
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aspects of extraction and biological evaluation of naturally occurring sugar-mimicking sulfonium-ion and their synthetic analogues as potent α-glucosidase inhibitors from Salacia: a review
Priyanka Bagri
a,
Karishma Chester
b,
Washim Khan
a and
Sayeed Ahmad
 *a
*a
aSchool of Pharmaceutical Education and Research, Bioactive Natural Product Laboratory, Department of Pharmacognosy and Phytochemistry, Jamia Hamdard, New Delhi, India. E-mail: sahmad_jh@yahoo.co.in; Fax: +91-011-26059663; Tel: +91-9891374647
bDepartment of Pharmacy, Banasthali University, P.O. Banasthali Vidyapith, Tonk, Rajasthan 304022, India
First published on 6th June 2017
Abstract
Human maltase-glucoamylase (MGAM) and sucrase-isomaltase (SI) are membrane bound small intestinal α-glucosidase enzymes which are responsible for the hydrolysis of terminal starch products and sugars into glucose. Each enzyme contains a catalytic N- and C-terminal subunit, ntMGAM, ctMGAM, ntSI, and ctSI, respectively. Inhibition of MGAM and SI is an important strategy for treatment of type-2 diabetes. In drug discovery, several plants from the Salacia species have been used for many years to treat diabetic conditions. Attempt to identify the source of the antidiabetic phytoconstituents in water-soluble fraction of this plant extract stimulated interest in its phytochemistry. This led to the identification of several novel class of sulfonium-ion. So far, eight sulfonium-ion glucosidase inhibitors have been isolated i.e. salacinol, kotalanol, ponkoranol, salaprinol and four of their corresponding de-O-sulfonated compounds. Due to both the intriguing structure and strong α-glucosidase inhibitory activity, much attention has been focused on salacinol and related compounds. Structural modification of sulfonium-ion represents a promising approach in the search for novel antidiabetic drugs. Several papers have described the synthetic route of sulfonium ion-based inhibitors. In this context, this review covers up to date information on sulfonium-ion from 1997 to till date and discusses the role of Salacia as antidiabetic drug as well as the extraction, isolation, structural elucidation, biological evaluation and structure–activity relationship studies of sulfonium-ion as potent α-glucosidase inhibitors coupled with crystallographic analysis and docking studies. We highlighted all the selective inhibitory activities of compounds against the intestinal glucosidases and also summarise the structural features which are important for its effective inhibition. The structural refinement of the sulfonium-ion generates inhibitors that selectively inhibit the activity of mucosal α-glucosidases. Further, the approach of toggling has been reviewed in aspects for controlling starch digestion and glucose release to the body and thus provides insight for designing most potent inhibitors for each subunit with low nanomolar inhibitory activities. This strategy will give a lead from the perspective of management of diabetes and associated metabolic disorders. The knowledge gained from this article will be helpful to a search for new potent inhibitors of human MGAM and SI. A summary of the inhibition data of all the compounds covered in this review have also been incorporated in tabular form as an Appendix.
 Priyanka Bagri | Priyanka Bagri received her M. Pharmacy and Ph.D. degrees in Pharmacognosy and Phytochemistry from Jamia Hamdard University, New Delhi, India and completed her doctoral thesis on standardization, extraction and isolation of bioactive antidiabetic principles from natural drugs in 2010. In 2014 she joined bioactive natural product laboratory under the supervision of Dr Sayeed Ahmad as a women scientist for postdoctoral researcher to work on the metabolomics and bioactivity guided isolation of antidiabetics phytoconstituents from herbal drugs. Her research interests concentrate on the phytochemical, metabolomics and chromatographic analysis of medicinal plants for quality control. She is also working on Chemoprofiling of oils using GC-MS as well as for in vitro/in vivo evaluation of plant drugs for antidiabetic, anti-inflammatory, antimicrobial and hepatoprotective activities. |
 Karishma Chester | Karishma Chester obtained her M. Pharmacy degree in Pharmaceutical Biotechnology from Jamia Hamdard University in 2011 which was followed as SRF in CCRUM project showing HIV potential of some herbal plants. She joined the Ph.D. program in 2013 in Pharmaceutical Sciences under the supervision of Prof. Sarvesh Paliwal, from Banasthali University, Rajasthan. She received P.D. Sethi award for best paper. Her research has focused on molecular analysis mainly DNA isolation, RAPD, phytochemical, metabolomics and chromatographic analysis of herbal drugs. She is currently working on druggability of metabolite enriched fractions for in vitro/in vivo screening of hepatoprotective and antidiabetic drugs. |
 Washim Khan | Mr Washim Khan received M. Pharm from Jamia Hamdard, and currently, he is doing Ph.D. under the Supervision of Dr Sayeed Ahmad, Department of Pharmacognosy and Phytochemistry, Faculty of Pharmacy, Jamia Hamdard (Hamdard University), New Delhi. He has been honoured with PD Sethi award in year 2016 for best publications in HPTLC, Best Poster award from Phytochemistry/Phytochemistry letters, awarded with Merit Scholarship from All India Council for Technical Education (AICTE) for the year 2010–2012 and from West from West Bengal Government for the year 2007–2010, qualified Graduate Pharmacy Aptitude Test (GPAT), Graduate Record Examinations (GRE). He has to his credit 23 research publications, participated in more than 30 conferences, presented 19 posters in India and Abroad. He was associated with the development of special issue of ‘Journal of Pharmaceutical and Bioallied Sciences’, published in October 2015 and of ‘Drug Development and Therapeutics’ published in January 2016 on Herbal Drugs and Botanicals edited by Dr Sayeed Ahmad. His field of interests in research isdrug discovery from natural sources, targeted and untargeted metabolomics through HPTLC, HPLC, GC-MS, and LC-MS and with modern statistical tools. He is also working in the biological screening of Indian traditional formulation to support their claims. |
 Sayeed Ahmad | Dr Sayeed Ahmad, a known researcher in the field of herbal drugs and natural products, received his Ph.D. degree in 2005 from Department of Pharmacognosy and Phytochemistry, Jamia Hamdard University, New Delhi, India. Since 2005 he has been working as a faculty member at the same university. He did post-doctoral research at Albert Einstien College of Medicine New York, USA (2011) and was a Visiting Scientist in Sultan Qaboos University, Muscat, Oman (2012) His research interests span the study of quality control, chromatography and metabolomics analysis. Dr Ahmad has been associated with more than 26 sponsored research projects from different funding agencies in addition to some industry-based consultancy projects. He is a recipient of University Gold Medal (2002), DST Young Scientist Award (2007), CST-UP Young Scientist Award (2008–09), DST BOYSCAST (2010), AICTE Career Award (2010), UGC Research Award (2016) and Society for Ethnopharmacology Young Ethnopharmacologist award (2017) in addition to some Best Paper Awards such as P. D. Sethi Award (2013 and 2015), Al-Ameen College of Pharmacy Award (2014 and 2015) as well as Prof M. L. Khurana Memorial Award (2015). |
1. Introduction
Glucosidases are considered to be an influential therapeutic target since they are the key enzymes involved in catalyzing the cleavage of glycosidic bonds in oligo- or polysaccharides to release free glucose. Glucosidases (α-glucosidase and β-glucosidase) causes hydrolysis of α and β glycosidic linkages of carbohydrates.1 Among them, α-glucosidases increase the postprandial blood glucose level and thus it is the major research topic of interest to pharmaceutical researchers.2 Recently, glucosidase-inhibiting sugar mimics are arousing great interest in the field of chemical and medicinal research owing to their promising therapeutic potential against some diseases such as diabetes, metastatic cancer, hepatitis B and human immunodeficiency virus (HIV) infection.3 Inhibition of α-glucosidase enzymes provides a new approach in the management of diabetes as it helps to maintain normal blood glucose level by slowing down the carbohydrate digestion, extend the carbohydrate digestion time and thus reduces the rate of glucose absorption.In humans, two major categories of enzymes namely α-amylase and α-glucosidase are responsible for the metabolism of dietary saccharides and starches into glucose. Maltase-glucoamylase (MGAM) and sucrase-isomaltase (SI) are the two human intestinal α-glucosidase enzymes, present in the brush border epithelial cell of small intestine, involved in glucose formation. Catalytic C and N-terminal subunit are present in both enzymes and these are ctMGAM and ntMGAM for MGAM whereas, ctSI and ntSI for SI.4–6 Inhibitors of α-glucosidase need to bind to all of these subunit for the hydrolysis of disaccharides and thus MGAM and SI are eye-catching targets for the drug discovery to control blood glucose levels in individuals with type-2 diabetes.7
In order to understand the pattern of structure–activity relationships for mimicking the enzyme-transition state and biochemical pathways, glucosidase inhibitors are the feature targets. Disaccharides, carbasugars, non-sugar, iminosugars and thiosugars derivatives have received great attention as ideal glucosidase inhibitors. Acarbose (1), miglitol (2) and voglibose (3) (Fig. 1) are well-known α-glucosidase inhibitors used in diabetes therapy.8–10 Acarbose (1) possess mainly α-amylase inhibition potential, while the other two compounds inhibit intestinal α-glucosidase. Since after the discovery of acarbose (1), a lots of synthetic11–13 and natural molecules14–16 are reported for the management of type-2 diabetes. The severe gastrointestinal side effects like abdominal discomfort, diarrhea, flatulence and hepatotoxicity17–19 are associated with acarbose and hence to improve the patient's quality of life, a novel class of α-glucosidase inhibitors having less side effects are required.
 | ||
| Fig. 1 Chemical structures of the well-known antidiabetics drugs. | ||
In view of aforementioned complications, the research continues to find other problem solving alternatives to obviate the effects. In support of the attention on natural and synthetic glucosidase inhibitors as prospective therapeutic agents and also as valuable tools to understand biochemical processes, this review gives a comprehensive overview of the chemical structure diversity of the α-glucosidase inhibitors that comprises thiosugars (replacement of the oxygen atom in the ring of a carbohydrate by sulfur). A novel class of sulfonium-ion compounds (salacinol, kotalanol, ponkoranol, salaprinol and their corresponding de-O-sulfonated derivatives), remarkable members of the thiosugars, isolated from Salacia sp. with unique structural features and potent inhibitory activities against the intestinal α-glucosidases, provides opportunities for the development of new drug candidates for treatment of type-2 diabetes.20,21 A systematic literature search was conducted to identify the up to date information on sulfonium-ion and their role in glucosidase inhibition. Comprehensive studies of sulfonium inhibitors reflected that the previously reported reviews mainly discussed only one or two sulfonium compounds from the class of sulfonium-ion α-glucosidase inhibitors family. Mohan and Pinto in 2007 reported a review article focused mainly on the syntheses of salacinol and related analogues.22 However, an update on the isolation, glucosidase inhibitory activities and synthesis of salaprinol and kotalanol analogue as well as the structure elucidation of kotalanol is presented in separate review article by the same authors in 2009 and 2010.23,24 Whereas, in 2013, a mini-review on sulfonium compounds has been reviewed by Xie et al.25 A review on further progress in kotalanol, ponkoranol and their synthetic analogues have appeared in 2014 by Mohan et al.26 Following this search, an effort to compile the literature on all compounds of sulfonium family has been done in this paper. In this account, we summarize the role of Salacia as antidiabetic drug as well as the aspects of extraction, structural elucidation and biological evaluation of naturally occurring sulfonium-ion and their synthetic analogues with particular focusing on their selective inhibitory activities against the intestinal glucosidases along with crystal structure, docking and the toggling studies. The insight gathered in this review provides a wealth of information about sulfonium-ion α-glucosidase inhibitors. This knowledge will be useful in deriving more potent antidiabetic candidates from the bioactive natural molecule.
2. Salacia: a promising class of potential antidiabetic drug
Literature survey revealed that, Salacia sp. plays an important role in the management of diabetes. The genus Salacia, a member of Celastraceae family, are woody climbing plants which consist of 407 species and have almost 108 Generas.27,28 Salacia sp. are widely spread in India, China, Sri Lanka and Southeast Asian countries such as Thailand, Indonesia, and also in South American torrid zone areas such as Brazil.21,29Salacia has been officially listed in the Ayurvedic Pharmacopeia, its aerial parts and roots are extensively used in Ayurvedic system of medicine, traditional Indian medicine, and Unani as a precise remedy for early-stage diabetes and for treating asthma, ear diseases, gonorrhoea, rheumatism, itching, leukemia and inflammations.29 Presently, Salacia sp. are extensively consumed in Japan and US and different countries as a food supplement formulation for the treatment of diabetes and have also been studied widely for their potential value in diabetes management.30,31 Ayurvedic medicine prescribes the herbal extract, obtained by storing water overnight in a mug made from the root of S. reticulata and other members of the Salacia genus, such as S. oblonga, S. prinoides, and S. chinensis, as a remedy for type-2 diabetes. Earlier people with diabetes use to drink water from the mugs made from Salacia wood.24,32 Since 1990, Salacia sp. have been used in traditional system of medicine as effective drug targets for the management of diabetes. Inspired by this traditional and medicinal use, several types of research have been carried out by researchers to find out the antidiabetic phytoconstituents responsible for the activity. Earlier studies on this plant have resulted in the isolation of several potent sulfonium inner salts, salacinol,33,34 kotalanol,35–37 and ponkoranol,38 responsible for the antidiabetic activity. These sulfonium phytoconstituents act as α-glucosidases inhibitor by competitively inhibiting α-glucosidase activity and thus helps to prevent the fast breakdown of oligosaccharides into monosaccharides and thereby control the normal blood sugar level (Fig. 2).39
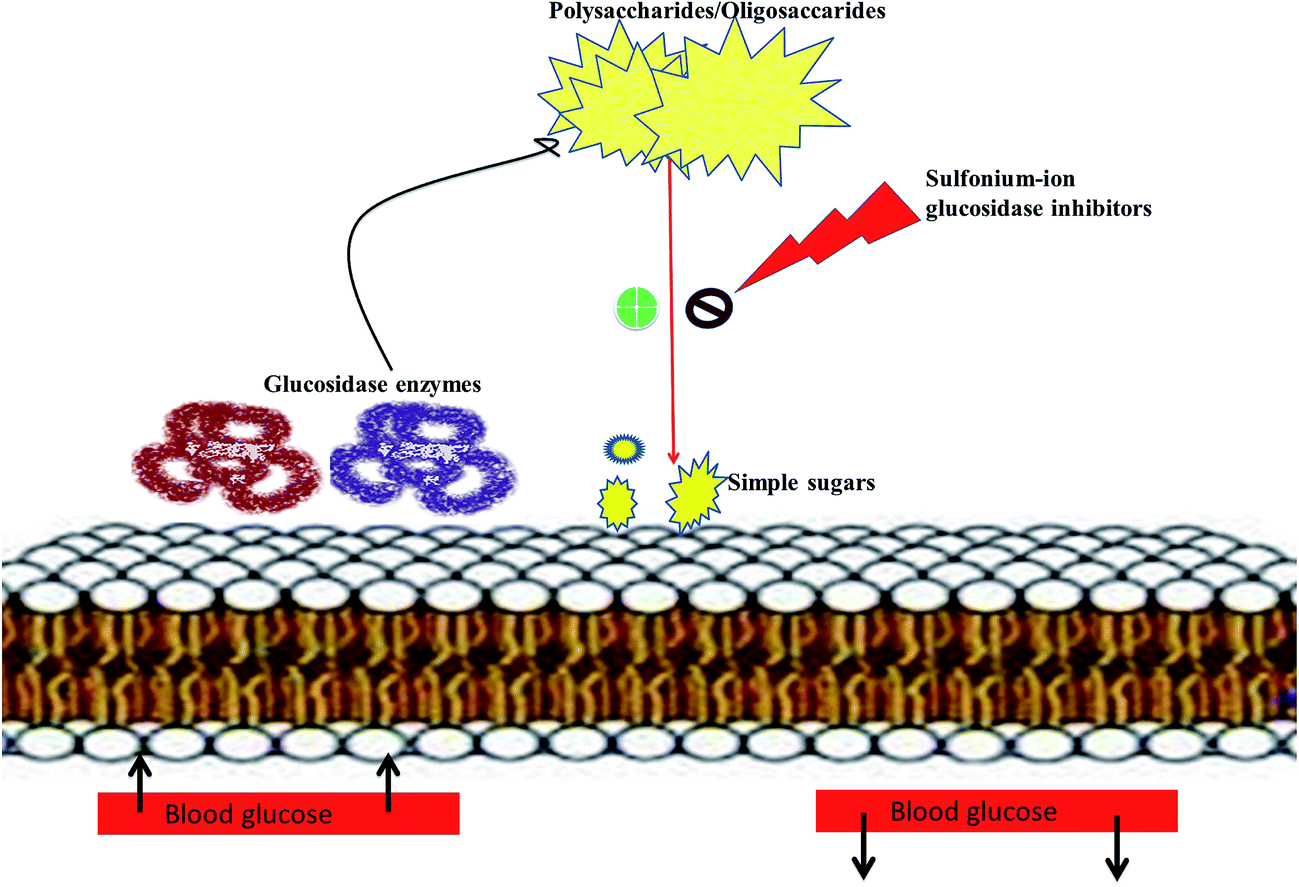 | ||
| Fig. 2 Pictorial depiction showing mechanism of sulfonium-ion as α-glucosidase inhibitors. | ||
Literature information strongly validated the approach that the glucosidase inhibitory activities of the aforesaid sulfonium compounds are very potent and have been revealed to be as strong as those of acarbose (1) and voglibose (3).33,35,38 A number of reports on animal and human have been published that reinforced the reported reductions in glycemia as well.40–42 Various studies declare the role of Salacia sp. as an effective antidiabetic drug.43,44 Table 1 illustrate the potential of S. oblonga,40,45–47 S. reticulata,41,48,49 S. prinoides,50 S. macrosperma51 and S. chinensis52 in diabetes therapy. Based on this evidence, it is apparent that Salacia sp., as a rich source for sulfonium ions, can be used as important nutraceuticals or as adjuvants for the management of diabetes.
| Plant sp. | Biological activity | Extract type/compound used | Model system | References |
|---|---|---|---|---|
| S. chinensis | Antidiabetic activity | Aqueous extract of stem | KK-Ay mice model (10–300 mg kg−1) | 52 |
| S. chinensis | α-Glucosidase inhibitory activity | Whole plant extract | In vitro enzyme inhibition assay (100 µg mL−1) | 53 |
| S. oblonga | Antidiabetic activity | Polyherbal combo with S. oblonga (G-400) | Human (1000 mg day−1 for 8 weeks) | 54 |
| S. chinensis | Antidiabetic activity | Mangiferin from aqueous solution of root | Male adult Wistar rats (40 mg kg−1 body wt per day) | 55 |
| S. chinensis | Antidiabetic activity | Mangiferin from aqueous solution of root | Wistar male albino rats (40 mg kg−1) | 56 |
| S. reticulate | Antidiabetic activity | Aqueous extract from leaves and root bark | Human (500 mg day−1) | 57 |
| S. oblonga | Antidiabetic activity | Mehani (polyherbal formulation) | Wistar strains of male albino rats (2 g/60 kg) | 58 |
| S. oblonga | Antidiabetic activity | Hydroalcoholic root extract | Streptozotocin induced diabetic Wistar rat (50 and 100 mg kg−1) | 59 |
| S. oblonga | Antidiabetic activity | Lipo polysaccharide tea | KK-Ay/TaJcl type-II diabetic mice model (200 and 300 mg) | 60 |
| S. reticulate | Antidiabetic and α-glucosidase inhibitory activity | Aqueous extracts of bark or core root | In vitro assay Wistar male alloxan (50 mg kg−1) | 61 |
| S. oblonga | Antidiabetic activity | Aqueous extract | L6 rat-myotubes model | 62 |
| S. reticulate | Antidiabetic activity | Aqueous extract | KK-Ay genetically diabetic mice | 63 |
| S. fruticosa | Antihyperglycemic activity | Methanolic extract | Wistar albino adult male rats (125 mg kg−1 and 250 mg kg−1) | 64 |
| S. reticulate | Antidiabetic activity | Aqueous extract | Maltose or sucrose loaded mice (1.0 mg) | 65 |
| S. reticulate | Antidiabetic activity | Aqueous extract and salacinol | Maltose or sucrose loaded Wistar rats (50 and 75 mg kg−1; 2.50 and 5.00 mg kg−1) | 66 |
| S. reticulate | α-Glucosidase inhibitory activity | Water soluble fraction | Sucrose or malt sugar loaded rats (25–100 mg kg−1) | 38 |
| S. oblonga | Antidiabetic activity | Aqueous extract | Human (240 and 480 mg) | 67 |
| S. reticulate | Antihyperglycemic activity | Hydroalcoholic extract | Hydrocortisone induced hypoglycemic rat model (500 mg kg−1) | 68 |
| S. reticulate | Antidiabetic activity | Tea | Human (200 mg) | 69 |
| S. reticulate | Antidiabetic activity | Bark powder | Human (2 g per day for 90 days) | 70 |
| S. oblonga | Antidiabetic activity | Aqueous extract | Obese Zucker rats (100 mg kg−1) | 71 |
| S. oblonga | Antidiabetic activity | Aqueous extract | Obese Zucker rats (100 mg kg−1) | 72 |
| S. chinensis | α-Glucosidase inhibitory activity | Methanolic extract | Sucrose or malt sugar loaded rats (50, 100, 200 mg kg−1) | 73 |
| S. reticulate | α-Glucosidase inhibitory activity | Water soluble fraction | Sucrose or malt sugar loaded rats (25–100 mg kg−1) | 74 |
| S. oblonga | Antidiabetic and antioxidant activity | Petroleum ether extract of the root bark | Streptozotocin induced diabetic rats (250 mg kg−1) | 45 |
| S. oblonga | Antidiabetic activity | Methanolic extract | Sucrose and maltose loaded male Wistar rats (0, 100, 200 mg kg−1) | 39 |
| S. oblonga | Antidiabetic study | Petroleum ether extract of the root bark | Streptozotocin induced diabetic albino rats (250 mg kg−1, orally) | 40 |
| S. macrosperma | Antidiabetic activity | Root extract | Rabbits and alloxan induced albino rats (200 mg kg−1) | 51 |
| S. reticulate | Antidiabetic activity | Aqueous extract | Sprague-Dawley rats | 41 |
| S. prinoides | Antidiabetic activity | Root bark extract | Alloxan induced diabetic rats | 50 |
3. Extraction, isolation and structural elucidation of sulfonium-ion from Salacia sp.
Salacinol, the first member of the naturally occurring sulfonium-ion class of glucosidase inhibitors, was originally isolated as one of the active principal constituents from the aqueous extracts of S. reticulate through bioassay guided separation using α-glucosidase inhibitory activities and afterward from other plant species of the Salacia genus, S. oblonga39 and S. chinensis.73 In the course of the search for antidiabetic phytoconstituents from Salacia, Yoshikawa et al. in 1997 discovered that a water-soluble fraction (25–100 mg kg−1) prepared from the roots and stems of S. reticulate strongly inhibited elevations in serum glucose levels in rats after oral administration of sucrose or maltose, but not glucose. To authenticate this activity, they conducted a bioassay guided separation assay, in which they isolated a novel five-membered sugar analogue, salacinol having an internal sulfate counterion.33,75Fig. 3 summarizes the extraction and fractionation scheme of Salacia sp. Under reflux condition, the dried root and stems of Salacia sp. were extracted using methanol. The prepared methanolic extract was mixed with water and fractionated by ethyl acetate. The enzyme inhibitory effect and amelioration of serum glucose level in sucrose-loaded rats were found in water soluble fraction whereas the ethyl acetate fraction did not show any affect. Further, a bioactivity guided fractionation was carried out for the isolation of the α-glucosidase inhibitor from the water soluble fraction. This water soluble fraction was suspended in methanol and then filtered to give a methanol-soluble phase and a residue. The methanol-soluble phase was subjected to ordinary-phase silica gel chromatography to give eight fractions. The active fractions were subjected to ODS and NH column chromatography and finally HPLC to furnish salacinol. However, some modifications in extraction method can be possible for the isolation of salacinol.
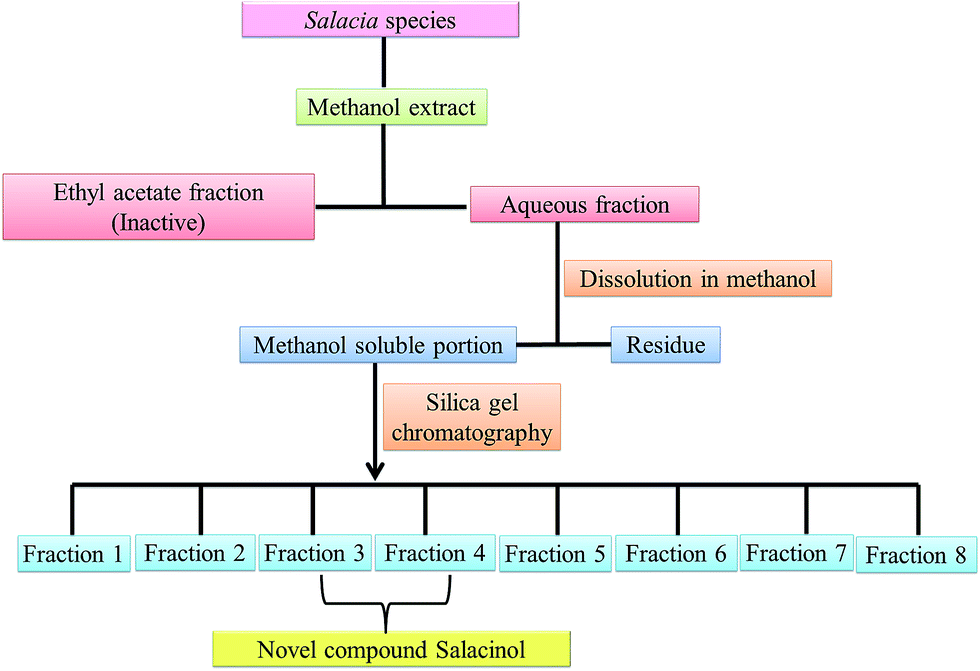 | ||
| Fig. 3 Schematic representation of extraction of salacinol from Salacia species. | ||
The stereostructure of salacinol have been elucidated on the basis of various chemical reactions and spectroscopic analyses including 1D and 2D NMR experiments and a potassium rhodizonate test (a positive test that confirmed the presence of a sulfate moiety) and suggested the presence of an unusual zwitterionic sulfonium-sulfate structure. Based on the spectral and X-ray crystallographic analyses, Yoshikawa et al.33 established the initial stereostructure of salacinol as an inner-salt sulfonium structure (4) that was composed of a 1,4-anhydro-4-thio-L-arabinitol unit alkylated at sulfur by a 1-deoxy-D-erythritol-3-sulfate unit (Fig. 4).33 The isolated salacinol showed competitive inhibition as equivalent to acarbose against maltase and sucrase in in vitro. However, the inhibitory activity of salacinol against isomaltase was found to be greater than that of acarbose (Table 2).
