
Oxygen activation by copper camphor complexes†
Alexandra P. S.
Roseiro
a,
Pedro
Adão
a,
Adelino M.
Galvão
a,
João
Costa Pessoa
a,
Ana M.
Botelho do Rego
b and
M. Fernanda N. N.
Carvalho
*a
aCentro de Química Estrutural, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, 1049-001 Lisboa, Portugal. E-mail: fcarvalho@ist.utl.pt
bCentro de Química-Física Molecular (CQFM) and Institute of Nanoscience and Nanotechnology (IN), Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, 1049-001 Lisboa, Portugal
First published on 23rd September 2015
Abstract
Cleavage of the carboximide CN bond promoted by reaction of camphor carboximide hydrochlorides (OC10H15CONHCH2COOLi(H2O)2·2HCl (2) or OC10H15CONHCH(CH2Ph)COOLi(H2O)·HCl (3)) with CuCl2 leads to the corresponding amino acid complexes ([Cu(H2NCH2COO)2] (I) and [Cu{H2NCH(CH2Ph)COO}2]·(H2O) (II)), and the camphor carboxylate residue (OC10H15COO−) which is catalytically oxidized to camphorquinone by oxygen from air. The process is mediated by coordination of the camphor carboxylate to copper. The reaction of the camphor carboximide hydrochloride (OC10H15CONH(CH2)2COOLi(H2O)·HCl) (4) with CuCl2 follows a different trend. In this case the CN camphor carboximide bond remains intact and the complexes [CuCl{OC10H15CONH(CH2)2COOLi(H2O)}] (III) and [{CuCl2}2{OC10H15CONH(CH2)2COOLi(H2O)}] (IV) form. However, a redox process also occurs, since formation of III requires Cu(II) → Cu(I) reduction as confirmed by X-ray photoelectron spectroscopy and cyclic voltammetry. A related reduction process was identified in the formation of [CuCl(OC10H15COOLi)] (V) from CuCl2 upon reaction with lithium camphor carboxylate (OC10H15COOLi) under nitrogen. The above results show that electron transfer is highly facilitated in the copper camphor carboximide/carboxylate system. Such an ability was used to activate oxygen from air and promote the oxidation of ethylacetoacetate to pyruvate using V as the catalyst.
Introduction
Copper compounds play a role in areas so diverse as catalysis,1–8 biological applications,9–15 food and materials related processing,16,17 waste treatment18 or agriculture (e.g. CuSO4 in vineyards). Such a wide scope of applications impelled us to synthesize and study the properties of copper camphor complexes, with the focus on Cu(I) complexes.19–21 Cu(I) to Cu(II) oxidation by air is a common drawback when doing Cu(I) chemistry. However, in reactions of CuCl with some camphor derivatives the process was found to enable oxidation of organic substrates. Such findings challenged further studies in particular because enzymes such as tyrosinases, laccases or catechol oxidases activate oxygen from air forming peroxo or oxo Cu(II) intermediates in processes that range from oxygen release as H2O22 to oxidative oxygen transfer to organic substrates.23 The activation of molecular oxygen by copper compounds in biological processes was recently reviewed by Solomon et al.24 The challenge for non-enzymatic systems to enable activation of oxygen from air is to find ligands that are coordinative and electronically versatile to stabilize the metal site in different oxidation states to allow coordination and transfer of oxygen to organic substrates without decomposition of the catalyst.From previous studies we knew that camphor derived ligands are able to stabilize transition metals (Pd, Pt, Zn)25–28 towards electron reorganization in CC triple bonds, thus a step forward was to design camphor based ligands to obtain copper complexes that promote Cu(I) → Cu(II) → Cu(I) electron transfer at potentials suitable for oxygen activation. Herein the results show that the camphor carboximide and carboxylate copper systems fulfill the above requirements and catalytically enable oxygen activation and transfer to substrates.
Results and discussion
The synthesis of the lithium camphor carboximides hydrochlorides OC10H15CONHCHRCO2Li(H2O·HCl)x (R = H, x = 2; (2); R = CH2Ph, x = 1 (3)) and OC10H15CONH(CH2)2CO2Li(H2O·HCl) (4) is based on ketone and amine condensation upon reaction of lithium camphor carboxylate (1) with α-amino acids (H2NCH(R)COOH)(R = H or CH2Ph) or β-alanine (eqn (1)). In contrast, no condensation of amino acids (glycine, L-phenylalanine or β-alanine) with camphor carboxylic acid (OC10H15COOH, 5) was observed, neither did condensation of amino acids with camphor or camphorquinone, in agreement with high relevance of the lithium ion for the processes.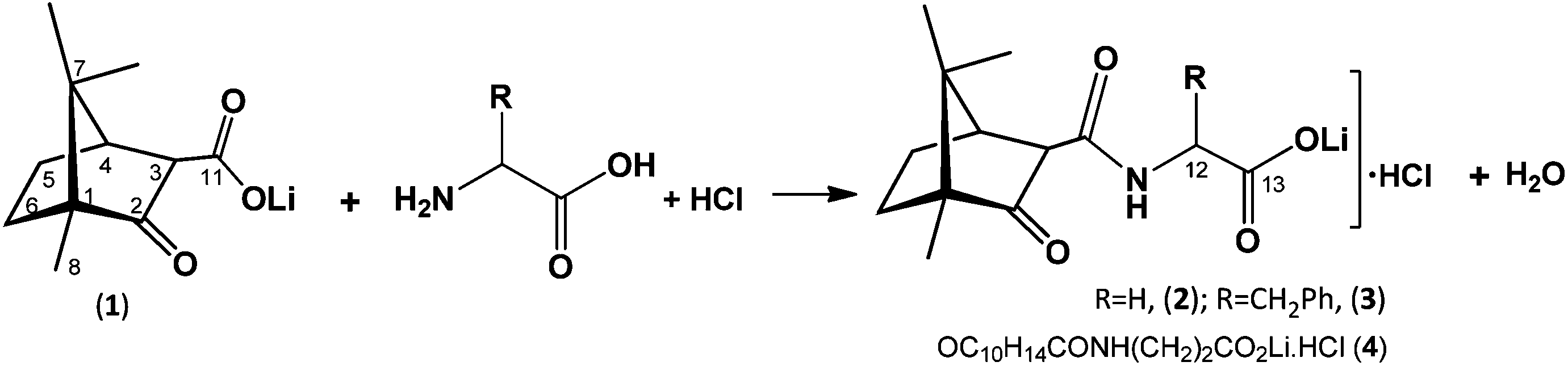 | (1) |
The new compounds (2, 3 and 4) were formulated based on the elemental analysis, FTIR and NMR (1H, 13C, DEPT, 2D). Characteristic camphor (ν(CO)ca. 1730 cm−1) carboxylate bands (and ν(COO)ca. 1600 cm−1) were observed by FTIR. Data from NMR confirms the condensation of the camphor carboxylate (OC10H15COOLi, 1) with the α-amino acids (glycine R = H, L-phenylalanine R = CH2Ph) or β-alanine (H2NCH2CH2COOH) (see the Experimental section).
The structural characteristics of the camphor carboximides (2–4) favor coordination to transition metals through one, two or more atoms enabling different hapticities which render them high attractive ligands to stabilize transition metals in different oxidation states.
