, Pessac 33607 Cedex, France
First published on 31st January 2017
Abstract
To understand the complex nanoscale dehydration process during the lower critical solution temperature (LCST) based inverse phase transition of a class of thermoresponsive biopolymers, diblock elastin-like polypeptides (ELPs) were investigated by spin probing continuous wave electron paramagnetic resonance (CW EPR) spectroscopy. The diblock copolymers composed of a hydrophobic block and a hydrophilic block showed different mechanisms of a temperature-driven phase transition. While the phase transition temperature is a function of the hydrophobic mass fraction of the diblock ELPs, the hydrophilic block length determines the molecular structure of the polymer aggregates formed above the transition temperature. When the weight ratio of hydrophilic block length to hydrophobic block length is greater than or equal to 0.3, the polymer aggregates consist of a hydrophobic core and a hydrophilic corona. The interface of these two regions become permeable at temperatures above the transition temperature. In case of smaller ratios, the aggregating hydrophobic parts of the polymer enclose the hydrated hydrophilic blocks, that are too small to form a hydrophilic corona, leading to bigger and less dense aggregates of higher polarity.
1 Introduction
Thermoresponsive polymers showing a lower critical solution temperature (LCST) phase transition behaviour like elastin-like polypeptides (ELPs) are of particular interest e.g. for use in tissue engineering applications.1,2 Moreover, these polymers can host small molecules to serve as drug delivery systems,3–5 due to their temperature-triggered encapsulation or release properties.6ELPs are high-precision peptide polymers based on the hydrophobic domain of tropoelastin.7 They are composed of the amino acid sequence (VPGXG)n (∀ X\P)8 and can in a broader sense also be viewed as model polymers for intrinsically disordered proteins (IDPs).9,10 These polymers gain an advantage over synthetic LCST polymers through their precisely controllable sequence and chain length by use of recombinant DNA- and protein-engineering techniques.11,12 Molecular parameters like the number of repeats of the pentapeptide or the guest residue control the LCST behaviour of ELPs.13 The LCST phase behaviour of ELPs is driven by changes in the molecular conformation and hydration state of the peptide–polymers.14 ELPs are soluble in aqueous solutions below their transition temperature, water molecules hydrate the polymer chains. Increasing the temperature and reaching the transition point leads to the phase transition into polymer-rich and water-rich phases, as water becomes a poor solvent for the peptide chains.15 The transition is characterised by thermal disruption of the water network, the loss of hydration of the polymer and the aggregation of polymer chains.8 Secondary structures like β-turns are now favoured compared to a solubilisation by water.15,16
Continuous wave (CW) electron paramagnetic resonance (EPR) spectroscopy on reporter molecules (spin probes) as a non-invasive, intrinsically local technique can help to understand the temperature-dependent phase transition. The sensitive time scale of EPR ranges from 10 ps to 1 μs.17 This makes EPR a powerful tool to investigate the inverse phase transition since the exchange processes of amphiphilic spin probes between polar and apolar regions of the LCST polymers coincide with the time scale of EPR.14 Incorporating amphiphilic nitroxide spin probes into the aqueous polymer system by self-assembly yields information about dynamics and polarity inside the aggregated hydrophobic regions of the ELP. The CW EPR spectrum of a nitroxide radical in the fast tumbling regime (fast spin probe rotation on timescales of τc < 1 ns) shows three peaks. The hydrophobic collapse at the transition temperature is indicated by a change in the isotropic hyperfine coupling (a), which is reflected in a splitting of the high-field line that stems from spin probes that reside in the dehydrated, polymer-rich regions, which sense less polar environments.
During the temperature-induced aggregation of thermoresponsive polymers, nanoscale inhomogeneities occur.17 Kurzbach described them as nanoscopic clusters of collapsed polymer segments, which represent an apolar, hydrophobic environment compared to a fully solvated polymer. Nanoscale inhomogeneities cannot be separated from the inverse phase transition of thermoresponsive polymers, since a few local dynamic inhomogeneities on a nanometre scale already lead to a transition that is macroscopically observable by turbidity measurements.18 Investigations of nanoscale inhomogeneities by spin probing CW EPR revealed three types:17
(1) Static inhomogeneities: the spin probe is incorporated inside the inhomogeneity and a dynamic exchange is impossible at any temperature. This type is characterised in the EPR spectra by a splitting of the high-field line of the nitroxide radical (spin probe), which is partitioned between two environments of different hydrophilicity. The line splitting remains constant during a change in temperature. Only the relative weight of the two spectral components changes.
(2) Dynamic inhomogeneities (I): in this case, a spin-probe exchange between polymer-rich and water-rich nanophases is featured at intermediate temperatures slightly above the initial formation of hydrophobic aggregates. The high-field peak of the EPR spectra is split characteristically and continually the two peaks diverge with temperature, due to a decreasing guest-exchange with increasing temperature.
(3) Dynamic inhomogeneities (II): the spin-probe exchange is featured at high temperatures. The EPR spectra show two lines merging with increasing temperature, due to an increasing spin-probe exchange with increasing temperatures.