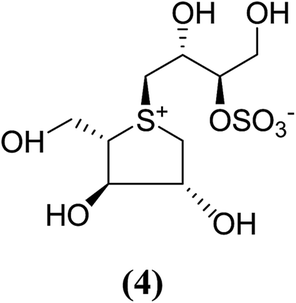 | ||
| Fig. 4 Previously proposed structure of salacinol. | ||
| Inhibitor | Ki (µg mL−1) | ||
|---|---|---|---|
| Maltase | Sucrase | Isomaltase | |
| Acarbose (1) | 0.12 | 0.37 | 75 |
| Salacinol (5) | 0.31 | 0.32 | 0.47 |
Another potent sulfonium ion α-glucosidase inhibitor, kotalanol (1,2,3-trihydroxy-propyl-salacinol) was isolated from the same plant in 1998 by the same group by performing another bioassay-guided separation study. They reported that this inhibitor possesses stronger inhibitory activity against certain glucosidase enzymes. Soon after, from Salacia genus, ponkoranol, salaprinol and de-O-sulfonated derivatives of all four parent compounds were also isolated.35
4. Structural features and structure–activity relationship studies of naturally occurring sulfonium-ion and their synthetic analogues as potent α-glucosidase inhibitors
Several drug discovery research based on these natural sulfonium-ion glucosidase inhibitor compounds have been carried out. Thus far, researchers have isolated eight sulfonium-ion glucosidase inhibitors (Fig. 5) from Salacia sp.: salacinol (5), kotalanol (6), ponkoranol (7), salaprinol (8) and four of their corresponding de-O-sulfonated compounds such as de-O-sulfonated salacinol; neosalacinol (9), de-O-sulfonated kotalanol; neokotalanol (10), de-O-sulfonated ponkoranol; neoponkoranol (11) and de-O-sulfonated salaprinol; neosalaprinol (12).22 The salacinol structure was assigned to be the enantiomer (5) of the previous stereostructure (4) as the plant origin and biosynthetic pathways of salacinol (5) and kotalanol (6) are same.34,39 Yoshikawa et al.35 revise the initial structure of salacinol (4) by performing degradation studies of kotalanol (6).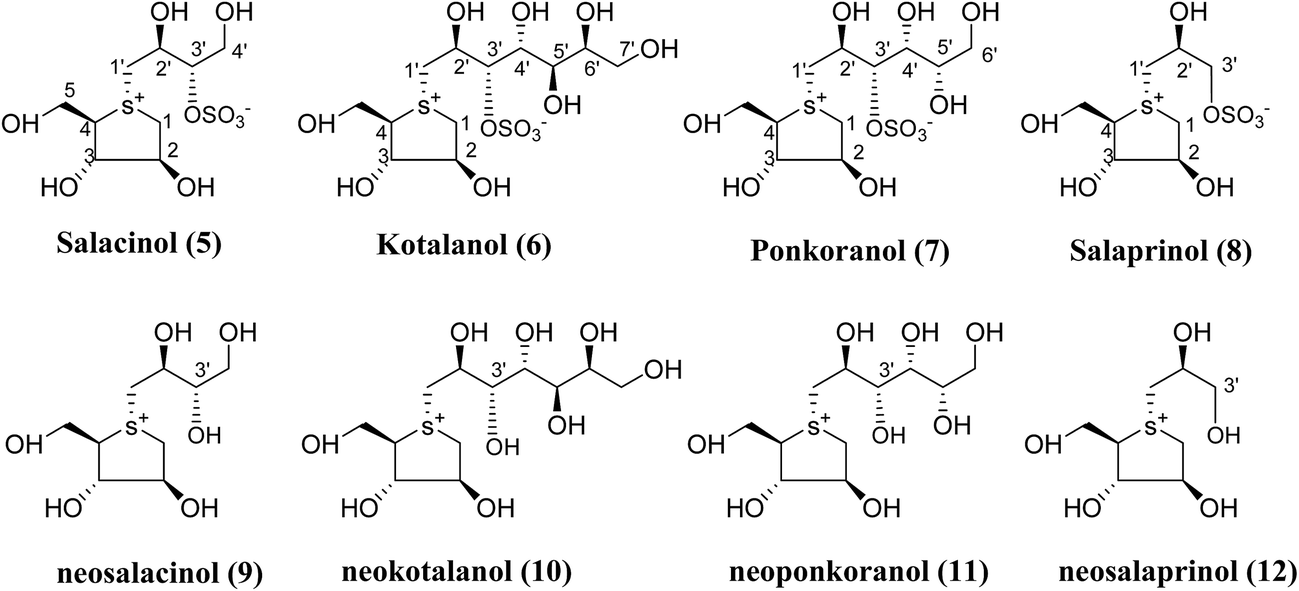 | ||
| Fig. 5 Major sulfonium constituents (5–12) of Salacia species. | ||
At the time of isolation, the absolute stereostructure for sulfonium compounds, except salacinol (5), was not resolved but synthetic work has shown the way to their stereochemical structure elucidation. In the case of parent compounds, salacinol (5), kotalanol (6), ponkoranol (7), and salaprinol (8), C-3′ carry a sulfate group which provides a unique sulfonium-sulfate inner salt structure. This particular characteristic of sulfonium ion inhibitor, having permanent positive charge carried by the sulfur, is supposed to bind in the active site of glucosidase in the same manner as a protonated amine inhibitor.76–78 The sulfonium compounds vary only in the length of the polyhydroxylated side chain. For example, salacinol (5) has a side chain of four-carbon with defined stereochemistry, whereas, kotalanol (6) contains a seven-carbon side chain, with unspecified stereochemistry at the stereogenic centers and salaprinol (8) has a three-carbon side chain.
It is remarkable that the configurations up to the C-3′ position in the side chain and configurations around the heterocyclitol moiety are preserved in all of these sulfonium compounds. Altering the configurations at any of these positions, including that at the stereogenic sulfur atom, give rise to complete or partial loss of inhibitory activities against the human intestinal α-glucosidase enzymes.22 Usually, the de-O-sulfonated analogues are more active than the corresponding original sulfates against the intestinal α-glucosidases. Various studies revealed that, on a comparison of the inhibitory activities of de-O-sulfonated salacinol (9) vs. salacinol (5) and de-O-sulfonated kotalanol (10) vs. kotalanol (6) against rat intestinal glucosidase (maltase, sucrase, and isomaltase), the desulfonated analogues were either equivalent or better inhibitors than the parent compounds.21,38,79 Data of inhibitory activity of these sulfonium compounds (5–12),38,80 tested against rat small intestinal α-glucosidase in vitro and compared with clinically used antidiabetics, acarbose (1) and voglibose (3), are given in Table 3.
| Inhibitor | IC50 (µM) | ||
|---|---|---|---|
| Maltase | Sucrase | Isomaltase | |
| a Values in parentheses indicate inhibition (%) at the corresponding concentrations (µM). | |||
| Acarbose (1) | 2.0 | 1.7 | 155 |
| Voglibose (3) | 1.2 | 0.2 | 2.1 |
| Salacinol (5) | 5.2 | 1.6 | 1.3 |
| Kotalanol (6) | 7.2 | 0.75 | 5.7 |
| Ponkoranol (7) | 3.2 | 0.29 | 2.6 |
| Salaprinol (8) | >329 (42)a | >329 (23)a | 15 |
| Neosalacinol (9) | 8.0 | 1.3 | 0.3 |
| Neokotalanol (10) | 4.8 | 4.5 | 1.8 |
| Neoponkoranol (11) | 5.1 | 1.0 | 1.4 |
| Neosalaprinol (12) | >384 (34.5)a | 90 | 6.5 |
Thus, because of the intriguing structure and high α-glucosidase inhibitory activities, great interest has been focused on sulfonium compounds and comprehensive studies on the structure–activity relationships of this new class of α-glucosidase inhibitors family has been reported.
4.1. Sulfonium-ion analogues as α-glucosidase inhibitors
Apart from extraction from natural sources, a non-naturally sulfonium-ion could be produced synthetically from readily accessible starting materials. The compounds belong to this group are represented by the general formula:where X is selected from the group consisting of S, Se and NH. R1, R2 and R3 are the same or different and are consist of H, OH, SH, NH2 and halogens. Whereas, R4 is chosen from the group consisting of:
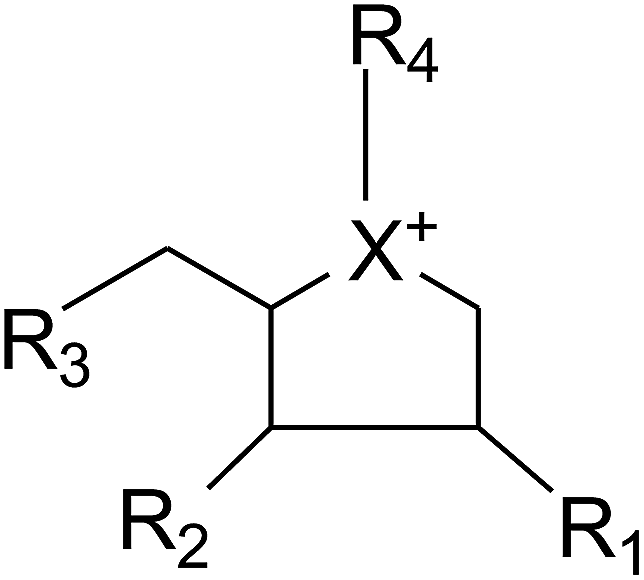
(1) a polyhydroxylated acyclic alkyl chain comprising an anionic sulfate, carboxylate or phosphate moiety.
(2) A lipophilic alkyl chain between 2 and 20 carbons in length with an external counterion.
These derivatives display interesting selectivity for glucosidase enzymes from various sources and act as active glucosidase inhibitors.
In a search for potential glucosidase inhibitors, various synthetic analogues of salacinol have been synthesized by different researchers for structure–activity studies with different glucosidase enzymes. Miglitol (2) and salacinol analogues selectively target intestinal glucosidases and is a poor inhibitor of α-amylases.83 In contrast, α-amylase is inhibited more strongly by acarbose (1) (Ki = 15 nM).84 Salacinol (5, Table 4) and some of its derivatives revealed an interesting selectivity for tested glucosidase enzymes from various sources like glucoamylase G2, porcine pancreatic α-amylase (PPA), barley α-amylase (AMY1), human pancreatic α-amylase (HPA), recombinant human maltase glucoamylase (MGAM), glucosidase from rice and baker's yeast and compared with salacinol (5).22
| α-Glucosidase | Salacinol (5) |
|---|---|
| NOI = no inhibition. | |
| Glucoamylase G2 Ki = (mM) | 1.71 |
| PPA Ki (mM) | 0.01 |
| AMY1 Ki (mM) | 0.015 |
| MGAM Ki (µM) | 0.19 |
| HPA Ki (mM) | 0.075 |
| Rice IC50 (mM) | 1.1 × 10−3 |
| Baker's yeast IC50 (mM) | NOI |
Total synthesis of salacinol (5) was accompanied by two groups.85–87 Yuasa et al.85 and Ghavami et al.86 independently reported the synthesis of salacinol (5) and its enantiomer (4) and diastereomers (13) (Fig. 6) and provided conclusive proof of structure of the natural product. Yuasa et al.88 have also investigated the glucosidase inhibitory activities of compounds (5) and (13) and showed that although salacinol (5) is better inhibitor (IC50 = 1.1 µM) of rice α-mannosidase than its diastereomer (13, IC50 = 0.38 mM), the inhibitory activities are comparable for almond α-glucosidase (IC50 = 2.1 mM for 5 and 3.6 mM for 13). In the case of almond β-glucosidase, (13) (IC50 = 3.4 mM) is a better inhibitor than (5) which showed no activity.
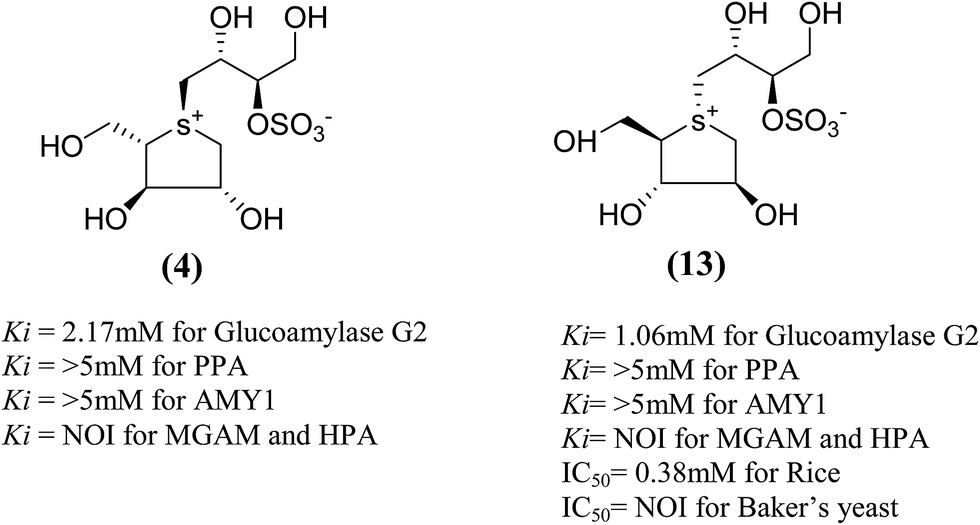 | ||
| Fig. 6 Stereoisomers of salacinol (4 and 13) and their inhibitory activity against α-glucosidases. | ||
In order to develop more effective glucosidase inhibitors, the azacyclic version of salacinol was synthesized in which the thiosugar sulfur was replaced by nitrogen to give compounds, ghavamiol (14) and (15) (Fig. 7), as potential glucosidase inhibitors.89 Inhibition assays using intestinal α-glucosidase indicated that ghavamiol (14) was less potent against maltase, sucrase and isomaltase, compared to salacinol (5). Salacinol (5) inhibits maltase, sucrase and isomaltase with IC50 values of 9.6 µM, 2.5 µM and 1.8 µM, respectively, whereas compound (14) inhibits these enzymes with IC50 values of 306 µM, 44 µM and 136 µM, respectively. Thus, the inhibitory activities of ghavamiol (14) decreased considerably against these enzymes when compared to salacinol (5).90 On comparison of inhibitory activity of ghavamiol (14) and (15) against three different glucosidase enzymes, it was noticed that despite the low activity of the natural sulfonium ion, salacinol (5), towards glucoamylase (K1 = 1.7 mM), nitrogen analogues (14) and (15) exhibited a 10-fold higher inhibition value than salacinol (5). Furthermore, the compounds (14) and (15) showed not active against AMY1 and PPA at concentrations of 5 mM. In contrast, salacinol (5) showed stronger inhibition against AMY1 and PPA in the micromolar range, with Ki values of 15 ± 1 µM and 10 ± 2 µM, respectively. Screening of ammonium compounds against HPA showed that ghavamiol (14) was not active against HPA whereas the compound (15) inhibited the enzyme with Ki value of 0.4 mM compared to salacinol that showed stronger inhibition with a Ki value of 75 µM.
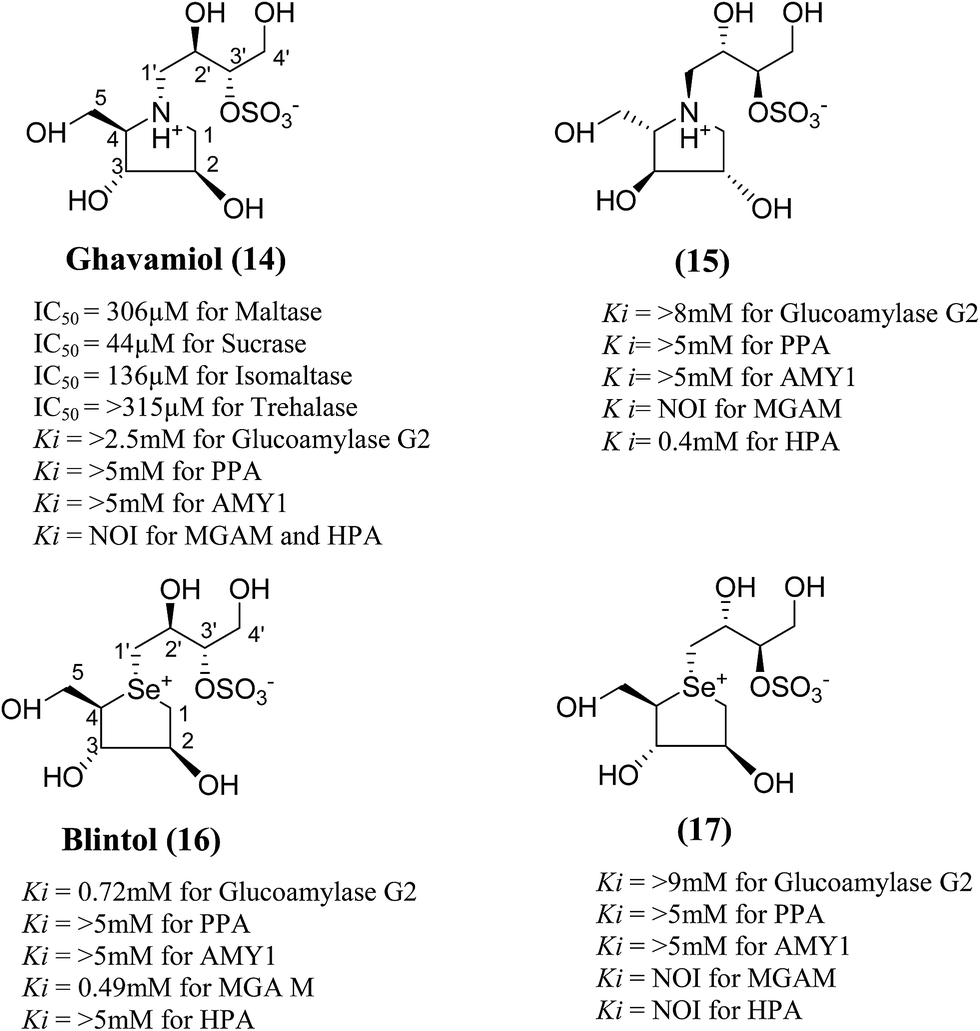 | ||
| Fig. 7 Nitrogen (14 and 15) and selenium (16 and 17) heteroatom analogues of salacinol and their inhibition potential against α-glucosidase enzymes. | ||
In the same manner, substitution of the sulfonium ion of salacinol (5) by selenium produced a derivative, blintol (16) (selenosalacinol) and its steroisomers (17) (Fig. 7). After enzyme inhibition studies on blintol (16), it was found that blintol (16) was a weak inhibitor of glucoamylase G2 (Ki = 0.72 mM) and showed no significant inhibition against AMY1, HPA, and PPA. The compound (17) showed no significant inhibition of glucoamylase, AMY1, HPA, and PPA. The blintol (16) was shown to be an effective treatment for regulating blood glucose levels in rats after a carbohydrate meal, thus act as a lead candidate for the treatment of type-2 diabetes.89,91–93
The search for potent glucosidase inhibitors has led to the preparation of several salacinol analogues with varying stereochemistry at one or more stereogenic centers by modifying substituents and ring size or by replacing the sulfur in the sulfonium-ion by nitrogen or selenium. Ghavami et al.94 explored five membered ring analogues of salacinol (5) with different sugar stereochemistry, an anhydro-thio-D-xylitol (18), its nitrogen congener (19) were synthesized (Fig. 8) in which stereochemistries at C-2 and C-3 were both inverted compared to salacinol (5). Compound (18) and (19) were tested for their inhibition of three glycosidase enzymes, namely glucoamylase G2, PPA and AMY1. Glucoamylase G2 was weakly inhibited by salacinol (5) (Ki = 1.7 mM) whereas compound (18) and (19) showed no significant inhibition of glucoamylase. The sulfonium ion (18) inhibited AMY1 and PPA, with Ki values of 109 ± 11 and 55 ± 5 µM, respectively, as compared to salacinol (5), with Ki values of 15 ± 1 and 10 ± 2 µM, respectively. The ammonium ion analogue (19) showed no significant inhibition of either AMY1 or PPA. Thus, analogues (18) and (19) and salacinol (5) showed discrimination or selectivity for certain glucosidase enzyme.
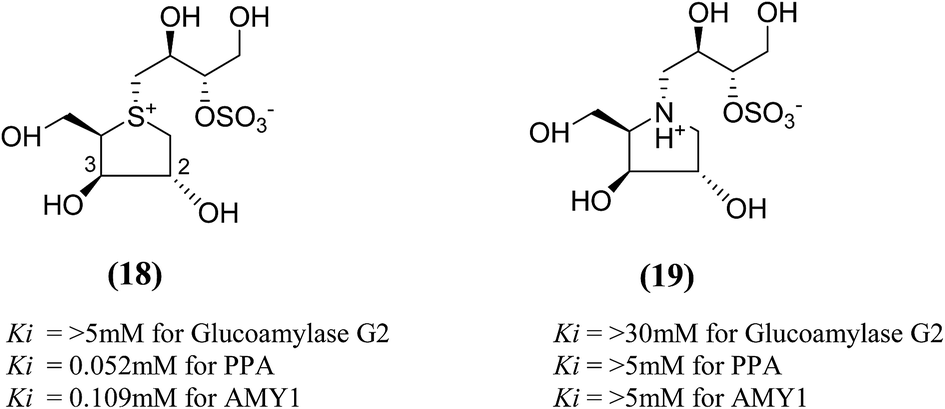 | ||
| Fig. 8 Salacinol analogues containing anhydrothio-D-xylitol (18) moiety, its nitrogen congener (19) and their inhibitory activity against glucosidase enzymes. | ||
Other analogues of salacinol, anhydrothio-D-lyxitol (20), and anhydrothio-D-ribitol (21) have also been reported (Fig. 9) in which the stereochemistries at C-3 and C-2, respectively, are inverted compared to salacinol (5).95 Enzyme inhibition assay of (20) and (21) against MGAM indicated that they were not effective inhibitors against MGAM. While salacinol inhibited this enzyme with a Ki value of 0.19 µM.96 Therefore, an important conclusion derived from the enzyme inhibition data that the D-arabinitol configuration in the heterocyclic ring as in salacinol is critical for its inhibitory activity.
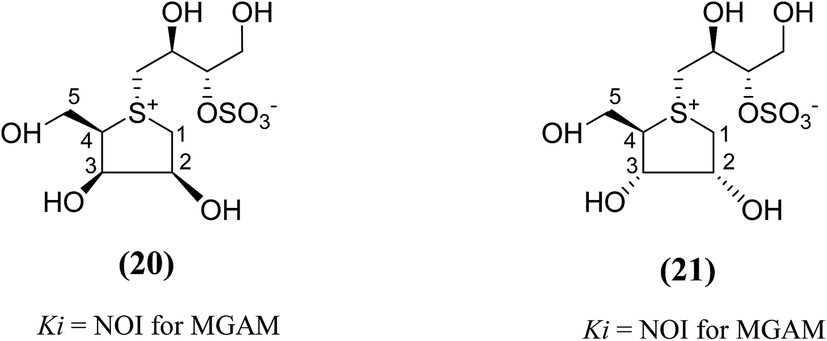 | ||
| Fig. 9 Salacinol analogues containing anhydrothio-D-lyxitol (20) and anhydrothio-D-ribitol (21) moiety and their inhibitory activity against MGAM. | ||
To discuss the importance of the heterocyclic ring size on glucosidase inhibitory activity, several six membered ring analogues (22–26, Fig. 10) of salacinol have been reported. Szczepina et al.97 synthesized a series of pyranosil compounds (22a–26a) and their corresponding isomers (22b–26b), having a cyclic alditol structure, in which an L-erythritol sulfated side chain was attached to the ring nitrogen, sulphur, or selenium atom. The objective was to investigate if changes to this group, acting as a counterion to ammonium or selenium salt analogues, might increase the in vivo stability or biomembrane permeability. The enzyme inhibition data indicated that these salacinol analogues with six-membered rings against glucoamylase G2 were either weak or absent, indicating the importance of the five-membered ring incorporated in salacinol (5).97 Hence, it could be inferred that the five-membered ring of salacinol (5) is necessary and key structural determinant of sulfonium sulfate glucosidase inhibitors and their analogues for this particular enzyme.86,94
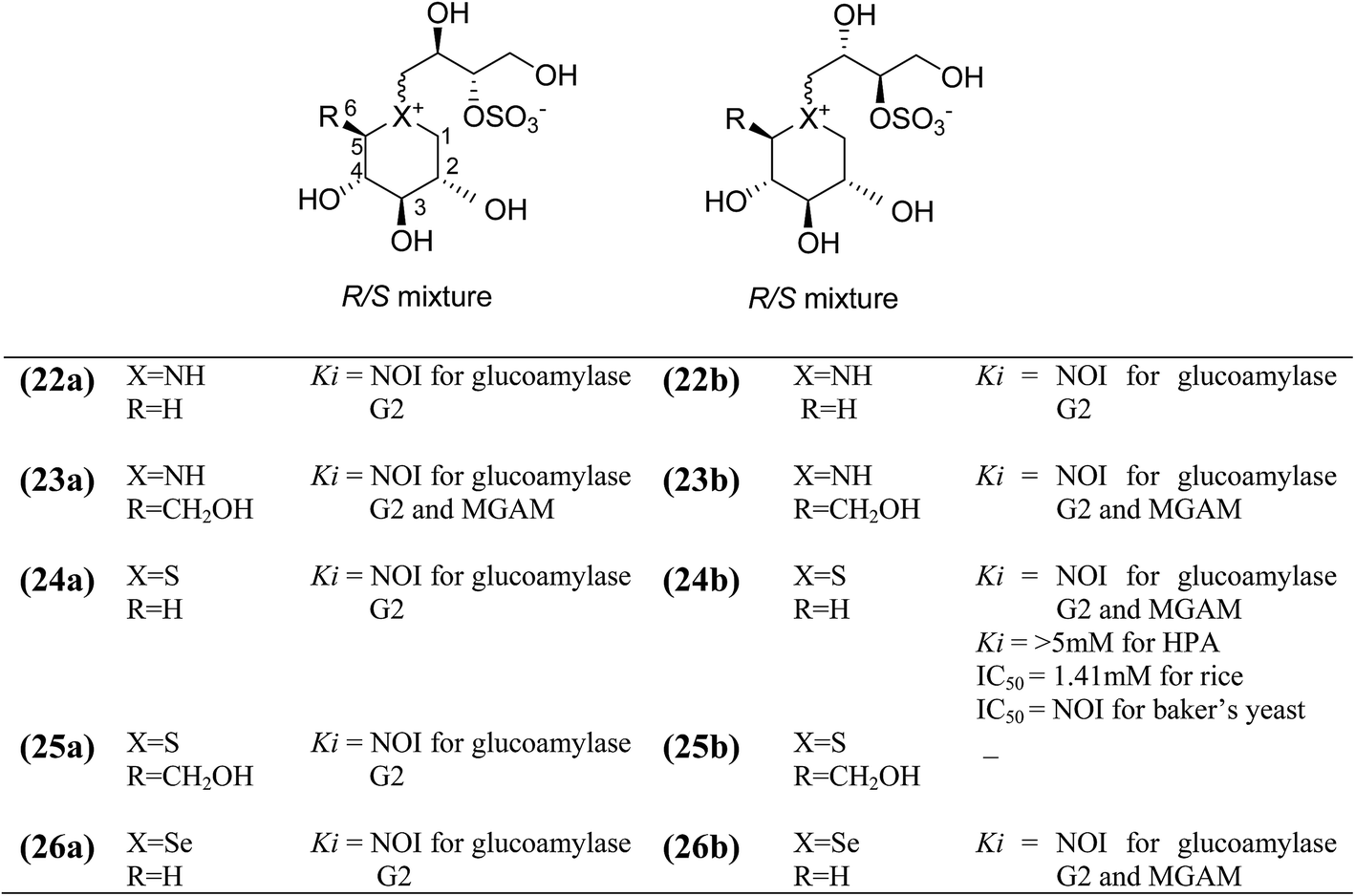 | ||
| Fig. 10 Structure of salacinol analogues having six-membered ring (22–26) and their inhibitory activity against glucosidase enzymes. | ||
In order to provide additional information about the structure requirements for anti-glucosidase activity, Gallienne et al.98 reported two-series of five (27–32, Fig. 11) and six-membered ring (33, 34, Fig. 12) analogues having thioanhydro alditol moieties with erythro, D,L-threo, xylo, ribo, D-arabino, and D-manno configurations (27–36), including previously reported 24b six membered ring analogue. These compounds (27–36 and 24b) were tested against six different commercial glucosidases including the rice-derived α-glucosidase. All compounds from five-membered ring series (27–32) that are missing the hydroxymethyl group at C-4 when compared to salacinol were inactive against the α-glucosidase from rice, whereas, salacinol (5) had been shown previously to exhibit a strong inhibitory effect toward this enzyme (IC50 = 1.1 × 10−3 mM).88 As a consequence, it was revealed that the hydroxymethyl group of salacinol is essential for its inhibitory activity toward this particular enzyme.
