
Assembly of photoluminescent [CunIn] (n = 4, 6 and 8) clusters by clickable hybrid [N,S] ligands†
Shi-Qiang
Bai
*a,
Lu
Jiang
a,
Ai Lin
Tan
b,
Sing Chen
Yeo
b,
David James
Young
ac and
T. S.
Andy Hor
*ab
aInstitute of Materials Research and Engineering, A*STAR (Agency for Science, Technology and Research), 3 Research Link, Singapore 117602, Republic of Singapore. E-mail: bais@imre.a-star.edu.sg; andyhor@imre.a-star.edu.sg; Tel: +(65) 65141515
bDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, Singapore 117543, , Republic of Singapore. E-mail: andyhor@nus.edu.sg
cFaculty of Science, Health, Education and Engineering, University of the Sunshine Coast, Locked Bag 4, Maroochydore DC, Queensland 4558, Australia
First published on 7th September 2015
Abstract
Three new 1,2,3-triazole-based NS ligands, 2-((4-((benzylthio)methyl)-1H-1,2,3-triazol-1-yl)methyl)pyridine (L1), 2-((4-(2-(cyclopentylthio)ethyl)-1H-1,2,3-triazol-1-yl)methyl)pyridine (L2) and 2-((4-(2-(cyclopentylthio)ethyl)-1H-1,2,3-triazol-1-yl)methyl)quinoline (L3) and the corresponding copper(I)-iodide complexes [Cu4I4(L1)2] (1), [Cu6I6(L2)2] (2) and [Cu6I6(L3)2] (3A) have been prepared and characterized by single-crystal X-ray diffraction (XRD), powder XRD, photoluminescence spectroscopy and thermogravimetric analysis. Complexes 1, 2 and 3A exhibit stair-step [CunIn] (n = 4 and 6) cluster structures with supporting ligands L1, L2 and L3, respectively. Ligand L1 coordinates with a bidentate/monodentate binding mode in the [Cu4I4] cluster complex 1 using the pyridyl–triazole moiety and with a pendant –CH2SCH2Ph group. Increasing the length of the bridge from –CH2– in L1 to –C2H4– in L2 and L3 engages the S donor and these ligands coordinate using a bidentate/monodentate/monodentate mode supporting larger [Cu6I6] cluster complexes 2 and 3A. A kinetic product [Cu8I8(L3)2(CH3CN)2] (3B) was isolated from the reaction of L3 with CuI in CH3CN and the single-crystal X-ray structure indicates a rare discrete stair-step [Cu8I8] core supported by two L3 and two coordinated CH3CN solvates. The structure is further stabilized by intermolecular π⋯π stacking interactions in the lattice. Isolation of 1–3 provides a good demonstration of the use of multidentate and multifunctional hybrid ligands in supporting [CunIn] clusters of different sizes (n = 4, 6, 8). Ligands L1–L3 are blue emissive molecules. The corresponding complexes display strong blue (1 and 2) or remarkable yellow (3A) emissions between 500 and 700 nm in the solid state. The structures of sulfur-containing ligands and their copper-iodide complexes are described and discussed.
Introduction
Metal cluster complexes, with unique structures and properties blending mononuclear complexes and bulk solids, have attracted considerable attention in view of their myriad of structures and potential applications as conductive, catalytic, magnetic or optical materials.1–4 Copper(I)-iodide based complexes are particularly appealing because of their diverse cluster motifs and rich photoluminescence behaviour.5–8 Many copper(I) luminescent complexes with various supporting ligands have been reported, but the precisely controlled synthesis of such species is yet unrealized because of the numerous structural possibilities of copper(I)-iodide clusters.9–13 Systematic design and synthesis of supporting ligands continues to be an important strategy towards controlled assembly of functional coordination complexes and our tool of choice is the copper-catalyzed azide–alkyne cycloaddition (CuAAC) click reaction. The major benefit is ready access to hybrid 1,2,3-triazole ligands under mild conditions. The excellent tolerance of this reaction towards diverse alkyne and azide substrates permits access to a range of functional molecules.14–18 Sulfur-containing ligands have been applied to the construction of copper(I) iodide complexes with interesting crystal structures and promising luminescence properties.19–22 We have contributed to this field with a systematic study of luminescent and/or catalytically active copper(I) iodide complexes supported by hybrid ligands including pincer SNS and chelate-bridging NS-N ligands.23–31 In the present work, three new NS ligands with different substituent groups have been synthesized using CuAAC click reactions and then used in the tunable assembly of emissive copper(I) iodide complexes (Scheme 1). The sulfur donor groups can be pendant or coordinating, thus making them adaptive in supporting neutral stair-step [Cu4I4], [Cu6I6] and [Cu8I8] cluster complexes (1–3B).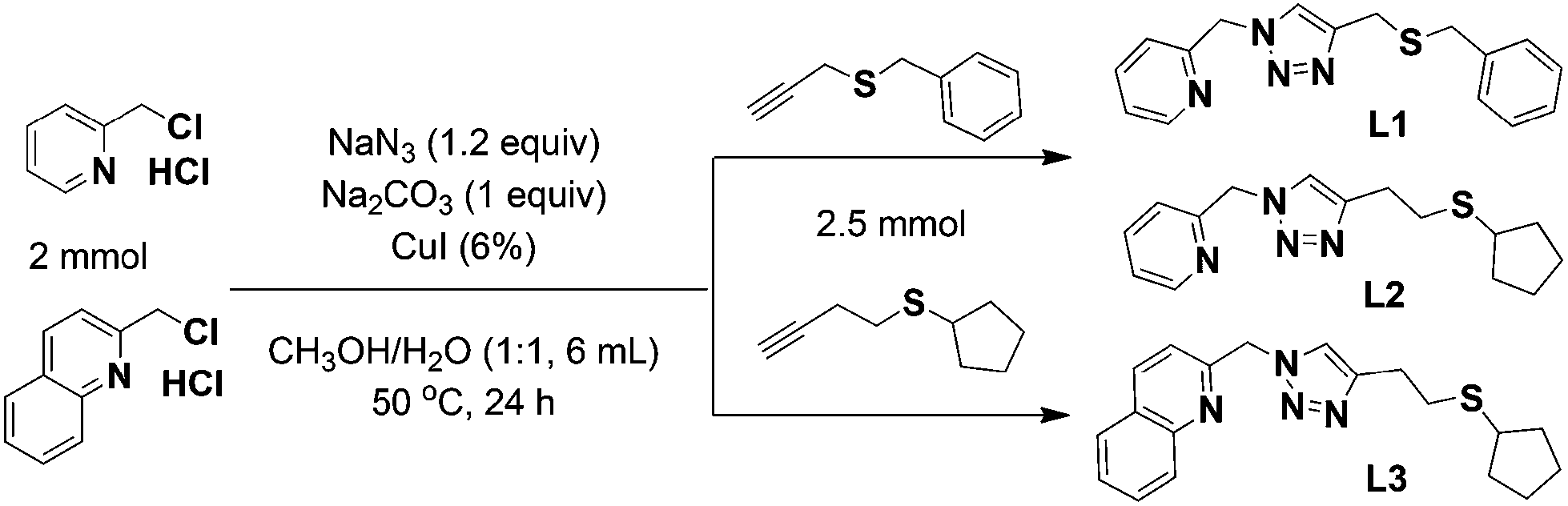 | ||
| Scheme 1 Synthesis of ligands L1–L3 from CuAAC reactions. | ||
Experimental
Materials and instrumentation