Reactions of lithium 3-camphor carboximides with CuCl2
The lithium camphor carboximides (OC10H15CONHCH(R)CO2Li·HCl) (2 and 3) react with CuCl2 (in EtOH, under air) affording the amino acid complexes [Cu(NH2CHRCOO)2]·xH2O (R = H, I; R = CH2Ph, II). In the process, cleavage of the carboximide CN bond and lithium camphor carboxylate (1) release occur, concomitant to the formation of the amino acid complexes (eqn (2)).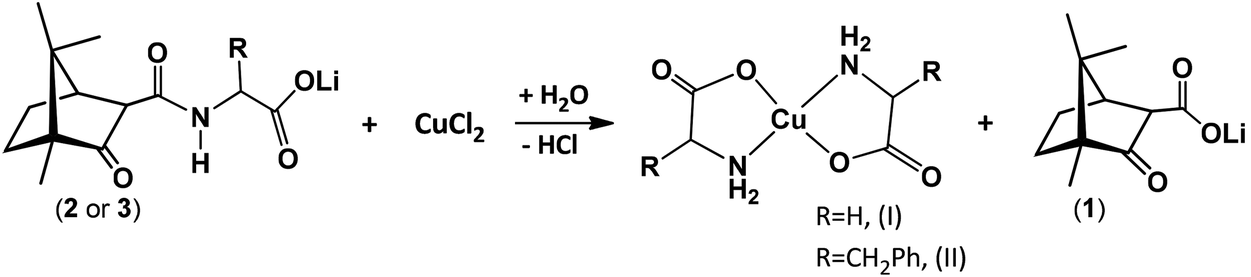 | (2) |
A first observation is that the reaction proceeds in the absence of an external base (e.g. NaOH) due to the basic character of 1, in contrast with the typical syntheses of Cu(II) amino acid complexes that require the presence of a strong base (e.g. NaOH). A second and more relevant aspect is that the camphor carboxylate (OC10H15COO−) is oxidized to camphorquinone (6, OC10H14O) (eqn (3)) without the presence of an oxidizer other than oxygen from air.
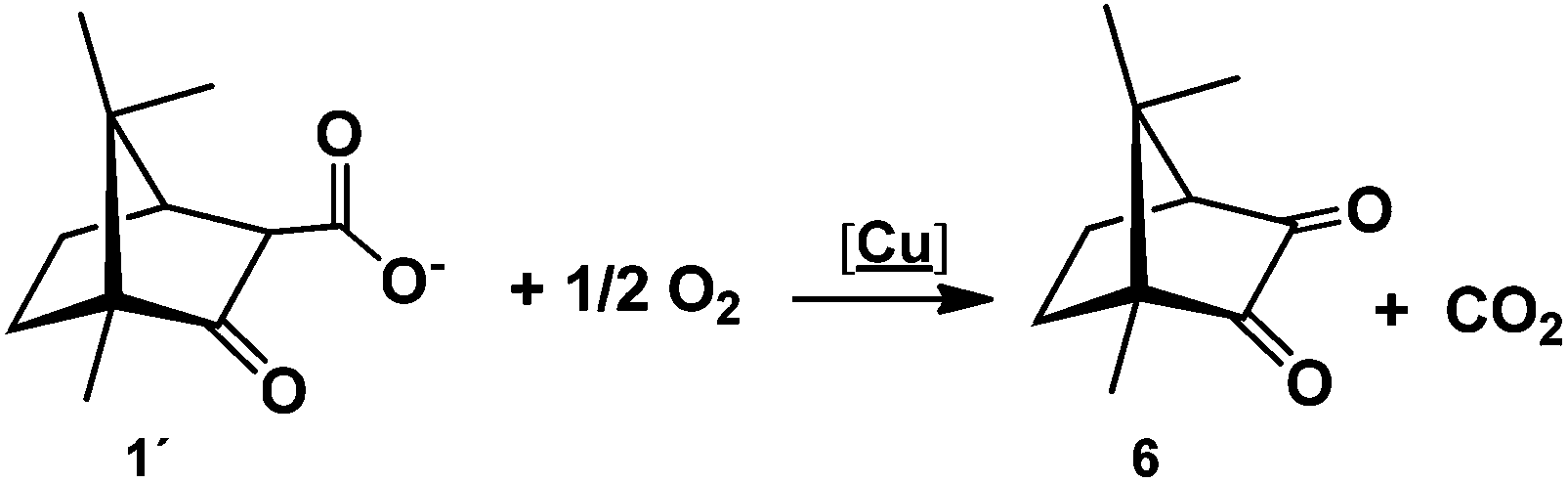 | (3) |
The amount of camphorquinone increases with increase of the excess of the camphor carboximide (2, 3) or camphor carboxylate suggesting a catalytic process (Scheme 1).
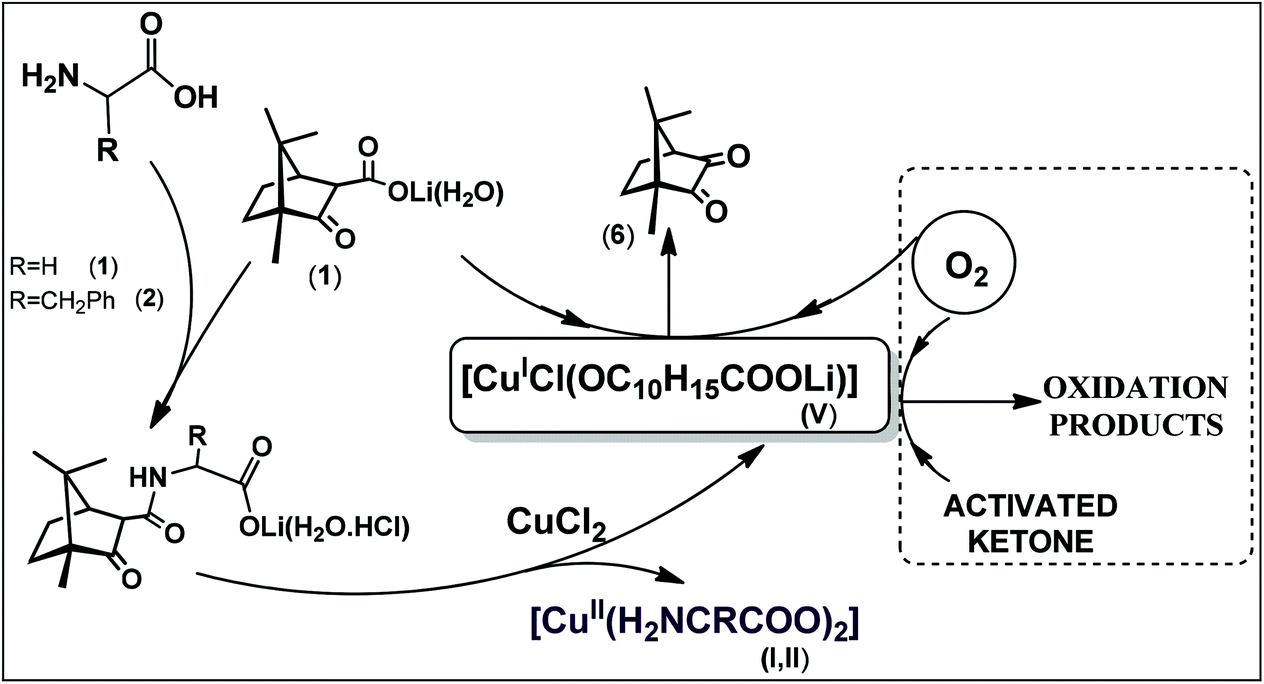 | ||
| Scheme 1 Proposed outline for formation of V and products of catalytic activation of oxygen. | ||
The analytical and spectroscopic data obtained for complexes I and II as well as for camphorquinone agree well with the former published data,29–32 thus corroborating their formulation.