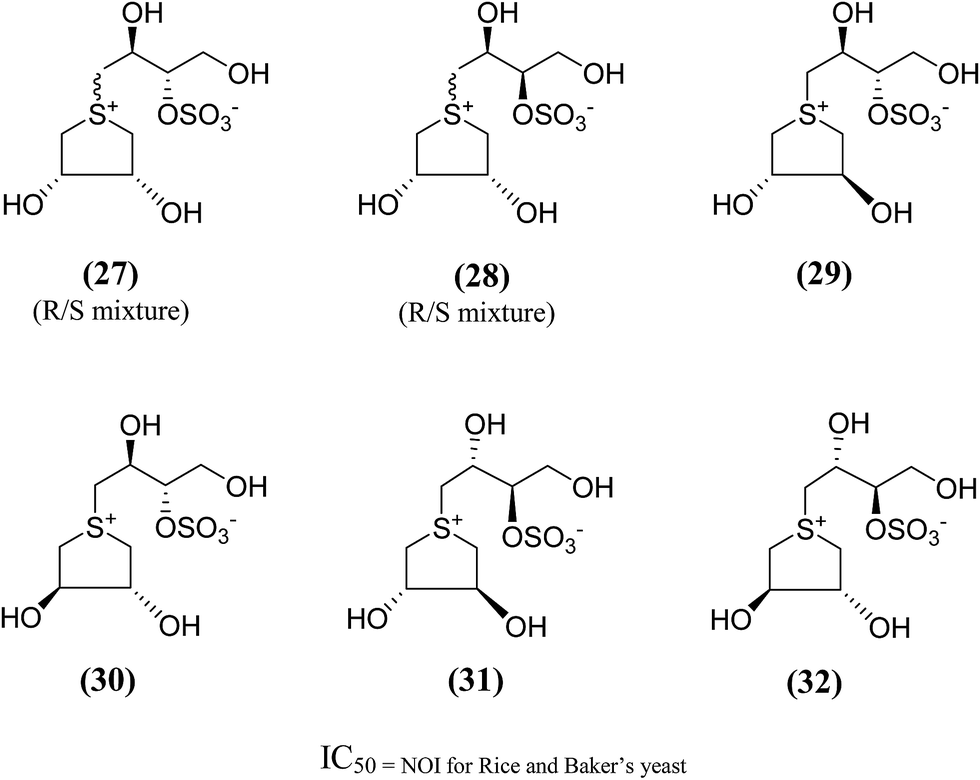 | ||
| Fig. 11 Five membered ring analogues (27–32) of salacinol synthesized by Gallienne et al.98 and their inhibitory profile against α-glucosidase enzymes. | ||
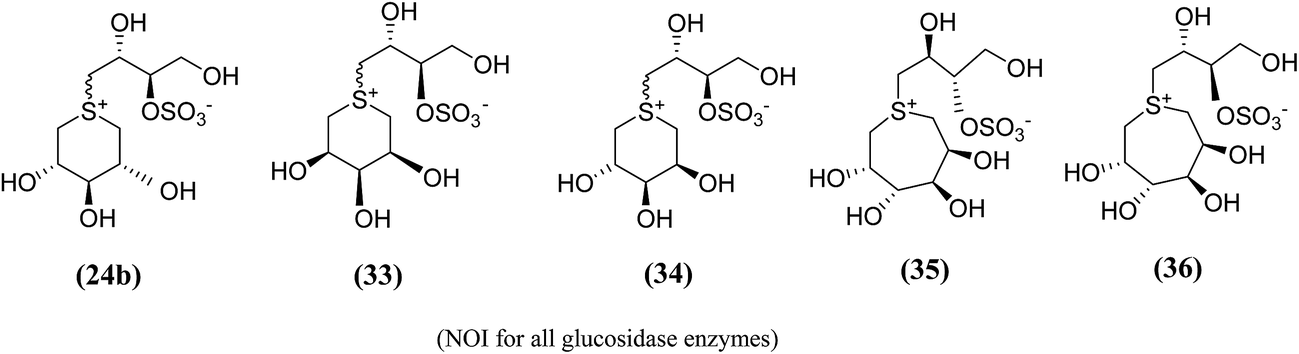 | ||
| Fig. 12 Six (33, 34) and seven (35, 36) membered ring analogue of salacinol synthesized by Gallienne et al.98 and their inhibitory activity against α-glucosidase enzymes. | ||
The six- and seven-membered ring analogues (33–36, Fig. 12) showed only very weak activities or no activity against the tested glucosidase enzymes. However, the xylitol analogue (24b) (R/S mixture) produced an inhibitory effect against β-glucosidase from almond. Based on observation of this study and previous study on various six-membered ring analogues (22–26),97 it was concluded that the ring expansion of the heterocycle exhibit in salacinol leads to loss of inhibitory activity.
Amusingly, this conclusion was corroborated, when Gallienne et al.99 synthesized a new series of salacinol analogues (37–40, Fig. 13) containing six-membered ring nitrogen heterocycles and tested them against glucosidases. As experimental with the other six-membered ring analogues, none of the synthesized ammonium salts were active against the tested six commercial glucosidases including rice α-glucosidase and baker's yeast α-glucosidase.
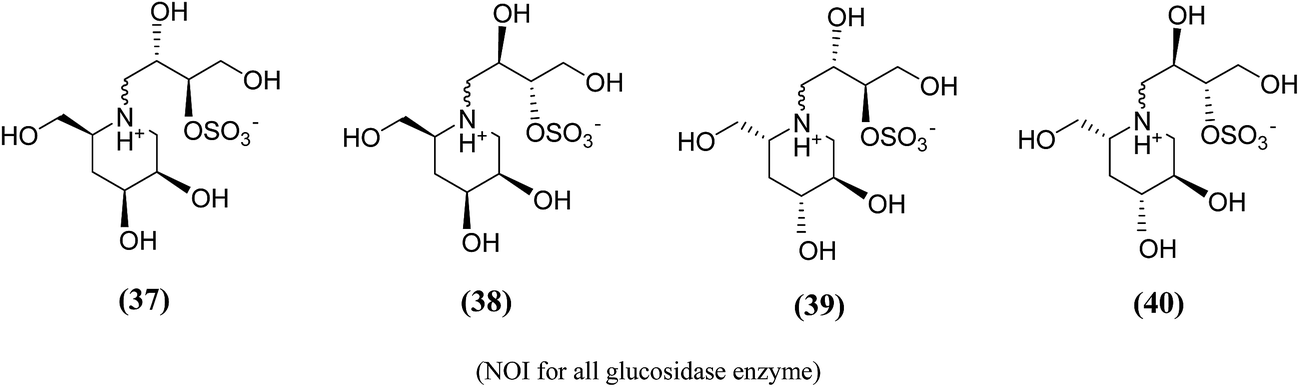 | ||
| Fig. 13 Six membered ring analogue (37–40) of salacinol synthesized by Gallienne et al.99 containing nitrogen heterocycle and their inhibitory profile against α-glucosidase enzymes. | ||
Several chain-modified analogues of salacinol have also been synthesized for evaluation as inhibitors of glucosidase enzymes. These modifications incorporate the extension of the acyclic chain, the change in the position or complete removal of the sulfate group, and the introduction of acyclic chains that are missing one or two hydroxyl groups when compared to salacinol (5). Some of these compounds displayed inhibitory activities in a low micromolar range against MGAM. The stereochemistries at the different stereogenic centres on the side chain play significant roles, and structure–activity studies revealed an interesting variation in the inhibitory power of these compounds (Table 5). Based on the information, kotalanol (6), with a longer polyhydroxylated side chain, has been reported to acquire greater inhibitory action against certain glucosidase enzymes when compared to salacinol (5).35 So, a lot of interest has been directed toward the synthesis of acyclic chain modified analogues of salacinol.
| Inhibitor | Stereochemistry at the stereogenic centres in the acyclic side-chain | Ki (µM) | References | |||
|---|---|---|---|---|---|---|
| C-2′ | C-3′ | C-4′ | C-5′ | |||
| NOI = no inhibition. | ||||||
| Salacinol (5) | S | S | — | — | 0.19 ± 0.02 | 96 |
| Ghavamiol (14) | S | S | — | — | NOI | 102 |
| Blintol (16) | S | S | — | — | 0.49 ± 0.05 | 96 |
| Sulphur analogue (41) | S | R | S | — | NOI | 100 |
| Sulphur analogue (42) | S | R | R | S | 0.25 ± 0.02 | 100 |
| Sulphur analogue (43) | S | S | R | S | 0.26 ± 0.02 | 100 |
| Sulphur analogue (44) | S | S | R | — | 0.17 ± 0.02 | 100 |
| Selenium analogue (45) | R | S | R | R | 41.0 ± 7.0 | 101 |
| Sulphur analogue (46) | R | S | R | R | NOI | 101 |
| Sulphur analogue (51) | S | S | R | R | 0.65 ± 0.10 | 102 |
| Selenium analogue (52) | S | S | R | R | 0.14 ± 0.03 | 102 |
| Selenium analogue (53) | S | R | R | S | 0.10 ± 0.03 | 13 |
| Selenium analogue (54) | S | S | R | S | 0.10 ± 0.01 | 13 |
To test this assumption and to afford new candidates for the treatment of type 2 diabetes, chain-extended homologues of salacinol (41–44, Fig. 14) containing polyhydroxylated, acyclic chains of 5- and 6-carbons differing in stereochemistry at the stereogenic centers were synthesized.96,100 The enzyme inhibition studies showed that the compound (41) did not inhibit the MGAM activity, while the other compounds (42–44) inhibited MGAM and showed Ki values of 0.25 ± 0.02, 0.26 ± 0.02, and 0.17 ± 0.02 µM, respectively. By comparison of the stereochemistry of the stereogenic centers in the extended acyclic chain and the observed inhibitory properties of the four compounds, it is clear that the R stereochemistry of the C-4′ stereogenic center is important for activity.
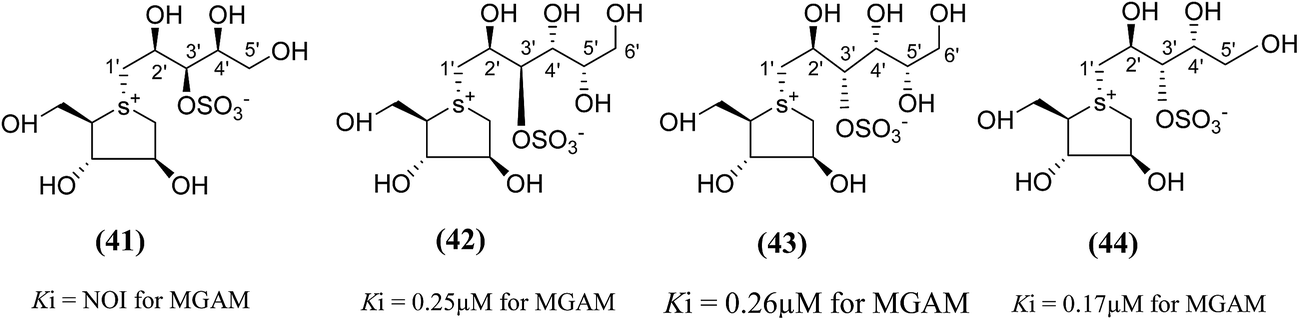 | ||
| Fig. 14 Chain extended salacinol homologues (41–44) containing polyhydroxylated, acyclic chain of 5- and 6-carbons and their inhibition profile against MGAM. | ||
A series of six chain extended analogues (45–50, Fig. 15) with heteroatom variations having polyhydroxylated, extended acyclic chains of 6-carbons, was also reported.101 The α-glucosidase inhibition data against MGAM revealed that the sulfur analogue (46) with the D-arabinitol configuration in the heterocyclic ring did not inhibit MGAM, while the corresponding selenium (45) and nitrogen (47) analogues inhibited MGAM with Ki values of 41.0 ± 7.0 and 26 ± 0.02 µM, respectively. It is pertinent to mention that the sulfur and nitrogen congeners with the L-arabinitol configuration in the heterocyclic ring, (49) and (50), were also active, with Ki values of 25 and 5 µM, respectively.
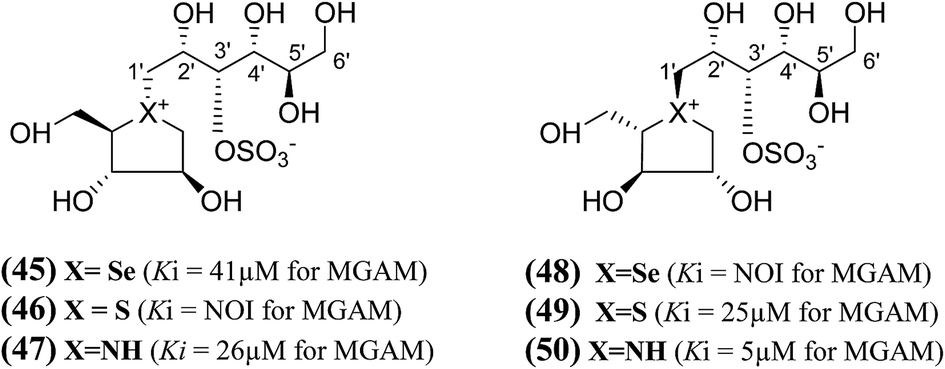 | ||
| Fig. 15 Salacinol analogues (45–50) with an extended chain, heteroatom variations and polyhydroxylated, acyclic chain of 6-carbon and their inhibition profile against MGAM. | ||
Another acyclic, chain-modified, sulfur (51) and selenium (52) analogues (Fig. 16), with an extended polyhydroxylated acyclic chain of 6-carbons, have also been reported.102 Enzyme inhibition observed from these compounds showed that the sulfonium ion (51) with the enantiomeric configuration at C-5′ to (44) has a Ki value of 0.65 ± 0.10 µM. In contrast, the corresponding selenonium ion (52) with the enantiomeric configuration at C-5′ to (54) is just as active, with a Ki value of 0.14 ± 0.03 µM, respectively.
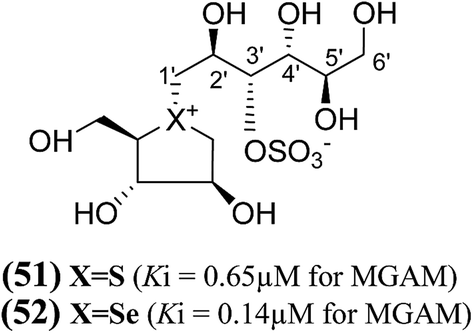 | ||
| Fig. 16 Chain extended sulfur (51) and selenium (52), analogues of salacinol containing polyhydroxylated, acyclic chains of 6-carbons and their inhibition potential against MGAM. | ||
In another study, a different synthetic path to the active chain-extended analogues (42) and (43) have developed.100 In addition, their corresponding selenium analogues (53) and (54) and nitrogen analogues (55) and (56) were also synthesized (Fig. 17) and the enzyme inhibition studies of above analogues showed that the selenium analogues (53) and (54) inhibited MGAM with Ki values of 0.10 ± 0.03 and 0.10 ± 0.01 µM, respectively.13 In this series, compounds (53) and (54) represent the most active glucosidase inhibitors. Both of the nitrogen analogues (55) and (56) were less active compared to the corresponding sulfur and selenium analogues, with Ki values of 35 ± 2 and 8 ± 1 µM, respectively. These data support the earlier conclusion with other derivatives (42–44 and 51, 52)100,102 that is, the configuration at C-3′ does not appear to be critical for inhibitory activity since all pairs with enantiomeric configurations at C-3′, (53) and (54); (55) and (56) shows similar Ki values in the same range.
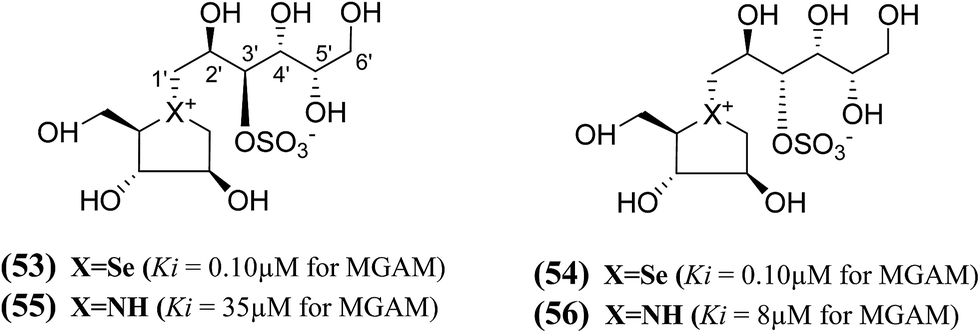 | ||
| Fig. 17 Selenium (53 and 54) and nitrogen (55 and 56) analogues of chain extended salacinol and their inhibitory activity against MGAM. | ||
In summary, the structure–activity relationships predict, comparison of the Ki values of the sulfonium and selenonium chain-extended analogues (41–44, 45, 46, 51, 52, 53, and 54) against MGAM, that the S configuration at C-2′ and the R configuration at C-4′ are essential for activity (highlighted in bold in Table 5). The S configuration at C-3′, bearing the sulfate group, was found to be unimportant. The configurational requirement at C-5′ was not clear because the trend was found to depend on the heteroatom. Thus, it is clear that the stereochemistry at the different stereogenic centers on the side chain plays a significant role in biological activity. It appears that the compounds containing the S configuration at C-2′, the R configuration at C-4′, and the S configuration at C-5′ are the most active in the sulfur series of compounds; however, that in the selenium series the activities of the selenium analogues, (52) and (53), suggest that the stereochemistry at C-5′ could be R. The stereochemistry at C-3′ was judged to be unimportant but can be fixed as S to reflect a presumed common biosynthetic pathway as salacinol. However, the overall conclusion was that the effect of the acyclic chain extension did not confer any dramatic change on inhibitory properties for this particular enzyme, because the Ki values of the active chain-extended analogues were in a similar range to those of salacinol (5) (0.19 µM) and blintol (16) (0.49 µM).
Three new class of deoxy-salacinols analogues lacking one (57 and 58) or two polar (59) (Fig. 18) substituents in the side chain of salacinol (5) were reported by Muraoka et al.103 All these analogues showed less inhibitory activity as compared to salacinol (5), and proved the importance of cooperative role of the polar substituents for the α-glucosidase inhibitory activity. Compounds (58) and (59) having acyclic chains that lack the hydroxyl group at C-2′ were not efficient inhibitors of intestinal α-glucosidases in vitro as compared to the acyclic chain of salacinol (5). However, compound (57), lacking a hydroxymethyl group at C-3′, maintain some inhibitory activity against sucrase (IC50 = 780 µM). These results recommended that both the hydroxymethyl group at C-3′ and the hydroxyl group at C-2′ with an S configuration are critical for α-glucosidase inhibitory activity.
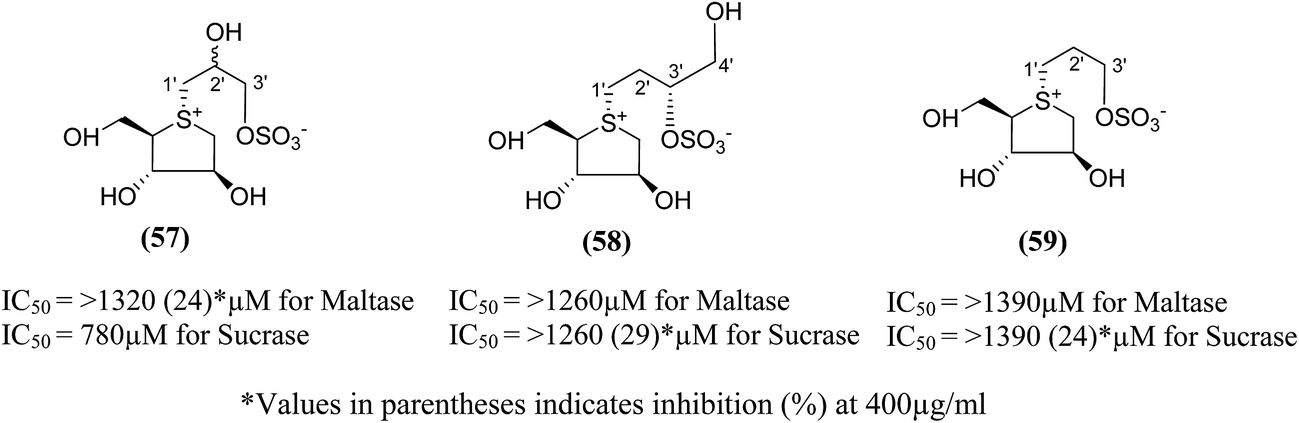 | ||
| Fig. 18 Deoxy-salacinol analogues lacking one (57 and 58) or two polar (59) substituents in the side chain and their inhibitory activity against disaccharidases. | ||
In order to develop more effective glucosidase inhibitors, additional salacinol derivatives were synthesized and tested for inhibitory activity including chain-extended and chain-modified embodiments, where the sulfate moiety is frame-shifted. Nasi et al.104 described a frame shifted chain-extended and chain-modified (60–63, Fig. 19) analogues of salacinol (5) and blintol (16) where the sulfate moiety was shifted from C-3′ to the C-4′ location. The inhibitory activities of compounds (60–63) against recombinant human MGAM was determined. Compounds (60) and (61), with the same configuration at the stereogenic centers in the acyclic chain, have Ki values of 20 ± 4 and 53 ± 5 µM, respectively. These compounds are less active than salacinol (5) and blintol, (16) with Ki values of 0.19 ± 0.02 and 0.49 ± 0.05 µM, respectively.96 Interestingly, the analogue (41), in which the sulfate moiety is located at the C-3′ and not the C-4′ position, is inactive.100 A second 5-carbon chain-extended compound with the same configuration at C-3′ and C-4′ as (44), but with the opposite configuration at C-2′ and in which the sulfate moiety is located at the C-3′ and not the C-4′ position, has a Ki value of 0.17 ± 0.03 µM. In contrast, compounds (62) and (63) were not active against MGAM.104
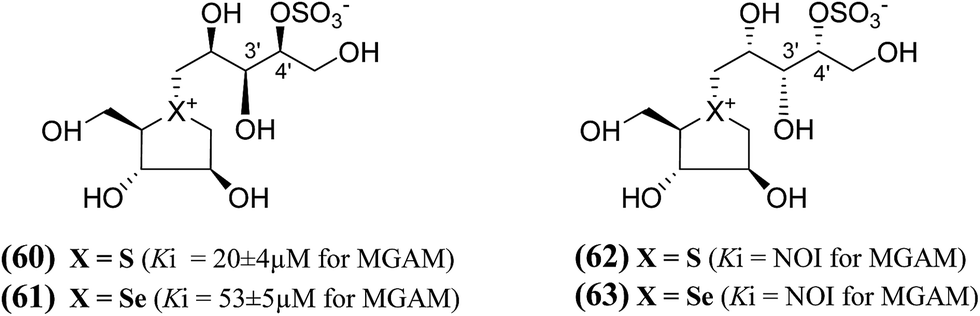 | ||
| Fig. 19 Chain-modified frame-shifted analogues (60–63) of salacinol showing the position of the sulfate moiety which is shifted from C-3′ to C-4′ and their inhibitory activity against MGAM. | ||
Tanabe et al.105 have reported the synthesis of de-O-sulfonated analogues (64, X = CH3OSO3 and 65, X = Cl, Fig. 20) of salacinol with monomethyl sulfate and chloride as external counter anions and their inhibitory activity were examined to discover the role of the sulfate anion moiety in the acyclic chain of salacinol (5). These analogues (64 and 65) had almost equal inhibitory activities to salacinol (5) against intestinal α-glucosidase in vitro. These studies indicated that the internal sulfate counterion or O-sulfonate anion moiety of salacinol is not essential for inhibitory activity as the de-O-sulfonated analogues (64 and 65) retained equal inhibitory activities against intestinal α-glucosidases when compared to salacinol (5).105
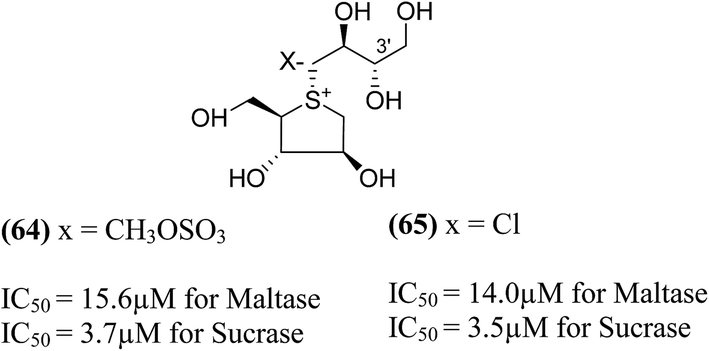 | ||
| Fig. 20 De-O-sulfonated analogues (64 and 65) of salacinol and their inhibitory activity against disaccharidases. | ||
An important new class of compounds comprising salacinol analogues containing sulfonium or ammonium ion-inner carboxylate structure were also discovered. The acyclic side chain of salacinol (5) and kotalanol (6) includes a sulfate group which is believed to be important to the inhibitory activity of these compounds. Exploring the glucosidase inhibitory activity, Chen et al.106 prepared carboxylate analogues in which the sulfate moiety has been substituted with a carboxylate moiety (Fig. 21). Target compounds (66) and (67) comprise a thioarabinitol having a polyhydroxylated side chain containing a carboxylate residue. The inhibitory activity of target compounds (66) and (67) was assessed in respect of recombinant human MGAM. Only compound (66), with the D-arabinitol configuration in the heterocyclic ring displayed by salacinol (5), and was found to be active, with a Ki value of 10 µM whereas compound (67) did not show significant inhibition against MGAM.
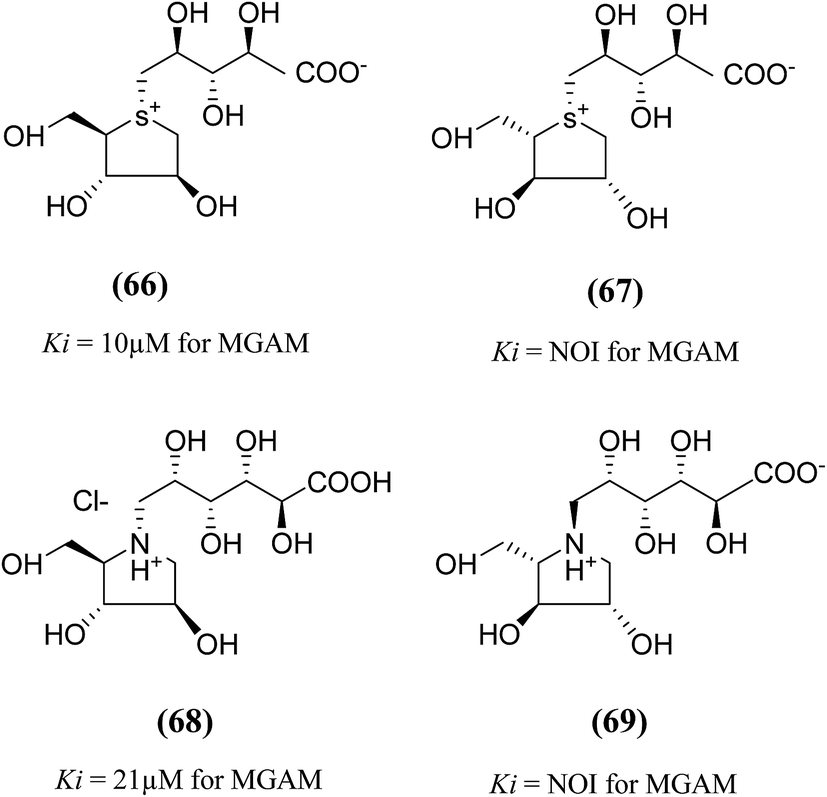 | ||
| Fig. 21 Carboxylate analogues (66 and 67) of salacinol and ghavamiol (68 and 69) and their inhibitory activity against MGAM. | ||
To understand the role of the sulfate group present in the acyclic chain of ghavamiol (nitrogen analogues of salacinol 14), an unusual class of amino acids (68) and (69) were synthesized (Fig. 21). These target analogues were considered to be the carboxylate analogues of ghavamiol (14), having a polyhydroxylated side chain containing a carboxylate residue.107 Compound (68), inhibits recombinant human MGAM with a Ki value of 21 µM whereas salacinol (5) itself has a Ki value of 0.2 µM. In addition, this compound was also active against Drosophila melanogaster Golgi α-mannosidase II (dGMII) with an IC50 of 0.3 mM. This is a significant improvement (25-fold) over the inhibition measured for salacinol (5) and kotalanol (6) as well as other salacinol analogues such as blintol (16) which all inhibited dGMII with an IC50 of approximately 7.5 mM.104 Compound (69) was not active on either enzyme MGAM or dGMII; similar results were obtained for other salacinol analogues derived from anhydro-L-heteroarabinitol moieties and presumably reflect minimal contacts of the enantiomeric five-membered rings in the enzyme active sites.108 The crystal structure of (68) bound in the active site of dGMII indicated that the hydroxyl groups from the acyclic chain and also the carboxylic acid group form extensive contacts with both side chains and water molecules in the active site. Previously, the role of the sulfate group on glucosidase inhibitory activity was inferred by Yuasa et al.109 It was reported that the docking of salacinol (5) into the binding site of glucoamylase indicated close contacts between the sulfate ion and Arg305.