All starting chemicals were used as received. Elemental analyses were performed on a thermo electron corporation flash EA 1112 series analyzer. Infrared spectra were obtained on a Perkin Elmer Spectrum 2000 FT-IR spectrometer from samples in KBr discs. Electrospray ionization mass spectra (ESI-MS) were recorded in the positive ion mode using a Shimadzu LCMS-IT-TOF mass spectrometer. NMR spectra were measured at room temperature using a JEOL 500 MHz NMR spectrometer. Powder X-ray diffraction data were collected on a Bruker AXS GADDS X-ray diffractometer with Cu-Kα radiation (λ = 1.54056 Å). Thermogravimetric analyses (TGA) were carried out under an air stream using a TA Instruments TGA Q500 analyzer with a heating rate of 20 °C min−1. Photoluminescence was measured using a Perkin Elmer LS55 luminescence spectrometer. An attenuation of 1% T was used for complexes 1–3A due to their strong emission.Ligand synthesis
Sodium azide is potentially explosive. Only micro-scaled reactions may be performed, and all operations must be handled with necessary precautionary measures. The alkyne precursors benzyl(prop-2-yn-1-yl)sulfane and but-3-yn-1-yl(cyclopentyl)sulfane were prepared following literature procedures for thioether formation.23 These crude alkynes benzyl(prop-2-yn-1-yl)sulfane (for L1) or but-3-yn-1-yl(cyclopentyl)sulfane (for L2 and L3) were placed in a round bottom flask containing 2-(chloromethyl)pyridine hydrochloride (328 mg, 2 mmol, for L1 and L2) or 2-(chloromethyl)quinoline hydrochloride (428 mg, 2 mmol, for L3), Na2CO3 (210 mg, 2 mmol), NaN3 (156 mg, 2.4 mmol), CuI (23 mg, 0.12 mmol) and CH3OH/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v:v, 6 mL) (Scheme 1). The reaction was stirred at 50 °C for 24 h. The residue was extracted with ethyl acetate (3 × 30 mL) and the combined organic layers were washed with water (3 × 10 mL) and dried over anhydrous Na2SO4. The product was purified on silica gel.
:1 v:v, 6 mL) (Scheme 1). The reaction was stirred at 50 °C for 24 h. The residue was extracted with ethyl acetate (3 × 30 mL) and the combined organic layers were washed with water (3 × 10 mL) and dried over anhydrous Na2SO4. The product was purified on silica gel.
Complex synthesis
Complexes 1–3 were prepared by mixing a CH3CN (2 mL) solution of the NS ligand (L1–L3, 0.1 mmol) with a CH3CN (5 mL) solution of CuI (0.2 mmol, 38 mg for 1; 0.3 mmol, 57 mg for 2 and 3) in a test tube, respectively. These tubes were then sealed and left to stand at room temperature to give crystals of Cu(I) complexes 1–3 within two weeks. Two kinds of crystals were observed in the reaction of L3 with CuI. The majority were yellow plate crystals of 3A (thermodynamic product) with a smaller amount of block crystals of 3B (kinetic product). The latter was characterized by single-crystal XRD characterization but limited sample amount did not permit other types of measurements.[Cu4I4(L1)2] (1), yield: 30 mg, 45%. Anal. Calcd for C32H32Cu4I4N8S2 (1354.54): C, 28.37; H, 2.38; N, 8.27%. Found: C, 28.35; H, 2.20; N, 8.29%. Main IR bands (cm−1): 3119(m), 3025(m), 2960(m), 2916(m), 1632(m), 1600(m), 1536(m), 1494(m), 1477(m), 1452(m), 1417(m), 1349(m), 1306(m), 1235(m), 1155(m), 1146(m), 1099(m), 1070(m), 1042(m), 1016(m), 935(m), 920(m), 898(m), 817(m), 759(s), 732(m), 705(m), 648(m), 562(m), 466(m) and 416(m). ESI-MS (m/z, %): [L1 + H]+ (297, 100) and [Cu(L1)2]+ (655, 12).
[Cu6I6(L2)2] (2), yield: 42 mg, 49%. Anal. Calcd for C30H40Cu6I6N8S2 (1719.46): C, 20.96; H, 2.34; N, 6.52%. Found: C, 20.65; H, 2.23; N, 6.46%. Main IR bands (cm−1): 3125(m), 2955(m), 2863(m), 1634(m), 1597(m), 1544(m), 1476(m), 1440(m), 1421(m), 1377(m), 1346(m), 1304(m), 1242(m), 1226(m), 1153(m), 1102(m), 1071(m), 1053(m), 1013(m), 966(m), 936(m), 889(m), 834(m), 798(m), 757(s), 724(m), 670(m), 637(m), 603(m), 458(m) and 412(m). ESI-MS (m/z, %): [Cu(L2)2]+ (639, 100).
[Cu6I6(L3)2] (3A), yield: 52 mg, 57%. Anal. Calcd for C38H44Cu6I6N8S2 (1819.65): C, 25.08; H, 2.44; N, 6.16%. Found: C, 25.24; H, 2.34; N, 6.18%. Main IR bands (cm−1): 3119(m), 3063(m), 2955(m), 2864(m), 1620(m), 1598(m), 1567(m), 1544(m), 1509(m), 1422(m), 1380(m), 1350(m), 1311(m), 1246(m), 1221(m), 1148(m), 1126(m), 1072(m), 1044(m), 973(m), 946(m), 904(m), 869(m), 826(m), 803(s), 778(m), 755(m), 733(m), 652(m), 627(m), 496(m), 482(m) and 427(m). ESI-MS (m/z, %): [L3 + H]+ (339, 7) and [Cu(L3)2]+ (739, 100).
X-ray crystallography
All measurements were conducted on a Bruker AXS APEX diffractometer equipped with a CCD area-detector using Mo-Kα radiation (λ = 0.71073 Å).(Table 1) The collecting frames of data, indexing reflection and determination of lattice parameters and polarization effects were performed with the Bruker SMART programs.32 The integration of intensity of reflections and scaling was carried out using Bruker SAINT.32 The empirical absorption correction was performed using SADABS.33 The space group determination, structure solution and least-squares refinements on |F|2 were carried out using Bruker SHELXL.34 The structures were solved by direct methods to locate the heavy atoms, followed by difference maps for the light non-hydrogen atoms. For complex 1, the –SCH2Ph group was disordered into two positions with an occupancy ratio of 50:50. Restraints in bond lengths and thermal parameters were applied on the disordered atoms. Anisotropic thermal parameters were refined for the rest of the non-hydrogen atoms. Hydrogen atoms were placed geometrically and refined isotropically. CCDC reference numbers: 1038927 (1), 1038928 (2), 1038929 (3A) and 1038930 (3B).