In contrast with 2 and 3, lithium camphor carboximide hydrochloride 4 reacts with CuCl2 affording [CuCl{OC10H15CONH(CH2)2COOLi(H2O)}] (III) and [{CuCl2}2{OC10H15CONH(CH2)2COOLi(H2O)}] (IV) that keep intact the camphor carboximide ligand. Formation of III (low yield) requires Cu(II) → Cu(I) reduction, a process which, in the absence of a reducing agent, was unexpected. Oxidation of the chloride co-ligand (Cl−/Cl2) (as found in other copper systems33) or amino acid oxidation (Strecker's type) could be the source electrons.
The Cu(I) character of III enables acquisition of the 1H NMR spectrum (in the usual region, see the Experimental section) that was further confirmed by XPS and cyclic voltammetry (see below).
In IV, the metal site behaves as Cu(II). However, exposure to air yields a gummy solid with a strong smell, from which the Cu(I) complex [CuCl(OC10H15COOLi)]·Et2O (V·Et2O) was obtained. The fate of the amino acid residue (not found in the isolated products) conceivably are the volatile smelly products (aldehyde, ammonia) formed through a Strecker's type amino acid degradation process.34–36 In this case, oxidation of the amino acid, chloride or both provide the electrons for Cu(II) → Cu(I) reduction. This process was not further studied. Compound V·Et2O is not stable in air for long periods. The related compound [CuCl(OC10H15COOLi)]·THF (V·THF) that differs from (V·Et2O) in the solvent, is straightforwardly obtained upon reduction of CuCl2 by the lithium camphor carboxylate (1) in THF. Under nitrogen, V·Et2O remains stable for long periods. In air CuCl2 reacts with 1 to form the Cu(II) compound [CuCl2(OC10H15COOLi)]·2H2O (VI). Differences between V (pale green) and VI (blue) include color and spectroscopic properties displayed by IR [V, 1732 (νCO), 1623 (νCOO) cm−1; VI, 1712 (νCO), 1587 (νCOO) cm−1], EPR (VI typical spectra, V EPR silent) and NMR (V typical spectra, VI no spectra).
Reactions of lithium 3-camphor carboximides with CuCl
To check whether the camphor carboximide Cu(I) complexes could be obtained directly from CuCl, the reactions with camphor carboximides (3 and 4) were undertaken in THF under nitrogen. From 3, [CuCl{OC10H15CONHCH(CH2Ph)COOLi}] (VII) was obtained; this is formally similar to III obtained from the reaction of 4 with CuCl2 (see above). From the reaction of CuCl with 4, the copper amino acid complex [{CuCl}2(H2NCH2CH2COOH)] (VIII) was obtained that requires breaking of the CN carboximide bond, such as in the cases of complexes I and II obtained from CuCl2. Complex VIII differs from I and II in what concerns the number of copper atoms per ligand which is two and one, respectively. The analytical composition of VIII suggests a zig-zag 1D-coordination polymer structure such as [{CuCl}2(YNC10H14O)] (Y = NMe2, NH2, NH2C6H4) obtained from the reaction of camphor hydrazones with CuCl.37Exposure of VIII to air (solution in DMSO) afforded blue paramagnetic crystals that were structurally characterized as trans-[Cu(H2NCH2CH2COO)2(H2O)2]·4H2O (IX, Fig. 1) by single crystal X-ray diffraction (XRD). It is worthy to note that VIII and IV (see above) that have in common the metal to ligand ratio (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), rearrange to mononuclear species.
:1), rearrange to mononuclear species.
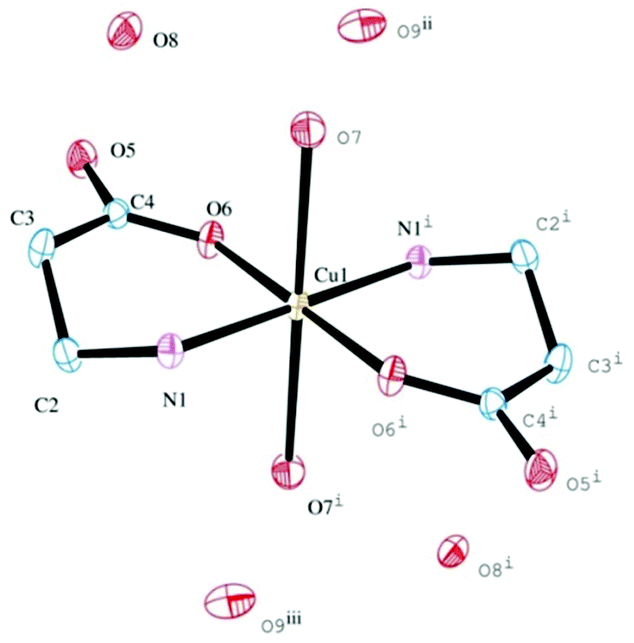 | ||
| Fig. 1 ORTEP drawing for IX, showing labeling scheme. Ellipsoids are drawn at 50% probability. Hydrogen atoms are omitted for clarity. Cu(1)–O(6) 1.9831(6); Cu(1)–N(1) 2.0010(6); N(1)–C(2) 1.480(1); C(2)–C(3) 1.517(2); C(3)–C(4) 1.514(1); C(4)–O(5) 1.256(1); C(4)–O(6) 1.272(1); O(6)–Cu(1)–N(1) 93.35(3); C(2)–N(1)–Cu(1) 116.21(5). | ||
A structural comparison between IX and the former reported L-alanine Cu(II) complex38 shows that an extended hydrogen bonding exists in IX (not observed before) which is responsible for a supramolecular arrangement formed by dyads of water molecules linking four molecules of the complex (Fig. 2, top). Crystal packing is displayed in Fig. 2 (bottom).
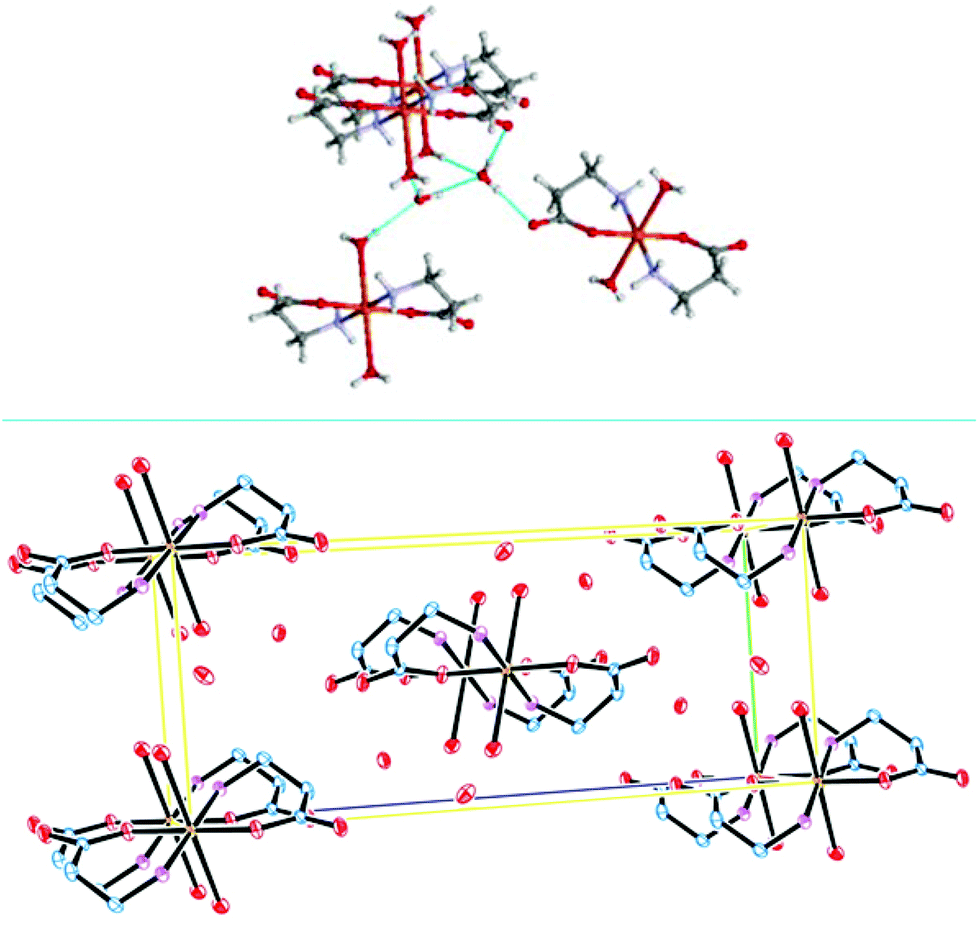 | ||
| Fig. 2 Crystal packing showing water dyads in the lattice of IX (bottom); supramolecular arrangement, showing hydrogen bonding (top). | ||
The camphor carboxylate (1) reacts with CuCl affording [{CuCl(OC10H15COOH)}] (X), which differs from V in the carboxylic residue (–COOX: X = H (X), X = Li (V). The higher acidity of CuCl solutions conceivably is responsible for Li/H exchange and isolation of X. Besides their apparently similar formula, the compounds have considerably distinct solubility (lower for V) and spectroscopic properties, namely, the IR stretching frequencies (νCOO: 1623 cm−1 (V); 1580 cm−1 (X)) as well as the 13C NMR resonances (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O: 215.0 ppm (V); 204.4 ppm (X), see the Experimental section for details).
O: 215.0 ppm (V); 204.4 ppm (X), see the Experimental section for details).
XPS
X-ray photoelectron spectroscopy (XPS) was used to ascertain the composition of selected compounds (II, III and VI) focusing on unraveling the oxidation state of the metal atom. The task may not be easy for complexes with transition metal sites other than copper, as the photoelectron binding energy is not enough to identify a specific oxidation state.39 However for copper the task becomes simplified since Cu(II) displays a Cu 2p region where a multiplet structure (due to spin–spin coupling) exists which is absent in the Cu(I) compounds. Fig. 3 displays the XPS Cu 2p region for the complexes under study. It is clear from this that the oxidation state of copper is one (+1) in III as attested by the absence of the multiplet structure. A peak width increase and appearance of the characteristic Cu(II) multiplet was observed upon exposure of III to air (Fig. 3, insert), in agreement with the Cu(I) → Cu(II) oxidation.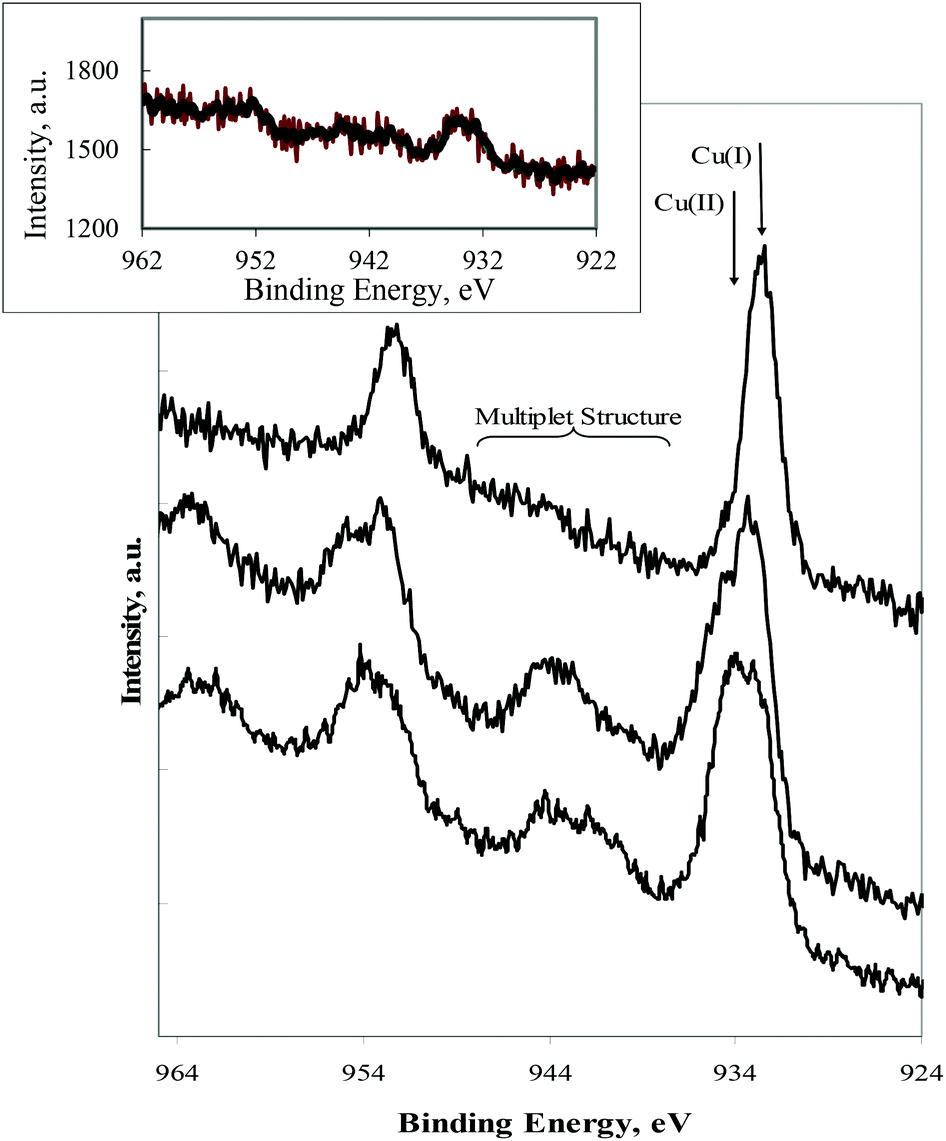 | ||
| Fig. 3 XPS Cu 2p (from bottom to top) for samples: II, VI, III (as prepared). Arrows indicate the Cu 2p3/2 doublet component assigned to the different features. Insert: XPS Cu 2p of III (upon exposure to air). | ||
The data obtained from complexes II and VI (undoubtedly Cu(II)) surprisingly display a combination of Cu(I) and Cu(II) oxidation states, attested by the large increase of the photoelectron peak width. The Cu(I) signals were attributed to Cu(II) → Cu(I) reduction by X-ray photoelectron radiation during data acquisition, a fact was also observed (the Cu(I) component increases) during acquisition of data from a sample of III intentionally oxidized. This aspect was not further explored, although a rather similar reduction process, from V(IV) to V(III), was recently reported by using intense synchrotron radiation during characterization of VIVO(dipicolinato)2–lysozyme adducts.40
EPR
Since no structural characterization could be made by single crystal XRD for most of the above mentioned complexes, EPR was used to get information on the structural arrangement and geometry of the selected Cu(II)-complexes (II and VI). Data measured in ethanol solutions confirmed the Cu(II) character of II and VI (Table 1) and structural information was also obtained from the calculated gz/Az ratio, which is considerably low for II (128.3 cm) in agreement with the amino acid acting as a N,O ligand bound to a square-planar Cu(II) metal center and considerably high for VI (184.1 cm) as expected for an O-ligand coordinated to a Cu(II) tetrahedral metal site.41| Complex | g x , gy | A x , Ay × 104 cm−1 | g z | A z × 104 cm−1 | g z /Az cm |
|---|---|---|---|---|---|
| a EPR spectra (measured at 77 K) were obtained from solutions of the complexes prepared under air at room temperature. b In ethanol. c In DMSO. d In ethanol/ethyleneglycol (1/1). | |||||