The substitution of phosphate functional groups for sulfate groups in biologically important molecules also continues to attract much interest in bioorganic and medicinal chemistry. Therefore, in a same manner, phosphate derivatives (phosphorylated heteroalditols, 70 and 71, Fig. 22) were prepared, in which the sulfate moiety on the acyclic side chain has been substituted with a phosphate moiety and after screening, it was found to not effective against MGAM.110
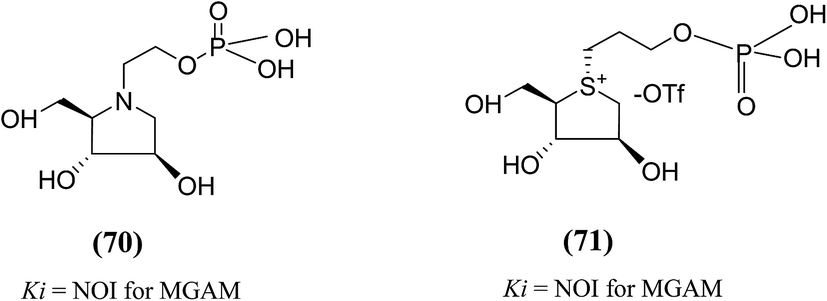 | ||
| Fig. 22 Phosphate analogues (70 and 71) of salacinol and their inhibitory activity against MGAM. | ||
New salacinol analogues (72–79, Fig. 23) based on novel anhydroseleno- and anhydrothio-allitols, derived from D-gulono-γ-lactone and L-ascorbic acid, have also been synthesized. Enzyme inhibition assays of these analogues indicated that they were not active against MGAM.111
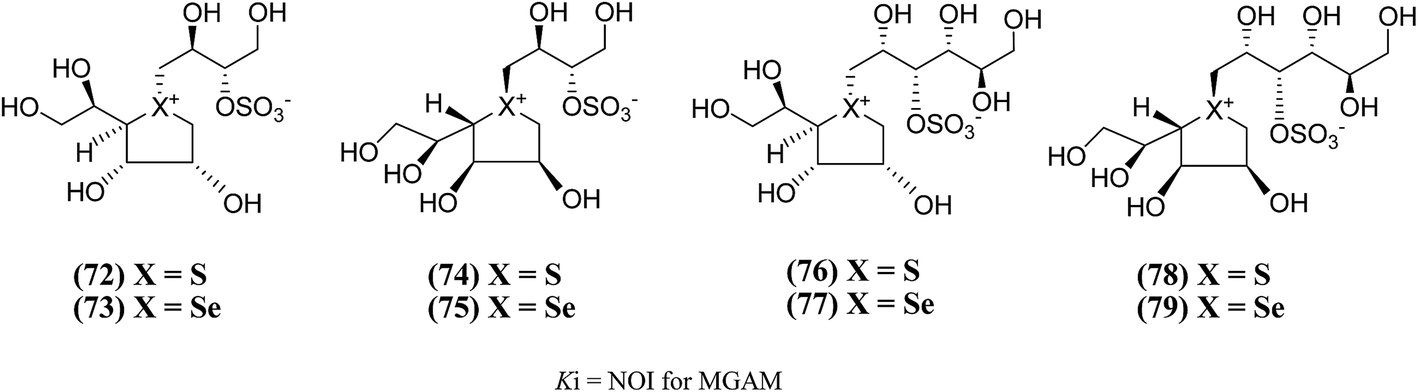 | ||
| Fig. 23 Analogues of salacinol related to D-gulono-γ-lactone and L-ascorbic acid and their inhibitory activity against MGAM. | ||
In another synthetic approach, salacinol related compounds were synthesized bearing a permanent positive charge of the sulphur atom and including a lipophilic alkyl chain which may be necessary for substrate recognition. These target compounds (80–88, Fig. 24) include the 1,4-anhydro-4-thio-D-arabinitol moiety.112 The inhibitory properties of target compounds were examined against recombinant human MGAM. All of the lipophilic compounds were inhibitors of MGAM with Ki values ranging from 6 to 75 µM. Increases in the alkyl chain lengths have significant, albeit not pronounced, effects on the enzyme inhibitory properties of these compounds. There is a 6.7-fold increase in inhibitory activity upon extension of the alkyl chain from four carbons to eighteen carbons. However, these compounds are less active than salacinol (5) which was previously shown to have a Ki value of 0.19 ± 0.02 µM against MGAM.
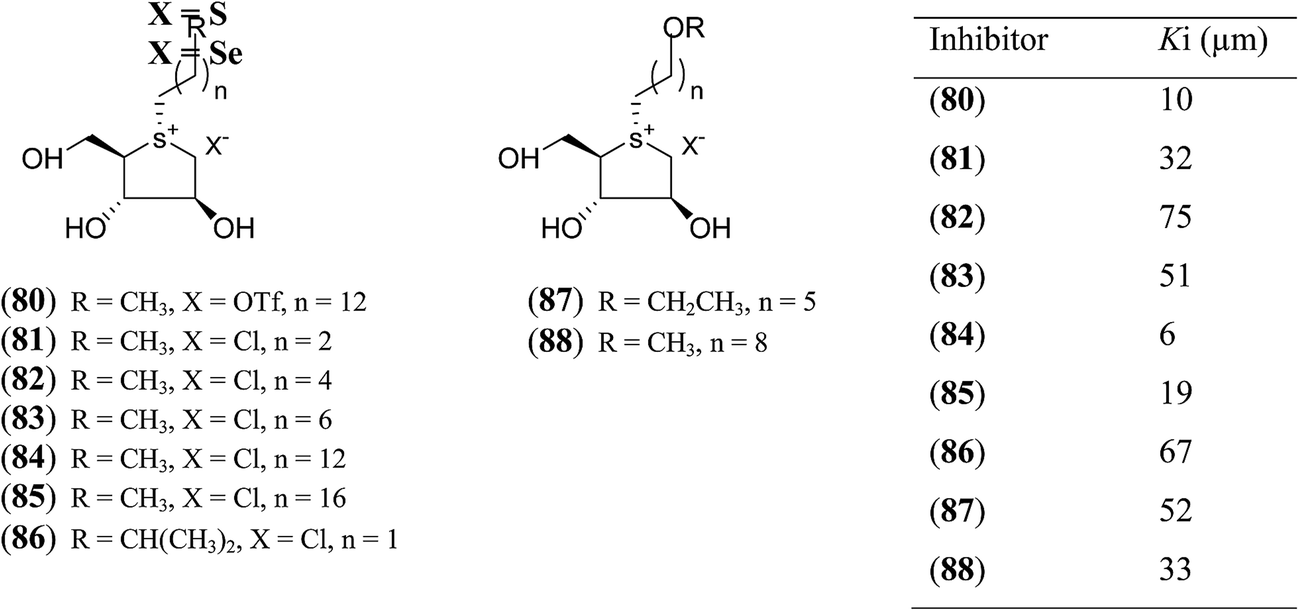 | ||
| Fig. 24 S-Alkylated sulfonium ions with different alkyl chains (80–88) and their inhibitory activity against MGAM. | ||
Tanabe et al.113 synthesized a series of four 3′-O-alkylated salacinol analogues (89–92, Fig. 25) having different hydrophobic groups (OCH3, OCH2H5, OC13H27, or OCH2Ph) at C-3′ of the salacinol side chain. Inhibition assays against rat intestinal glucosidases revealed that all of these synthesized compounds showed equal or higher inhibitory activities as compared to salacinol (5). Compound (92) having benzyloxy group at C-3′ was the most potent inhibitor of maltase (IC50 = 0.44 µM) and isomaltase (IC50 = 0.14 µM), and compound (90) with an ethoxy group at C-3′ showed highest inhibitory activity against sucrase (IC50 = 0.12 µM).
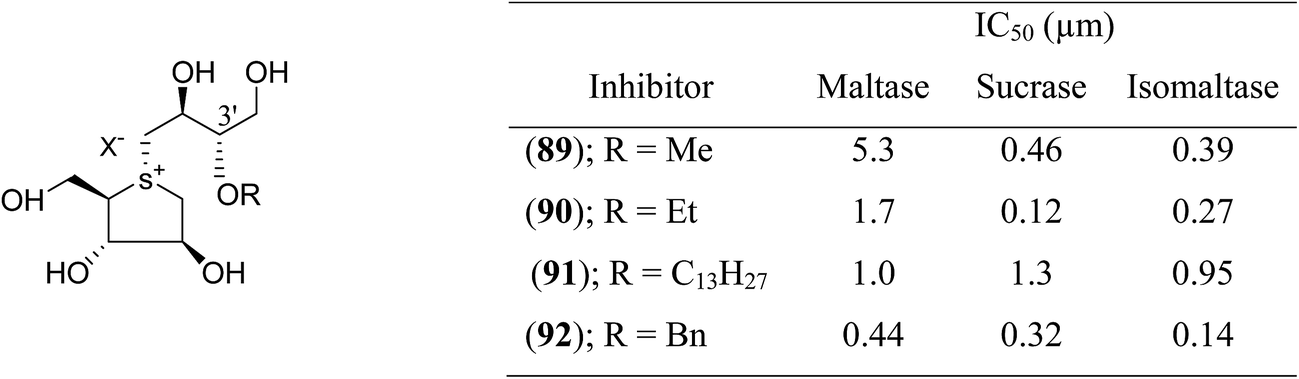 | ||
| Fig. 25 3′-O-Alkylated analogues (89–92) of salacinol showing different hydrophobic groups and their IC50 (µm) values against disaccharidases. | ||
With the aid of the in silico method, Tanabe et al.114 designed 3′-O-benzyl derivative of salacinol (92) and developed twelve analogues (93–103, Fig. 26) having four different substituents (methyl, chloro, trifluoromethyl, and nitro) at different positions (meta or para or ortho) of the phenyl ring. By comparison of enzyme inhibition activity, all the sulfonium salts (93–103) showed better inhibitory activities than the salacinol (5). In general, ortho substituted compounds were more active than the corresponding para and meta substituted compounds. Compound (102), with an ortho-nitrophenyl group, was found to be the most potent inhibitor of all three rat intestinal glucosidases, maltase (IC50 = 0.13 µM), sucrase (IC50 = 0.042 µM), and isomaltase (IC50 = 0.21 µM).
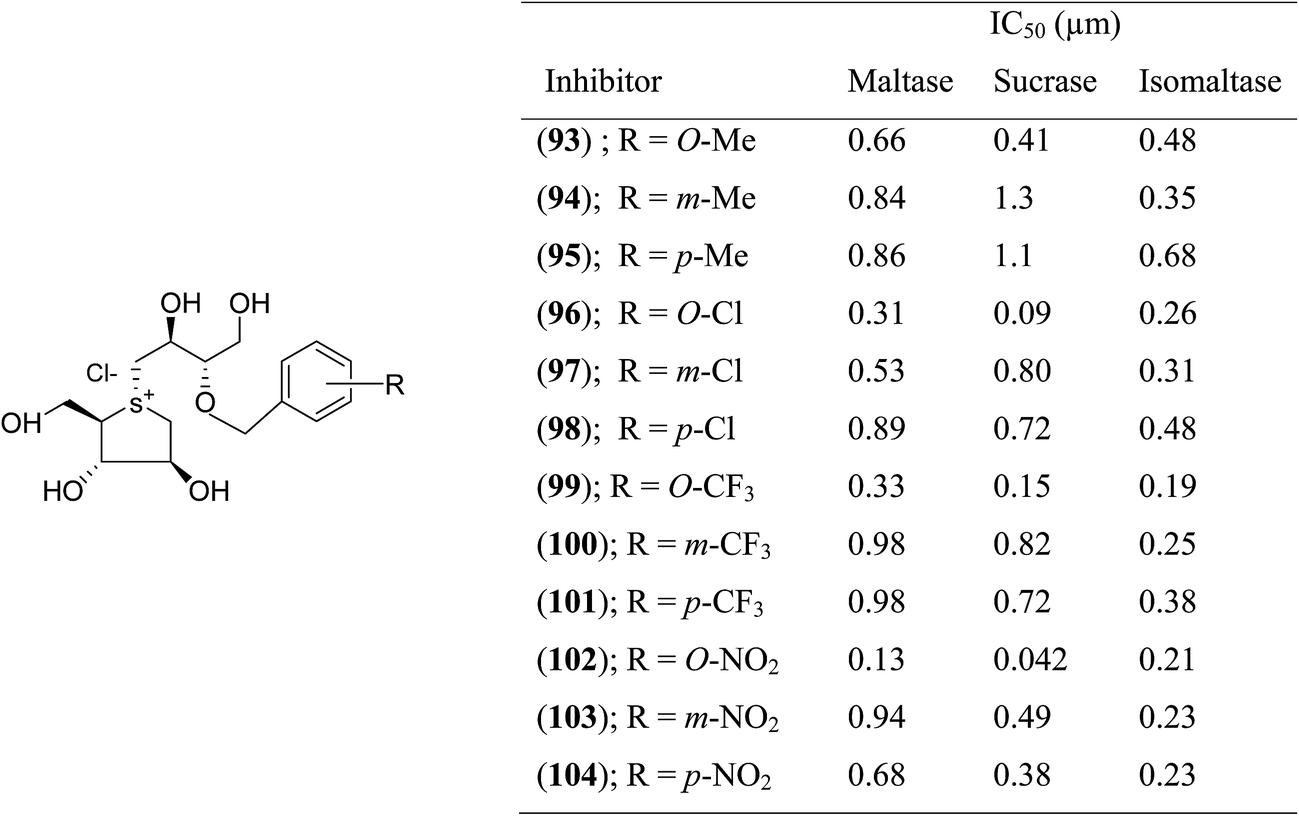 | ||
| Fig. 26 Synthetic analogues (93–104) of 3′-O-benzyl derivatives of salacinol and their IC50 (µm) values against disaccharidases. | ||
Thus, structure–activity relationship studies towards salacinol (5) and related analogues enabled us to conclude the best inhibitors belonging to the salacinol (5) class have Ki values in the low micromolecular range (i.e., 0.10–0.19 µM). The salacinol inhibitors have been revealed to be a relatively poor inhibitor of HPA (Ki = 75 µM) and hence, they are selective against the intestinal glucosidases.22 Structural drawing of synthetic analogues of salacinol showing important structural features needed for the inhibitory activity is given in Fig. 27.
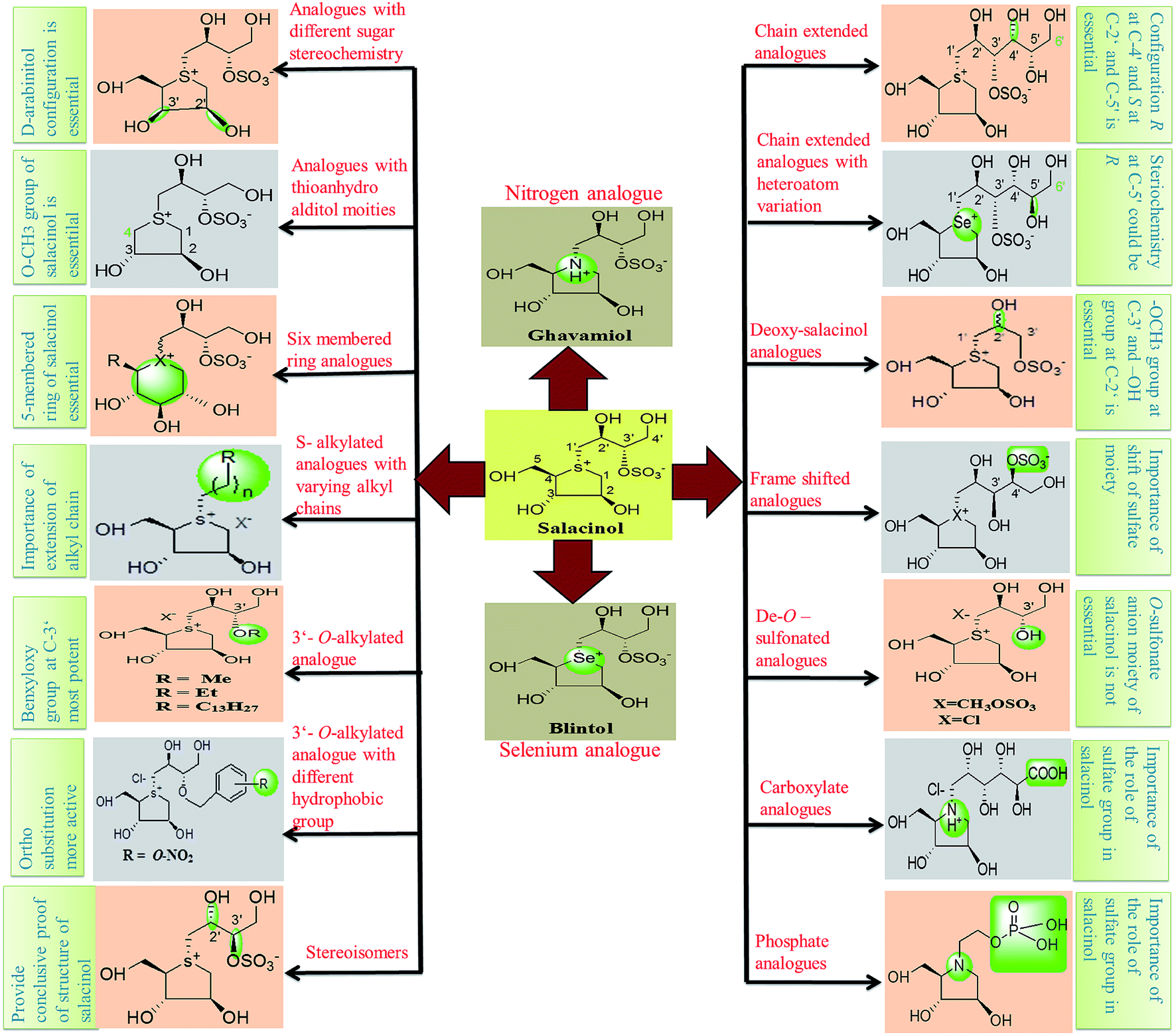 | ||
| Fig. 27 Structural drawing of salacinol analogues showing important structural features required for the inhibitory activity. | ||
4.1.1.1. Crystallographic analysis and molecular docking studies of salacinol and its analogues. With the aim of gaining information on structure–activity relationships, various studies on salacinol derivatives are vigorously reported.38,115 The structure–activity studies have revealed a remarkable variation in the inhibitory power of these compounds against glucosidase enzymes of different origin.13 The molecular basis for this selectivity is being investigated through structural studies of the enzyme-bound inhibitors, using molecular modeling in conjuction with conformational analysis by STD-NMR techniques116 and X-ray crystallography.108 Crystallographic analysis of the interactions of Drosophila melanogaster Golgi α-mannosidase II (dGMII) with salacinol (5) and its analogues shows that the sulfate group does interact with residues in the enzyme active site.108 The X-ray crystal structure analysis of ntMGAM or ntSI complex of these compounds has also been performed.78,117 In addition, docking of salacinol (5) into the binding site of glucoamylase showed close contacts between the sulfate ion with Arg305.109 As salacinol (5) has superior inhibitory activity and specificity against α-glucosidases than the methyl sulfonium ion revealed that the sulfate moiety of the side chain is functionally important.
A study on docking stimulation between salacinol derivatives 5a–5c, 9a, 9b (Fig. 28), and ntMGAM have conducted by Nakamura et al.118 The predicted binding mode of salacinol (5) and its derivatives to the α-glucosidase from the perspective of protein–ligand interaction elaborates the structure–activity relationships of salacinol derivatives.
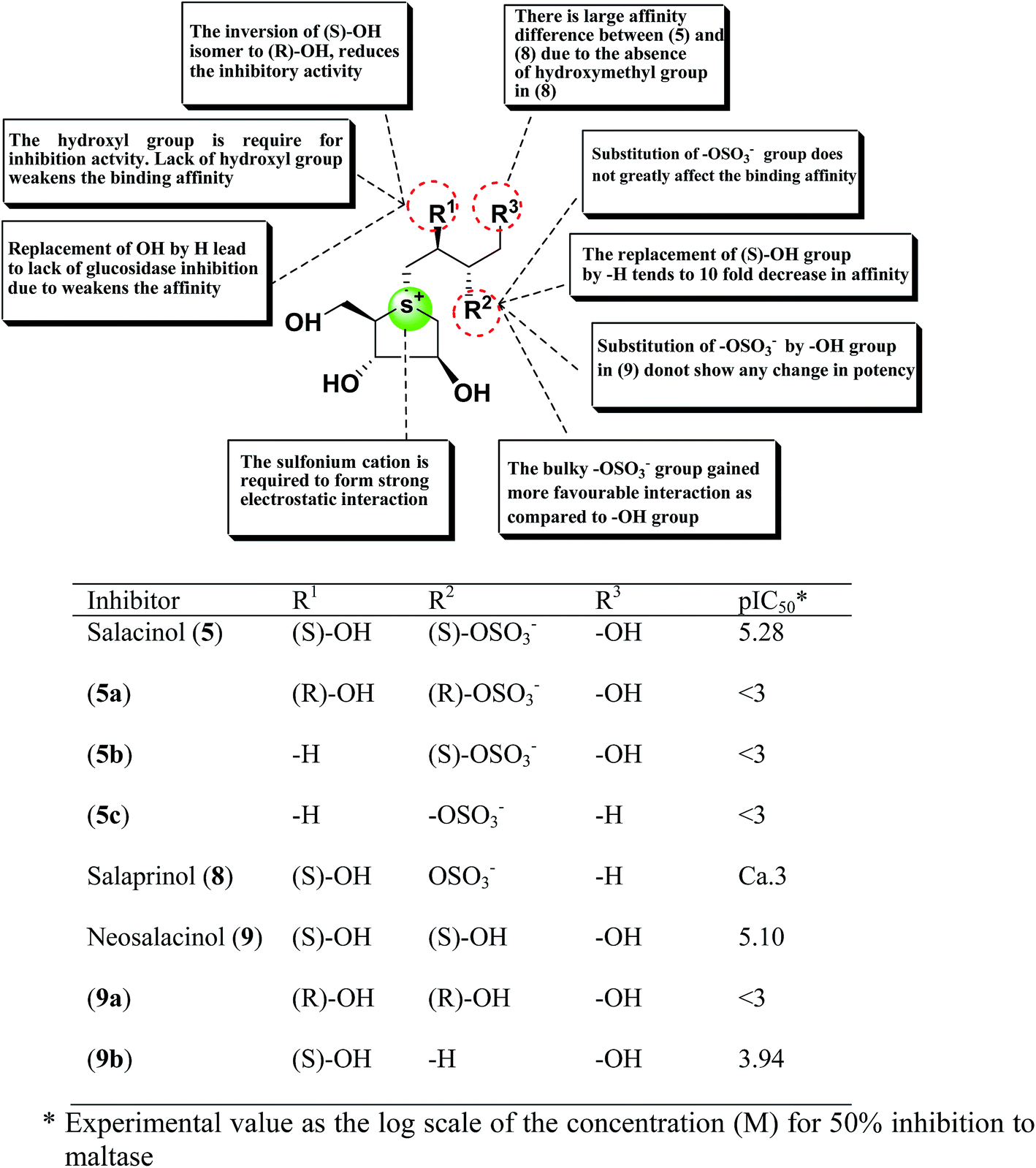 | ||
| Fig. 28 Schematic representation of chemical structures, structure–activity relationship and potency of salacinol derivatives. | ||
In the predicted binding mode of salacinol (5) and its derivative by using docking protocol, the C2 hydroxy group of salacinol (5) interacts with Asp542 directly by a hydrogen bond. Two hydroxy groups at C2′ and C4′ on the methylene chain interacted with Asp203 and Arg526 by hydrogen bonding. These interactions are considered to reinforce the binding affinity. On the other hand, the sulfate group did not interact significantly with protein. The sulfonium cation interacts with Asp443 by the strong electrostatic interaction similar to intramolecular salt bridge between the sulfonium cation and the sulfate anion in the crystal state (Fig. 29).
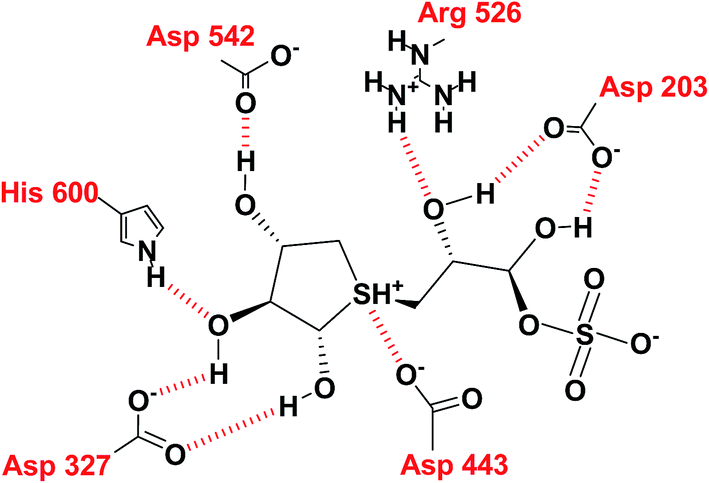 | ||
| Fig. 29 Interaction diagram of predicted binding mode of salacinol with amino acid residues. | ||
Nakamura et al.119 also performed the homology modeling for ntMGAM, ntSI, ctMGAM and ctSI to predict the binding mode of salacinol (5) and its derivative for each subunit. So far, several compounds have been discovered that enhanced the inhibitory activities.113
Structure–activity-relationship studies of kotalanol analogues exposed that the inhibitory activities against ntMGAM improve significantly upon de-O-sulfonation.122 De-O-sulfonated kotalanol (10) (Ki = 0.03 ± 0.01 µm) was found to be a ∼sevenfold better inhibitor of ntMGAM than kotalanol (6) (Ki = 0.19 ± 0.03 µm).78 The X-ray crystallographic studies of ntMGAM in complex with kotalanol (6), de-O-sulfonated kotalanol (10) and several salacinol derivatives revealed interesting features that de-O-sulfonated kotalanol (10) from the salacinol class of compounds is a ∼30-fold stronger inhibitor compared to miglitol (2) and an ∼2000-fold better inhibitor than the acarbose (1). The crystal structures revealed that in the case of kotalanol (6), the sulfate group at C-3′ occupies a hydrophobic cavity causing a positional constraint, while in the case of de-O-sulfonated kotalanol (10), the positional constraint is removed and allows the rest of the polyhydroxylated side chain to make optimal contacts with the ntMGAM active site.78
To discuss the role of the side chain stereochemistry to the inhibitory activity against MGAM, various synthetic studies directed towards the stereochemical determination of kotalanol (6), have been carried out.123 The stereoisomer of kotalanol (105, Fig. 30), with the opposite stereochemistry at the C-6′ stereogenic centre, inhibited the ntMGAM with a Ki value of 0.20 ± 0.02 µM,115 this compares to Ki value for kotalanol (6) of 0.19 ± 0.03 µM.78 The results indicated that the configuration at C-6′ is inconsequential for inhibitory activity against this enzyme as the C-6′ epimer (105) of kotalanol showed equal inhibitory activity against ntMGAM to that of kotalanol (6).