| Complex | 1 | 2 | 3A | 3B |
|---|---|---|---|---|
| Formula | C32H32Cu4I4N8S2 | C30H40Cu6I6N8S2 | C38H44Cu6I6N8S2 | C42H50Cu8I8N10S2 |
| M W | 1354.54 | 1719.46 | 1819.57 | 2282.56 |
| T/K | 100(2) | 100(2) | 100(2) | 100(2) |
| Crystal size/mm3 | 0.17 × 0.16 × 0.10 | 0.50 × 0.25 × 0.03 | 0.30 × 0.20 × 0.05 | 0.15 × 0.12 × 0.05 |
| Crystal system | Triclinic | Triclinic | Triclinic | Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P |
P |
| a/Å | 9.1116(5) | 8.712(2) | 8.7035(7) | 9.3044(6) |
| b/Å | 9.1187(5) | 9.519(2) | 9.4819(8) | 12.4409(8) |
| c/Å | 13.5556(7) | 14.794(4) | 15.507(1) | 26.221(2) |
| α/° | 76.481(2) | 84.748(5) | 86.797(2) | 103.477(1) |
| β/° | 85.819(2) | 80.363(6) | 82.541(2) | 95.743(2) |
| γ/° | 60.208(1) | 65.779(5) | 71.277(2) | 96.496(1) |
| V/Å3 | 949.09(9) | 1102.8(5) | 1201.6(2) | 2907.4(3) |
| Z | 1 | 1 | 1 | 2 |
| D calc/g cm−3 | 2.370 | 2.589 | 2.515 | 2.607 |
| μ/mm−1 | 5.610 | 7.168 | 6.587 | 7.227 |
| θ range /° | 2.579–28.318 | 1.397–30.703 | 1.325–33.240 | 0.805–26.410 |
| Reflections collected | 23795 |
38504 |
58285 |
89730 |
| Independent reflections [Rint] | 4709 [0.0276] | 6701 [0.0438] | 9129 [0.0365] | 11909 [0.0508] |
| Parameters | 270 | 235 | 271 | 631 |
| GOF | 1.058 | 1.196 | 1.060 | 1.045 |
| R 1(I > 2σ(I)) | 0.0217 | 0.0452 | 0.0281 | 0.0564 |
| wR2(all data) | 0.0414 | 0.1181 | 0.0714 | 0.1530 |
Results and discussion
Synthesis and characterization
Ligands L1–L3 were synthesized from one-pot CuAAC reactions. The Cu(I) complexes 1–3 were prepared by the reaction of the corresponding ligand (L1–L3) and CuI in CH3CN solvent. The bulk powder samples of 1–3A were collected from filtration under vacuum and washed sequentially with CH3CN, ethanol and diethyl ether. The crystal and powder samples of complexes 1–3A were measured and confirmed by single-crystal and powder XRD characterization and microanalyses. All complexes 1–3A are thermally stable below 220 °C in air.Crystal structures
Ligand L1 coordinates to the stair-step [Cu4I4] cluster in 1 using a bidentate/monodentate binding mode of the pyridyl–triazole moiety with the S atom remaining unbound. Ligands L2 and L3 exhibit the more common NS mode of bidentate/monodentate (for N) and monodentate (for S) coordination in complexes 2 and 3 (3A and 3B). Both complexes 2 and 3A are stair-step [Cu6I6] cluster structures, while 3B is a stair-step [Cu8I8] cluster. Complexes 1–3 crystallize in the triclinic crystal system with a space group of P. All of the Cu(I) centers in complexes 1–3 are four-coordinate tetrahedral. The [CunIn] (n = 4, 6 and 8) stair-stepped core structures are supported by two ligands in 1–3B and by additional coordinating CH3CN molecules in 3B.
The asymmetric unit of complex 1 contains two crystallographically independent Cu(I) centers, two I− and one L1 ligand. The Cu1 is coordinated by two N donors from the pyridyl–triazole moiety of ligand L1, one μ2-I and one μ3-I bridges, giving a [CuN2I2] environment (Fig. 1). The Cu2 is coordinated by the 3′-NTri donor of ligand L1, one μ2-I and two μ3-I bridges, forming a [CuNI3] environment. The geometry index35τ4 for Cu1 and Cu2 is 0.80 and 0.84, respectively, which indicates the distortion of the tetrahedron. The Cu1⋯Cu2 and Cu2⋯Cu2 distances are 2.62 and 2.78 Å, respectively. The shortest intermolecular Cu⋯Cu distance is 6.2 Å in 1.
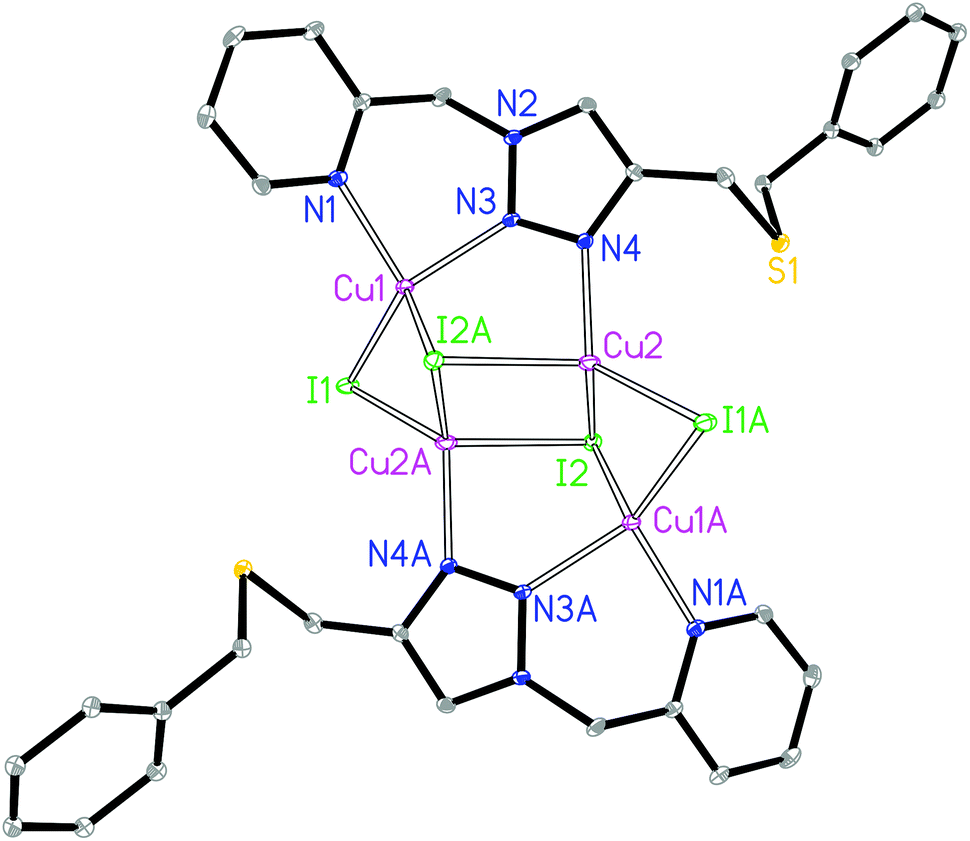 | ||
| Fig. 1 Molecular structure of 1. | ||
The asymmetric unit of 2 contains three crystallographically independent Cu(I) centers, three I− ions and one ligand L2. The Cu1 is coordinated by two N donors from the pyridyl–triazole moiety of ligand L2, one μ2-I and one μ3-I bridges, giving a [CuN2I2] environment (Fig. 2). The Cu2 is coordinated by one S donor from another ligand L2, one μ2-I and two μ3-I bridges, forming a [CuSI3] environment. The Cu3 is coordinated by one 3′-NTri atom of ligand L2 and three μ3-I bridges, forming a [CuNI3] environment. The tetrahedral geometry index35τ4 for Cu1, Cu2 and Cu3 is 0.82, 0.90 and 0.89, respectively. The Cu1⋯Cu2, Cu2⋯Cu3 and Cu3⋯Cu3 distances are 2.65, 3.08 and 2.64 Å, respectively. The shortest intermolecular Cu⋯Cu distance is 7.0 Å in 2.