| II | 2.041, 2.060 | 20.3, 5.4 | 2.264 | 176.5 | 128.3 |
| VI | 2.076 | 5.7 | 2.375 | 129.0 | 184.1 |
| III ox | |||||
| (Major) | — | — | 2.315 | 162.0 | 143.0 |
| (Minor) | 2.270 | 130.0 | 174.6 | ||
| V ox | |||||
| (Major) | — | — | 2.311 | 161.8 | 142.8 |
| (Minor) | 2.370 | 130.0 | 182.3 | ||
Complexes III and V are diamagnetic, thus EPR silent. Nevertheless, there was curiosity to know how solutions of III and V would behave upon being exposed to air, due to their ability to activate oxygen from air. EPR spectra were then measured from solutions of III and V prepared under air. The first observation was that, as expected, the solutions did not remain EPR silent and at least two Cu(II) species formed in solution. A second observation is that the pattern of the hyperfine coupling constant Az in the two products of oxidation of III and V does not differ significantly. The more abundant species exhibit Az values (161.8 to 162.0 × 10−4 cm−1) considerably higher than the less abundant species (Az ≈ 130 × 10−4 cm−1) in both solutions (Table 1).
Unfortunately, the signals are too broad to allow unambiguous determination of the gx, and gy values. The relatively high gz/Az ratios (>140 cm, Table 1) in conjunction with their representation in the Peisach–Blumberg Az/gz correlation plots37 indicate a tetrahedral distortion in the Cu(II) metal centers both in the major and the minor oxidized species. In addition, the high gz/Az ratio (>170 cm) calculated for the major component of the oxidized solutions of III and V supports Cu(II) coordination of O-containing ligands to significantly distorted tetragonal metal centers. Data from EPR for the major component of the oxidized solutions of III and V does not differ appreciably from that obtained for VI suggesting similar compounds.
Cyclic voltammetry
The redox properties of Cu(I) (III, V) and Cu(II) (I, II, VI) complexes were studied by cyclic voltammetry under an inert atmosphere. Representative cyclic voltammograms are displayed in Fig. 4. For comparative purposes the behavior of CuCl2 was also studied. The cyclic voltammograms of III (Eox1/2 = 0.40 V, Fig. 4a) and V (Eox1/2 = 0.48 V) display quasi-reversible anodic waves consistent with Cu(I) → Cu(II) oxidation processes, followed by irreversible waves (Eoxp = 1.48 V, III; Eoxp = 1.60 V, V) at higher potentials in the range of potential of the second oxidation wave displayed by CuCl2 (Cu(II) → Cu(III), Eoxp = 1.60 V).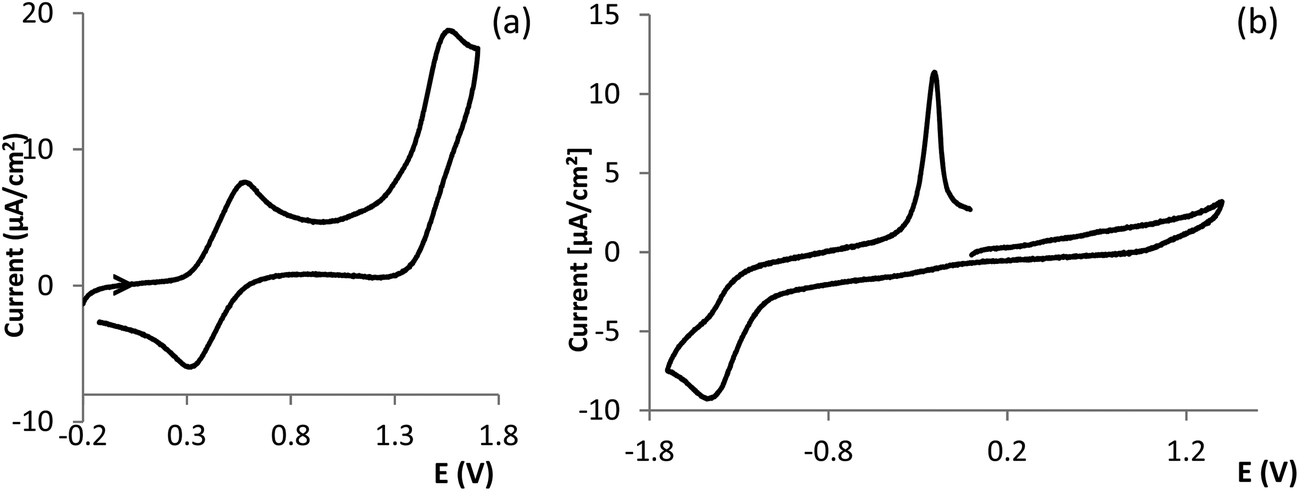 | ||
| Fig. 4 Cyclic voltammograms of complexes III (a) and VI (b) obtained under nitrogen in CH3CN. | ||
No anodic processes in the range of Cu(I) → Cu(II) oxidation were detected for complexes I, II, VI which display irreversible cathodic waves (Eredp = −0.60 V, I; Eredp = −0.57 V, II and Eredp = −1.43 V, VI) consistent with Cu(II) → Cu(I) reduction processes. The wide dispersion of potentials (Δ = 860 mV) is consistent with different geometries and binding atoms. The low reduction potential measured for VI (Fig. 4b) corroborates the tetrahedral distorted geometry of the copper center as ascertained by EPR and the considerably higher potential confirms the square planar geometry for complex II. In Cu(II) complexes the redox potentials may be used as a sensitive indication for geometry42 and structural characteristics (e.g. N or O coordinating atoms).43,44
It is worth highlighting that the potential of the reduction Cu(II) → Cu(I) in CuCl2 is just slightly higher than that of the Cu(I) → Cu(II) oxidation in complexes III or V, pointing to facile electron transfer between them.
Catalysis – oxidation of 1,3-dicarbonyls
The oxidation of ethylacetoacetate was used as a model to extend to substrates other than camphor carboxylate, the ability of [CuCl(OC10H15COOLi)] (V) to catalyze the oxidation of 1,3-dicarbonyls using oxygen from air. The rationale behind this study is that the camphor carboxylate or camphor carboximide compounds used as ligands throughout this work are formally 1,3-dicarbonyls which are easily enolizable under the appropriate conditions and prone to oxidation at the α-carbon45–49 forming α-dicarbonyls or α-hydroxycarbonyls.50 Formation of such types of products from ethylacetoacetate by reaction with V, in the absence of oxidizing agents other than air, was used to corroborate the catalytic activity of V towards oxidation processes using molecular oxygen as the oxygen source. In the case of ethylacetoacetate the expected products are pyruvate (CH3COCOO− formed by α-oxygenation) or hydroxyacetone (CH3COCH2OH, which could be further oxidized to pyruvate). In fact, upon addition of ethylacetoacetate to a solution of V it changed from pale green to orange-brown and back to green upon quenching with acetic acid. This color alteration suggests copper oxidation state exchange in solution. The evolution of gas upon addition of acetic acid to quench the reaction mixture is attributed to CO2 formed by decarboxylation of acetoacetate. The outcome of the reaction was analyzed by 1H NMR, 13C NMR and HSQC. By 1H NMR a signal at 2.18 ppm (not observed in the reagents) evidences the formation of a new compound, that was assigned to pyruvate (CH3COCOO−, ca. 55% yield) according to integration of the methyl groups. The presence of hydroxyacetone (CH3COCH2OH) or methylglyoxal (CH3COCHO) was excluded, since no correlation signals were observed in the HSQC spectrum attributable to CH2 or CH groups respectively.Formation of pyruvate from ethylacetoacetate thus corroborates molecular oxygen activation catalyzed by V as found in the case of oxidation of camphor carboxylate to camphorquinone (Scheme 1).
Oxidases, such as tyrosinases activate molecular oxygen towards phenol to quinone oxidation1,2 through coordination to copper, such coincidence is a challenge to pursue the study of copper camphor carboxylate/carboximide systems.
Conclusions
A variety of complexes were obtained from the reaction of lithium camphor carboximide hydrochlorides (2, 3 and 4) with Cu(II) and Cu(I) chlorides. In reactions with CuCl2, two distinct situations were found. In one, the CN carboximide bond (–C(O)NHCHRCOOLi; 2, 3) breaks and copper amino acid complexes [Cu(NH2CHRCOO)2] (I, II) and camphorquinone are obtained. In the other the camphor carboximide ligand (–C(O)NHCHRCOOLi; 4) remains intact and the Cu(I) complex [CuCl{OC10H15CONH(CH2)2COOLi(H2O)}] (III) unexpectedly forms, in addition to the dinuclear complex [{CuCl2}2{OC10H15CONH(CH2)2COOLi(H2O)}] (IV) where no reduction of the metal is evident. However, upon exposure of IV to air bad smelling vapors evolve (aldehyde, NH3, other) due to oxidation of the amino acid ligand (Strecker type) with concomitant formation of [CuCl(OC10H15COOLi)] (V) where the metal has a Cu(I) character.The Cu(II) → Cu(I) reduction and the camphorquinone formation by oxidation of camphor carboxylate are processes that require metal mediated electron transfer which are believed to be facilitated by the close potentials measured for the oxidation of camphor carboximide (III, Eox1/2 = 0.40 V) or camphor carboxylate complexes (V, Eox1/2 = 0.48 V) and reduction of CuCl2 (Ered1/2 = 0.50 V).
In camphorquinone formation, oxygen from air is activated by the copper camphor carboxylate/carboximide system as corroborated by formation of pyruvate from ethylacetoacetate catalyzed by [CuCl(OC10H15COOLi)] (V) in the absence of oxidizers other than oxygen from air.
From reactions of CuCl with lithium camphor carboxylate or lithium camphor carboximides, Cu(I) or Cu(II) complexes were obtained, although activation of oxygen from air was inhibited, since the reactions were carried out under an inert atmosphere to avoid copper oxide formation.
Experimental section
Complexes were prepared under a nitrogen atmosphere using vacuum/Schlenk techniques. In some cases, solutions obtained upon separation of the complexes by filtration were exposed to air. Anhydrous CuCl2 was purchased from Merck. CuCl was synthesized from CuCl2 by reduction with sodium sulphite.51 Compound 1 was prepared from camphor (Aldrich) following the procedure in ref. 52. The solvents (p.a. grade) were from Sigma-Aldrich or Panreac (EtOH). EtOH was placed over molecular sieves. IR spectra were obtained from KBr pellets or Nujol mulls using a JASCO FT/IR 4100 spectrometer. NMR spectra (1H, 13C, DEPT, HSQC and HMBC) were obtained from D2O or DMSO-d6 solutions using Bruker Avance II+ (300 or 400 MHz) spectrometers. NMR chemical shifts are referred to TMS (δ = 0 ppm). ESI mass spectra were obtained using a 500-MS Ion Trap (Varian Inc., Palo Alto, CA, USA) mass spectrometer operating in the positive ion mode. The XPS data was obtained using a Kratos XSAM800 equipment and the electron paramagnetic resonance (EPR) spectra were obtained from frozen samples at 77 K using a DPPH radical as a reference and a Bruker ESP 300E X-band spectrometer. The measured 1st derivative X-band EPR was simulated with the EPR simulation software developed by Rockenbauer and Korecz.53 Cyclic voltammograms (CV) were obtained from solutions in CH3CN of Bu4NBF4 (0.1 M) using a three compartment cell equipped with a glassy carbon working electrode interfaced with a VoltaLab PST050 equipment. The potentials were measured at 200 mV s−1 in volts (±10 mV) versus SCE being used as internal reference, [(Fe(η5-C5H5)2)] (E1/2red = 0.382 V (CH3CN)).Synthesis of amino acid camphor derivatives
Synthesis of copper complexes
From the solution upon exposure to air yellow crystals precipitated that were identified as camphorquinone comparing the IR and NMR spectra with the published data.54 (Yield, ca. 15%, based on 2.)
Alternative procedure: Glycine (0.10 g, 1.3 mmol) and Cu(CH3COO)2 (0.12 g, 0.63 mmol) were dissolved in EtOH (10 mL) and stirred overnight. A blue precipitate formed that was filtered, washed with ethanol and dried under vacuum. Yield: 66%.
Alternative procedure: Phenylalanine (0.28 g, 1.7 mmol) and CuCl2 (0.10 g, 0.77 mmol) were dissolved in deoxygenated water (2 mL) and stirred for 1½ h. The blue precipitate was filtered, washed with cold water and dried under vacuum. Yield: 32%.