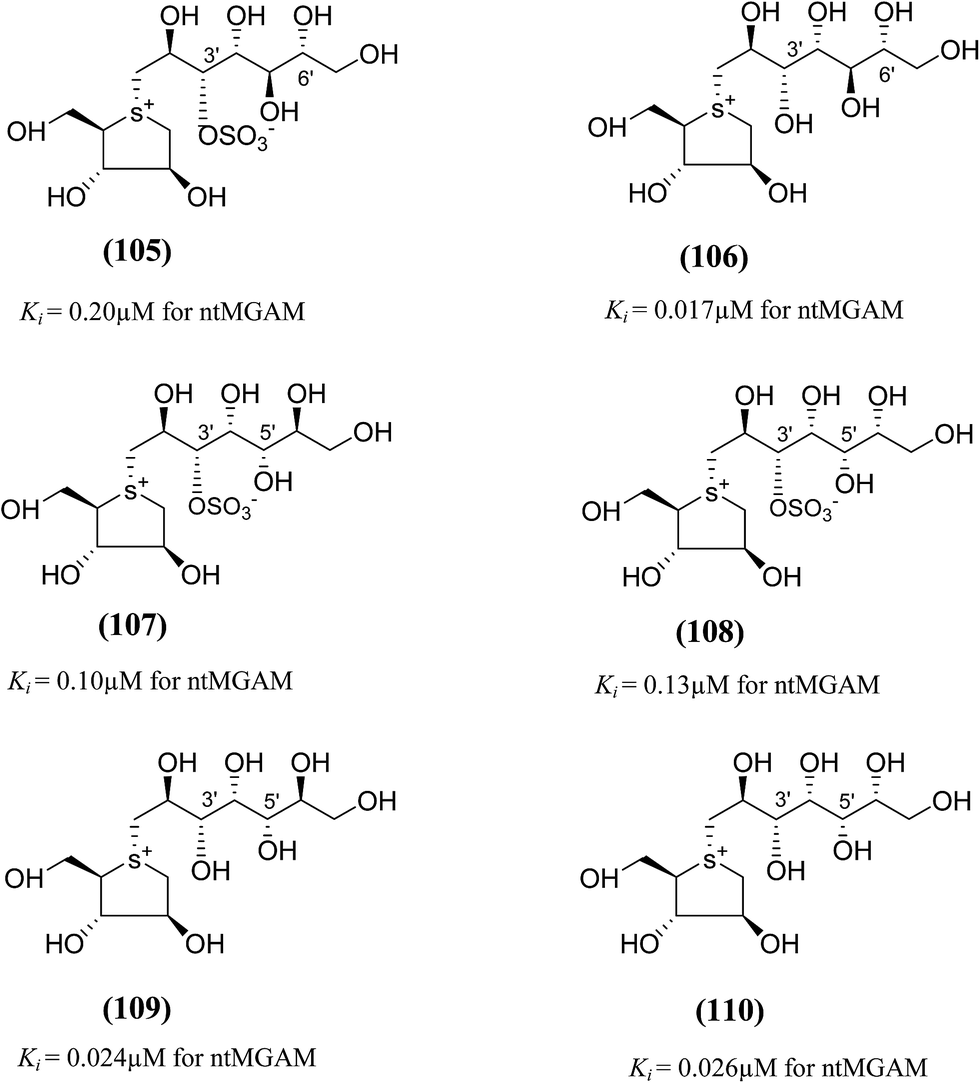 | ||
| Fig. 30 Diastereomers of kotalanol (105, 107, 108) and de-O-sulfonated kotalanol (106, 109, 110) and their inhibitory activity against ntMGAM. | ||
The compound (106), diastereomers of de-O-sulfonated kotalanol (10), is the most potent inhibitor of ntMGAM (Ki = 17 ± 1 nM) in vitro.36 The inhibitory values of (105) are comparable to the inhibitory activities of other diastereomers of kotalanol (107) and (108) with opposite configuration at C-5′ (Ki = 0.10 ± 0.02 µM and 0.13 ± 0.02 µM, respectively, Fig. 30).123 The de-O-sulfonated derivatives (109, 110) derived from biologically active C-5′ diastereomers of kotalanol, (107) and (108), were found to be more active against ntMGAM than the parent compounds, supporting the general proposition that de-O-sulfonation leads to an increase in the inhibitory activity against ntMGAM compared to the parent sulfated compounds.22
To study the effect of heteroatom substitution on the inhibitory activities of kotalanol (6) and de-O-sulfonated kotalanol (10), Mohan et al.122 designed their nitrogen (111–112) and selenium analogues (114–115, Fig. 31). Screening of these analogues against ntMGAM indicated that, except for the nitrogen analogue of kotalanol (111), all of the compounds synthesized showed greater inhibitory activities than acarbose (1). As discussed above, de-O-sulfonation leads to an increase in inhibitory activity compared to the parent sulfated compounds. Interestingly, in the case of the nitrogen analogue of kotalanol, de-O-sulfonation resulted in a very large increase in inhibitory activity (compare Ki values of compound (111) and (114)). Additionally, selenium substitution doesn't confer any significant advantage on the inhibitory activities of both kotalanol (X = Se, Ki = 80 ± 6 nM; X = S, Ki = 190 ± 30 nM) and de-O-sulfonated kotalanol (X = Se, Ki = 20 ± 3 nM; X = S, Ki = 30 ± 10 nM). In contrast, nitrogen substitution (111) drastically decreased the inhibitory activity in the case of kotalanol (Ki = 90 µM), whereas it does not have any significant change in the case of de-O-sulfonated kotalanol (114) (Ki = 61 ± 5 nM). This trend was also seen in the case of the nitrogen analogue of salacinol (14) (ghavamiol IC50 = high mM range) resulted in a remarkable decrease in inhibitory activity against MGAM (compare the Ki value of salacinol (5), 0.19 µM), whereas substitution of the ring sulfur by selenium (16) (blintol Ki = 0.49 µM) didn't affect its inhibitory activity significantly.89 Based on crystallographic work with salacinol and kotalanol derivatives,78 that the positioning of the sulfate anion of nitrogen analogue of kotalanol (111) in a hydrophobic pocket in the active site is more sterically compromised than in the sulfur congener (6). Relief of this steric interaction by de-O-sulfonation to give (114) apparently relieves this interaction and gives a compound that is just active as its sulfur congener (10). Compound (113) bearing S configuration, differs from (112) only with respect to the configuration at the stereogenic selenium center, was converted into the corresponding de-O-sulfonated compound (116). Inhibitory activities of compounds (112) and (115) are considerably less than those of their corresponding diastereomers (113 and 116) with R configuration. As R configuration at the stereogenic heteroatom center, as exhibited by all of the natural compounds isolated so far, is essential for inhibitory activity. The undesired diastereomers (113 and 116, Fig. 31) bearing the S configuration at the stereogenic selenium center, showed significant decrease in inhibitory activities than those of their corresponding diastereomers with R configuration.122
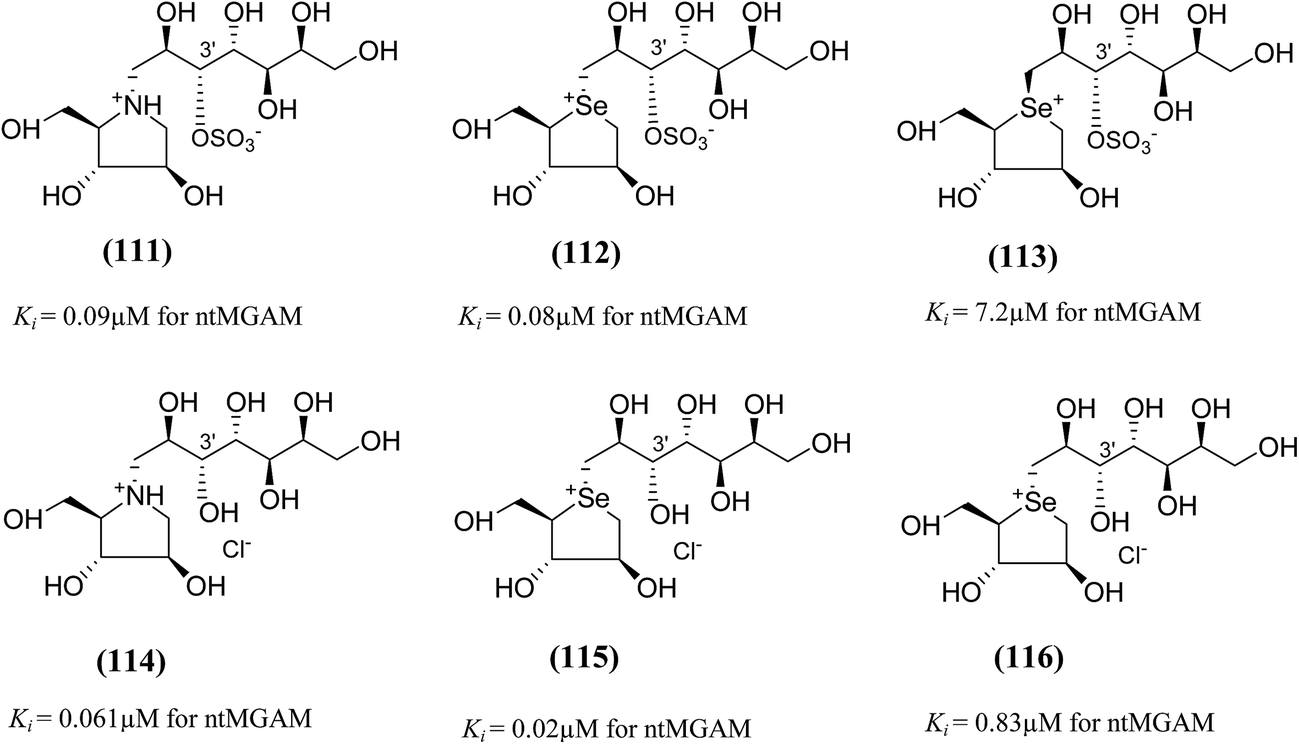 | ||
| Fig. 31 Nitrogen, selenium and diastereomers of selenium analogues of kotalanol (111, 112 and 113) and de-O-sulfonated kotalanol (114, 115 and 116) and their inhibitory activity against ntMGAM. | ||
In order to investigate the active-site requirements of ntMGAM, two six-membered ring nitrogen analogues (117 and 118, Fig. 32) were synthesized.124 The target compounds are hybrids of kotalanol (6) and miglitol (2) and consist of the six-membered ring of miglitol (2) and the 7-carbon side chains of kotalanol (6) and de-O-sulfonated kotalanol (10). The basis of these hybrid molecules was twofold: (1) to improve the binding interactions of miglitol (2) in the +1 binding site of ntMGAM by replacing the N-hydroxyethyl side chain with the polyhydroxylated side chain of kotalanol (6) or de-O-sulfonated kotalanol (10), and (2) to probe the purported transition state mimicry by the kotalanol class of molecules. Inhibition studies of these hybrid compounds against ntMGAM indicated that they have comparable activities (Ki = 2.3 ± 0.6 and 1.4 ± 0.5 µM, respectively) to that of miglitol (2) (Ki = 1.0 ± 0.1 µM). However, they are less active compared to kotalanol (6) (Ki = 0.19 ± 0.03 µM).78,124
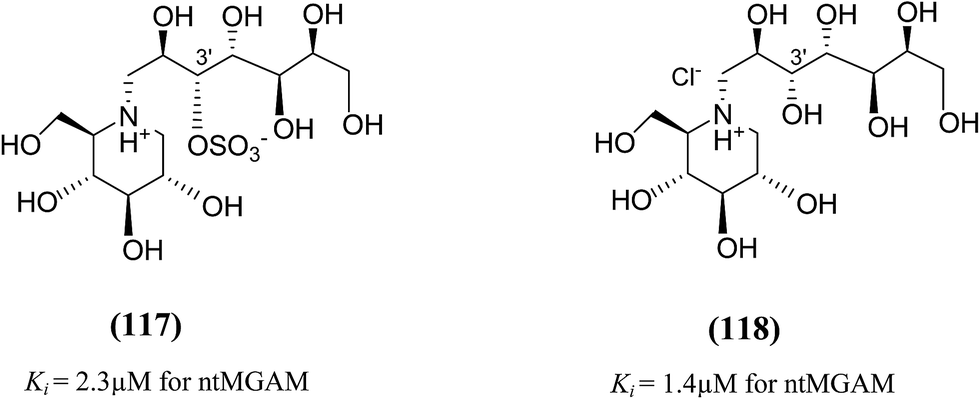 | ||
| Fig. 32 Six-membered ring nitrogen analogues of kotalanol (117) and de-O-sulfonated kotalanol (118) and their inhibitory activity against ntMGAM. | ||
These outcomes undoubtedly signify the importance of the five-membered ring thiocyclitol moiety with a permanent positive charge and thus provide further support for transition state mimicry by these sulfonium-ion glucosidase inhibitors. The inhibitory activities of all synthesized kotalanol analogues (105–118) against the maltase activity of recombinant, ntMGAM are summarized in Table 6.
| Inhibitor | Chemical structure | ntMGAM (µM) | References |
|---|---|---|---|
| Kotalanol (6) |  |
0.19 ± 0.03 | 78 |
| De-O-sulfonated kotalanol (10) | 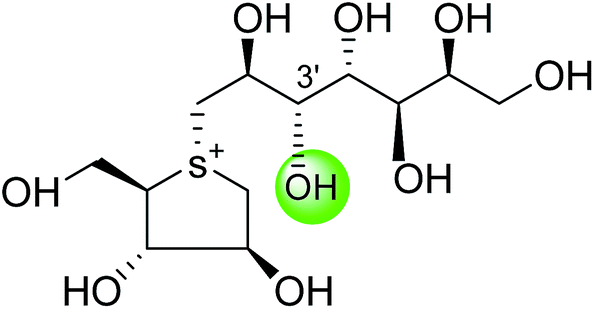 |
0.03 ± 0.01 | 78 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||
| Diastereomers of kotalanol and de-O-sulfonated kotalanol | |||
| C-6′ epimer of kotalanol (105) | 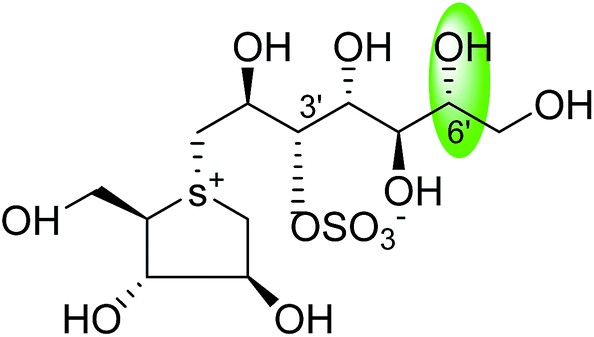 |
0.20 ± 0.02 | 115 |
| C-6′ epimer of de-O-sulfonated kotalanol (106) | 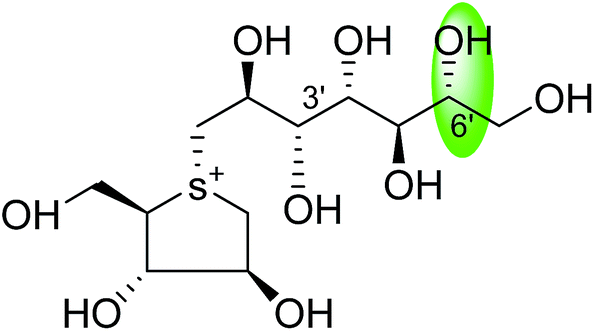 |
0.017 ± 0.01 | 36 |
| C-5′ diastereomers of kotalanol (107) | 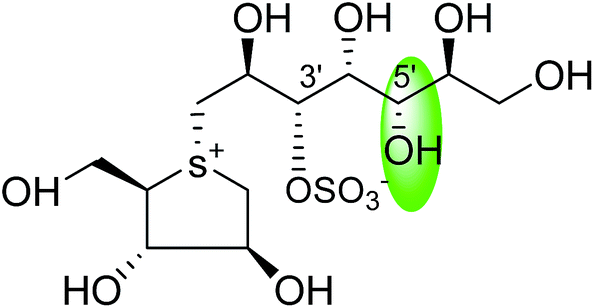 |
0.10 ± 0.02 | 123 |
| C-5′ diastereomers of kotalanol (108) | 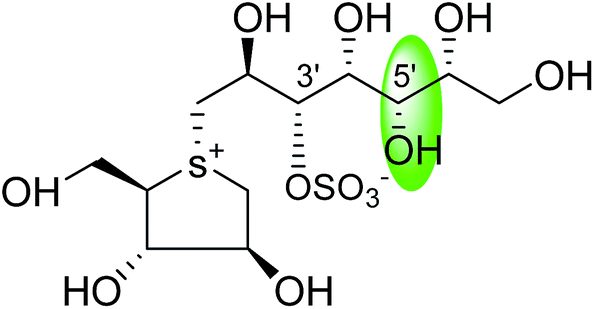 |
0.13 ± 0.02 | 123 |
| C-5′ diastereomers of de-O-sulfonated kotalanol (109) | 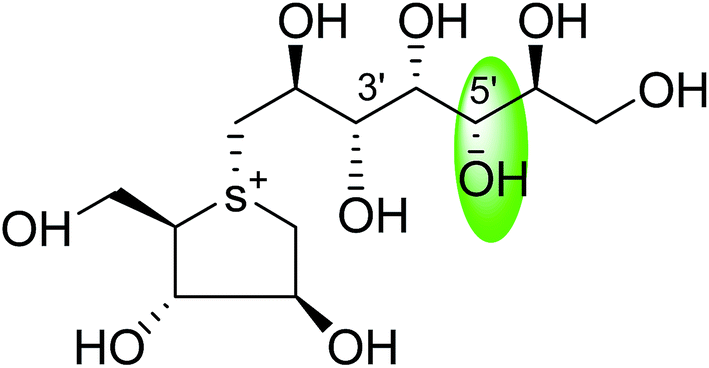 |
0.024 ± 0.02 | 123 |
| C-5′ diastereomers of de-O-sulfonated kotalanol (110) | 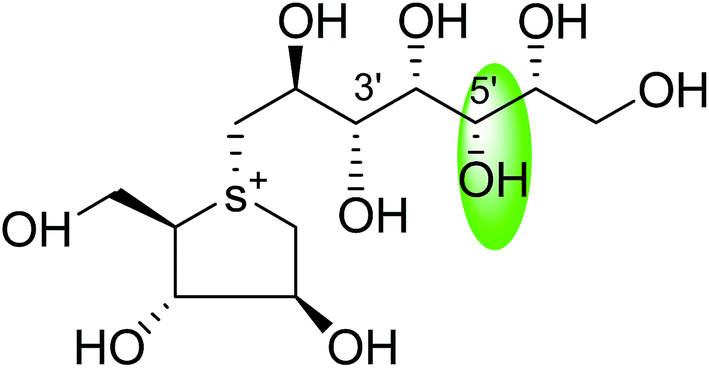 |
0.026 ± 0.02 | 123 |
|
|||
| Heteroatom analogues of kotalanol | |||
| Nitrogen analogue of kotalanol (111) | 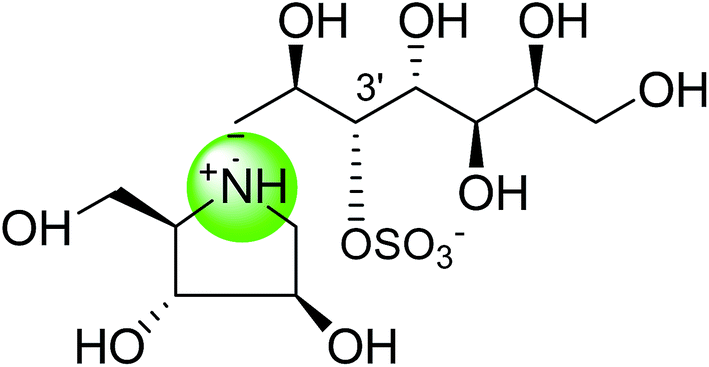 |
0.09 ± 0.06 | 122 |
| Selenium analogue of kotalanol (112) | 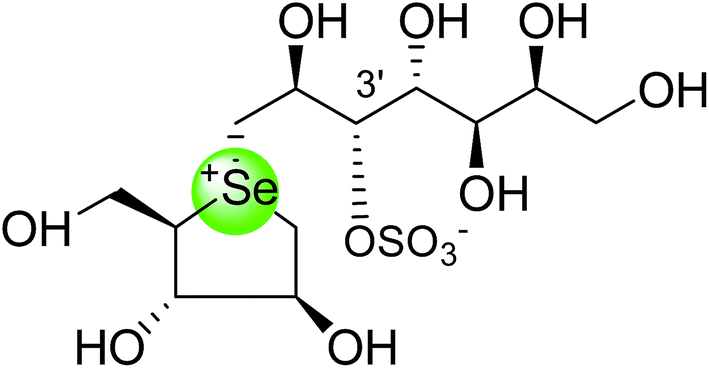 |
0.08 ± 0.06 | 122 |
| Diastereomer of selenium analogue (113) | 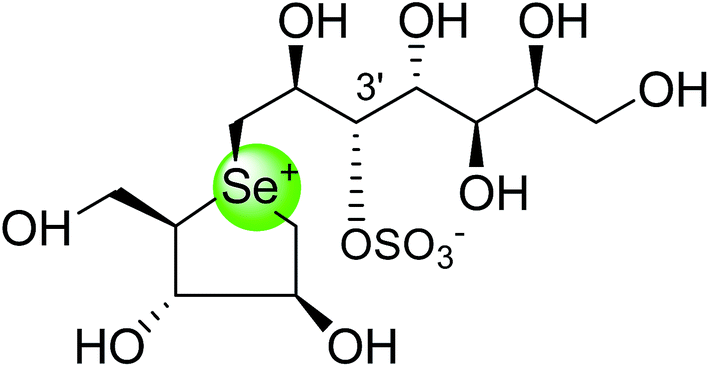 |
7.2 ± 0.7 nm | 122 |
|
|||
| Heteroatom analogues of de-O-sulfonated-kotalanol | |||
| Nitrogen analogue of de-O-sulfonated-kotalanol (114) | 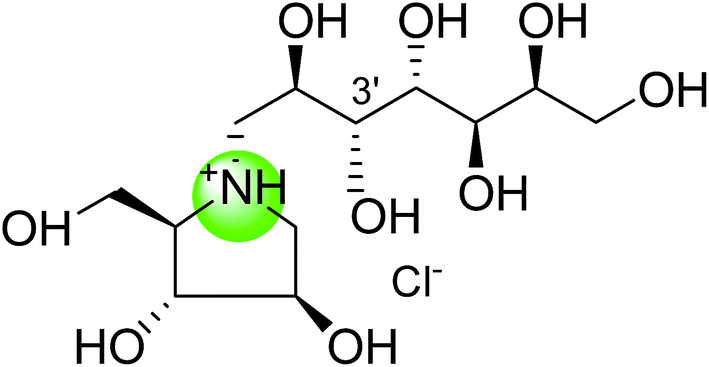 |
0.061 ± 0.05 | 122 |
| Selenium analogue of de-O-sulfonated-kotalanol (115) | 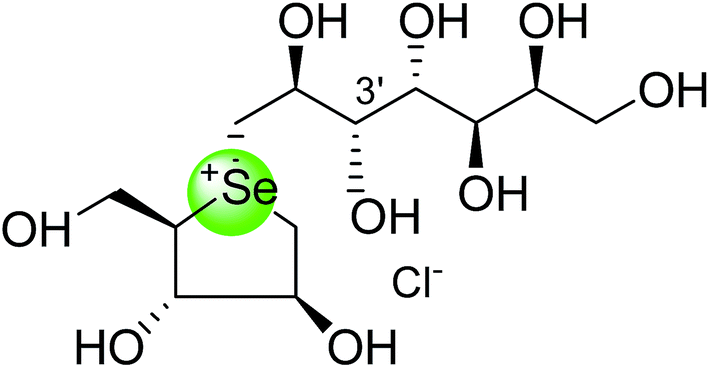 |
0.02 ± 0.03 | 122 |
| Diastereomer of selenium analogue of de-O-sulfonated kotalanol (116) | 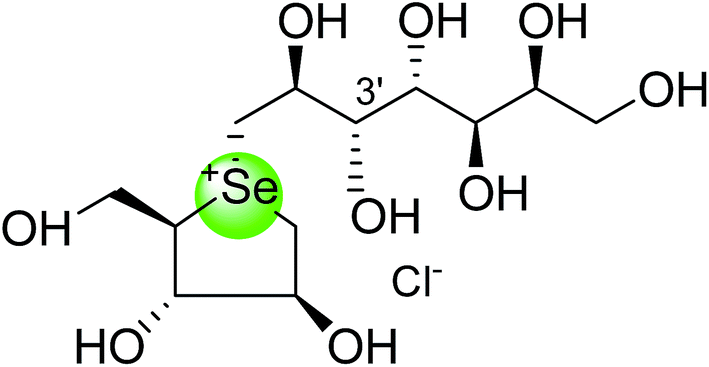 |
0.83 ± 0.07 nm | 122 |
|
|||
| Six membered ring analogues of kotalanol and de-O-sulfonated-kotalanol | |||
| Six membered ring nitrogen analogue of kotalanol (117) | 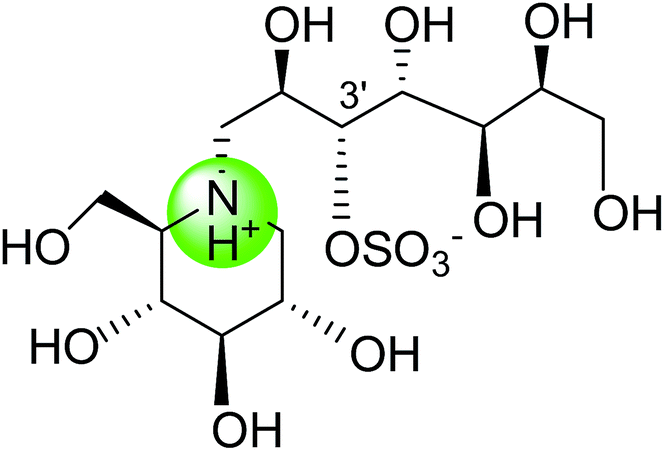 |
2.3 ± 0.6 | 124 |
| Six membered ring nitrogen analogue of de-O-sulfonated-kotalanol (118) | 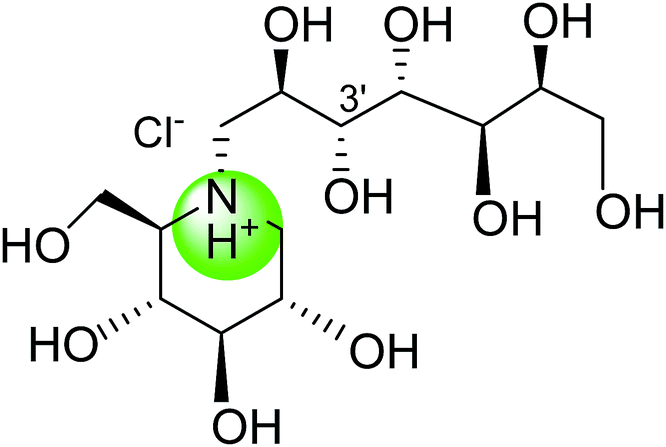 |
1.4 ± 0.5 | 124 |
The synthesis, evaluation and structure–activity relationship studies on four kotalanol diastereomers (119–122, Fig. 33), having opposite configurations at C-3′ and C-4′, were described by Xie et al.80 to understand the role of the side chain stereochemistry on the inhibitory activity. Out of the four analogues examined, compared to kotalanol (6) (IC50 values for sucrase = 0.75 µM; maltase = 7.2 µM; and isomaltase = 5.7 µM), all of these diastereomers showed higher IC50 values against maltase, sucrase, and isomaltase (see Fig. 33 for comparison of IC50 values). Except compound (120), a considerable loss of inhibitory activity (IC50 values > 236 µM) was observed against both maltase and sucrase, showing the importance of the R configuration at C-4′.