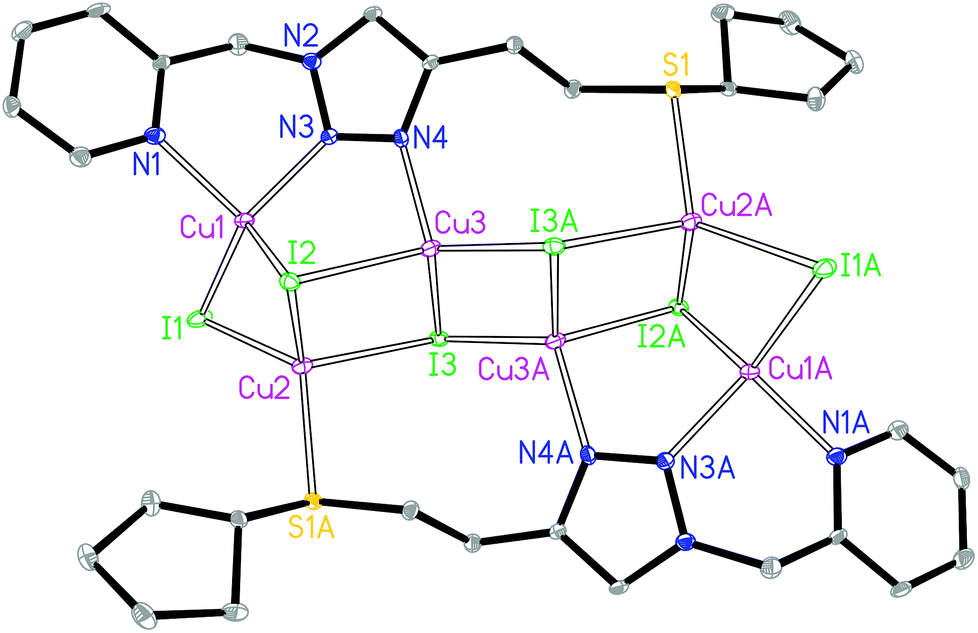 | ||
| Fig. 2 Molecular structure of 2. | ||
The asymmetric unit of 3A contains three crystallographically independent Cu(I) centers, three I− and one L3 ligand. The Cu1 is coordinated by two N donors from the quinolyl–triazole moiety, one μ2-I and one μ3-I bridges, giving a [CuN2I2] environment (Fig. 3). The Cu2 is coordinated by one S donor from another ligand L3, one μ2-I and two μ3-I bridges, forming a [CuSI3] environment. The Cu3 is coordinated by one 3′-NTri donor of ligand L3 and three μ3-I bridges, forming a [CuNI3] environment. The tetrahedral geometry index35τ4 for Cu1, Cu2 and Cu3 is 0.81, 0.88 and 0.92, respectively. The Cu1⋯Cu2, Cu2⋯Cu3 and Cu3⋯Cu3 distances are 2.72, 2.93 and 2.77 Å, respectively. The shortest intermolecular Cu⋯Cu distance is 6.9 Å in 3A.
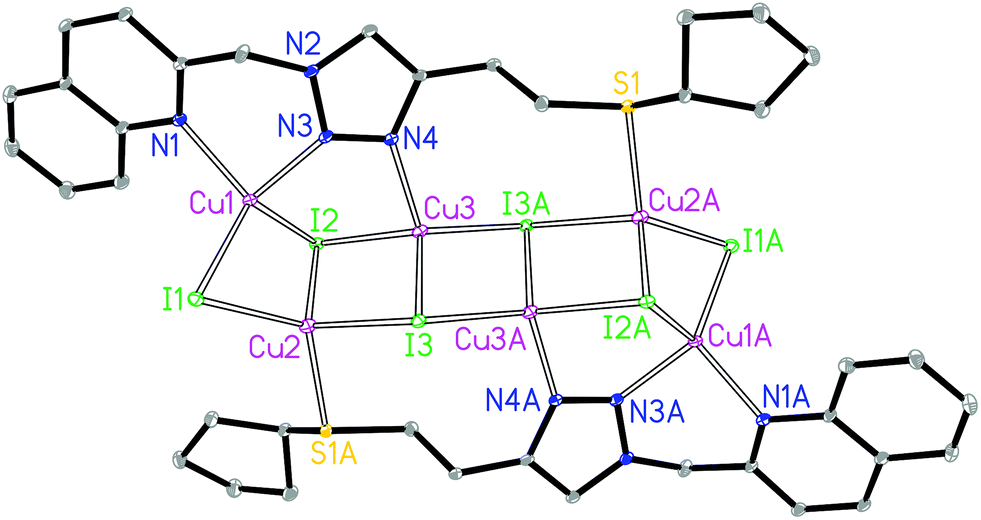 | ||
| Fig. 3 Molecular structure of 3A. | ||
The asymmetric unit of 3B contains eight crystallographically independent Cu(I) centers, eight I− ions, two L3 ligands and two coordinating CH3CN molecules. Both Cu1 and Cu8 are coordinated by two N donors from the quinolyl–triazole moiety, one μ2-I and one μ3-I bridges, giving a [CuN2I2] environment (Fig. 4). Both Cu2 and Cu7 are coordinated by one N donor from the CH3CN molecule, one μ2-I and two μ3-I bridges, forming a [CuNI3] environment. Both Cu3 and Cu6 are coordinated by one 3′-NTri donor from ligand L3 and three μ3-I bridges, forming a [CuNI3] environment. Both Cu4 and Cu5 are coordinated by one S donor and three μ3-I bridges to form a [CuSI3] core. This is a rare example of a [M8] (M = Cu) open cluster assembled by two multidentate hybrid ligands (with bridging iodides and coordinating CH3CN molecules providing additional support). The tetrahedral geometry index35τ4 for Cu1, Cu2, Cu3, Cu4, Cu5, Cu6, Cu7 and Cu8 is 0.82, 0.94, 0.91, 0.93, 0.93, 0.91, 0.91 and 0.86, respectively. The [Cu8I8] stair-stepped core is supported by two bidentate/monodentate/monodentate ligand L3 and two coordinating CH3CN molecules. The Cu1⋯Cu2, Cu2⋯Cu3, Cu3⋯Cu4, Cu4⋯Cu5, Cu5⋯Cu6, Cu6⋯Cu7 and Cu7⋯Cu8 distances are 2.67, 3.04, 2.91, 2.89, 2.94, 3.23 and 2.70 Å, respectively. There are intermolecular π⋯π interactions between the quinoline groups of neighbouring ligands with a centroid–centroid distance of 3.6 Å. The shortest intermolecular Cu⋯Cu distance is 6.7 Å in 3B.