By taking the solution to dryness a precipitate was obtained that was washed with EtOH (2 × 3 mL) and dried to afford [{CuCl2}2{OC10H15CONH(CH2)2COOLi(H2O)}] (IV) as a green-blue powder. Yield: 46%; IR (nujol) (cm−1): 1733 (νCO), 1627, 1597 (νCOO). Elemental analysis (%) for Cu2Cl4C14H20NO4Li·H2O·½EtOH, Found: C 30.5, N 2.0, H 4.7; Calc.: C 30.9, N 2.4, H 4.3. Exposure of IV to air, undergoes formation of a smelly gummy solid that was pumped dry (under vacuum) and washed with Et2O, affording the blue precipitate [CuCl(OC10H15COOLi)]·Et2O (V·Et2O). (IR (cm−1): 1733 (νCO), 1626 (νCOO) and elemental analysis (%) for CuClC11H15O3Li·Et2O, Found: C 48.8, H 6.5; Calc.: C.48.9, H 6.8.
A progressive disappearance, during spectra acquisition (MeOH-d4) of the hydrogen atom at position 3, due to deuteration and the broadening of atom C11 (see eqn (3) for labeling) precluding detection by 13C NMR (as in V) is a characteristic behavior of 1.40 Nevertheless, no doubts exist concerning C-11 in the molecule, since the carboxylate stretch (νCOO, 1623 cm−1) is observed by FTIR.
Blue crystals were obtained from DMSO solution under air that were filtrated and dried. Yield 50%. ESI-MS (MeOH/DMSO): m/z 308, [(IX-4H2O)+ H + MeOH]+. IR (cm−1): 3403 (νOH), 3308 (νNH), 1589, 1555 (νCO). Structural analysis by single crystal X-diffraction confirmed trans-[Cu(H2NCH2CH2COO)2(H2O)2]·4H2O (IX) was obtained.
Catalysis
The typical procedure was: ethyl acetoacetate (1 mmol) was diluted with a mixture (1:1) of ethanol/water (4 mL) and triethylamine (2 mmol) was added to favour the enolization. Then the catalyst was added (1%, V) and the mixture stirred at 40 °C in a vented screw-cap glass vessel, under air for 24 h. Then, the reaction was quenched with 2 mmol of acetic acid and the mixture stirred for an additional 24 h. The reaction products of were analyzed by NMR (1H, 13C, HSQC), using toluene as the internal standard.
XPS analysis
Samples were irradiated with the unmonochromatic Al Kα radiation. Experimental conditions and details about data treatment were the same as described elsewhere.33 Sensitivity factors used here were: C 1s: 0.25; O 1s: 0.66; N 1s: 0.42; Cu 2p3/2: 4.2; Cl 2p: 0.61.Single-crystal X-ray diffraction analysis
Single crystal XRD analysis on [Cu(H2NCH2CH2COO)2(H2O)2]·4H2O (IX) was performed at 150(2) K on a Bruker AXS-KAPPA APEX II area detector apparatus using graphite-monochromated Mo Kα (λ = 0.71073 Å) and were corrected for Lorentz, polarization and empirically for absorption effects. The structure was solved by direct methods using SHELX9755 and refined by full matrix least squares against F2 using SHELX97 all included in the suit of programs WinGX v1.70.01 for Windows.56 Non-hydrogen atoms were refined anisotropically and H atoms were inserted in idealized positions and allowed refine riding on the parent carbon atom. Crystal data and refinement parameters are summarized in Table 2. Illustrations of the molecular structures were made with ORTEP3.57| Empirical formula | C6H24N2O10Cu |
| Formula weight | 347.81 |
| Crystal system | Monoclinic |
| Space group | P21/c |
| Unit cell dimensions | |
| a/Å | 5.5238(2) |
| b/Å | 7.5320(3) |
| c/Å | 17.7276(7) |
| α/° | 90 |
| β/° | 90.499(2) |
| γ/° | 90 |
| Volume (Å−3) | 737.53(5) |
| Z, Dcal (g cm−3) | 2, 1.566 |
| Absorption coefficient (mm−1) | 1.526 |
| F(000) | 366 |
| Crystal size (mm3) | 0.2 × 0.3 × 0.3 |
| θ range for data collection (°) | 2.94 to 38.62 |
| Index ranges | — |
| Reflections collected/unique | 9 ≤ h ≤ 9, −13 ≤ k ≤ 13, −30 ≤ l ≤ 1 |
| Data/restraints/parameters | 9 |
| Final R (observed) | 16343/4168 [R(int) = 0.0407] |
| 4168/0/136 | |
| R 1 = 0.025 wR2 = 0.065 |
CCDC 1030294 contains the supplementary crystallographic data for IX.
Acknowledgements
Financial support by FCT-Fundação para a Ciência e Tecnologia (UID/QUI/00100/2013, RECI/QEQ-MED/0330/2012 and RECI/QEQ-QIN/0189/2012) and the NMR and MS Networks (IST-UTL Nodes) for facilities.References
- M. Remko, D. Fitz, R. Broer and B. M. Rode, J. Mol. Model, 2011, 17, 3117–3128 CrossRef CAS PubMed.
- P. Adão, S. Barroso, F. Avecilla, M. C. Oliveira and J. Costa Pessoa, J. Organomet. Chem., 2014, 760, 212–223 CrossRef PubMed.
- M. S. Aljahdali, A. T. Abedelkarim, A. A. El-Sherif and M. M. Ahmed, J. Coord. Chem., 2014, 67, 870–790 CrossRef CAS PubMed.
- A. A. El-Sherif and M. S. Aljahdal, J. Coord. Chem., 2013, 66, 3423–3468 CrossRef CAS PubMed.
- Y. I. Slyvka, A. V. Pavlyuk, B. R. Ardan, N. T. Pokhodilo, E. A. Goreshnik and P. Y. Demchenko, Russ. J. Inorg. Chem., 2012, 57, 815–821 CrossRef CAS.
- M. Yoshida, T. Shimada, T. Ishida and T. Kogane, Polyhedron, 2013, 66, 75–80 CrossRef CAS PubMed.
- S. Meghdali, M. Amirnasr, Z. Azarkamanzad, K. S. Joβ, F. Fadaee, A. Amiri and S. Abbasi, J. Coord. Chem., 2013, 66, 4330–4343 CrossRef PubMed.
- R. P. Houser, Z. Wang, D. Powell and T. J. Hubin, J. Coord. Chem., 2013, 66, 4080–4092 CrossRef CAS PubMed.
- M. Rolff, J. Schottenheim, H. Decker and F. Tuczek, Chem. Soc. Rev., 2011, 40, 4077–4098 RSC.
- L. Yang, J. Guoxiang, N. Jungfeng, W. Ying and H. Ljuan, Progr. Chem., 2009, 21, 2028–2036 Search PubMed.
- M. Patil, R. Hunoor and K. Gudasi, Eur. J. Med. Chem., 2010, 45, 2891–2986 Search PubMed.
- X. Qiao, Z.-Y. C.-Z. MaXie, F. Xue, Y.-W. Zhang, J.-Y. Xu, Z.-Y. Qiang, J.-S. Lou, G.-J. Chen and S.-P. Yan, J. Inorg. Biochem., 2011, 105, 728–737 CrossRef CAS PubMed.
- S. Thalamuthu, B. Annaraj and M. A. Neelakantan, Spectrochim. Acta, Part A, 2014, 118, 120–129 CrossRef CAS PubMed.
- A. Stănilă, C. Braicu and S. Stănilă, Pop, R.M. Not. Bot. Horti Agrobo, 2011, 39, 124–129 Search PubMed.
- A. Ganguly, P. Chakraborty, K. Banergee and S. K. Choudhuri, Eur. J. Pharm. Sci., 2014, 51, 96–109 CrossRef CAS PubMed.
- G. Faccio, K. Kruus, M. Saloheimo and L. Thöny-Meyer, Proc. Biochem., 2012, 47, 1749–1760 CrossRef CAS PubMed.
- P. Demarche, C. Junghanns, R. R. Nair and S. N. Aghatos, Biotechnol. Adv., 2012, 30, 933–953 CrossRef CAS PubMed.
- N. Durán, M. A. Rosa, A. D́Annibale and L. Gianfreda, Enzyme Microb. Technol., 2002, 31, 907–931 CrossRef.
- M. F. N. N. Carvalho, M. T. Duarte, T. A. Fernandes, A. M. Galvão and A. M. Botelho do Rego, Inorg. Chem., 2010, 49, 10330–10337 CrossRef CAS PubMed.
- T. A. Fernandes, M. F. N. N. Carvalho, A. M. Galvão, N. A. G. Bandeira, M. J. Calhorda and A. M. Botelho do Rego, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1102–1110 CrossRef CAS PubMed.
- T. A. Fernandes, F. Mendes, A. P. S. Roseiro, I. Santos and M. F. N. N. Carvalho, Polyhedron, 2015, 87, 215–219 CrossRef CAS PubMed.
- S. Kakuda, R. L. Peterson, K. Ohkubo, K. D. Karlin and S. Fukuzumi, J. Am. Chem. Soc., 2013, 135, 6513–6522 CrossRef CAS PubMed.
- C. Zhang, P. Feng and N. Jiao, J. Am. Chem. Soc., 2013, 135, 15257–15262 CrossRef CAS PubMed.
- E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. Kieber-Emmons, C. H. Kjaergaard, R. G. Hadt and L. Tian, Chem. Rev., 2014, 114, 3659–3853 CrossRef CAS PubMed.
- M. F. N. N. Carvalho, A. S. D. Ferreira and A. M. Galvão, Inorg. Chim. Acta, 2010, 363, 1767–1772 CrossRef CAS PubMed.
- M. F. N. N. Carvalho, A. M. Galvão and A. S. D. Ferreira, J. Organomet. Chem., 2009, 694, 2061–2068 CrossRef CAS PubMed.
- M. F. N. N. Carvalho, F. M. T. Almeida, A. M. Galvão and A. J. L. Pombeiro, J. Organomet. Chem., 2003, 679, 142–149 CrossRef.
- T. A. Fernandes, A. M. Ferraria, A. M. Galvão, A. M. Botelho do Rego, A. C. M. Suárez and M. F. N. N. Carvalho, J. Organomet. Chem., 2014, 760, 186–196 CrossRef CAS PubMed.
- http://wwwchem.uwimona.edu.jm/lab_manuals/c31lex1.html .
- A. Stănilă, A. Marcu, D. Rusu, M. Rusu and L. David, J. Mol. Struct., 2007, 834–836, 364–368 CrossRef PubMed.
- K. Tomita and I. Nitta, Bull. Chem. Soc. Jpn., 1961, 34, 286–291 CrossRef.
- H. C. Freeman and M. R. Snow, Acta Crystallogr., 1964, 17, 1463–1470 CrossRef CAS.
- Md. M. Hossain, M.-C. Tseng, C.-R. Lee and S.-G. Shyu, Eur. J. Inorg. Chem., 2014, 36–40 CrossRef CAS PubMed.
- G. P. Rizzi, Food Rev. Int., 2008, 24, 416–435 CrossRef CAS PubMed.
- J. Strecker, Liebigs Ann. Chem., 1862, 123, 363–365 CrossRef PubMed.
- A. Schönberg and R. Moubasher, Chem. Rev., 1952, 50, 261–227 CrossRef.
- M. F. N. N. Carvalho, M. T. Duarte, T. A. Fernandes, A. M. Galvão and A. M. Botelho do Rego, Inorg. Chem., 2010, 49, 10330–10337 CrossRef CAS PubMed.
- A. Dijkstra, Acta Crystallogr., 1966, 20, 588–590 CrossRef CAS.
- A. M. Ferraria, A. M. Botelho do Rego and A. P. Carapeto, Vacuum, 2012, 86, 1988–1991 CrossRef CAS PubMed.
- M. F. A. Santos, I. Correia, A. R. Oliveira, E. Garribba, J. Costa Pessoa and T. Santos-Silva, Eur. J. Inorg. Chem., 2014, 3293–3297 CrossRef CAS PubMed.
- U. Sakaguchi and A. W. Addison, J. Chem. Soc., Dalton Trans., 1979, 600–608 RSC.
- G. Tabbi, A. Giuffrida and R. P. Bonomo, J. Inorg. Biochem., 2013, 128, 137–145 CrossRef CAS PubMed.
- S. Meghadi, K. Mereiter, V. Langer, A. Amiri, R. S. Erami, A. A. Massoud and M. Amirnasr, Inorg. Chim. Acta, 2012, 385, 31–38 CrossRef PubMed.
- G. T. Musie, X. Li and D. R. Powell, Inorg. Chim. Acta, 2004, 357, 1134–1136 CrossRef CAS PubMed.
- S.-H. Caoa and M. Shi, Tetrahedron: Asymmetry, 2010, 21, 2675–2680 CrossRef PubMed.
- D. Li, K. Schröder, B. Bitterlich, M. K. Tse and M. Beller, Tetrahedron Lett., 2008, 49, 5976–5979 CrossRef CAS PubMed.
- A. P. Rodrigues, L. Marcos da Fonseca, O. M. de Faria Oliveira, I. L. Brunetti and V. F. Ximenes, Biochim. Biophys. Acta, 2006, 1760, 1755–1761 CrossRef CAS PubMed.
- T. Watanabe and T. Ishikawa, Tetrahedron Lett., 1999, 40, 7795–7798 CrossRef CAS.
- F. A. Davis, H. Liu, B.-C. Chen and P. Zhou, Tetrahedron, 1998, 10481–10492 CrossRef CAS.
- N. Durán, M. A. Rosa, A. D'Annibale and L. Gianfreda, Enzyme Microb. Technol., 2002, 31, 907–931 CrossRef.
- J. Tanaka and S. L. Suib, Experimental Methods in Inorganic Chemistry, Prentice Hall, Inc., London, U.K., 1999, 231 Search PubMed.
- A. S. D. Ferreira, J. Schulz, A. M. Galvão, A. P. S. Roseiro, P. Štěpnička, L. F. Veiros and M. F. N. N. Carvalho, J. Organomet. Chem., 2014, 760, 108–114 CrossRef CAS PubMed.
- A. Rockenbauer and L. Korecz, Appl. Magn. Reson., 1996, 10, 29–43 CrossRef CAS.
- http://sdbs.db.aist.go.jp/sdbs/cgi-bin/direct_frame_top.cgi .
- G. M. Sheldrick, SHELX-97- Programs for Crystal Structure Analysis (release 97–2), Institüt für Anorganische Chemie der Universität, Tammanstrasse 4, D-3400 Göttingen, Germany, 1998 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837 CrossRef CAS.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
Footnote |
| † CCDC 1030294. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qi00064e |
| This journal is © the Partner Organisations 2015 |