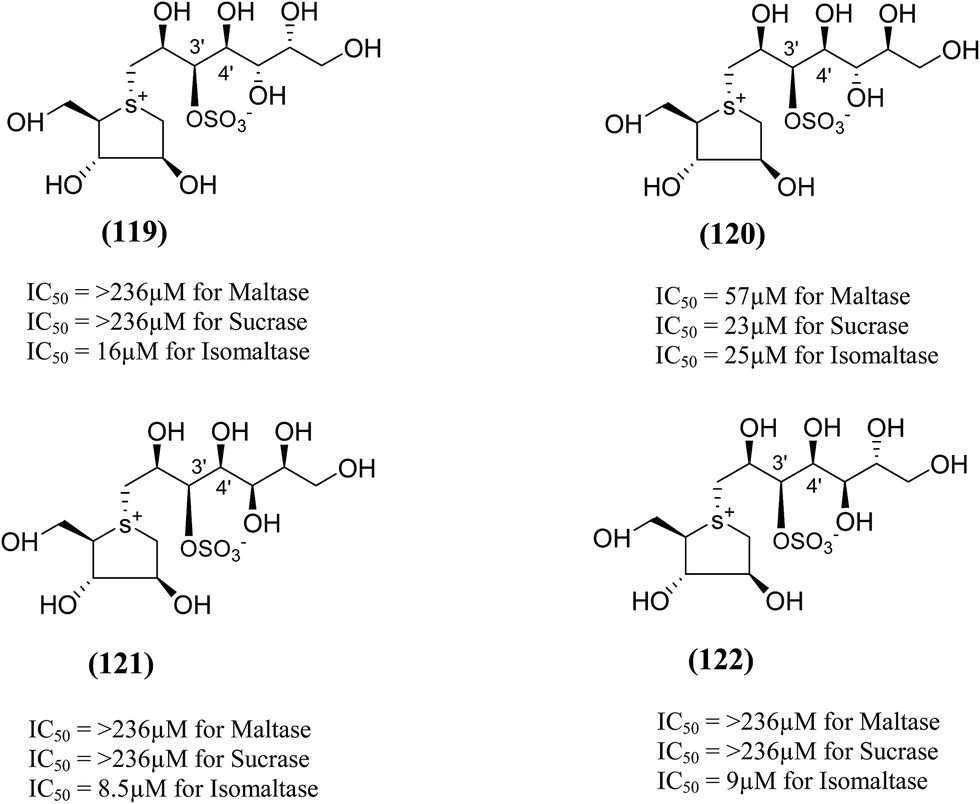 | ||
| Fig. 33 Diastereomers of kotalanol (119–122) synthesized by Xie et al.80 and their inhibitory activity against rat intestinal disaccharidases. | ||
An efficient synthetic route to de-O-sulfonated ponkoranol (11) and its 5′-stereoisomer (123) (Fig. 34) was reported by Eskandari et al.125 and showed that they are very potent inhibitors of the ntMGAM. As observed previously with the de-O-sulfonated analogues of kotalanol (10), the de-O-sulfonated ponkoranol (11) and its C-5′ epimer (123) showed significantly higher inhibitory activities with Ki values of 43 ± 3 and 15 ± 1 nM, respectively,125 against ntMGAM compared to the parent sulfated compound, ponkoranol (7) (Ki = 170 ± 30 nM).100 Thus, it would appear that de-O-sulfonation is beneficial. Comparison of the Ki values for (11) and (123) to that of Ki value for de-O-sulfonated kotalanol (10) of 30 ± 1 nM,78 led to the finale that the configuration at C-5′ is not important for dictating enzyme inhibitory activity against ntMGAM and further expansion of the acyclic side chain beyond six carbons is not favorable.
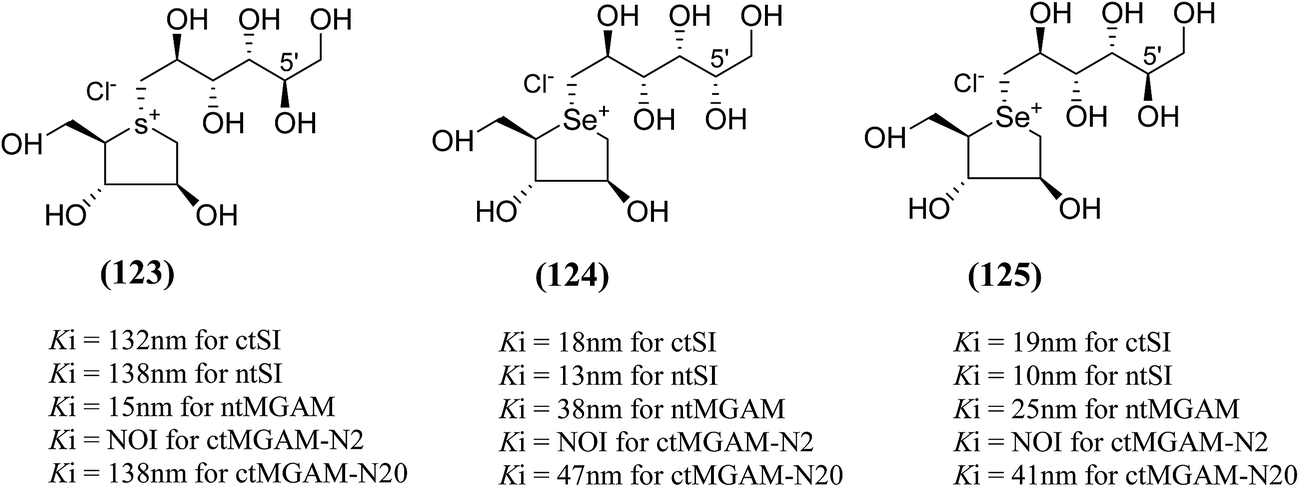 | ||
| Fig. 34 5′-Stereoisomer (123) of de-O-sulfonated ponkoranol and selenium analogues of de-O-sulfonated ponkoranol (124) and its C-5′ epimer (125) and their inhibitory profile against MGAM and SI. | ||
After applying the similar protocol, the selenium analogues of de-O-sulfonated ponkoranol (124) and its C-5′ epimer (125) (Fig. 34) were synthesized126 and tested against the four subunits, ntMGAM, ctMGAM, ntSI, and ctSI, and two spliceforms of ctMGAM, ctMGAM-N2, and ctMGAM-N20.118,127 The catalytic activities of the ctMGAM-N2 and ctMGAM-N20 are slightly different, as revealed by the kinetic analysis for maltose as a substrate; ctMGAM-N2 (Km = 1.59 ± 0.18 mM) and ctMGAMN20 (Km = 1.91 ± 0.23 mM).127 Compared to the parent compounds (11), (ntSI, Ki = 302 nM; ctSI, Ki = 103 nM) and (123) (ntSI, Ki = 138 nM; ctSI, Ki =132 nM, respectively), substitution of the ring sulfur atom with selenium (124 and 125) increased the enzyme inhibitory activities against both ntSI (Ki = 13 and 10 nM, respectively) and ctSI (Ki = 18 and 19 nM, respectively), while it did not have much effect in the case of ntMGAM and ctMGAM-N20. Surprisingly, all four compounds, both sulfur (11 and 123) and selenium (124 and 125) analogues did not inhibit the ctMGAM-N2 spliceform.128 These results suggested that only slight changes in the structure of these compounds could selectively inhibit one or a combination of subunits.
The installation of a hydrophobic methoxy group in place of a sulfate group might increase inhibitory activity against ntMGAM. A new synthetic analogue, 3′-O-methylponkoranol (126) (Fig. 35), was synthesized and screened against ntMGAM. Disappointingly, the Ki value of 3′-O-methylponkoranol (126) was higher than that of de-O-sulfonated ponkoranol (11) and was less active (Ki = 0.50 ± 0.04 µM) against ntMGAM than de-O-sulfonated ponkoranol (11, Ki = 43 nM)129 showing that the hydrophobic interactions between the methyl group and the hydrophobic residues in the active site of ntMGAM are not as optimal as the interactions of the latter groups with the rest of the polyhydroxylated chain in the absence of the methyl ether. However, compound (126) showed some interesting selectivity for the four subunits and two spliceforms of ctMGAM.130 It showed significant inhibitory activity against ctSI (Ki = 0.007 ± 0.002 µM), which is approximately 70 times higher when compared to ntMGAM (Ki = 0.50 ± 0.04 µM). The compound (126) also revealed an inhibitory activity against ntSI (Ki = 0.035 ± 0.013 µM), ctMGAM-N2 (Ki = 0.060 ± 0.015 µM), and ctMGAMN20 (Ki = 0.055 ± 0.014 µM) in a nanomolar range. Comparison with the inhibition report of de-O-sulfonated ponkoranol (11) against all four subunits clearly support that the installation of the methoxy group at C-3′ is beneficial except in the case of ntMGAM where the activity dropped by an order of magnitude. The favorable effect of a methoxy group at C-3′ (126) is remarkable, giving the most active inhibitor of ctSI with a Ki value of 0.007 ± 0.002 µM (de-O-sulfonated ponkoranol (11) had a Ki value of 0.103 ± 0.037 µM), and also showing an order of magnitude increased inhibitory activity against ntSI compared to (11). Based on the potent inhibitory activity of the 3′-O-methyl derivative (126) against ctSI, it is envisaged that ctSI will have a hydrophobic pocket in the catalytic site that better accommodates the methyl group and provides favorable hydrophobic interactions relative to the other subunits. Such information is essential in designing inhibitors against ctSI, particularly in the lack of its crystal structure.
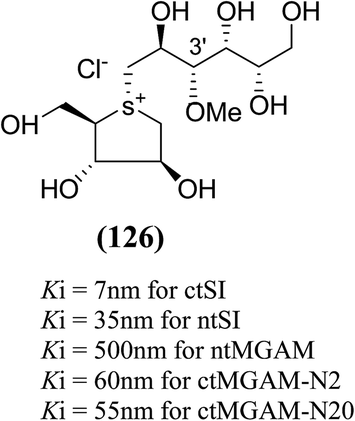 | ||
| Fig. 35 Chemical structure of 3′-O-methylponkoranol analogue and their inhibitory profile against MGAM and SI. | ||
Xie et al.131 reported the isolation, structure identification and structure–activity relationship studies of de-O-sulfonated ponkoranol (11), de-O-sulfonated salaprinol (12) and their epimers (Fig. 36) against rat small intestinal α-glucosidase in vitro. Neo-ponkoranol (11) showed potent inhibitory activity against three enzymes tested, while the neosalaprinol (12) inhibited both sucrase and isomaltase more effectively than its sulfate (8). As mentioned, upon de-O-sulfonation, all the resulting sulfonium salts showed improved inhibitory activities against isomaltase. The configuration of the chiral center at C′3 and/or C′5 positions showed no significant influence on the inhibitory activity while the 2′-epimerization caused drastic decrease in its inhibitory activity against isomaltase, all the four compounds in ponkoranol series i.e. ponkoranol (7), neoponkoranol (11), 3′-epi-ponkoranol (127), and 5′-epi-ponkoranol (128) showed potent inhibitory activities irrespective of existence of the sulfate moiety at C3′ and also of stereochemistry at 3′ and/or 5′. 3′-epi-ponkoranol (127) showed almost the same inhibitory abilities as voglibose (3) against all enzymes and was found to be the most potent inhibitor against rat small intestinal α-glucosidases. Epimerization at C′2-position in 2′-episalaprinol (130) resulted in losing completely the activity even against isomaltase.
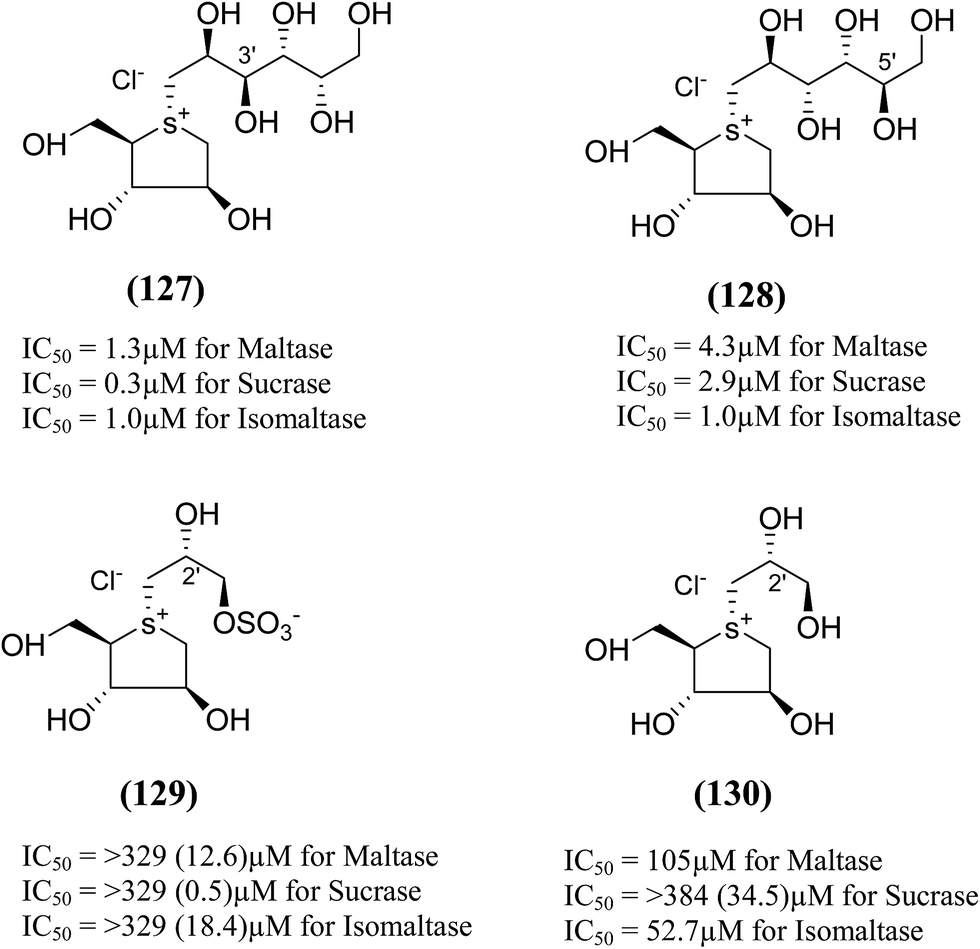 | ||
| Fig. 36 Epimers of de-O-sulfonated ponkoranol (127 and 128), salaprinol (129) and de-O-sulfonated salaprinol (130) synthesized by Xie et al.131 and their inhibitory activity against rat intestinal disaccharidases. | ||
5. Toggling of the mucosal α-glucosidases by sulfonium-ion inhibitors
The concept of toggling of the enzymes i.e. differential inhibition of individual mucosal α-glucosidase activities is considered to be one of the effective approaches for monitoring blood glucose in type-2 diabetes. Through toggling of enzymes, the rate of starch digestion and glucose release to the body can be controlled and thus, slow glucose delivery is possible.26 Natural molecules have been derivatized through structural activity relationship to make it more potent inhibitors that have the ability to inhibit activities of α-glucosidase enzymes four subunits together with two alternative spliceforms of ctMGAM. Thus, the optimization of structural changes yielded the most potent inhibitors, known to have for each subunit. Various synthetic inhibitors reported to block the activity of mucosal α-glucosidases and can be used to switch off selectively the different enzyme activities.In a study, a series of inhibitors, including acarbose (1), miglitol (2), salacinol (5), kotalanol (6), de-O-sulfonated kotalanol (10) and blintol (16), were investigated to regulate the activities of four subunits including two spliceforms of ctMGAM.127 This catalytic domain selectivity study helps to differentiate the active site requirements of each catalytic subunits. It was found that acarbose (1) was a poor inhibitor of ntMGAM (Ki = 0.009 µm) and ntSI (Ki = 14 µm) and a 1000-fold better inhibitor of two spliceforms ctMGAM-N2 and ctMGAM-N20 and 100-fold better inhibitor of ctSI. Salacinol (5) and kotalanol (6) showed 4 to 6-fold selectivity for inhibiting ctMGAM and ctSI, as compared to N-terminal MGAM and SI units. In comparison to salacinol (5), blintol (16) effectively inhibited ct-MGAM-N2 with little effect on the other enzymes. Miglitol (2) showed a small difference between the catalytic subunits. This study reflects a small change in the structure of these compounds can have brought a significant difference in inhibiting the selectivity of one enzyme over the others and showed different biochemical and structural properties of these enzyme units, despite their overall similarity in amino acid sequence and tertiary structure.26,127 An example of toggling effect, as acarbose (1) is better inhibitor of C-terminal domains of MGAM and SI but a weak inhibitor of N-terminal domains of MGAM and SI. To understand the further role of ctMGAM in the terminal starch digestion process, X-ray crystallography of the ctMGAM in complex with acarbose (1) was studied by Ren et al.132
To understand the individual roles of the MGAM and SI domains in the course of terminal starch digestion, an advance modification had proposed to the ponkoranol (7) scaffold to toggle their activities on and off with domain specific inhibitors. Two ponkoranol-based compounds, 3′- and 5′-β-maltose-extended de-O-sulfonated ponkoranol (131 and 132, Fig. 37), was described by Eskandari et al.133 and their inhibitory activities were tested against all four subunits, using maltose as a substrate.
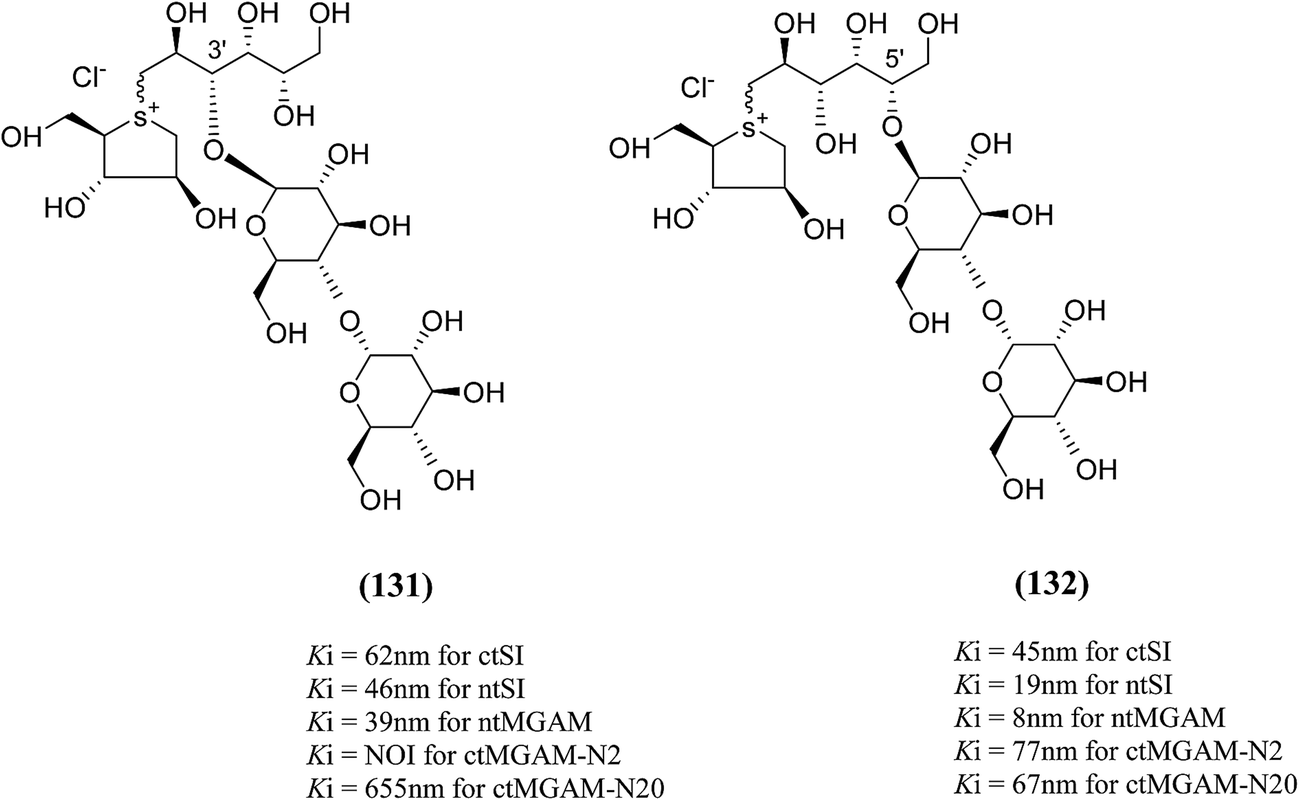 | ||
| Fig. 37 3′-O-β-Maltose-extended de-O-sulfonated ponkoranol (131) and 5′-β-maltose-extended de-O-sulfonated ponkoranol (132) and their inhibitory profile against MGAM and SI. | ||
These sulfonium compounds have shown some striking selectivities on inhibition of all four subunits. Compound (132) inhibit all of the catalytic subunits very well and is the most potent inhibitor of ntMGAM (Ki = 0.008 µm) to date. In contrast, compound (131), showed selectivity and differentiated ctMGAM from the others is a poor inhibitor of ctMGAM-N20 (Ki = 0.655 µM) and showed no inhibition against ctMGAM-N2; while, the other subunits showed similar inhibition (Ki = 0.039 µM for ntMGAM; 0.046 µM for ntSI and 0.062 µM for ctSI). As a result, with compound (131), it is feasible to maintain the ctMGAM activity on and dampen the others. The most potent inhibitor known to date for each subunit is shown in Fig. 38. Enzyme inhibition values and selectivity profile of some of the potent inhibitors, described in this review, against the four subunits i.e. ntMGAM, ctMGAM, ntSI and ctSI are given in Table 7.