A recent study by Garanger and co-workers19 investigated diblock ELPs composed of a hydrophobic block and a hydrophilic block with different hydrophobic block size by light and neutron scattering techniques (absorbance spectroscopy, DLS, SLS and SANS). Their study yielded that once above the first transition temperature the hydrophobic block has been desolvated independently from the hydrophilic block, the ELP block copolymers form aggregates with a core of hydrophobic blocks and a corona of hydrophilic blocks. The aggregates are first strongly hydrated and then continue to evolve thermally. A further increase in temperature leads to a temperature-driven desolvation and compaction of the core. As a consequence the aggregates expel water. At a second transition temperature macroscopic aggregates are built through aggregation of the globular aggregates through their hydrophilic coronas.
Since these diblock ELPs are possibly applicable as a drug delivery system, it is important to investigate what small molecules sense in their environment and how they are expelled or taken up during the phase transition. Hence, spin probing EPR is used in the present study to investigate the temperature-triggered self-assembly process of the diblock ELPs of Garanger and co-workers on the molecular scale at a constant molar concentration. Different diblock ELPs were examined and compared with regard to the influence of the length of the hydrophobic block on the one hand and of the length of the hydrophilic block on the other hand. Moreover, the effect of the guest residue in the hydrophilic block without significantly changing its hydrophilicity is considered.
2 Experimental section
The diblock ELPs investigated in this study were cloned and produced in Escherichia coli and their synthesis, expression, and purification is described in ref. 19. We received them from Elisabeth Garanger et al. (Université de Bordeaux), who already described their synthesis, expression and purification.19All ELPs are composed of a hydrophobic block (VPGVG)x1 at the N-terminal end of the polypeptide and a hydrophilic block (VPGXG)x2 located at the C-terminal end. The so-called leader sequence MGCGWPG was encoded at the N-terminus and the respective trailer sequence PGGS at the C-terminus (see ref. 19). In case of a constant hydrophilic block with the guest residue X = A, G (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and x2 = 60, the hydrophobic block lengths were varied within x1 = 40, 80, 120, 200. Two additional diblocks were investigated with a hydrophobic block length of x1 = 200 and a hydrophilic block length of x2 = 30: one with the previous guest residue X = A, G (1:1) and the second one with X = S. These diblock compositions lead to hydrophilic mass fractions between 0.12 and 0.56 (Table 1). Following the established nomenclature, it will be referred to the diblock ELPs as ‘ELP x1–x2’ with the hydrophobic block length x1 and the hydrophilic block length x2. In case of serine as the guest residue of the hydrophilic block the polymer is called ‘ELP 200–S30’.
:1) and x2 = 60, the hydrophobic block lengths were varied within x1 = 40, 80, 120, 200. Two additional diblocks were investigated with a hydrophobic block length of x1 = 200 and a hydrophilic block length of x2 = 30: one with the previous guest residue X = A, G (1:1) and the second one with X = S. These diblock compositions lead to hydrophilic mass fractions between 0.12 and 0.56 (Table 1). Following the established nomenclature, it will be referred to the diblock ELPs as ‘ELP x1–x2’ with the hydrophobic block length x1 and the hydrophilic block length x2. In case of serine as the guest residue of the hydrophilic block the polymer is called ‘ELP 200–S30’.
| x 1 | x 2 | f hydrophilic | T t/EPR in °C |
|---|---|---|---|
| 200 | 30 | 0.12 | 22 |
| 200 | S30 | 0.13 | 22 |
| 200 | 60 | 0.21 | 24 |
| 120 | 60 | 0.31 | 26 |
| 80 | 60 | 0.4 | 28 |
| 40 | 60 | 0.56 | 38 |
For the measurement with CW EPR the polymers were dissolved in PBS (pH 7.4) cooled by ice to avoid an early phase transition. The amphiphilic spin probe 16-DSA (16-DOXYL stearic acid), dissolved in ethanol, was added. Due to the octanol/water partition coefficient of 16-DSA, the spin probe has a strong preference for apolar environments (logPoctanol/water = 4.49).20 Different concentrations of polymer and spin probe were tested to find the most suitable approach. The final samples contained 300 μM polymer and 1 mM 16-DSA. Micropipettes (BLAUBRAND intraMARK, Wertheim, Germany) were filled with about 15 μl of the solution and sealed with CRITOSEAL (Leica) for subsequent EPR measurements.
CW EPR spectra were detected using a MiniScope MS400 (Magnettech, Berlin, Germany) spectrometer with a frequency of approximately ν = 9.4 GHz and a magnetic field sweep of 15 mT centred around 336 mT. Temperatures were adjusted with the Temperature Controller H03 (Magnettech) with an accuracy of ±0.2 °C and a heating rate of 1 °C min−1. The exact frequency was recorded using a frequency counter (Racal Dana 2101, Neu-Isenburg, Germany).
EPR spectra were analysed by spectral simulation with MATLAB R2014a using the program package EasySpin for EPR spectroscopy, which applies the Schneider–Freed model to solve the Schrödinger equation for slow tumbling nitroxides.21 The simulation parameters diffusion tensor D (rotational correlation time τ = ⅙(DxxDyyDzz)−1/3) and hyperfine splitting tensor A (hyperfine splitting constant a = ⅓(Axx + Ayy + Azz)) of the spin probe as well as the fractions of 2–3 components of the nitroxide spectra were extracted in an iterative manual procedure.
To distinguish the effects of the temperature on the probe from the effects of the polymer aggregates on the probe, reference spectra of 1 mM 16-DSA in pure buffer were measured.
3 Results and discussion
3.1 Spectral changes
The CW EPR spectra detected during heating of the polymer solution are shown in Fig. 1(a) and (b). At small T, the EPR spectra show no interaction between probe and polymer (see Fig. S5, ESI† for 10 and 20 °C). The probe senses merely a slightly higher viscosity. The interaction starts with the aggregation of the polymer. At temperatures above the transition temperature the spectra exhibited temperature-dependent changes that differ from the reference measurement of 16-DSA in pure buffer (see ESI† for reference spectra). The spectra are composed of different components of the spin probe. Three components were taken into account for the simulation of the spectra (Fig. 1(c)):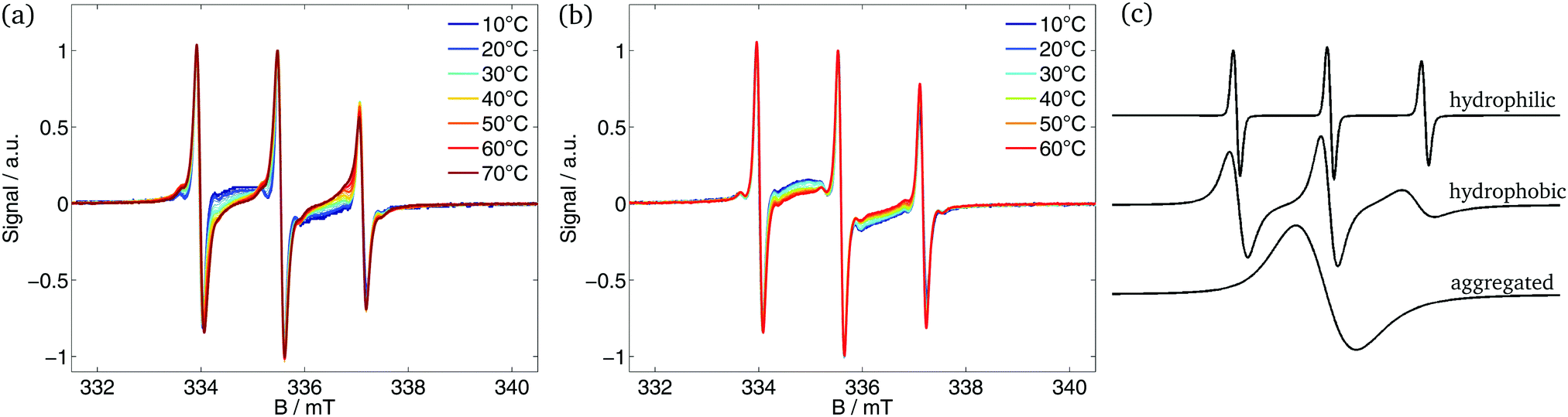 | ||
| Fig. 1 CW EPR spectra of 16-DSA in polymer buffer solution. (a) Measured spectra of the diblock ELP 200–30, (b) measured spectra of the diblock ELP 40–60 and (c) simulated components of which the EPR spectra are composed. | ||
(A) Hydrophilic component: 16-DSA molecules freely rotating in buffer and showing no interaction with the polymer. The rotational correlation time is in the order of magnitude of 10−10 s. The probe senses a polar environment leading to a high hyperfine splitting constant a.
(B) Hydrophobic component: 16-DSA molecules located in hydrophobic and apolar regions of the polymer. The probe rotates slower and senses a smaller polarity leading to higher τ (≈10−9 s) and smaller a.
(C) Aggregated component: 16-DSA molecules forming aggregates (e.g. micelles) in buffer. This leads to highly frequent spin probe collisions, which results in high Heisenberg spin exchange frequencies, which as its hallmark shows a collapse of the three peaks into one broad peak with lower amplitude. This component vanishes partly during the increase of temperature and since the fraction of component C is not reproducible, the fractions of the 16-DSA aggregates were subtracted out of the fraction of the depicted hydrophobic component for the sake of comparability.
At temperatures below the transition temperature the spectra are a superposition of the three-peak spectrum of the freely rotating nitroxide molecules and the broad one-peak spectrum of 16-DSA aggregates. In this temperature region the spectra of 16-DSA in polymeric solution are similar to the reference spectra of 16-DSA in pure buffer: no interaction of the spin probe with the polymer is detectable. With reaching the transition temperature of the respective polymer the hydrophobic component appears in the EPR spectra, indicating the formation of polymer-rich nanophases that 16-DSA as an amphiphilic molecule partitions into a large degree. Due to a smaller hyperfine splitting constant and the shift of the hydrophobic component spectrum towards smaller magnetic (B-)fields, the appearance of the hydrophobic component becomes visible next to the high-field peak at smaller B-field values than the third peak of the hydrophilic component. The relative weight of these two spectral contributions changes with increasing temperature. In case of smaller hydrophilic mass fractions (e.g. ELP 200–30, Fig. 1(a)) the appearance of the hydrophobic component in the spectra is more distinct than for higher hydrophilic mass fractions (e.g. ELP 40–60, Fig. 1(b)).
3.2 Microscopic phase transition
The transition temperature Tt/EPR of the diblock ELPs is marked by the onset of appearance of the hydrophobic component in the EPR spectra. Since spin probing EPR reports on the first occurrence of polymer-rich, apolar cavities in the polymeric system, the here stated transition temperatures (Table 1) have to be discerned from absorbance spectroscopic data. The latter may identify the transition only when much larger aggregates are present to scatter the transmitted light.Fig. 2 shows the transition temperatures of all measured diblock ELPs as a function of the hydrophilic mass fraction f. The transition temperatures derived from EPR measurements as well as the data of absorbance spectroscopy19 (ESI†) shows an exponential dependence from the hydrophilic mass fraction.
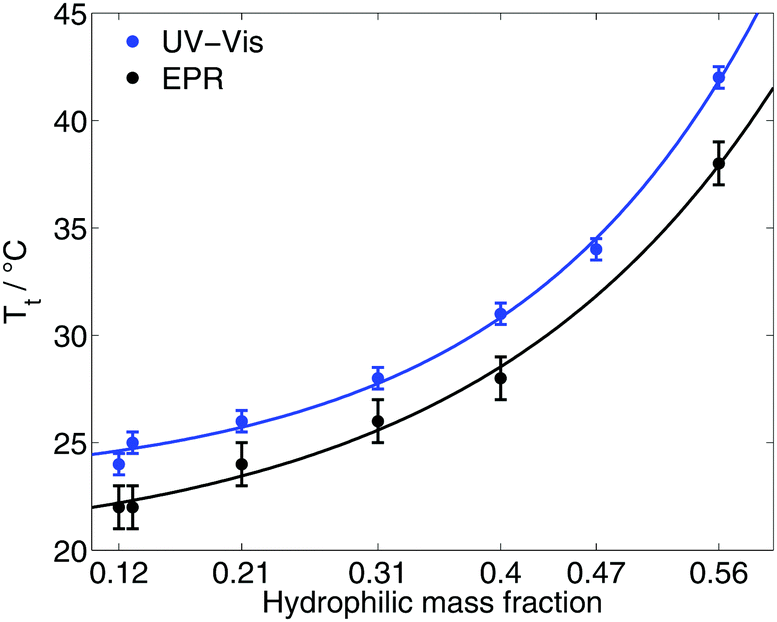 | ||
| Fig. 2 Transition temperatures as a function of the hydrophilic mass fraction of all diblock ELPs as measured by EPR and UV-Vis spectroscopy19 (ESI†). | ||
Taking the exponential regression (for equations see ESI†) into account, a transition temperature can be determined for f = 0 corresponding to the ELP motif (VPGVG). Table 2 compares the theoretical values for the transition temperature of (VPGVG) determined by EPR, absorbance spectroscopy and turbidity.22 Urry et al. assumed a linear dependence for Tt = f(f), but considered only hydrophilic mass fractions of f < 0.5. The results of EPR show slightly lower T-values than those of Garanger19 and Urry.22 This could have two reasons: 16-DSA sees smaller inhomogeneities earlier (at lower temperatures) than absorbance spectroscopy and turbidity and/or higher concentrations of the polymer lead to smaller transition temperatures. But the former appears less likely because 16-DSA is too big to enter small hydrophobic cavities. The smaller but more hydrophilic spin probe TEMPO (2,2,6,6-tetramethylpiperidine-1-oxyl) was also tested but did not show any interaction with the diblock ELPs (ESI†). Hence, the slightly lower transition temperature is most likely due to the employed higher polymer concentration.
|
T
t/EPR in °C
c = 300 μM |
T
t/CMT in °C
c = 25 μM |
T
t/Urry in °C
c = ? |
|
|---|---|---|---|
| f = 0 (VPGVG)x1 | 21.15 | 23.26 | 24 (ref. 22) |
The other extremum, f = 1, leading to the motif (VPGAG–VPGGG) would lead to far too high transition temperatures compared to Urry, who stated Tt = 45 °C for (VPGAG) and Tt = 55 °C for (VPGGG). In conclusion the exponential model is an empirical model to estimate the transition temperature for a given hydrophilic mass fraction f, but it collapses at high hydrophilic mass fractions.
Since the appearance of the hydrophobic component in the EPR spectra of the diblock ELPs marks the temperature of the inverse phase transition on the nanoscale, the fraction of this component mirrors the progression of the transition process. At temperatures near Tt the fraction of 16-DSA molecules located in the polymeric aggregates increases rapidly (Fig. 3) indicating a sharp transition of the diblock ELPs. The smaller the hydrophilic mass fraction, the faster the increase of the hydrophobic fraction after their appearance. At higher temperatures, the slope of the fraction becomes smaller for all diblock ELPs showing an asymptotic behaviour.
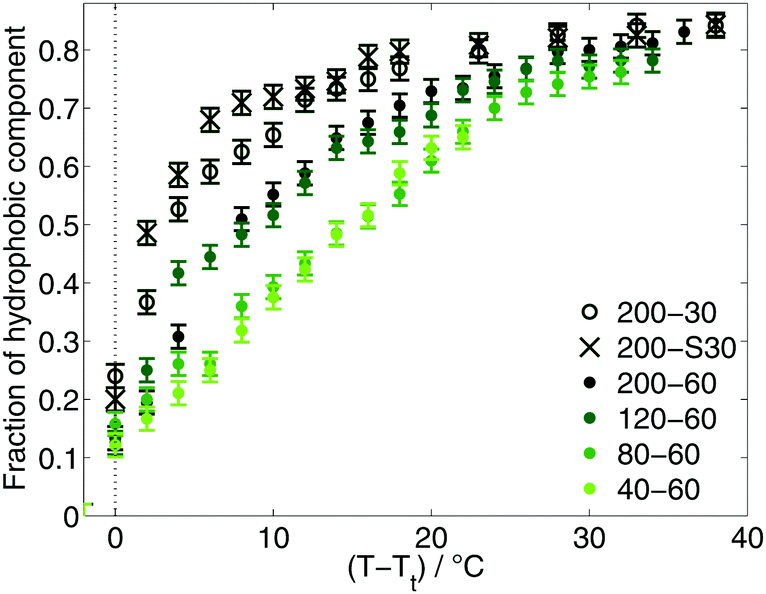 | ||
| Fig. 3 Fraction of hydrophobic component. | ||
A higher hydrophilicity of a polymer leads to a higher hydration of the polymer chains and less hydrophobic aggregated regions. As a consequence, less 16-DSA molecules are located near to hydrophobic regions. This leads to smaller fractions of hydrophobic component in the spectra and a smaller slope of the fraction increase, since more water molecules around the not-aggregated hydrophilic parts of the polymer make it less likely that the spin probe molecules favouring more non-polar regions actually enter the aggregated regions.
3.3 Type of inhomogeneities
With spin probing EPR spectroscopy, we could only detect one transition. Garanger et al.19 described two transitions for the diblock ELPs x1–60. If the loss of water and the densification of the polymer aggregates that they reported between the transitions, could have been detected by EPR, we would have seen the occurrence of static inhomogeneities (see Introduction, case 1). In this case, the 16-DSA molecules would stay inside the hydrophobic regions of the polymer sensing the loss of water but not exchanging with the surrounding water-rich phase. The loss of water of the aggregates would be characterised by an increase of the rotational correlation times with increasing temperatures as a consequence of the restricted mobility of the spin probe in the densified aggregate and by a decrease of the polarity at higher temperatures in the spin probes environment.The simulation results do not support these considerations. As can be seen in Fig. 4, the rotational correlation times decrease and the hyperfine splitting constant increases during the temperature rise. The increase of mobility of the spin probe molecules and the increase of polarity in their environment characterises the polymer aggregates as dynamic inhomogeneities. The gained mobility due to the temperature rise allows the 16-DSA molecules to diffuse between hydrophobic and hydrophilic regions. The detected values of τ and a are the average over both regions. Due to different a-values of the hydrophobic and the hydrophilic component, the high-field peak of the EPR spectra is separated (Fig. 1(a)) at temperatures just above Tt. Way above the transition temperature of the polymer further increase of temperature leads to an increasing spin-probe exchange. This becomes visible over the merge of the separated high-field peak of the spectra.
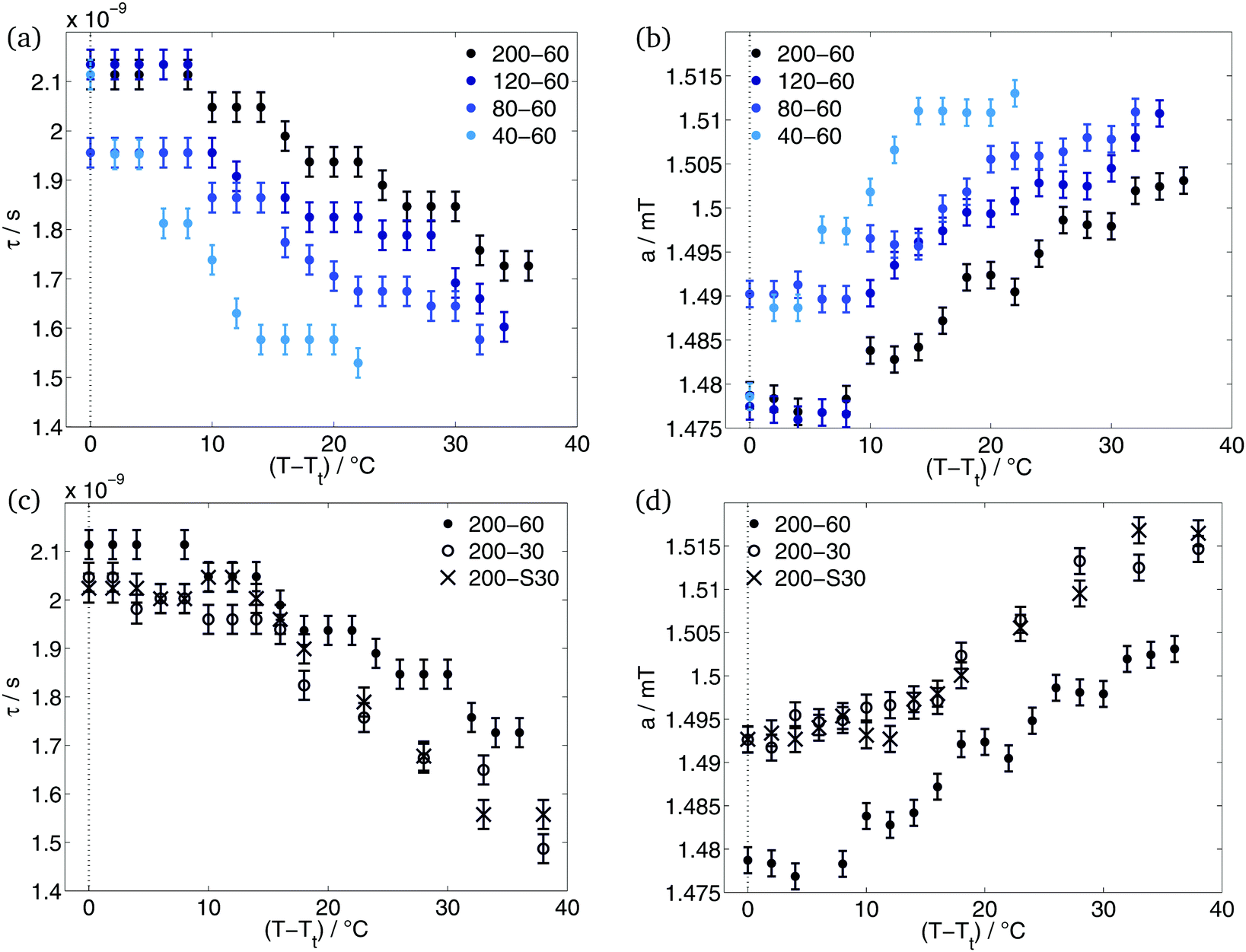 | ||
| Fig. 4 Results of simulation: rotational correlation times (a and c) and hyperfine splitting constant (b and d) of the hydrophobic component for different hydrophobic block lengths (a and b) and different hydrophilic blocks (c and d). | ||
After the hydrophobic component arises in the EPR spectra, a stays constant for some degrees. The increase of temperature leads to an increase of a only after temperature reached Tt + 8 °C in case of ELP 200–x2, 120–60 and 80–60. ELP 40–60 showed an increase of polarity already at Tt + 4 °C. At and above these temperatures, the interface between polymeric aggregate of hydrophobic blocks and the corona of hydrophilic blocks becomes permeable by our spin probes. This and the higher mobility allows the spin probe molecules to diffuse between these two regions. According to Kurzbach et al.17 the polymer aggregates and the collapse mechanism represent dynamic inhomogeneities of type II.
3.4 Influence of the hydrophobic block
Diblock ELPs with different lengths of the hydrophobic block (VPGVG)x1 were investigated by spin probing EPR. Their block length x1 amounted to 40, 80, 120 and 200, while the hydrophilic block stays constant with the guest residues X = A, G (1:1) and a constant length x2 = 60. Higher x1 cause smaller transition temperatures due to smaller hydrophilic mass fractions f (Table 1).
Simulation of the CW EPR spectra yielded information about the environment of the different spin probe components. Since the hydrophilic component represents the 16-DSA molecules freely tumbling in water, only the results of the spin probe molecules located in the hydrophobic polymer aggregates are plotted here (Fig. 4).
As can be seen in Fig. 4(a), diblock ELPs with higher x1 show higher rotational correlation times τ inside the polymer aggregates. This means that the here located 16-DSA molecules have a smaller mobility and rotate slower than the spin probes in polymer aggregates of ELPs with smaller x1. A dependence of the hydrophobic block length is also visible in the progression of the hyperfine splitting constant with increasing temperature (Fig. 4(b)). Smaller x1 cause higher hyperfine splitting constants. Higher values of a indicate a higher polarity in the environment of the spin probe, often speaking for a higher amount of water.
The temperature-dependent progression of both – the rotational correlation time and the hyperfine splitting constant – can be interpreted such that the 16-DSA molecules sense more water inside polymeric aggregates of more hydrophilic polymers. As already stated, the more hydrophilic an ELP is, the more water molecules surround this polymer due to its higher hydration. Hence, a spin probe inside hydrophobic regions of ELP with higher hydrophilicity show higher values of a and smaller values of τ.
3.5 Influence of the hydrophilic block
The hydrophilic block of the diblock ELPs was varied with regard to block length x2 and guest residue in the hydrophilic motif of (VPGXG). x2 was chosen as either 60 and 30 with the guest residue X = A, G (1:1), in each case with the hydrophobic block (VPGVG)200. Another diblock ELP was composed of (VPGVG)200 and (VPGSG)30.
As can be expected, the effect of the number of repeat units of the hydrophilic block x2 on the transition temperature is reverse to the effect of the number of repeat units of the hydrophobic block x1. The diblock ELP with x2 = 60 shows a higher transition temperature than the diblock ELPs with x2 = 30 (Table 1). This is consistent with the influence of x1 on the transition temperature, since a higher x2 causes a larger hydrophilic mass fraction.
The comparison of the diblock ELPs with different guest residues in the hydrophilic block shows no significant differences. The fraction (Fig. 3), rotational correlation time and hyperfine splitting constant (Fig. 4(c) and (d)) of the hydrophobic component of the EPR spectra of ELP 200–30 and 200–S30 show similar results for both polymers. Considering only the hydrophobicity of the different pentapeptides, the similarity in behaviour is not surprising. According to the hydrophobicity scale of Urry et al.22 the transition temperature of (VPGAG) amounts to 45 °C, (VPGGG) to 55 °C and (VPGSG) to 50 °C. Since the transition temperatures are a measure for hydrophobicity and hydrophilicity, respectively, the hydrophilicity of the diblock ELPs 200–30 and 200–S30 is identical within experimental error. The additional OH-group of serine seems to have no effect on the aggregation behaviour of the diblock copolymer.
ELPs 200–30 and 200–S30 have similar hydrophilic mass fractions (Table 1). Due to their x2 = 30, their hydrophilic mass fraction is smaller than of all other investigated diblock copolymers. According to the former subsection, it was expected that the hydrophobic component of the ELPs 200–30 and 200–S30 shows higher rotational correlation times and smaller hyperfine coupling constants. Less hydration of the polymers with x2 = 30 gives rise to the presumption, that the spin probe senses less water molecules than in case of x2 = 60. However, the results (Fig. 4(c) and (d)) are contradictory to this consideration. The rotational correlation times of the spin probe molecules located in the aggregated regions of the ELPs 200–30 and 200–S30 are slightly smaller than of 200–60. Concomitantly, the polarity in the probes environment is higher for smaller x2 observable on higher values of a of diblock copolymers with x2 = 30. As the studies of Garanger19 on diblock ELPs with a hydrophilic chain length of x2 = 60 showed, the hydrophobic parts of these diblock ELPs collapses into aggregates and the hydrophilic blocks are located around these micelles. This is also in agreement with our EPR-spectroscopic measurements of the ELP x1–60 copolymers. Apparently, this does not apply for ELPs with shorter hydrophilic block length of x2 = 30. The short hydrophilic blocks seem not to be capable of forming a nanophase-separated corona and are rather incorporated into the hydrophobic regions during their aggregation. Due to the hydration of the hydrophilic part of the polymer, more water molecules are also incorporated into the polymer aggregates. The higher water content inside the aggregates leads to a higher mobility of the here located spin probe molecules accompanied by smaller rotational correlation times. Moreover, the spin probes sense a more polar environment explaining the higher values of a for ELP 200–30 and 200–S30.
4 Conclusion
The inverse phase transition behaviour of diblock ELPs with different hydrophobic block length and different hydrophilic blocks were investigated by self-assembly spin probing CW EPR spectroscopy. This study gained new insights into the temperature-responsive transition of the block copolymer that is driven by the hydrophobic block. The thermoresponsive phase behaviour proceeds via structural inhomogeneities on the nanometre scale, similarly to what was found for synthetic and ELP homopolymers. The here observed polymeric aggregates are dynamic inhomogeneities featuring a spin-probe exchange between polymer-rich and water-rich nanophases at specific temperatures above the transition temperature. These specific temperatures are 8 °C and 4 °C above the transition temperature, respectively, depending on the relative hydrophilic mass fractions. At the onset of exchange, the spin probe molecules so far located in the hydrophobic regions diffuse through the permeable interface between hydrophobic aggregate and hydrophilic regions. Kurzbach et al.14 already found this type of inhomogeneities for synthetic block copolymers.A second transition driven by aggregation of the hydrophilic blocks as well as a loss of water leading to the densification of the polymer aggregates was not visible at the molecular level with our EPR-spectroscopic characterisation. Remarkably, EPR spectroscopy becomes a helpful tool if this process is not observed by scattering techniques. As seen here in the diblock copolymers with only 30 repeat units in the (short) hydrophilic block, these hydrophilic blocks cannot form a hydrophilic corona (Fig. 5) but these short chains are rather incorporated into the hydrophobic aggregates formed at the transition temperature of the polymer. This is in contrast to diblock ELPs with long (60 repeat units) hydrophilic blocks, which are located around the hydrophobic aggregates building a corona (Fig. 5). This explains why Garanger and co-workers did not observe a second temperature transition and hence nanophase-separation of the diblocks with hydrophilic block length of 30, yet they did observe it at block lengths of 60.
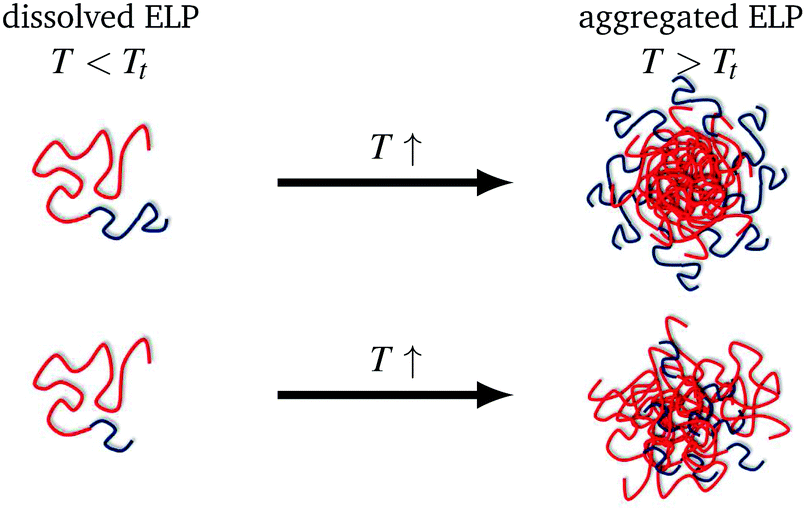 | ||
| Fig. 5 Schematic depiction of the different aggregation mechanisms for diblock ELP 200–60 (top) and ELP 200–30 (bottom), blue: hydrophilic polymer block, red: hydrophobic polymer block. | ||
The knowledge of the phase transition behaviour obtained by spin probing EPR is gained in an indirected manner. One has to deduce the behaviour of a host-polymer from the guest molecules' behaviour and one can use EPR spectroscopy to characterise the temperature-induced nanophase-separation and the processes at the interface of polymer- and water-rich nanophase.
Acknowledgements
We gratefully acknowledge financial support from the Deutsche Forschungsgemeinschaft (DFG) in collaborative research center SFB-TRR 102.References
- D. L. Nettles, A. Chilkoti and L. A. Setton, Adv. Drug Delivery Rev., 2010, 62, 1479–1485 CrossRef CAS PubMed.
- T. Kowalczyk, K. Hnatuszko-Konka, A. Gerszberg and A. K. Kononowicz, World J. Microbiol. Biotechnol., 2014, 30, 2141–2152 CrossRef CAS PubMed.
- D. E. Meyer, B. C. Shin, G. A. Kong, M. W. Dewhirst and A. Chilkoti, J. Controlled Release, 2001, 74, 213–224 CrossRef CAS PubMed.
- J. R. McDaniel, D. J. Callahan and A. Chilkoti, Adv. Drug Delivery Rev., 2010, 62, 1456–1467 CrossRef CAS PubMed.
- R. J. Mart, R. D. Osborne, M. M. Stevens and R. V. Ulijn, Soft Matter, 2006, 2, 822–835 RSC.
- B. Helms and J. M. J. Fréchet, Adv. Synth. Catal., 2006, 348, 1125–1148 CrossRef CAS.
- D. W. Urry, T. L. Trapane and K. U. Prasad, Biopolymers, 1985, 24, 2345–2356 CrossRef CAS PubMed.
- D. W. Urry, J. Protein Chem., 1988, 7, 1–34 CrossRef CAS PubMed.
- D. Kurzbach, G. Platzer, T. C. Schwarz, M. A. Henen, R. Konrat and D. Hinderberger, Biochemistry, 2013, 52, 5167–5175 CrossRef CAS PubMed.
- S. Roberts, M. Dzuricky and A. Chilkoti, FEBS Lett., 2015, 589, 2477–2486 CrossRef CAS PubMed.
- J. C. M. van Hest and D. A. Tirrell, Chem. Commun., 2001, 1897–1904 RSC.
- A. Chilkoti, M. R. Dreher and D. E. Meyer, Adv. Drug Delivery Rev., 2002, 54, 1093–1111 CrossRef CAS PubMed.
- J. A. MacKay, D. J. Callahan, K. N. FitzGerald and A. Chilkoti, Biomacromolecules, 2010, 11, 2873–2879 CrossRef CAS PubMed.
- D. Kurzbach, W. Hassouneh, J. R. McDaniel, E. A. Jaumann, A. Chilkoti and D. Hinderberger, J. Am. Chem. Soc., 2013, 135, 11299–11308 CrossRef CAS PubMed.
- N. K. Li, F. G. Quiroz, C. K. Hall, A. Chilkoti and Y. G. Yingling, Biomacromolecules, 2014, 15, 3522–3530 CrossRef CAS PubMed.
- D. Schmaljohann, Adv. Drug Delivery Rev., 2006, 58, 1655–1670 CrossRef CAS PubMed.
- D. Kurzbach, M. J. N. Junk and D. Hinderberger, Macromol. Rapid Commun., 2013, 34, 119–134 CrossRef CAS PubMed.
- M. J. N. Junk, W. Li, A. D. Schlueter, G. Wegner, H. W. Spiess, A. Zhang and D. Hinderberger, Angew. Chem., Int. Ed., 2010, 49, 5683–5687 CrossRef CAS PubMed.
- E. Garanger, S. R. MacEwan, O. Sandre, A. Brûlet, L. Bataille, A. Chilkoti and S. Lecommandoux, Macromolecules, 2015, 48, 6617–6627 CrossRef CAS.
- A. N. Cimato, L. L. Piehl, G. B. Facorro, H. B. Torti and A. A. Hager, Free Radicals Biol. Med., 2004, 37, 2042–2051 CrossRef CAS PubMed.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
- D. W. Urry, D. C. Gowda, T. M. Parker, C.-H. Luan, M. C. Reid, C. M. Harris, A. Pattanaik and R. D. Harris, Biopolymers, 1992, 32, 1243–1250 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sm02427k |
| ‡ Present address: Institute for Molecular Engineering, University of Chicago, Chicago, Illinois 60637, USA. |
| This journal is © The Royal Society of Chemistry 2017 |