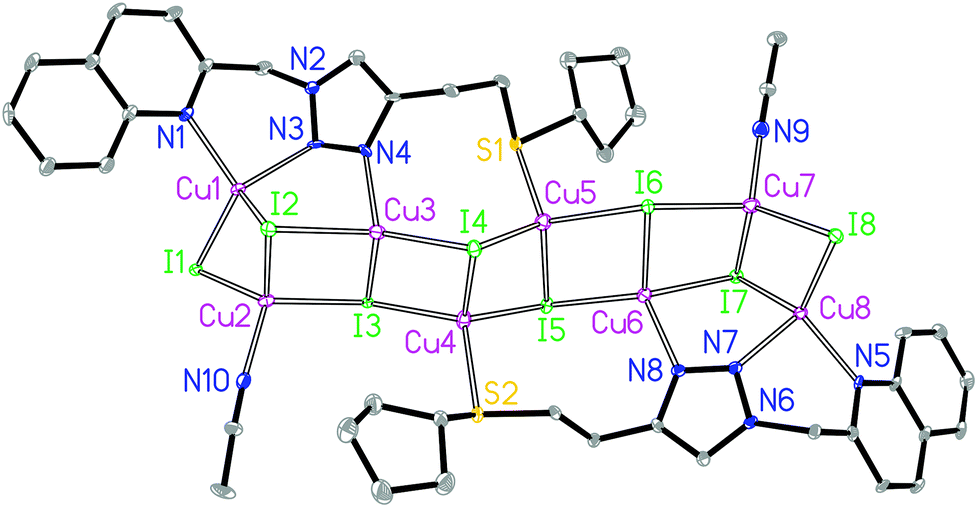 | ||
| Fig. 4 Molecular structure of 3B. | ||
Structural discussion
Many types of sulfur-containing ligands have been synthesized for the construction of copper(I)-iodide complexes, because the soft sulfur donor has an affinity for soft metals. In our previous study, ligand 1-(2-picolyl)-4-(2-(methylthio)-pyridine)-1H-1,2,3-triazole was used to support a [Cu6I6]-cluster based complex (I) and all of the potential N atoms were involved in coordination, while the S atom of the 2-(methylthio)-pyridine substituent was not needed (Scheme 2).29 This contrasts a similar S donor ligand 2-(4-pyridylsulfanylmethyl)pyridine that is coordinated to Cu(I) in a 2-D coordination polymer.27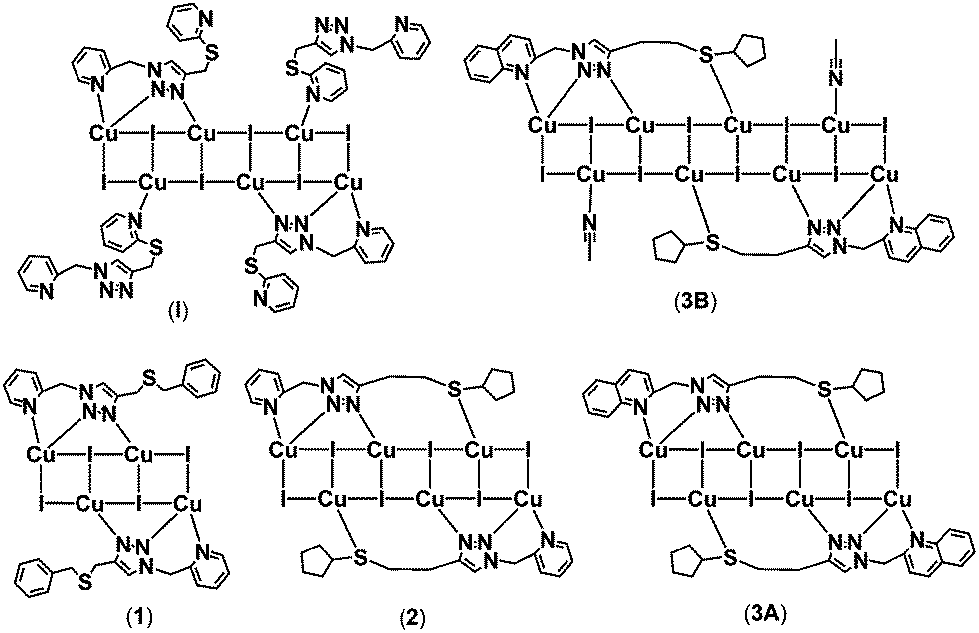 | ||
| Scheme 2 Coordination mode of triazole NS ligands and the resultant chemical structures of copper(I)-iodide complexes. | ||
Other sulfur-containing ligands similarly display a variety of coordination modes in copper(I)-iodide metal complexes. Tetrahydrothiophene, for example, exhibits μ2-bridging in cluster-based coordination polymers and mono-coordination in [Cu4] and [Cu2] clusters, depending on the temperature and substrate content.36 Very recently, dialkyl sulfide ligands displaying mono-coordination and μ2-bridging modes were used in the construction of copper(I)-iodide clusters and coordination polymers.37 The anionic sulfur donor of ligand 1-(4-pyridyl)-4-thiopyridine displays a μ2-bridging mode in a cluster-based coordination polymer.38 Likewise, the podal ligand 1,12-diphenyl-5,8-dioxa-2,11-dithiadodecane shows bridges with two S donors to construct a [Cu4I4] cluster-based coordination polymer.39 The ditopic xylyl-bridged NO2S2 macrocycle ligand (1,4-bis((7,8,10,11,19,20-hexahydro-5H,9H,13H-dibenzo[e,p][1,4]dioxa[8,14]dithia[11]azacycloheptadecin-9-yl)methyl)benzene) forms a [Cu4I4]-based one-dimensional coordination polymer.40 The asymmetric dithioether ligands (2-(benzylthio)-1-thiomorpholinoethan-1-one) and (2-((cyclohexylmethyl)thio)-1-thiomorpholinoethan-1-one) adopt bridging modes using their sulfur donors in [Cu2I2] and [Cu4I4]-cluster based coordination polymers.41 The bridging ligand 1,4-bis((cyclohexylthio)acetyl)piperazine using sulfur donors yields a ‘three-runged ladder’ [Cu3I3] subunit based coordination polymer.42 The potentially chelating calix[4]bis(thiacrown-5) instead assembles copper(I)-iodide cluster-based coordination polymers bridged by sulfur donors.43 Less complicated is the heterocyclic thione benz-1,3-imidazole-2-thione which forms a simple copper(I)-iodide coordination polymer using μ2-bridging mode.44 Dithioether ligands PhS(CH2)nSPh (n = 1–5) exhibit tunable bridging coordination in a similar system.19,22,45 Interestingly, tribenzo-macrocycle ligands yield endo- and exocyclic copper(I)-iodide supramolecular complexes,20 while tri- and di-thioethers tris(2-tert-butyl-4-methylphenylthiomethyl)amine and bis(2,4-dimethylphenylthio)methane exhibit chelating and bridging modes of binding, respectively.21
These previous studies indicate that both the assembly conditions (temperature, solvent and reaction stoichiometry) and nature of the ligand (stereogeometry and electron density) influence the structural outcome in these systems.
The present work illustrates the coordination flexibility of hybrid ligands. Ligand L1 with a benzyl substituent on S displays only N coordination. Increasing the flexibility of the substituent from –py to –CH2Ph is not sufficient to promote S coordination. However, increasing the length of the bridge from –CH2– to –C2H4– in L2 and L3 enables binding of the thioether.
Stairstep [Cu4I4] and [Cu6I6] structures are observed in the biimidazole (1,4-di(2-methyl-imidazol-1-yl)butane) supported 1-D and 2-D coordination polymers.46 The double-cubane [Cu8I8] cluster can be stabilized with support from ligand 4-(diphenylphosphino)-N,N-dimethylaniline.47 Quinolyl-triazoles promote [Cu4I4] and [Cu3I3] cluster-based copper(I)-iodide complexes.31 More interesting is that the subtle change from pyridyl in L2 to quinolyl in L3 enables the formation of a discrete stairstep [Cu8I8] cluster structure. This rare motif arises from the additional stability imparted by weak π⋯π stacking and solvate (CH3CN) coordination interactions that allow this (presumably) kinetic intermediate to be observed. Acetonitrile stabilization of [Cu4I4] structures has been observed previously.21
Powder XRD, TGA and photoluminescence
The powder X-ray diffraction patterns of complexes 1–3A showed good agreement with their simulated patterns derived from single-crystal XRD, supporting their phase purity (Fig. 5). The thermogravimetric analysis (TGA) curves of these complexes from room temperature to 600 °C are given in Fig. 6. Complexes 1–3A were stable in air up to ∼220 (for 1) and 250 °C (for 2 and 3A), followed by steady decompositions until reaching residual weights at ∼580, 550 and 560 °C in 1–3A, respectively. Complexes 2 and 3A exhibited slightly higher thermal stabilities (∼30 °C) than 1, pointing to the thermal robustness of a larger [Cu6I6] core.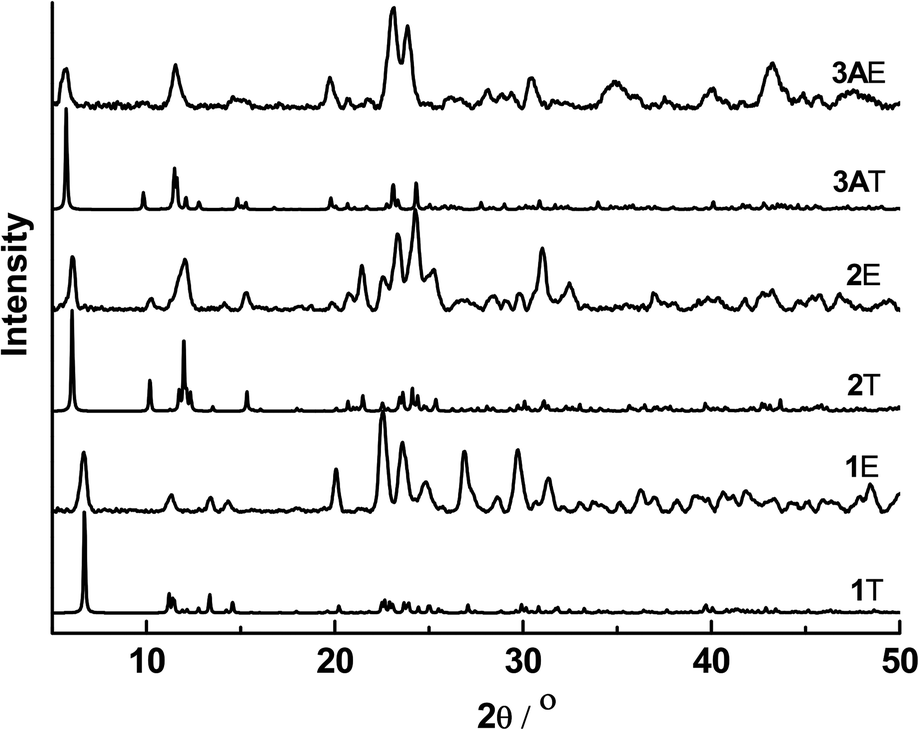 | ||
| Fig. 5 Powder XRD patterns of 1–3A (T = theoretical profile referenced to the experimentally determined structure by single-crystal XRD; E = experimental data). | ||
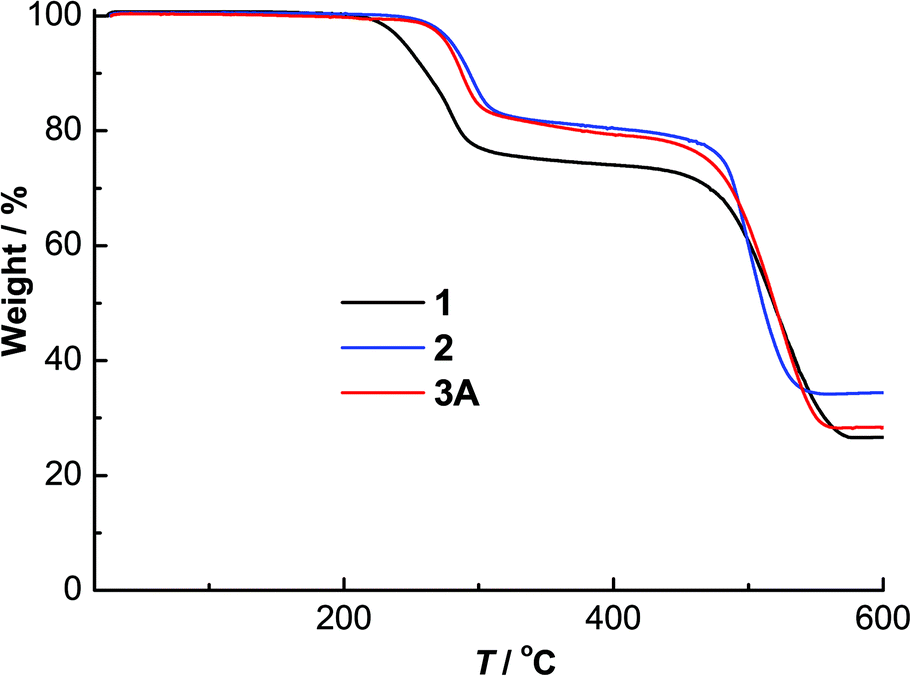 | ||
| Fig. 6 TGA curves of 1–3A. | ||
The excitation spectra of ligands L1–L3 (Fig. 7) contained absorption peaks between 327 and 400 nm with maxima at ∼345, 343 and 353 nm and broad emission spectra between 360 and 570 nm with maxima at ∼431, 433 and 443 nm, respectively. These emissions probably originate from intramolecular π–π* transitions. The excitation spectra of complexes 1–3A (Fig. 8) displayed two main absorption peaks between 325 and 420 nm with maxima at ∼326, 326 and 394 nm, respectively. Complexes 1 and 2 indicated blue emissions with maxima at ∼483 nm. Complex 3A exhibited a remarkable red-shift (∼101 nm) relative to 1 and 2 with broad yellow emission between 500 and 700 nm with maxima at 584 nm, which is comparable to the emissions (543–592 nm) in the quinolyl–triazole ligand supported copper(I)-iodide clusters and coordination polymers.31
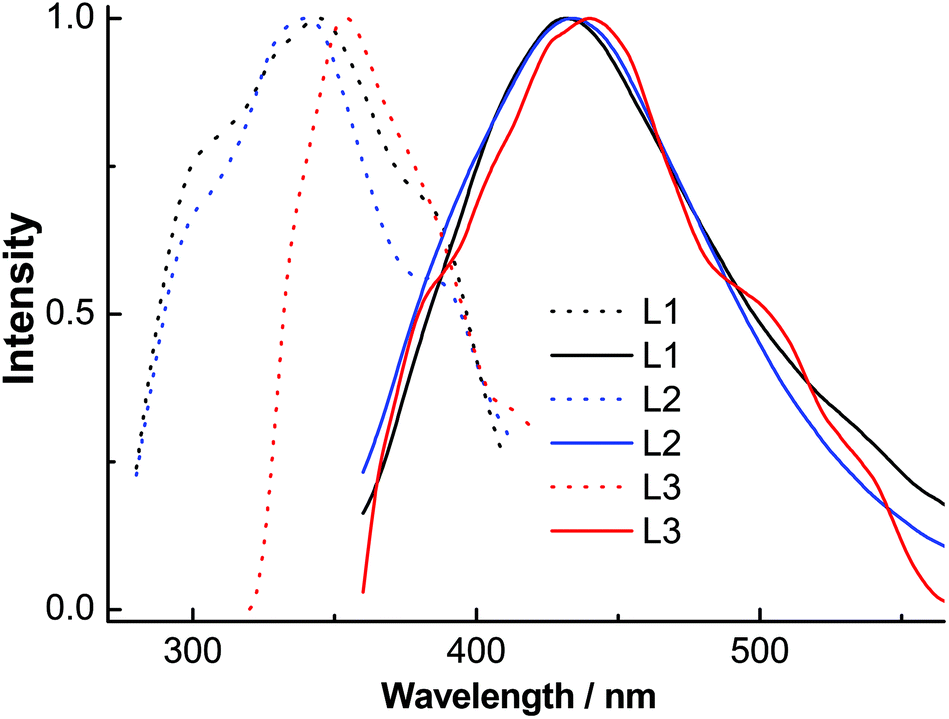 | ||
| Fig. 7 Normalized solution excitation (dotted line) and emission (solid line) spectra of ligands L1–L3. | ||
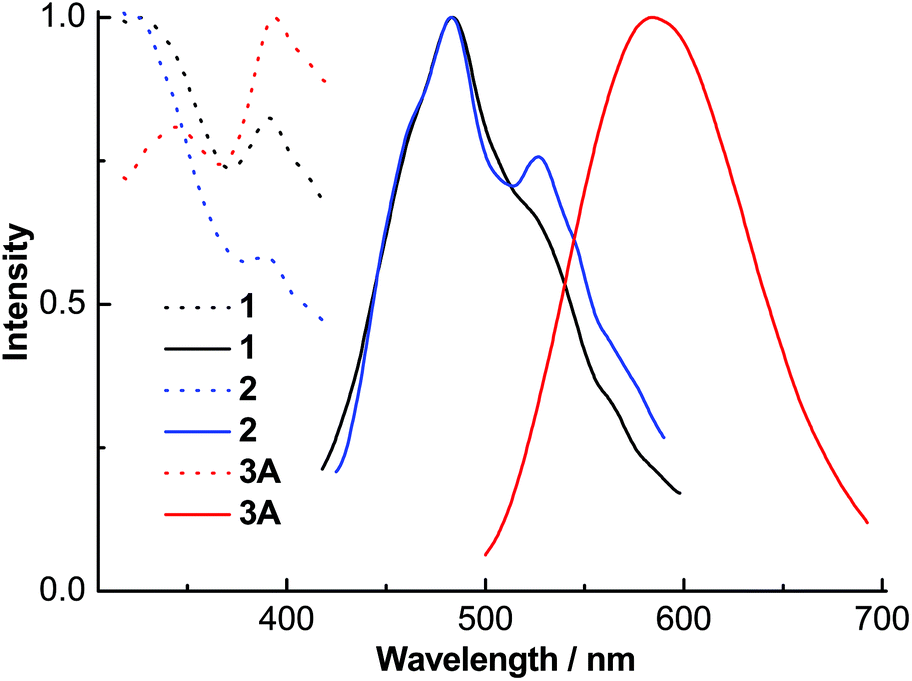 | ||
| Fig. 8 Normalized solid state excitation (dotted line) and emission (solid line) spectra of complexes 1–3A. | ||
It is reasonable to assign this difference to the extended conjugation of the quinolyl moiety in L3 compared to pyridyl. The origins of the emissions in complexes 1–3A can be assigned to triplet metal-to-ligand charge transfer (MLCT), iodide-to-ligand charge transfer (XLCT) and/or cluster-centered (CC) excited states.48,49 The different emission behaviours of complexes 1–3A are due to the combined substituent effects of the different supporting ligands, Cu(I) geometries, size difference of [CunIn] stair-step clusters and the different intramolecular Cu⋯Cu distances.
Conclusions
We have extended the application of CuAAC click chemistry to the preparation of hybrid NS ligands L1–L3 that provide diverse and stable coordination environments around Cu(I) centers in complexes 1–3. The variation of the substituent groups leads to stair-step cluster structures from a [Cu4I4] (1) to [Cu6I6] (2 and 3A) and a [Cu8I8] (3B) with remarkably different photoluminescence behavior between 3A and its pyridyl counterparts 1 and 2. Extending the bridge to the sulfur donor moiety brings this coordinating group into an active role in stabilizing large clusters. The isolation of complexes 3A and 3B raises the possibility for material growth and modification by introduction and integration of additional supporting interactions. It also raises the possibility of controlled and stepwise growth of metal clusters.Acknowledgements
We are grateful to Dr L. L. Koh, G. K. Tan and Y. M. Hong for X-ray diffractometry measurement assistance. We acknowledge the financial support (IMRE/12-1P0907, IMRE/14-1C0248 & IMRE/15-1C0202) provided by the Institute of Materials Research and Engineering, A*STAR and the National University of Singapore.Notes and references
- H. Xiang, J. Cheng, X. Ma, X. Zhou and J. J. Chruma, Chem. Soc. Rev., 2013, 42, 6128–6185 RSC.
- Y.-G. Huang, F.-L. Jiang and M.-C. Hong, Coord. Chem. Rev., 2009, 253, 2814–2834 CrossRef CAS PubMed.
- T. Wen, D.-X. Zhang, Q.-R. Ding, H.-B. Zhang and J. Zhang, Inorg. Chem. Front., 2014, 1, 389–392 RSC.
- S. Sculfort and P. Braunstein, Chem. Soc. Rev., 2011, 40, 2741–2760 RSC.
- V. W.-W. Yam and K. K.-W. Lo, Chem. Soc. Rev., 1999, 28, 323–334 RSC.
- M. Vitale and P. C. Ford, Coord. Chem. Rev., 2001, 219–221, 3–16 CrossRef CAS.
- A. J. Blake, N. R. Brooks, N. R. Champness, L. R. Hanton, P. Hubberstey and M. Schröder, Pure Appl. Chem., 1998, 70, 2351–2357 CrossRef CAS.
- R. Peng, M. Li and D. Li, Coord. Chem. Rev., 2010, 254, 1–18 CrossRef CAS PubMed.
- C. H. Arnby, S. Jagner and I. Dance, CrystEngComm, 2004, 6, 257–275 RSC.
- S. V. Pavlova, H. L. To, E. S. H. Chan, H.-W. Li, T. C. W. Mak, H. K. Lee and S. I. Chan, Dalton Trans., 2006, 2232–2243 RSC.
- Y. Chen, H.-X. Li, D. Liu, L.-L. Liu, N.-Y. Li, H.-Y. Ye, Y. Zhang and J.-P. Lang, Cryst. Growth Des., 2008, 8, 3810–3816 CAS.
- X.-C. Shan, F.-L. Jiang, D.-Q. Yuan, H.-B. Zhang, M.-Y. Wu, L. Chen, J. Wei, S.-Q. Zhang, J. Pan and M.-C. Hong, Chem. Sci., 2013, 4, 1484–1489 RSC.
- R. Visbal and M. C. Gimeno, Chem. Soc. Rev., 2014, 43, 3551–3574 RSC.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef PubMed.
- J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302–1315 RSC.
- W. S. Brotherton, H. A. Michaels, J. T. Simmons, R. J. Clark, N. S. Dalal and L. Zhu, Org. Lett., 2009, 11, 4954–4957 CrossRef CAS PubMed.
- J. D. Crowley and P. H. Bandeen, Dalton Trans., 2010, 39, 612–623 RSC.
- M. Knorr, F. Guyon, A. Khatyr, C. Däschlein, C. Strohmann, S. M. Aly, A. S. Abd-El-Aziz, D. Fortin and P. D. Harvey, Dalton Trans., 2009, 948–955 RSC.
- E.-J. Kang, S. Y. Lee, H. Lee and S. S. Lee, Inorg. Chem., 2010, 49, 7510–7520 CrossRef CAS PubMed.
- P. R. Martínez-Alanis, V. M. Ugalde-Saldívar and I. Castillo, Eur. J. Inorg. Chem., 2011, 212–220 CrossRef PubMed.
- M. Knorr, F. Guyon, A. Khatyr, C. Strohmann, M. Allain, S. M. Aly, A. Lapprand, D. Fortin and P. D. Harvey, Inorg. Chem., 2012, 51, 9917–9934 CrossRef CAS PubMed.
- S.-Q. Bai, L. L. Koh and T. S. A. Hor, Inorg. Chem., 2009, 48, 1207–1213 CrossRef CAS PubMed.
- S.-Q. Bai, S. Leelasubcharoen, X. Chen, L. L. Koh, J.-L. Zuo and T. S. A. Hor, Cryst. Growth Des., 2010, 10, 1715–1720 CAS.
- S.-Q. Bai, J. Y. Kwang, L. L. Koh, D. J. Young and T. S. A. Hor, Dalton Trans., 2010, 39, 2631–2636 RSC.
- S.-Q. Bai, A. M. Yong, J. J. Hu, D. J. Young, X. Zhang, Y. Zong, J. Xu, J.-L. Zuo and T. S. A. Hor, CrystEngComm, 2012, 14, 961–971 RSC.
- S.-Q. Bai, L. Jiang, J.-L. Zuo and T. S. A. Hor, Dalton Trans., 2013, 42, 11319–11326 RSC.
- L. Jiang, Z. Wang, S.-Q. Bai and T. S. A. Hor, CrystEngComm, 2013, 15, 10451–10458 RSC.
- L. Jiang, Z. Wang, S.-Q. Bai and T. S. A. Hor, Dalton Trans., 2013, 42, 9437–9443 RSC.
- S.-Q. Bai, L. Jiang, B. Sun, D. J. Young and T. S. A. Hor, CrystEngComm, 2015, 17, 3305–3311 RSC.
- S.-Q. Bai, L. Jiang, D. J. Young and T. S. A. Hor, Dalton Trans., 2015, 44, 6075–6081 RSC.
- SMART & SAINT Software Reference Manuals, Bruker AXS GmbH, Karlsruhe, Germany, 2000 Search PubMed.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3–10 CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef PubMed.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- K. M. Henline, C. Wang, R. D. Pike, J. C. Ahern, B. Sousa, H. H. Patterson, A. T. Kerr and C. L. Cahill, Cryst. Growth Des., 2014, 14, 1449–1458 CAS.
- M. Knorr, A. Bonnot, A. Lapprand, A. Khatyr, C. Strohmann, M. M. Kubicki, Y. Rousselin and P. D. Harvey, Inorg. Chem., 2015, 54, 4076–4093 CrossRef CAS PubMed.
- J. Wang, S.-L. Zheng, S. Hu, Y.-H. Zhang and M.-L. Tong, Inorg. Chem., 2007, 46, 795–800 CrossRef CAS PubMed.
- T. H. Kim, K. Y. Lee, Y. W. Shin, S.-T. Moon, K.-M. Park, J. S. Kim, Y. Kang, S. S. Lee and J. Kim, Inorg. Chem. Commun., 2005, 8, 27–30 CrossRef CAS PubMed.
- H. J. Kim, Y. Jin, J. Seo, J.-E. Lee, J. Y. Lee and S. S. Lee, Inorg. Chem. Commun., 2006, 9, 1040–1044 CrossRef CAS PubMed.
- T. H. Kim, Y. W. Shin, S. S. Lee and J. Kim, Inorg. Chem. Commun., 2007, 10, 11–14 CrossRef CAS PubMed.
- T. H. Kim, Y. W. Shin, J. S. Kim, S. S. Lee and J. Kim, Inorg. Chem. Commun., 2007, 10, 717–719 CrossRef CAS PubMed.
- J. Y. Lee, S. Y. Lee, J. Seo, C. S. Park, J. N. Go, W. Sim and S. S. Lee, Inorg. Chem., 2007, 46, 6221–6223 CrossRef CAS PubMed.
- C.-F. Yan, F.-L. Jiang, L. Chen, R. Feng, N. Li and M.-C. Hong, Inorg. Chem. Commun., 2010, 13, 191–194 CrossRef CAS PubMed.
- H. N. Peindy, F. Guyon, A. Khatyr, M. Knorr and C. Strohmann, Eur. J. Inorg. Chem., 2007, 1823–1828 CrossRef CAS PubMed.
- S. Yuan, H. Wang, D.-X. Wang, H.-F. Lu, S.-Y. Feng and D. Sun, CrystEngComm, 2013, 15, 7792–7802 RSC.
- X.-C. Shan, H.-B. Zhang, L. Chen, M.-Y. Wu, F.-L. Jiang and M.-C. Hong, Cryst. Growth Des., 2013, 13, 1377–1381 CAS.
- N. P. Rath, E. M. Holt and K. Tanimura, J. Chem. Soc., Dalton Trans., 1986, 2303–2310 RSC.
- G. F. Manbeck, W. W. Brennessel, C. M. Evans and R. Eisenberg, Inorg. Chem., 2010, 49, 2834–2843 CrossRef CAS PubMed.
Footnote |
| † CCDC 1038927–1038930. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qi00030k |
| This journal is © the Partner Organisations 2015 |