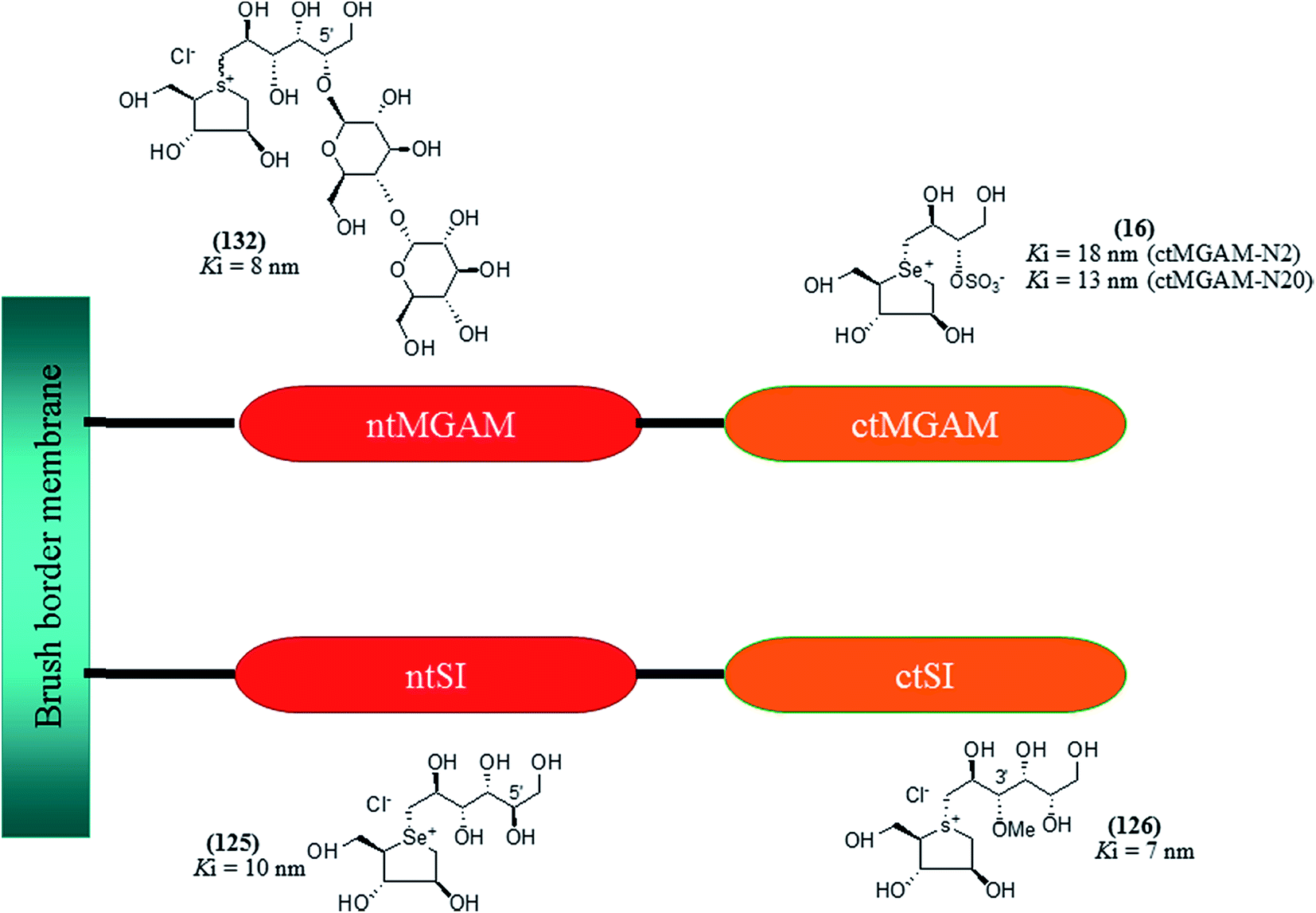 | ||
| Fig. 38 Schematic diagram of MGAM and SI indicating most potent sulfonium inhibitor of ntMGAM, ctMGAM, ntSI and ctSI. | ||
| Inhibitor | Structure | ctSI (nm) | ntSI (nm) | ntMGAM (nm) | ctMGAM-N2 (nm) | ctMGAM-N20 (nm) |
|---|---|---|---|---|---|---|
| a NOI = no inhibition. | ||||||
| Acarbose (1) |  |
246 | 14000 |
62000 |
9 | 28 |
| Miglitol (2) | 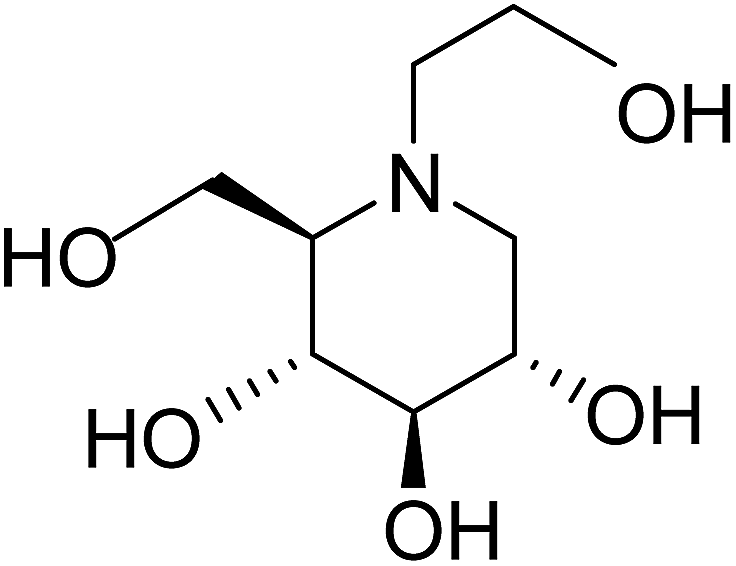 |
130 | 148 | 1000 | 211 | 230 |
| Salacinol (5) | 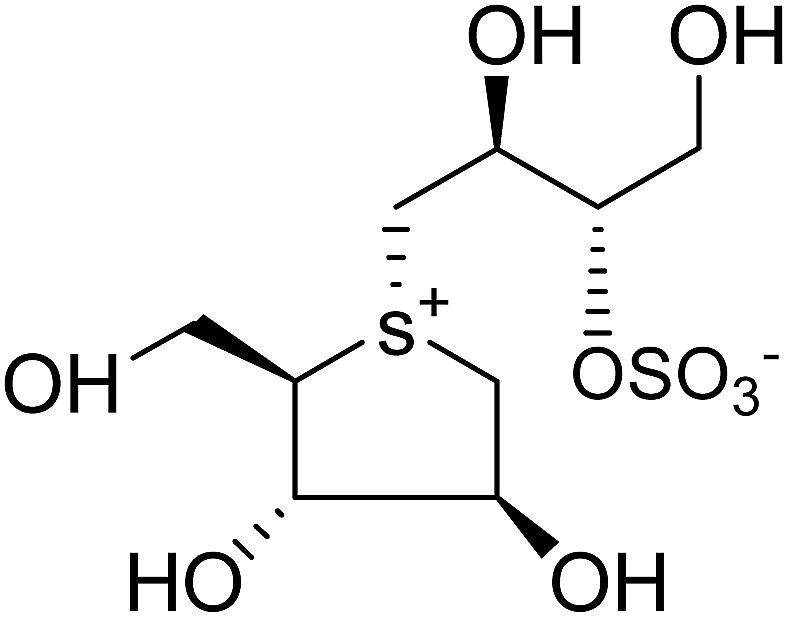 |
47 | 277 | 190 | 213 | 58 |
| Kotalanol (6) | 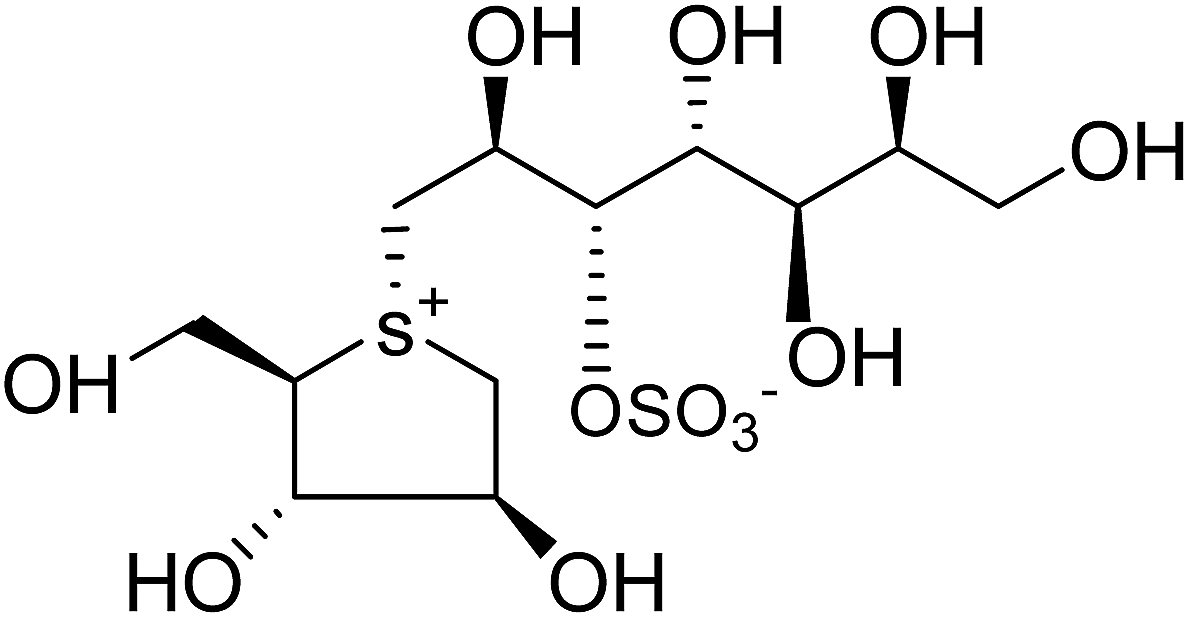 |
42 | 600 | 190 | 64 | 92 |
| Neo-kotalanol (10) | 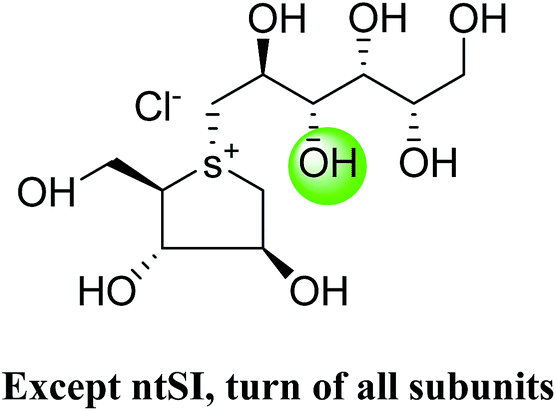 |
26 | 600 | 30 | 26 | 78 |
| Neo-ponkoranol (11) | 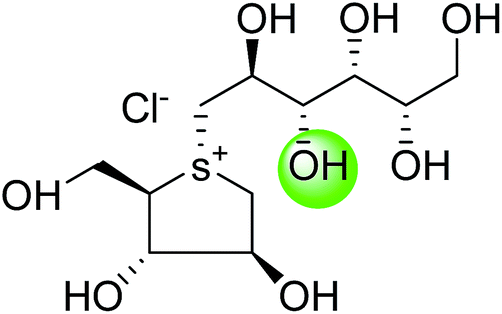 |
103 | 302 | 43 | NOI | 96 |
| Blintol (16) | 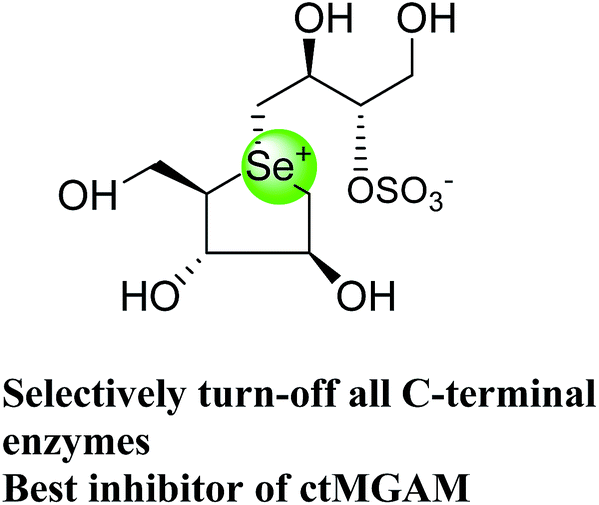 |
29 | 160 | 490 | 18 | 13 |
| C-5′ epimer of ponkoranol (123) |  |
132 | 138 | 15 | NOI | 138 |
| Selenium analogue of neo-ponkoranol (124) | 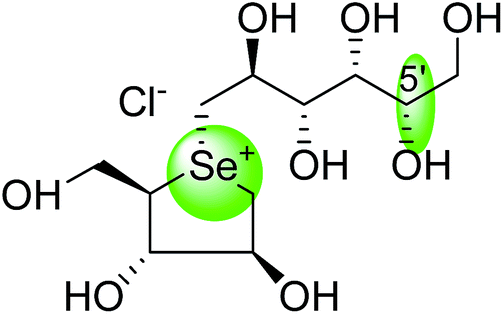 |
18 | 13 | 38 | NOI | 47 |
| Selenium analogue of C-5′ epimer of ponkoranol (125) | 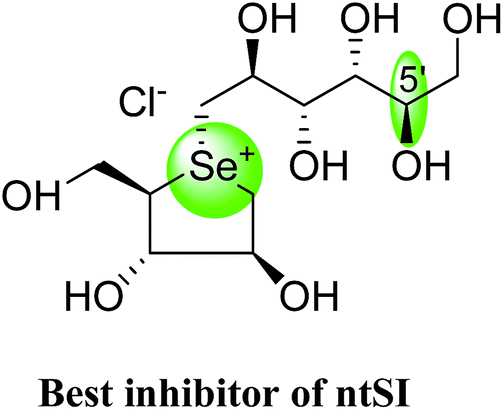 |
19 | 10 | 25 | NOI | 41 |
| 3′-O-Methyl ponkoranol (126) | 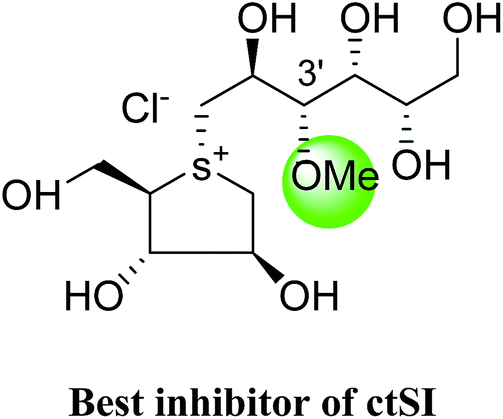 |
7 | 35 | 500 | 60 | 55 |
| 3′ β-Maltose-extended de-O-sulfonated ponkoranol (131) | 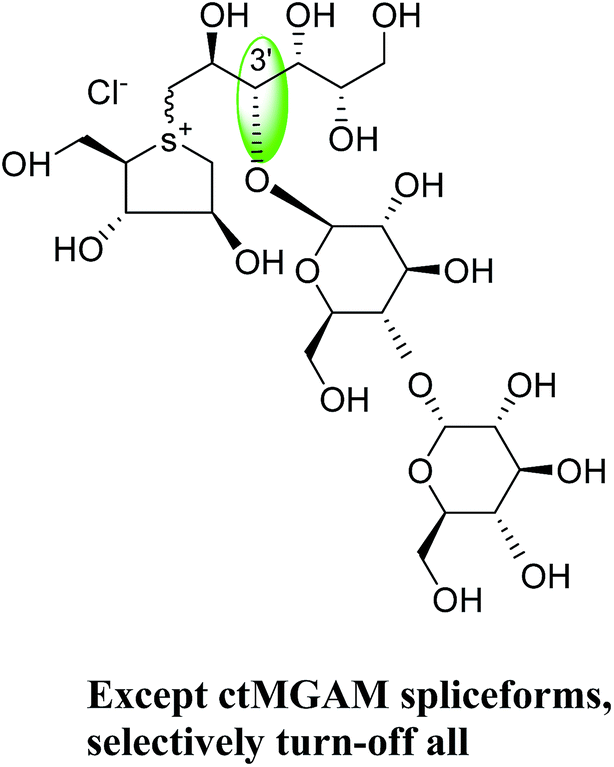 |
62 | 46 | 39 | NOI | 655 |
| 5′-β-Maltose-extended de-O-sulfonated ponkoranol (132) | 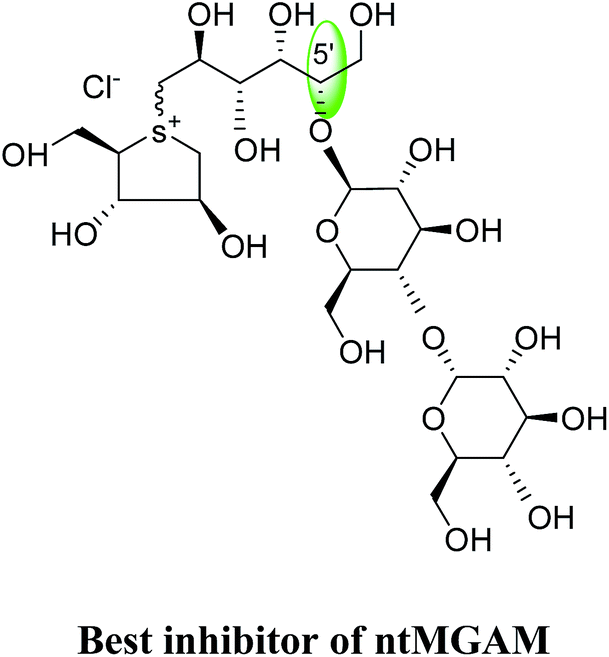 |
45 | 19 | 8 | 77 | 67 |
In another study, Lee et al.134 investigated the control of starch digestion through the concept of toggling of differential inhibition of MGAM and SI enzymes activities. Inhibitory profile of acarbose (1) and three sulfonium glucosidase inhibitors, de-O-sulfonated kotalanol (10) and the 3′-and 5′-β-maltose-extended de-O-sulfonated ponkoranol (131 and 132) were studied against all four subunits, using LM-αLDx (linear maltooligosaccharides, LM and α-limit dextrins, αLDx) as substrates, produced by the α-amylase reaction on waxy corn starch. Blood glucose level can be regulated by the regulation of activities of mucosal α-glucosidases, as MGAM and SI enzyme subunits are required to produce glucose from LM-αLDx mixture. Once again, this study showed that acarbose (1), effectively inhibited C-terminal enzymes (IC50 = 0.12 nM for ctMGAM and 0.4 nM for ctSI) as compared to N-terminal enzymes (IC50 = 62 nM for ctMGAM and 135 nM for ctSI). Inhibition of mucosal α-glucosidases by de-O-sulfonated kotalanol (10) showed 20% and 40% inhibition of ctMGAM and ctSI activity, respectively, while the N-terminal subunits were not inhibited at a concentration of 50 pmol. Thus, de-O-sulfonated kotalanol (10) could be used to toggle off ctSI subunit with sucrase activity, while having little effect on N-terminal subunits. The toggling effect by C-3′-β-maltose-extended de-O-sulfonated ponkoranol (131) on LM-αLDx hydrolysis, showed IC50 values in same range for ctMGAM (0.58 nM), ctSI (0.17 nM) and ntMGAM (0.69 nM). IC50 for ntSI (2.69 nM) was found to be ∼3–15 times higher as compared to other subunits. Thus, this derivative shows potential for selective inhibition of ctSI. On the other hand, C-5′-β-maltose-extended de-O-sulfonated ponkoranol (132) derivative did not inhibit any of the mucosal α-glucosidases. Thus, little variations in the structure of inhibitors can lead to different hydrolytic activity against LM-αLDx.
6. Conclusion
Glucosidase inhibitors from natural or synthetic sources can play biochemical tools for the evaluation of enzyme mechanism through kinetics parameters along with their variations in structural conformation. By virtue of this, a lead molecule can be discovered for therapeutic use. Now a days, α-glucosidase inhibitors have received a substantial attention for the development of antidiabetic drugs. This review highlighted the potent α-glucosidases inhibition potential by the sulfonium ion-based inhibitors, isolated from the various Salacia sp. and also describes the structural features which are important for effective inhibition of glucosidase enzyme. Mimicking in the design of sulfonium-ion could be a promising approach to the discovery of novel inhibitors. The intensive structure–activity relationship studies of naturally occurring sulfonium compounds, as well as their synthetic analogues, enable us to determine the more potent inhibitors. Subtle in structural modifications of molecules may improve the selectivity of inhibitory potential. Such structural information will be helpful for the designing of main structural moiety needed for the inhibitory activity and can be used for future generation drug design and for the synthesis of specific inhibitors of the α-glucosidase enzymes subunits. The insights gathered in this review provides a wealth of information about sulfonium-ion α-glucosidase inhibitors, this knowledge will be useful in deriving more potent antidiabetic candidates from the bioactive natural molecule for the clinical development of antidiabetic therapeutics and can be used as a model for researchers in the field of natural product chemistry. Similar efforts in structural-activity relationship based evaluation can be devoted for other natural molecules to make it more potent. Furthermore, the crystal structure and the concept of toggling will give a lead in terms of treatment of diabetes and other metabolic syndrome-associated diseases. Inhibition data for the compounds listed in this article and the type of each glucosidase linked with a specific inhibitor are incorporated in a tabular form as an Appendix.Appendix
| Inhibitor | α-Glucosidase inhibition | Other enzyme inhibition | References | |
|---|---|---|---|---|
| Ki | IC50 | IC50 | ||
| NOI: no inhibition. aα-glucosidase: rat intestinal maltase (Ki and IC50 values in µM); bα-glucosidase: rat intestinal sucrase (Ki and IC50 values in µM); cα-glucosidase: rat intestinal isomaltase (Ki and IC50 values in µM); dα-glucosidase: gluco-amylase G2 (Ki values in mM); eα-glucosidase: porcine pancreatic α-amylase (PPA) (Ki values in mM); fα-glucosidase: barley α-amylase (AMY1) (Ki values in mM); gα-glucosidase: recombinant human maltase glucoamylase (MGAM) (Ki values in µM); g1nt-MGAM: n-terminal maltase glucoamylase (Ki values in µM); g2ctSI: c-terminal sucrase isomaltase (Ki values in µM); g3ntSI: n-terminal sucrase isomaltase (Ki values in µM); g4ctMGAM-N2: spliceform of ctMGAM (Ki values in µM); g5ctMGAM-N20: spliceform of ctMGAM (Ki values in µM); hα-glucosidase: human pancreatic α-amylase (HPA) (Ki values in mM); iα-glucosidase: rice α-glucosidase (IC50 values in mM); jα-glucosidase: baker's yeast α-glucosidase (IC50 values in mM); kβ-glucosidase: β-glycase from almond (IC50 values in mM); lα-mannosidase: α-manase from jack beans (IC50 values in mM); l*α-mannosidase: α-manase from almond (IC50 values in mM); mα-glucosidase: rat intestinal trehalase (Ki values in µM); nβ-glactosidase: β-Galase from Aspergillus oryzae (IC50 values in mM); oα-galactosidase: α-galase from green coffee beans (IC50 values in mM); p40% inhibition; qDrosophila melanogaster Golgi mannosidase II; rKi values in mM; spreliminary determined with Km and one K′m; tIC50 values in µM. | ||||
| Salacinol (5) | 0.31a | 9.6a | NOIk | 33, 86 and 93 |
| 0.32b | 2.5b | 2.1l*,p ∼7.5q | ||
| 0.47c | 1.8c | |||
| 1.71d | 1.1 × 10−3i | |||
| 0.01e | NOIj | |||
| 0.015f | ||||
| 0.19g | ||||
| 0.075h | ||||
| Kotalanol (6) | 0.23a | — | — | 35 and 78 |
| 0.18b | ||||
| 1.8c | ||||
| 0.19g | ||||
| Ponkoranol (7) | 0.17g1 | — | — | 100 |
| De-O-sulfonated kotalanol (10) | 0.03g1 | — | — | 78 |
| De-O-sulfonated ponkoranol (11) | 0.043g1 | — | — | 125 |
|
||||
| Diastereomers of salacinol | ||||
| (4) | 2.17d | — | — | 81–84 |
| >5e | ||||
| >5f | ||||
| NOIg | ||||
| NOIh | ||||
| (13) | 1.06d | 0.38i | 3.4k | 85–88 |
| >5e | NOIj | 3.6l*, ∼7.5q | ||
| >5f | ||||
| NOIg | ||||
| NOIh | ||||
|
||||
| Heteroatom analogues of salacinol | ||||
| Ghavamiol (14) | >2.5d | 306a | ∼7.5l,q | 89 |
| >5e | 44b | |||
| >5f | 136c | |||
| NOIg | >315m | |||
| NOIh | ||||
| (15) | >8d | — | — | 89 |
| >5e | ||||
| >5f | ||||
| NOIg | ||||
| 0.4h | ||||
| Blintol (16) | 0.72d | — | ∼7.5l,q | 89 and 91–93 |
| >5e | ||||
| >5f | ||||
| 0.49g | ||||
| >5h | ||||
| (17) | >9d | — | ∼7.5l,q | 89 and 91–93 |
| >5e | ||||
| >5f | ||||
| NOIg | ||||
| NOIh | ||||
|
||||
| Five-membered ring analogues of salacinol with different sugar stereochemistry | ||||
| (18) | >5d | — | — | 94 |
| 0.052e | ||||
| 0.019f | ||||
| (19) | >30d | — | — | 94 |
| >5e | ||||
| >5f | ||||
| (20) | NOIg | — | — | 96 |
| (21) | NOIg | 96 | ||
| (27) | — | NOIi | NOIk | 98 |
| NOIj | NOIl | |||
| 0.853n,r | ||||
| NOIo | ||||
| (28) | — | NOIi | 2.47k,r,s | 98 |
| NOIj | NOIl | |||
| 0.716n,r | ||||
| NOIo | ||||
| (29 + 30) | — | NOIi | 1.87k,r,s | 98 |
| NOIj | NOIl | |||
| NOIn | ||||
| NOIo | ||||
| (31 + 32) | — | NOIi | NOIk | 98 |
| NOIj | NOIl | |||
| 1.29n,r,s | ||||
| NOIo | ||||
|
||||
| Six-membered ring analogues of salacinol | ||||
| (22) | NOId | — | — | 97 |
| NOIg | ||||
| (23) | NOId | — | — | 97 |
| (24a, 25, 26a) | NOId | — | — | 97 |
| NOIg | ||||
| (24b) | NOId | 1.41i,r,s | 0.016k,r | 97 |
| NOIg | NOIj | 0.467l,r | ||
| >5h | NOIn | |||
| NOIo | ||||
| (26b) | NOId | — | — | 97 |
| NOIg | ||||
| >5h | ||||
| (33) | — | NOIi | NOIk | 98 |
| NOIj | 1.34l,r,s | |||
| 0.85n,r,s | ||||
| — | ||||
| (34) | — | 1.32i,r,s | NOIk | 98 |
| NOIj | 3.59l,r,s | |||
| NOIn | ||||
| NOIo | ||||
| (37–40) | — | NOIi | NOIk | 99 |
| NOIj | NOIl | |||
| NOIn | ||||
| NOIo | ||||
|
||||
| Seven-membered ring analogues of salacinol | ||||
| (35) | — | NOIi | NOIk | 98 |
| NOIj | NOIl | |||
| NOIn | ||||
| NOIo | ||||
| (36) | — | NOIi | NOIk | 98 |
| 1.83l,s,t | ||||
| NOIn | ||||
| — | ||||
|
||||
| Chain-modified analogues of salacinol | ||||
| (41) | NOIg | — | — | 100 |
| (42) | 0.25g | — | — | 100 |
| (43) | 0.26g | — | — | 100 |
| (44) | 0.17g | — | — | 100 |
| (45) | 41g | — | — | 101 |
| (46) | NOIg | — | — | 101 |
| (47) | 26g | — | — | 101 |
| (48) | NOIg | — | — | 101 |
| (49) | 25g | — | — | 101 |
| (50) | 5g | — | — | 101 |
| (51) | 0.65g | — | — | 102 |
| (52) | 0.14g | — | — | 102 |
| (53) | 0.10g | — | — | 13 |
| (54) | 0.10g | — | — | 13 |
| (55) | 35g | — | — | 13 |
| (56) | 8g | — | — | 13 |
|
||||
| Deoxy-salacinol analogues of salacinol | ||||
| (57) | — | >1320a | — | 103 |
| 780b | ||||
| (58) | — | >1260a | — | 103 |
| >1260b | ||||
| (59) | — | >1390a | — | 103 |
| > 1390b | ||||
|
||||
| Frame-shifted analogues of salacinol | ||||
| (60) | 20g | — | — | 104 |
| (61) | 53g | — | — | 104 |
| (62) | NOIg | — | — | 104 |
| (63) | NOIg | — | — | 104 |
|
||||
| De-O-sulfonated analogues of salacinol | ||||
| (64) | 15.6a | — | 105 | |
| 3.7b | ||||
| (65) | 14.0a | — | 105 | |
| 3.5b | ||||
|
||||
| Carboxylate analogues of salacinol | ||||
| (66) | 10g | — | — | 106 |
| (67) | NOIg | — | — | 106 |
| (68) | 21g | — | 0.3l,q | 107 |
| (69) | NOIg | — | NOIl,q | 107 |
|
||||
| Phosphate analogues of salacinol | ||||
| (70) | NOIg | — | — | 110 |
| (71) | NOIg | — | — | 110 |
|
||||
| Anhydroseleno- and anhydrothio-allitols analogues of salacinol | ||||
| (72–79) | NOIg | 111 | ||
|
||||
| Alkylated alkyl analogues with varying alkyl chain of salacinol | ||||
| (80–88) | 6–7.5g | — | — | 112 |
|
||||
| 3′-O-Alkylated analogues of salacinol | ||||
| (89) | — | 5.3a | — | 113 |
| 0.46b | ||||
| 0.39c | ||||
| (90) | — | 1.7a | — | 113 |
| 0.12b | ||||
| 0.27c | ||||
| (91) | — | 1.0a | — | 113 |
| 1.3b | ||||
| 0.95c | ||||
| (92) | — | 0.44a | — | 113 |
| 0.32b | ||||
| 0.14c | ||||
|
||||
| 3′-O-Benzyl analogues of salacinol | ||||
| (93) | — | 0.66a | — | 114 |
| 0.41b | ||||
| 0.48c | ||||
| (94) | — | 0.84a | — | 114 |
| 1.3b | ||||
| 0.35c | ||||
| (95) | — | 0.86a | — | 114 |
| 1.1b | ||||
| 0.68c | ||||
| (96) | — | 0.31a | — | 114 |
| 0.09b | ||||
| 0.26c | ||||
| (97) | — | 0.53a | — | 114 |
| 0.80b | ||||
| 0.31c | ||||
| (98) | — | 0.89a | — | 114 |
| 0.72b | ||||
| 0.48c | ||||
| (99) | — | 0.33a | — | 114 |
| 0.15b | ||||
| 0.19c | ||||
| (100) | — | 0.98a | — | 114 |
| 0.82b | ||||
| 0.25c | ||||
| (101) | — | 0.98a | — | 114 |
| 0.72b | ||||
| 0.38c | ||||
| (102) | — | 0.13a | — | 114 |
| 0.042b | ||||
| 0.21c | ||||
| (103) | — | 0.94a | — | 114 |
| 0.49b | ||||
| 0.23c | ||||
| (104) | — | 0.68a | — | 114 |
| 0.38b | ||||
| 0.23c | ||||
|
||||
| Stereoisomer of kotalanol | ||||
| (105) | 0.20g1 | — | — | 115 |
| (106) | 0.017g1 | — | — | 36 |
| (107) | 0.10g1 | — | — | 123 |
| (108) | 0.13g1 | — | — | 123 |
| (109) | 0.024 1 | — | — | 123 |
| (110) | 0.026g1 | — | — | 123 |
| (111) | 0.09g1 | — | — | 122 |
| (112) | 0.08g1 | — | — | 122 |
| (113) | 7.2g1 | — | — | 122 |
| (114) | 0.061g1 | — | — | 122 |
| (115) | 0.02g1 | — | — | 122 |
| (116) | 0.83g1 | — | — | 122 |
|
||||
| Six-membered ring analogues of kotalanol | ||||
| (117) | 2.3g1 | — | — | 124 |
| (118) | 1.4g | — | — | 124 |
|
||||
| Other diastereomers of kotalanol with different side chain stereochemistry | ||||
| (119) | — | >236a | — | 80 |
| >236b | ||||
| 16c | ||||
| (120) | — | 57a | — | 80 |
| 23b | ||||
| 25c | ||||
| (121) | — | >236a | — | 80 |
| >236b | ||||
| 8.5c | ||||
| (122) | — | >236a | — | 80 |
| >236b | ||||
| 9c | ||||
|
||||
| Stereoisomer of ponkoranol | ||||
| (123) | 0.015g1 | — | — | 125 |
| 0.132g2 | ||||
| 0.138g3 | ||||
| NOIg4 | ||||
| 0.138g5 | ||||
|
||||
| Heteroanalogues of de-O-sulfonated ponkoranol | ||||
| (124) | 0.038g1 | — | — | 126 and 128 |
| 0.018g2 | ||||
| 0.013g3 | ||||
| NOIg4 | ||||
| 0.047g5 | ||||
| (125) | 0.025g1 | — | — | 126 and 128 |
| 0.019g2 | ||||
| 0.010g3 | ||||
| NOIg4 | ||||
| 0.041g5 | ||||
|
||||
| 3′-O-Methyl analogue of ponkoranol | ||||
| (126) | 0.500g1 | — | — | 129 and 130 |
| 0.007g2 | ||||
| 0.035g3 | ||||
| 0.060g4 | ||||
| 0.055 g5 | ||||
|
||||
| Epimers of de-O-sulfonated ponkoranol, salaprinol and de-O-sulfonated salaprinol | ||||
| (127) | — | 1.3a | — | 131 |
| 0.3b | ||||
| 1.0c | ||||
| (128) | — | 4.3a | — | 131 |
| 2.9b | ||||
| 1.0c | ||||
| (129) | — | >329 (12.6)a | — | 131 |
| >329 (0.5)b | ||||
| >329 (18.4)c | ||||
| (130) | — | 105a | — | 131 |
| >384 (34.5)b | ||||
| 52.7c | ||||
|
||||
| 3′-And 5′-β-maltose-extended de-O-sulfonated ponkoranol | ||||
| (131) | 0.039g1 | — | — | 133 |
| 0.062g2 | ||||
| 0.046g3 | ||||
| NOIg4 | ||||
| 0.655g5 | ||||
| (132) | 0.008g1 | — | — | 133 |
| 0.045g2 | ||||
| 0.019g3 | ||||
| 0.077g4 | ||||
| 0.067g5 | ||||
Acknowledgements
The author, Dr Priyanka Bagri, is supported by a grant from University Grants Commission, New Delhi, India under Postdoctoral fellowship for women (PDFSS-2013-14-DEL-SC-5938).References
- T. D. Heightman, T. Andrea and A. T. Vasella, Angew. Chem., Int. Ed., 1999, 38, 750–770 CrossRef CAS.
- J. H. Park, S. Ko and H. Park, Bull. Korean Chem. Soc., 2008, 29, 921–927 CrossRef CAS.
- E. Borges de Melo, A. da Silveira Gomes and I. Carvalho, Tetrahedron, 2006, 62, 10277–10302 CrossRef CAS.
- G. Semenza, Annu. Rev. Cell Biol., 1986, 2, 255–313 CrossRef CAS PubMed.
- E. H. Van Beers, H. A. Buller, R. J. Grand, A. W. Einerhand and J. Dekker, Crit. Rev. Biochem. Mol. Biol., 1995, 30, 197–262 CrossRef CAS PubMed.
- B. L. Nichols, S. Avery, P. Sen, D. M. Swallow, D. Hahn and E. Sterchi, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 1432–1437 CrossRef CAS PubMed.
- N. Asano, Glycobiology, 2003, 10, 93R–104R CrossRef PubMed.
- J. L. Chiasson, R. G. Josse, R. Gomis, M. Hanefeld, A. Karasik and M. Laakso, Lancet, 2002, 359, 2072–20777 CrossRef CAS.
- N. Katsilambros, P. Philippides, A. Toskas, J. Protopapas, D. Frangaki, M. Marangos, P. Siskoudis, K. Anastasopoulou, H. Xetteri and I. Hillebrand, Arzneimittelforschung, 1986, 36, 1136–1138 CAS.
- B. Goke, H. Fuder, G. Wieckhorst, U. Theiss, E. Stridde, T. Littke, P. Kleist, R. Arnol and P. W. Lucker, Digestion, 1995, 56, 493–501 CAS.
- H. W. Xu, G. F. Dai, G. Z. Liu, J. F. Wang and H. M. Liu, Bioorg. Med. Chem., 2007, 15, 4247–4255 CrossRef CAS PubMed.
- G. Tanabe, K. Yoshikai, T. Hatanaka, M. Yamamoto, Y. Shao, T. Minematsu, O. Muraoka, T. Wang, H. Matsuda and M. Yoshikawa, Bioorg. Med. Chem., 2007, 15, 3926–3937 CrossRef CAS PubMed.
- H. Liu, R. Nasi, K. Jayakanthan, L. Sim, H. Heipel, D. R. Rose and B. M. Pinto, J. Org. Chem., 2007, 72, 6562–6572 CrossRef CAS PubMed.
- J. G. Luo, X. B. Wang, L. Ma and L. Y. Kong, Bioorg. Med. Chem., 2007, 17, 4460–4463 CrossRef CAS PubMed.
- J. P. Saludes, S. C. Lievens and T. F. Molinski, J. Nat. Prod., 2007, 70, 436–438 CrossRef CAS PubMed.
- Z. Y. Du, R. R. Liu, W. Y. Shao, X. P. Maom, L. Ma, L. Q. Gu, Z. S. Huangm and A. S. C. Chan, Eur. J. Med. Chem., 2006, 41, 213–218 CrossRef CAS PubMed.
- L. K. Campbell, D. E. Baker and R. K. Campbell, Ann. Pharmacother., 2000, 34, 1291–1301 CrossRef CAS PubMed.
- R. J. Andrade, M. Lucena, J. L. Vega, M. Torres, F. J. Salmeron, V. Bellot, M. D. Garcia-Escano and P. Moreno, Diabetes Care, 1998, 21(11), 2029–2030 CrossRef CAS PubMed.
- S. H. Hsiao, L. H. Liao, P. N. Cheng and T. J. Wu, Ann. Pharmacother., 2006, 40, 151–154 CrossRef PubMed.
- J. P. C. Chandrasena, The Chemistry and Pharmacology of Ceylon and Indian Medicinal Plants, H&C Press, Colombo, Sri Lanka, 1935 Search PubMed.
- H. Matsuda, M. Yoshikawa, T. Morikawa, G. Tanabe and O. Muraoka, J. Tradit. Med., 2005, 22, 145–153 CAS.
- S. Mohan and B. M. Pinto, Carbohydr. Res., 2007, 342, 1551–1580 CrossRef CAS PubMed.
- S. Mohan and B. M. Pinto, Collect. Czech. Chem. Commun., 2009, 74, 1117–1136 CrossRef CAS.
- S. Mohan and B. M. Pinto, Nat. Prod. Rep., 2010, 27, 481–488 RSC.
- W. Xie, G. Tanabe, J. Xu, X. Wu, T. Morikawa, M. Yoshikawa and O. Muraoka, Mini-Rev. Org. Chem., 2013, 10, 141–159 CrossRef CAS.
- S. Mohan, R. Eskandari and B. M. Pinto, Acc. Chem. Res., 2014, 47, 211–225 CrossRef CAS PubMed.
- K. R. Kirtikar and B. D. Basu, Indian medicinal plants, CAB Direct, 1987 Search PubMed.
- K. M. Nandkarni, The Indian Materia Medica, Popular Prakashan Pvt. Ltd., Mumbai, 1993, vol. 1, p. 1089 Search PubMed.
- D. M. A. Jayaweera, Medicinal plants used in Ceylon, part 1, National Science Council of Sri Lanka, Colombo, 1981, p. 77 Search PubMed.
- A. Singh and S. Duggal, A Clinician's Journal, 2010, 9, 40–43 Search PubMed.
- D. Ramakrishna, A. T. Shashank, K. Shinomol George, S. Kiran and G. A. Ravishankar, International Journal of Pharmacognosy and Phytochemical Research, 2015, 7, 374–382 Search PubMed.
- P. S. Vaidyartanam, Indian Medicinal Plants: a compendium of 500 species, ed. P. K. Warrier, V. P. K. Nambiar and C. Ramankutty, Orient Longman, Madras, India, 1993 Search PubMed.
- M. Yoshikawa, T. Murakami, H. Shimada, H. Matsuda, J. Yamahara, G. Tanabe and O. Muraoka, Tetrahedron Lett., 1997, 38, 8367–8370 CrossRef CAS.
- M. Yoshikawa, T. Morikawa, H. Matsuda, G. Tanabe and O. Muraoka, Bioorg. Med. Chem., 2002, 10, 1547–1554 CrossRef CAS PubMed.
- M. Yoshikawa, T. Murakami, K. Yashiro and H. Matsuda, Chem. Pharm. Bull., 1998, 46, 1339–1340 CrossRef CAS PubMed.
- K. Jayakanthan, S. Mohan and B. M. Pinto, J. Am. Chem. Soc., 2009, 131, 5621–5626 CrossRef CAS PubMed.
- O. Muraoka, W. J. Xie, S. Osaki, A. Kagawa, G. Tanabe, M. F. A. Amer, T. Minematsu, T. Morikawa and M. Yoshikawa, Tetrahedron, 2010, 66, 3717–3722 CrossRef CAS.
- M. Yoshikawa, F. Xu, S. Nakamura, T. Wang, H. Matsuda, G. Tanabe and O. Muraoka, Heterocycles, 2008, 75, 1397–1405 CrossRef CAS.
- H. Matsuda, T. Murakami, K. Yashiro, J. Yamahara and M. Yoshikawa, Chem. Pharm. Bull., 1999, 47, 1725–1729 CrossRef CAS PubMed.
- K. T. Augusti, P. Joseph and T. D. Babu, Indian J. Physiol. Pharmacol., 1995, 39, 415–417 CAS.
- E. H. Karunanayake, J. Welihinda, S. R. Sirimanne and G. Sinnadorai, J. Ethnopharmacol., 1984, 11, 223–231 CrossRef CAS PubMed.
- M. Patricia, R. D. Heacock, R. Steven, R. D. Hertzler, A. Jennifer, M. P. H. Williams and W. Bryan Wolf, Curr. Res., 2005, 65–71 Search PubMed.
- S. J. Stohs and S. Ray, Phytother. Res., 2015, 29, 986–995 CrossRef CAS PubMed.
- P. S. Kushwaha, A. K. Singh, A. K. Keshari, S. Maity and S. Saha, Pharmacogn. Rev., 2016, 10, 109–114 CrossRef PubMed.
- K. Krishnakumar, K. T. Augusti and P. L. Vijayammal, Indian J. Physiol. Pharmacol., 1999, 43, 510–514 CAS.
- A. L Collene, S. R. Hertzler, J. A. Williams and B. W. Wolf, Nutrition, 2005, 21, 848–854 CrossRef PubMed.
- P. M. Heathcock, S. R. Hertzler, J. A. Williams and B. W. Wolf, J. Am. Diet. Assoc., 2005, 105, 65–71 CrossRef PubMed.
- S. Serasinghe, P. Serasinghe, H. Yamazaki, K. Nishiguchi, F. Hombhanje, S. Nakanishi, K. Sawa, M. Hattori and T. Namba, Phytother. Res., 1990, 4, 205–206 CrossRef.
- H. Shimoda, S. Kawamori and Y. Kawahara, J. Jpn. Soc. Nutr. Food Sci., 1998, 51, 279–287 CrossRef CAS.
- N. R. Pillai, C. Seshadri and G. Santhakumara, Indian J. Exp. Biol., 1979, 17(1), 1–14 Search PubMed.
- V. Venkateswarlu, C. K. Kokate, D. Rambhau and C. Veeresham, Planta Med., 1993, 59, 391–393 CrossRef CAS PubMed.
- T. Morikawa, J. Akaki, K. Ninomiya, E. Kinouchi, G. Tanabe, Y. Pongpiriyadacha, M. Yoshikawa and O. Muraoka, Nutrients, 2015, 7, 1480–1493 CrossRef CAS PubMed.
- A. Patwardhan, M. Pimputkar and R. Joshi, J. Biodivers. Biopros. Dev., 2014, 1, 107–111 Search PubMed.
- G. A. Kurian, V. Manjushi, S. S. Nair, T. Varghese and J. Padikkala, Nutrition, 2014, 30, 1156–1164 CrossRef PubMed.
- P. S. Sellamuthu, P. Arulselvan, S. Fakurazi and M. Kandasamy, Pak. J. Pharm. Sci., 2014, 27, 161–167 CAS.
- P. S. Sellamuthu, P. Arulselvan, B. P. Muniappan, S. Fakurazi and M. Kandasamy, J. Med. Food, 2013, 16, 719–727 CrossRef CAS PubMed.
- H. N. Shivaprasad, M. Bhanumathy, G. Sushma, T. Midhun, K. R. Raveendra, K. R. Sushma and K. Venkateshwarlu, J. Med. Food, 2013, 16, 564–568 CrossRef CAS PubMed.
- S. S. Basha, M. Baruah, A. Shaker, S. B. Kondaveeti and S. Narayana, Res. J. Pharm., Biol. Chem. Sci., 2012, 3, 239–246 Search PubMed.
- B. M. Bhat, C. V. Raghuveer, V. D'Souza and P. A. Manjrekaar, J. Clin. Diagn. Res., 2012, 6, 1685–1687 Search PubMed.
- K. Nakata, Y. Taniguchi, N. Yoshioka, A. Yoshida, H. Inagawa, T. Nakamoto, H. Yoshimura, S. Miyake, C. Kohchi, M. Kuroki and G. Soma, Nutr. Res. Pract., 2011, 5, 435–442 CrossRef CAS PubMed.
- C. N. Chandrashekar, S. Madhyastha, S. Benjamin, G. K. Krishna and K. K. Srinivasan, J. Physiol. Sci., 2009, 21, 48–57 Search PubMed.
- M. D. Giron, N. Sevillano, R. Salto, A. Haidour, M. Manzano, M. L. Jimenez, R. Rueda and J. M. López-Pedrosa, Clin. Nutr., 2009, 28, 565–574 CrossRef CAS PubMed.
- R. Im, H. Mano, T. Matsuura, S. Nakatani, J. Shimizu and M. Wada, J. Ethnopharmacol., 2009, 121, 234–240 CrossRef PubMed.
- E. A. Venkateshwarlu, R. N. A. Reddy, S. A. Sunder, G. Kiran, J. V. Rao and S. Madhusudhan, Drug Invent. Today, 2009, 1, 95–97 Search PubMed.
- K. Yoshino, Y. Miyauchi, T. Kanetaka, Y. Takagi and K. Koga, Biosci., Biotechnol., Biochem., 2009, 73, 1096–1104 CrossRef CAS PubMed.
- H. Oe and S. Ozaki, Biosci., Biotechnol., Biochem., 2008, 72, 1962–1964 CrossRef CAS PubMed.
- J. A. Williams, Y. S. Choe, M. J. Noss, C. J. Baumgartner and V. A. Mustad, Am. J. Clin. Nutr., 2007, 86, 124–130 CAS.
- S. I. Rabbani, M. Asad and S. M. B. Asdaq, Indian Drugs, 2006, 43, 844–847 Search PubMed.
- M. H. Jayawardena, N. M. de Alwis, V. Hettigoda and D. J. Fernando, J. Ethnopharmacol., 2005, 97, 215–218 CrossRef CAS PubMed.
- R. Radha and M. Amrithaveni, Adv. Life Sci., 2009, 29, 14–16 CAS.
- Y. Li, G. Peng, S. Wen, T. H. Huang, B. D. Roufogalis and J. Yamahara, Life Sci., 2004, 75, 1735–1746 CrossRef CAS PubMed.
- L. Yuhoo, Herbal Medicine Practical, The University of Sydney, 2004 Search PubMed.
- M. Yoshikawa, Y. Pongpiriyadacha, A. Kishi, T. Kageura, T. Wang, H. Morikawa and H. Matsuda, Yakugaku Zasshi, 2003, 123, 871–880 CrossRef CAS.
- M. Yoshikawa, Chem. Pharm. Bull., 2001, 49, 1503–1505 CrossRef.
- B. W. Wolf and S. E. Weisbrode, Food Chem. Toxicol., 2003, 41, 867–874 CrossRef CAS PubMed.
- B. M. Pinto, B. D. Johnston, A. Ghavami, M. G. Szezepina, H. Liu, K. S. Sadalapure, H. H. Jensen, N. S. Kumar and R. Nasi, US Pat., 20060247222A1, 2006.
- B. M. Pinto, N. S. Kumar, R. G. Bhat, H. Liu, R. Nasi, W. Chen and S. Mohan, US Pat., 20070244184A1, 2007.
- L. Sim, K. Jayakanthan, S. Mohan, R. Nasi, B. D. Johnston, B. Mario Pinto and D. R. Rose, Biochemistry, 2010, 49, 443–451 CrossRef CAS PubMed.
- S. Ozaki, H. Oe and S. Kitamura, J. Nat. Prod., 2008, 71, 981–984 CrossRef CAS PubMed.
- W. Xie, G. Tanabe, K. Matsuoka, M. F. A. Amer, T. Minematsu, X. Wu, M. Yoshikawa and O. Muraoka, Bioorg. Med. Chem., 2011, 19, 2252–2262 CrossRef CAS PubMed.
- Y. Minami, C. Kuriyama, K. Ikeda, A. Kato, K. Takebayashi, I. Adachi, G. W. J. Fleet, A. Kettawan, T. Okamoto and N. Asano, Bioorg. Med. Chem., 2008, 16, 2734–2740 CrossRef CAS PubMed.
- G. Tanabe, W. Xie, A. Ogawa, C. Cao, T. Minematsu, M. Yoshikawa and O. Muraoka, Bioorg. Med. Chem. Lett., 2009, 19, 2195–2198 CrossRef CAS PubMed.
- H. Bischoff, Eur. J. Clin. Invest., 1994, 24, 3–10 CrossRef CAS PubMed.
- C. Li, A. Begum, S. Numao, K. H. Park, S. G. Withers and G. D. Brayer, Biochemistry, 2005, 44, 3347–3357 CrossRef CAS PubMed.
- H. Yuasa, J. Takada and H. Hashimoto, Tetrahedron Lett., 2000, 41, 6615–6618 CrossRef CAS.
- A. Ghavami, B. D. Johnston and B. M. Pinto, J. Org. Chem., 2001, 66, 2312–2317 CrossRef CAS PubMed.
- A. Ghavami, K. S. Sadalapure, B. D. Johnston, M. Lobera, B. B. Snider and B. M. Pinto, Synlett, 2003, 1259–1262 CAS.
- H. Yuasa, J. Takada and H. Hashimoto, Bioorg. Med. Chem. Lett., 2001, 11, 1137–1139 CrossRef CAS PubMed.
- A. Ghavami, B. D. Johnston, M. T. Jensen, B. Svensson and B. M. Pinto, J. Am. Chem. Soc., 2001, 123, 6268–6271 CrossRef CAS PubMed.
- O. Muraoka, S. Ying, K. Yoshikai, Y. Matsuura, E. Yamada, T. Minematsu, G. Tanabe, H. Matsuda and M. Yoshikawa, Chem. Pharm. Bull., 2001, 49, 1503–1505 CrossRef CAS PubMed.
- B. D. Johnston, A. Ghavami, M. T. Jensen, B. Svensson and B. M. Pinto, J. Am. Chem. Soc., 2002, 124, 8245–8250 CrossRef CAS PubMed.
- B. M. Pinto, B. D. Johnston and A. Ghavami, US Pat., 20030191104A1, 2003.
- B. M. Pinto, B. D. Johnston, M. G. Szczepina, H. Liu, K. Sadalpure and A. Ghavami, US Pat., 20050065139A1, 2005.
- A. Ghavami, B. D. Johnston, M. D. Maddess, S. M. Chinapoo, M. T. Jensen, B. Svensson and B. M. Pinto, Can. J. Chem., 2002, 80, 937–942 CrossRef CAS.
- N. S. Kumar and B. M. Pinto, Carbohydr. Res., 2005, 340, 2612–2619 CrossRef CAS PubMed.
- E. J. Rossi, L. Sim, D. A. Kuntz, D. Hahn, B. D. Johnston, A. Ghavami, M. G. Szczepina, N. S. Kumar, E. E. Sterchi, B. L. Nichols, B. M. Pinto and D. R. Rose, FEBS J., 2006, 273, 2673–2683 CrossRef CAS PubMed.
- M. G. Szczepina, B. D. Johnston, Y. Yuan, B. Svensson and B. M. Pinto, J. Am. Chem. Soc., 2004, 126, 12458–12469 CrossRef CAS PubMed.
- E. Gallienne, M. Benazza, G. Demailly, J. Bolte and M. Lemaire, Tetrahedron, 2005, 61, 4557–4568 CrossRef CAS.
- E. Gallienne, T. Gefflaut, J. Bolte and M. Lemaire, J. Org. Chem., 2006, 71, 894–902 CrossRef CAS PubMed.
- B. D. Johnston, H. H. Jensen and B. M. Pinto, J. Org. Chem., 2006, 71, 1111–1118 CrossRef CAS PubMed.
- H. Liu, L. Sim, D. R. Rose and B. M. Pinto, J. Org. Chem., 2006, 71(8), 3007–3013 CrossRef CAS PubMed.
- R. Nasi, L. Sim, D. R. Rose and B. M. Pinto, J. Org. Chem., 2007, 72, 180–186 CrossRef CAS PubMed.
- O. Muraoka, K. Yoshikai, H. Takahashi, T. Minematsu, G. X. Lu, G. Tanabe, T. Wang, H. Matsuda and M. Yoshikawa, Bioorg. Med. Chem., 2006, 14, 500–509 CrossRef CAS PubMed.
- R. Nasi, L. Sim, D. R. Rose and B. M. Pinto, Carbohydr. Res., 2007, 342, 1888–1894 CrossRef CAS PubMed.
- G. Tanabe, K. Yoshikai, T. Hatanaka, M. Yamamoto, Y. Shao, T. Minematsu, O. Muraoka, T. Wang, H. Matsuda and M. Yoshikawa, Bioorganic Med. Chem., 2007, 15, 3926–3937 CrossRef CAS PubMed.
- W. Chen, L. Sim, D. R. Rose and B. M. Pinto, Carbohydr. Res., 2007, 3, 1661–1667 CrossRef PubMed.
- W. Chen, D. A. Kuntz, T. Hamlet, L. Sim, D. R. Rose and B. Mario Pinto, Bioorganic Med. Chem., 2006, 14, 8332–8340 CrossRef PubMed.
- D. A. Kuntz, A. Ghavami, B. D. Johnston, B. M. Pinto and D. R. Rose, Tetrahedron: Asymmetry, 2005, 16, 25–32 CrossRef CAS.
- H. Yuasa, C. Saotome and O. Kanie, Trends Glycosci. Glycotechnol., 2002, 14, 231–251 CrossRef CAS.
- R. G. Bhat, N. S. Kumar and B. M. Pinto, Carbohydr. Res., 2007, 342(12–13), 1934–1942 CrossRef CAS PubMed.
- H. Liu and B. M. Pinto, Can. J. Chem., 2006, 84, 1351–1362 CrossRef.
- S. Mohan, L. Sim, D. R. Rose and B. M. Pinto, Carbohydr. Res., 2007, 342, 901–912 CrossRef CAS PubMed.
- G. Tanabe, T. Otani, W. Cong, T. Minematsu, K. Ninomiya, M. Yoshikawa and O. Muraoka, Bioorganic Med. Chem. Lett., 2011, 21, 3159–3162 CrossRef CAS PubMed.
- G. Tanabe, S. Nakamura, N. Tsutsui, G. Balakishan, W. Xie, S. Tsuchiya, J. Akaki, T. Morikawa, K. Ninomiya, I. Nakanishi, M. Yoshikawa and O. Muraoka, Chem. Commun., 2012, 48, 8646–8648 RSC.
- R. Eskandari, K. Jayakanthan, D. A. Kuntz, D. R. Rose and B. Mario Pinto, Bioorganic Med. Chem., 2010, 18, 2829–2835 CrossRef CAS PubMed.
- X. Wen, Y. Yuan, D. A. Kuntz, D. R. Rose and B. M. Pinto, Biochemistry, 2005, 44, 6729–6737 CrossRef CAS PubMed.
- L. Sim, C. Willemsma, S. Mohan, H. Y. Naim, B. M. Pinto and D. R. Rose, J. Biol. Chem., 2010, 285, 17763–17770 CrossRef CAS PubMed.
- S. Nakamura, K. Takahira, G. Tanabe, T. Morikawa, M. Sakano, K. Ninomiya, M. Yoshikawa, O. Muraoka and I. Nakanishi, Bioorg. Med. Chem. Lett., 2010, 20, 4420–4423 CrossRef CAS PubMed.
- S. Nakamura, K. Takahira, G. Tanabe, O. Muraoka and I. Nakanishi, Open J. Med. Chem., 2012, 2, 50–60 CrossRef CAS.
- G. Tanabe, M. Sakano, T. Minematsu, H. Matsuda, M. Yoshikawa and O. Muraoka, Tetrahedron, 2008, 64, 10080–10086 CrossRef CAS.
- G. Tanabe, M. F. A. Amer, T. Minematsu, T. Morikawa and M. Yoshikawa, Tetrahedron, 2010, 66, 3717 CrossRef.
- S. Mohan, K. Jayakanthan, R. Nasi, D. A. Kuntz, D. R. Rose and B. M. Pinto, Org. Lett., 2010, 12, 1088–1091 CrossRef CAS PubMed.
- R. Nasi, B. O. Patrick, L. Sim, D. R. Rose and B. M. Pinto, J. Org. Chem., 2008, 73, 6172–6181 CrossRef CAS PubMed.
- S. Mohan, L. Sim, D. R. Rose and B. M. Pinto, Bioorg. Med. Chem., 2010, 18, 7794–7798 CrossRef CAS PubMed.
- R. Eskandari, D. A. Kuntz, D. R. Rose and B. M. Pinto, Org. Lett., 2010, 12, 1632–1635 CrossRef CAS PubMed.
- R. Eskandari, K. Jones, D. R. Rose and B. M. Pinto, Chem. Commun., 2011, 47, 9134–9136 RSC.
- K. Jones, L. Sim, S. Mohan, J. Kumarasamy, H. Liu, S. Avery, H. Y. Naim, R. Quezada- Calvillo, B. L. Nichols, B. M. Pinto and D. R. Rose, Bioorg. Med. Chem., 2011, 19, 3929–3934 CrossRef CAS PubMed.
- D. G. Naumoff, Mol. Biol., 2007, 41, 962–973 CrossRef CAS.
- R. Eskandari, K. Jones, D. R. Rose and B. M. Pinto, Bioorganic Med. Chem. Lett., 2010, 20, 5686–5689 CrossRef CAS PubMed.
- R. Eskandari, B. M. Pinto, K. Jones and D. R. Rose, Bioorganic Med. Chem. Lett., 2011, 21, 6491–6494 CrossRef CAS PubMed.
- W. Xie, G. Tanabe, J. Akaki, T. Morikawa, K. Ninomiya, T. Minematsu, M. Yoshikawa, X. Wu and O. Muraoka, Bioorganic Med. Chem., 2011, 19, 2015–2022 CrossRef CAS PubMed.
- L. Ren, X. Qin, X. Cao, L. Wang, F. Bai, G. Bai and Y. Shen, Protein Cell, 2011, 2, 827–836 CrossRef CAS PubMed.
- R. Eskandari, K. Jones, K. Ravinder Reddy, K. Jayakanthan, M. Chaudet, D. R. Rose and B. M. Pinto, Chem.–Eur. J., 2011, 17, 14817–14825 CrossRef CAS PubMed.
- B. H. Lee, R. Eskandari, K. Jones, K. R. Reddy, R. Quezada-Calvillo, B. L. Nichols, D. R. Rose, B. R. Hamaker and B. M. Pinto, J. Biol. Chem., 2012, 287, 31929–31938 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |