
Modulation of the Aβ peptide aggregation pathway by KP1019 limits Aβ-associated neurotoxicity†
Michael R.
Jones
,
Changhua
Mu
,
Michael C. P.
Wang
,
Michael I.
Webb
,
Charles J.
Walsby
* and
Tim
Storr
*
Department of Chemistry, Simon Fraser University, Burnaby, BC V5A1S6, Canada. E-mail: cwalsby@sfu.ca; tim_storr@sfu.ca
First published on 3rd November 2014
Abstract
Alzheimer's disease (AD) is a neurodegenerative disorder that is increasing worldwide due to increased life expectancy. AD is characterized by two pathological hallmarks in the brain: amyloid-β (Aβ) plaque deposits and neurofibrillary tangles. A focus of AD research has concentrated on either inhibiting Aβ peptide aggregation that leads to plaque formation or breaking down pre-formed Aβ peptide aggregates. An alternative approach is to modulate the Aβ aggregation profile by facilitating the formation of Aβ species that are off-pathway and non-toxic. Herein, we report the re-purposing of the widely studied Ru(III) anti-cancer complex KP1019, towards regulating the aggregation profile of the Aβ peptide. Using electron paramagnetic resonance (EPR) spectroscopy, we conclude that KP1019 binds to histidine residues, located at the N-terminus of the peptide, in a rapid and robust fashion. Native gels and transmission electron microscopy (TEM) analyses have provided insight into the species and structures that are generated by KP1019-Aβ interactions. Finally, incubation in an in vitro human neuronal cell model has demonstrated that the formation of KP1019-Aβ species rescues cell viability from Aβ-associated neurotoxicity. Modulation of the Aβ aggregation pathway via covalent interactions with small molecules is thus a promising AD therapeutic strategy.
Introduction
Increased life expectancy in many developed countries has resulted in a rapid rise in the prevalence of aging-associated diseases such as Alzheimer's disease (AD).1–6 The increased incidence of AD, and the lack of effective treatment strategies, presents a significant burden on healthcare systems worldwide. Two neuropathological hallmarks are invariably present in the AD brain; Aβ-plaques and neurofibrillary tangles (NFTs). NFTs are intracellular fibrillar aggregates of oxidatively-modified and hyperphosphorylated microtubule-associated protein tau. The formation of Aβ plaque deposits in the brain results from sequential enzymatic cleavage of the amyloid-precursor protein, leading to the release of the Aβ peptide, which can aggregate to form extracellular plaque deposits in the brain.7 This peptide predominantly exists as either the Aβ1–40 or Aβ1–42 amino acid isoforms; the latter is more prone to aggregation while the former is more abundant in the human brain.8,9 Under normal physiological conditions, Aβ processing is well controlled, however, Aβ homeostasis is compromised in AD.10,11 Monomeric Aβ dimerizes readily in solution,12 and can then follow many different aggregation pathways, leading to soluble prefibrillar13 or fibrillar14 oligomers, annular protofibrils,15 or disordered aggregates.16 While insoluble fibrillar structures (amyloid fibrils) are the end point for Aβ aggregation, more recent reports suggest that soluble oligomeric species are the most deleterious.17–21 Many therapeutic strategies attempt to inhibit the aggregation of Aβ,22 break down fibrillar aggregates using monoclonal antibodies23 or small molecule derivatives,24,25 or modulate aggregate formation by producing high molecular weight non-toxic off-pathway species.26 The first strategy is difficult to implement since AD symptoms are usually manifested following significant plaque deposition. Breaking down pre-formed amyloid plaques has shown promise,27–29 but this approach could lead to the formation of smaller molecular weight species such as toxic soluble oligomers. Thus, the development of a therapeutic compound that can regulate the Aβ aggregation pathway, leading to off-pathway non-toxic species, is of current interest.26,30,31Metal-based therapeutics have recently been identified as potential treatments for AD.32–43 The success of cis-platin for the treatment of ovarian and testicular cancer has highlighted the promise of metal complexes in medicine. In the context of AD, in 2008 Barnham et al. reported Pt(II) phenanthroline derivatives that bind to the Aβ peptide, limiting its aggregation and toxicity.36 More recently, the same group demonstrated that a Pt(IV) complex with improved bioavailability is able to decrease plaque formation in the brains of APP/PS1 mice.37 Emissive cyclo-metalated Ir and Rh complexes were recently shown to bind to the Aβ peptide, and alter its aggregation profile.38 A small number of Ru-based complexes have also been evaluated for their ability to bind to the Aβ peptide,39 inhibit aggregation, and limit Aβ-associated toxicity in cultured rat primary cortical neurons.41
Ru(III) complexes are leading candidates for the next generation of metal-based anti-cancer compounds.44,45 Central to this development has been the Ru(III) complex indazolium [trans-RuCl4(1H-indazole)2] (KP1019, Fig. 1),46 which has shown activity against a wide array of cancer cell lines47–52 and has also performed well in clinical trials.53,54 A notable property of KP1019 is its low toxicity,55 particularly in comparison to Pt chemotherapeutics. The mechanism of action of KP1019 and other related Ru(III) complexes remains under investigation. However, favourable ligand exchange rates, typical of Ru(III) complexes,43 and interactions with biomolecules have been implicated in the transport, uptake and anti-cancer activity of KP1019.56,57 Ru(III) complexes exhibit a high affinity for accessible histidine residues on biomolecules, and KP1019 has been shown to coordinate to human serum albumin, human serum transferrin and other proteins.56,58–60 Based on these results, and the potential for hydrophobic interactions of the axial indazole moieties with the Aβ peptide, we have investigated the interaction of KP1019 with the Aβ peptide and the effect of this Ru(III) complex on the Aβ aggregation profile and resulting in vitro toxicity. Herein, we describe how KP1019 regulates the Aβ aggregation pathway towards non-toxic, off-pathway aggregates using a combination of physical and biochemical methods.
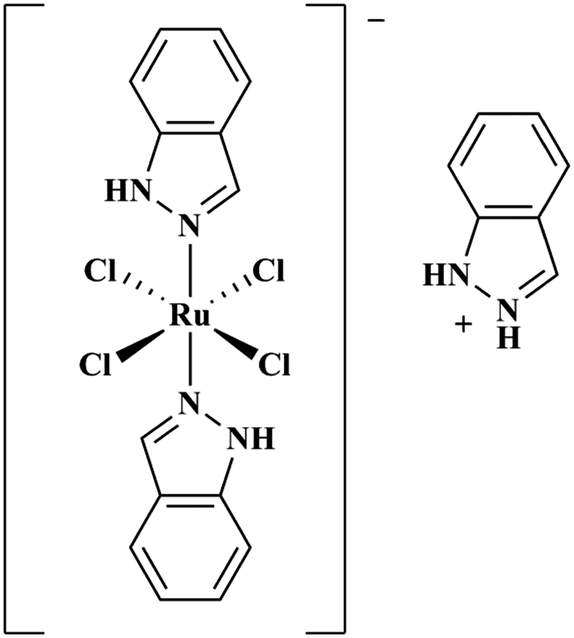 | ||
| Fig. 1 Chemical structure of KP1019. | ||
Results and discussion
Effect of KP1019 on Aβ peptide aggregation
The aggregation of the amyloid-beta (Aβ) peptide leading to the formation of soluble toxic oligomers and eventually plaque deposits has been linked to the pathology of AD.61,62 The fluorogenic dye Thioflavin-T (ThT) is commonly used to monitor Aβ fibril formation, exhibiting a marked fluorescence response (excitation wavelength (λex) = 404 nm, emission wavelength (λem) = 477 nm) upon binding to fibrils.63 We evaluated the concentration-dependent effect of KP1019 (0.25–2 eq. KP1019/Aβ) on Aβ fibril formation over a 48 hour period using ThT. Both the Aβ1–40 (Fig. S1, ESI†) and Aβ1–42 (Fig. 2) isoforms were evaluated to assess KP1019's ability to disrupt peptide fibrillization. We focus our discussion on the more aggregation prone Aβ1–42 isoform, however KP1019 exhibits the same concentration-dependent anti-aggregation effect on both peptides.64 When Aβ is incubated alone, significant fibrillization occurs as indicated by the increase in ThT fluorescence (Fig. 2 and Fig. S1, ESI†). At 0.25 eq. KP1019/Aβ1–42, Aβ1–42 fibril formation is significantly retarded even after 24 hours and continues after 48 hours (Fig. 2). At 0.5–2 eq. KP1019/Aβ1–42, no significant ThT fluorescence was observed. KP1019 at ≥0.5 eq. limits ThT fluorescence in a similar manner to Congo red, a known inhibitor of Aβ fibril formation.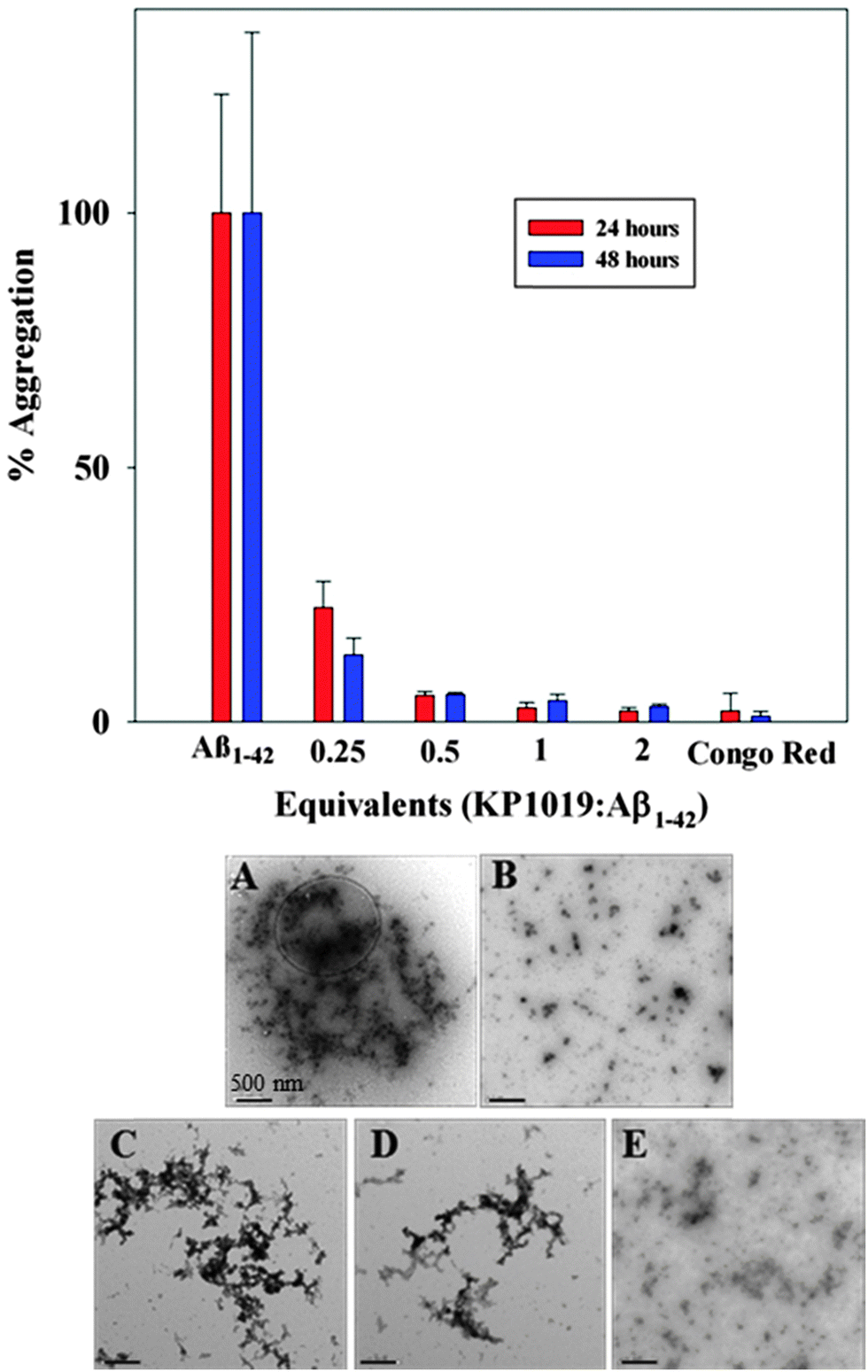 | ||
| Fig. 2 (top) 24 (red) and 48 (blue) hour aggregation profiles of Aβ1–42 (5 μM) analyzed by monitoring the ThT fluorescence (5 μM). Varying equivalents of KP1019/Aβ1–42 (0.25–2 eq.) were compared against a known inhibitor (Congo red, 2 eq. as a control) of Aβ peptide aggregation. (bottom) TEM images of Aβ1–42 morphology sampled from the ThT assay after 48 hours of incubation. (A) Aβ1–42 only; (B) Aβ1–42 + 2 eq. Congo red; (C) Aβ1–42 + 0.25 eq. KP1019; (D) Aβ1–42 + 0.5 eq. KP1019; (E) Aβ1–42 + 1 eq. KP1019. Conditions: PBS pH 7.4 buffer, 37 °C, constant agitation for 48 hours. Scale bars are 500 nm. | ||
Interestingly, addition of KP1019 to ThT-stained pre-formed Aβ1–42 aggregates leads to a ca. 50% decrease in fluorescence (Fig. S2, ESI†), indicating possible interference of the Ru(III) complex with this assay. The cause of the decreased ThT fluorescence upon addition of KP1019 could be due to an immediate change in aggregate morphology, KP1019-ThT interactions, or displacement of ThT from the pre-formed aggregates. We thus turned to TEM analysis to further understand the effect of KP1019 on Aβ1–42 aggregation (Fig. 2, bottom A–E). As expected, Aβ1–42 alone displays large heterogeneous aggregates consisting of a combination of amorphous and fibrillar structures (Fig. 2A),65 while treatment with Congo red demonstrates a significant reduction in aggregate size and no fibrillar structures were observed (Fig. 2B). Incubation in the presence of 0.25 eq. KP1019/Aβ1–42 results in smaller and less dense heterogeneous aggregates in comparison to peptide alone (Fig. 2C). Incubation in the presence of 0.5 eq. KP1019/Aβ1–42 results in smaller aggregate sizes in comparison to both Aβ1–42 alone and 0.25 eq. KP1019/Aβ1–42 (Fig. 2D). At 1 eq. KP1019/Aβ1–42, morphologies and aggregate sizes resemble those observed in the presence of the aggregation inhibitor Congo red (Fig. 2E). Based upon results obtained from the ThT assay and TEM analysis, KP1019 demonstrates a concentration-dependent effect on Aβ1–42 aggregation, and at ≥0.5 eq. KP1019 significant regulation of Aβ1–42 peptide aggregation is observed.
Aβ peptide coordination to KP1019
In order to delineate specific interactions of KP1019 with the Aβ peptide of relevance to aggregation, we investigated the time-dependent interaction of KP1019 with the Aβ1–28 peptide, a water soluble non-aggregating fragment containing the metal binding N-terminus. Ru(III) (d5, low spin, S = 1/2) is amenable to electron paramagnetic resonance (EPR) analysis and this technique has been used in previous studies to probe interactions of Ru(III) complexes with biomolecules such as human serum albumin.58,66,67 In the presence of serum proteins, KP1019 has been proposed to bind to the imidazole nitrogen of accessible histidine amino acid residues.58,60,66 The three histidine residues (His6, His13, and His14) at the N-terminus of the Aβ peptide are known to bind to biologically-relevant metal ions (Fe, Cu, Zn) with moderate to high affinity.68 To investigate the binding of KP1019 to Aβ peptide histidine residues, we incubated KP1019 with the shorter-length Aβ1–28 peptide and evaluated the EPR spectra at 2 and 24 hour time points (Fig. 3). Free KP1019 in solution exhibits a characteristic spectrum of overlapping axial (g⊥ = 2.64, g∥ = 1.20, linewidth⊥ = 120, linewidth∥ = 500 Gauss) and rhombic signals (g = [2.94, 2.31, 0.95], linewidths = [100, 200, 600] Gauss), which have been reported previously.58,66 Changes to the Ru(III) coordination environment via ligand exchange are reflected in the EPR spectra, providing clear evidence for peptide binding. KP1019, when in the presence of human serum albumin or human serum transferrin, exhibits a coordinate protein interaction detected by the appearance of a broad signal with g = [2.44, 2.24, 1.79], linewidths = [200, 220, 300] Gauss, assigned to histidine coordination.58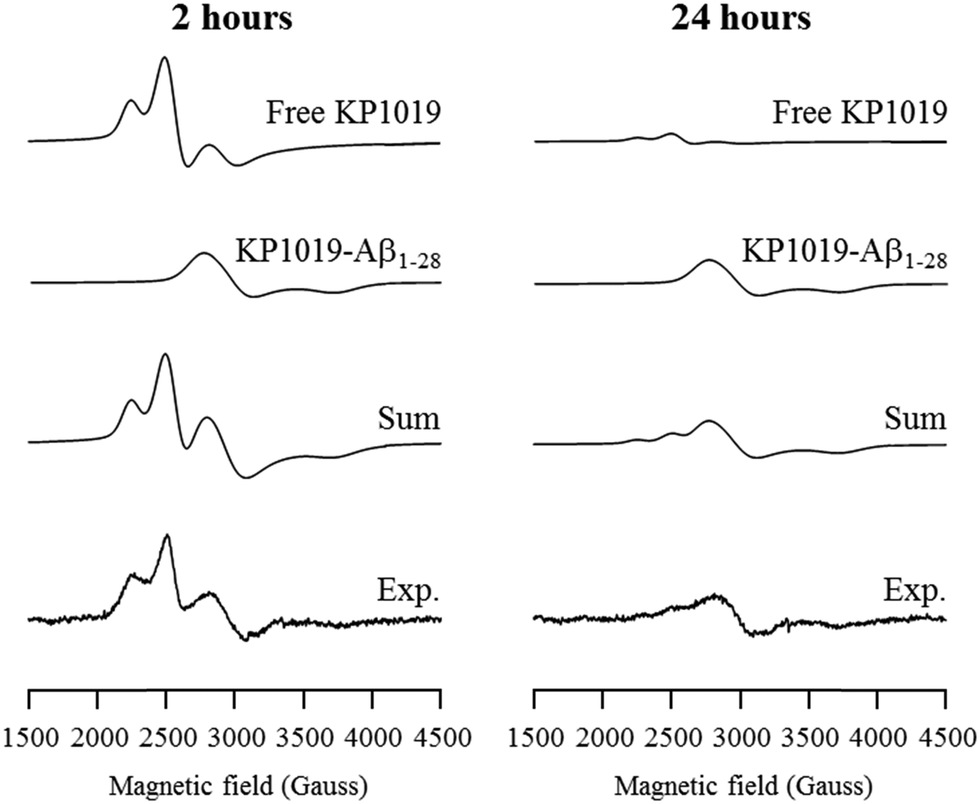 | ||
| Fig. 3 Frozen-solution EPR spectra of KP1019 (1.5 μM) and Aβ1–28 (0.5 μM) incubated for 2 hours (left) and 24 hours (right) in PBS pH 7.4 buffer. The experimental spectra were deconvoluted into KP1019, and Aβ1–28 coordinated KP1019 (KP1019-Aβ1–28) spectral components via simulation. Experimental conditions: frequency 9.38 GHz, microwave power 2.0 mW, time constant 40.96 ms, modulation amplitude 6 G, average of 10 scans of 2 minutes, temperature 20 K. | ||
The EPR spectra from KP1019 incubated with Aβ1–28 for 2 and 24 hours were simulated using the previously reported parameters for free KP1019 and protein-coordinated KP1019 (Fig. 3).58 After 2 hours, a significant amount of KP1019 is bound to the peptide, which is essentially unchanged after 24 hours. Excess free KP1019 undergoes ligand exchange of chloride for H2O, and the resulting neutral species displays low solubility,69 leading to a loss of EPR signal intensity, correlating with the decrease in free KP1019 signal from 2 hours to 24 hours in the experimental data (Fig. 3). Thus, the EPR data are consistent with binding of KP1019 to an N-terminus histidine of the Aβ peptide, of which there are 3 possible residues: His6, His13, or His14.
Time-course dot-blot assays were also performed to determine if increased KP1019 binding to the Aβ peptide would have an effect on antibody recognition at specific sites (i.e.: hydrophilic vs. hydrophobic regions) along the peptide chain (Fig. S3, ESI†). Two different primary antibodies were used that specifically recognize the hydrophilic N-terminus region (6E10; amino acids 4–9) and the hydrophobic region (4G8; amino acids 18–22) of the Aβ peptide.26,70 At a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of KP1019:Aβ1–42 the 6E10 antibody recognizes the N-terminus over the 24, 48, and 72 hour incubation period (Fig. S1, ESI†). An increased KP1019:Aβ1–42 ratio (2:1, 4:1, 10:1), however, results in decreased recognition by 6E10, likely due to saturation of the N-terminus with KP1019 (multiple binding events to His residues). In comparison to the results for 6E10, KP1019 exhibits little effect on 4G8 recognition in the dot blot assay over time. The dot blot assays thus provide further evidence for KP1019 binding to the N-terminus region of the Aβ peptide, likely targeting His residues (His6, His13, and His14) of the Aβ peptide.
:1 ratio of KP1019:Aβ1–42 the 6E10 antibody recognizes the N-terminus over the 24, 48, and 72 hour incubation period (Fig. S1, ESI†). An increased KP1019:Aβ1–42 ratio (2:1, 4:1, 10:1), however, results in decreased recognition by 6E10, likely due to saturation of the N-terminus with KP1019 (multiple binding events to His residues). In comparison to the results for 6E10, KP1019 exhibits little effect on 4G8 recognition in the dot blot assay over time. The dot blot assays thus provide further evidence for KP1019 binding to the N-terminus region of the Aβ peptide, likely targeting His residues (His6, His13, and His14) of the Aβ peptide.
Native gel electrophoresis/western blotting
To further evaluate the Aβ1–42 peptide aggregation process in the presence of KP1019, molecular weight separation by native gel electrophoresis followed by imaging using western blotting techniques was performed. The Aβ1–42 peptide with or without KP1019 (0–2 eq.) was incubated for 24 hours and the soluble fraction was then subjected to native gel electrophoresis and western blotting to determine relative abundance and the molecular weight of aggregates. In the absence of KP1019, primarily low molecular weight oligomeric species (ca. 10–20 kDa) are observed in the soluble fraction (Fig. 4, lane 5). Upon addition of 0.25 eq. KP1019 (lane 1), little change in the amount of oligomeric species is observed in comparison to Aβ1–42 alone, but soluble high molecular weight (>100 kDa) species are visible. At 0.5–2 eq. KP1019 (lanes 2–4), less oligomer formation is observed when compared to Aβ1–42 alone, and a concentration-dependent increase in soluble high molecular weight species is evident. Further analysis of these species was also completed by TEM (Fig. 4, right). Aβ1–42 alone displays large heterogeneous aggregates matching the ThT fluorescence assay (vide supra) (Fig. 2A). Incubation of Aβ1–42 with 0.25–1 eq. KP1019 results in a decrease in population and density of Aβ1–42 aggregates (Fig. 4B–D), also matching the results of the ThT experiment. Based on the native gel results, increased concentrations of KP1019 drive the formation of soluble high molecular-weight Aβ1–42 species, while the TEM images demonstrate that the overall size and abundance of insoluble aggregates decrease with increased KP1019 concentration. Morphologies observed by TEM correlate with how aggregates assemble and therefore it is important to describe the overall state of aggregation using TEM in combination with native gel results of the soluble fraction. These results demonstrate the ability of KP1019 to drive the formation of soluble high molecular weight aggregates while potentially limiting the formation of toxic oligomeric Aβ1–42 species.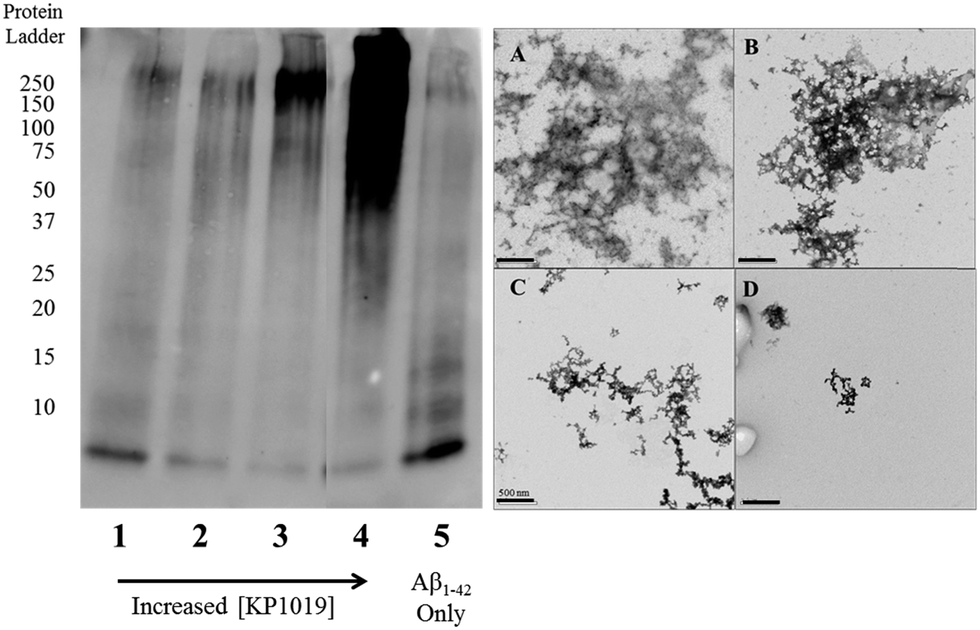 | ||
| Fig. 4 24 hour inhibition experiment of Aβ1–42 (25 μM) aggregation probed by native gel electrophoresis/western blotting using the 6E10 primary antibody and TEM imaging. (left) Lane 1 0.25 eq. KP1019/Aβ1–42; 2: 0.5 eq. KP1019/Aβ1–42; 3: 1 eq. KP1019/Aβ1–42; 4: 2 eq. KP1019/Aβ1–42; 5: Aβ1–42 only (right) (A) Aβ1–42 only; (B) 0.25 eq. KP1019/Aβ1–42; (C) 0.5 eq. KP1019/Aβ1–42; (D) 1 eq. KP1019/Aβ1–42. Scale bar = 500 nm. | ||
To determine the effect of KP1019 on pre-formed Aβ1–42 aggregates, Aβ1–42 was incubated for 24 hours under constant agitation in PBS pH 7.4 buffer, followed by a 24 hour treatment with 0–2 eq. KP1019 (Fig. 5). In the absence of KP1019, Aβ1–42 incubation affords primarily oligomeric species in the soluble fraction (Fig. 5, lane 5). Interestingly, in all cases where Aβ1–42 is treated with KP1019, a general decrease in low molecular weight oligomeric species (ca. 10–25 kDa) is observed along with an increase in the formation of soluble high molecular weight aggregates (>100 kDa, lanes 2–4). When comparing the native gel results with images obtained from TEM, an increase in KP1019 concentration leads to the formation of smaller, more diffuse aggregates (Fig. 5, right). In combination, the inhibition and disaggregation results demonstrate that KP1019 is not only able to modulate the Aβ1–42 aggregation pathway, but can also alter the assembly of pre-formed Aβ1–42 aggregates.
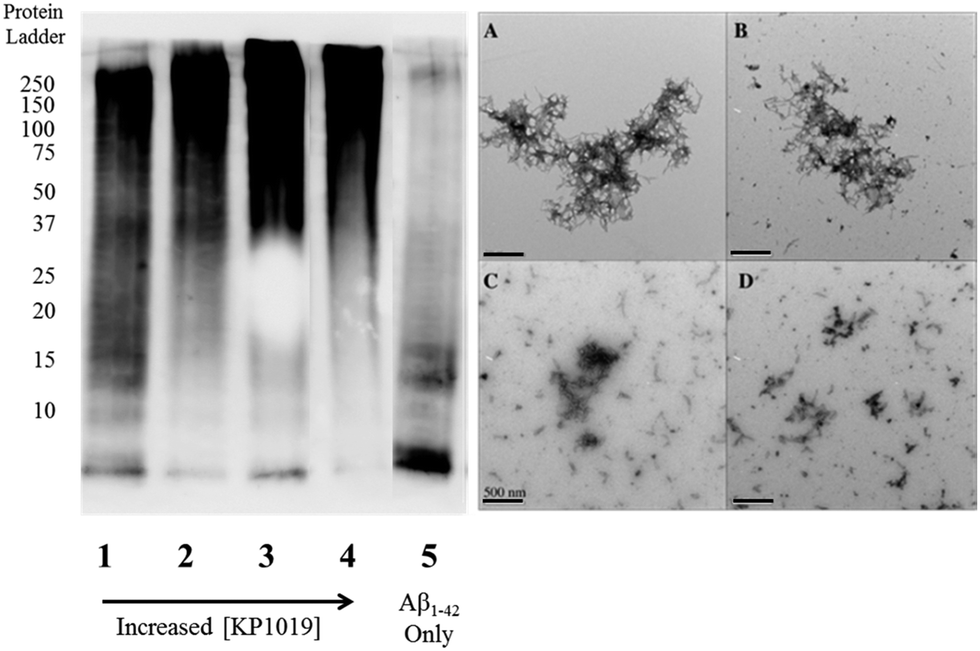 | ||
| Fig. 5 Native gel electrophoresis/western blotting of the Aβ1–42 disaggregation experiment. Aβ1–42 (25 μM) was incubated for 24 hours, followed by treatment with 0.25–2 eq. KP1019 for a further 24 hours. (left) Lane 1 0.25 eq. KP1019/Aβ1–42; 2: 0.5 eq. KP1019/Aβ1–42; 3: 1 eq. KP1019/Aβ1–42; 4: 2 eq. KP1019/Aβ1–42; 5: Aβ1–42 only (right) (A) Aβ1–42 only; (B) 0.25 eq. KP1019/Aβ1–42; (C) 0.5 eq. KP1019/Aβ1–42; (D) 1 eq. KP1019/Aβ1–42. Scale bar = 500 nm. | ||
KP1019 rescues differentiated SH-SY5Y cells from Aβ1–42-induced toxicity
Aβ1–42 neurotoxicity has been reported to stem from multiple factors including disrupted synaptic plasticity,18,62,71 increased inflammation72 and oxidative stress,71 dysregulation of ion homeostasis,73,74 and disrupted kinase/phosphatase activity.75 To probe if KP1019 could reduce the neurotoxicity of Aβ1–42, SH-SY5Y human neuroblastoma cells were cultured and differentiated for use as a neuronal cell model.26,76–78 Pre-formed Aβ1–42 oligomers, or those formed in situ, show concentration-dependent toxicity to SH-SY5Y cells,26 and recent work has correlated this toxicity to membrane pore formation.79–81 Varying equivalents of KP1019 (0.1, 0.5, 1 or 2 eq.) were combined with Aβ1–42 and incubated for a set period prior to administration to differentiated SH-SY5Y cells (Fig. 6). In line with previous reports,82,83 exposure of SH-SY5Y cells to Aβ1–42 for 24 hours results in a significant decrease in cell viability at >10 μM (Fig. S4, ESI†). The toxicity at 20 μM Aβ1–42 (52% ± 7% viability, Fig. 6) was found to be appropriate for the current work. When SH-SY5Y cells have been treated with incubated KP1019-Aβ1–42 species, a significant reversal of cell viability is observed at ≥1 equiv. KP1019, even at post 5 minute incubation (88% ± 4%) when compared to cells treated with Aβ1–42 only. Our results show that a 1:1 ratio of KP1019 to Aβ1–42 peptide is needed to rescue cell viability in this assay.
 | ||
| Fig. 6 Toxicity studies using differentiated SH-SY5Y human neuroblastoma cells. Cells were treated with Aβ1–42 (20 μM) or pre-incubated KP1019-Aβ1–42 for 24 hours and then the cell viability measured using an Alamar Blue assay. Varying equivalents of KP1019-Aβ1–42 were incubated for 5 minutes (yellow), 2 hours (orange), or 24 hours (red) and then exposed to cells for a further 24 hours. Alamar Blue reagent was then incubated with cells for 2 hours at 37 °C, followed by fluorescence measurements to determine cell viability. Data are means ± SEM; n = 4. The t-test analysis of experimental data compared to Aβ1–42 identifies values of p < 0.001 except for *p < 0.1. | ||
KP1019 alone exhibits slight toxicity at 1 mM in the SH-SY5Y cell line, outside the concentration range that was investigated in this work (Fig. S5, ESI†). Our in vitro results demonstrate that the interaction of KP1019 with Aβ1–42 reverses the neurotoxicity of the Aβ1–42 peptide in the SH-SY5Y cell line, likely by promoting the formation of non-toxic high molecular weight aggregates (vide supra). While outside the scope of this work, we speculate that the different morphology and/or charge of the KP1019-Aβ1–42 species that form limit cell membrane interactions and pore formation in the SH-SY5Y cell line.
Summary
This report highlights the ability of the well-known Ru(III) anti-cancer compound KP1019 to modulate the aggregation profile of the Aβ peptide associated with AD. KP1019 covalently binds to the Aβ peptide within 2 hours and remains bound for at least 24 hours, demonstrating promising kinetic and thermodynamic properties for regulating Aβ aggregation. Regardless of whether KP1019 is exposed to monomeric or pre-aggregated Aβ species, this Ru(III) complex facilitates the formation of soluble high molecular weight Aβ aggregates. In addition, KP1019 was shown to limit the cell toxicity of the Aβ1–42 peptide in the SH-SY5Y human neuroblastoma cell line. Our results suggest that Ru(III) complexes may be promising candidates for modulating the aggregation pathway, and associated toxicity of the Aβ peptide in AD.Acknowledgements
T. S. and C. J. W. thank the National Science and Engineering Research Council of Canada and T. S. thanks the Michael Smith Foundation for Health Research for their generous support. M. R. J. thanks the Alzheimer's Society of Canada for a doctoral award. The Klassen group at the University of Alberta is acknowledged for their analytical support. The authors appreciate the efforts of Neil Dobson for his continued support in maintaining the tissue culturing facility. Prof. Frank Lee from the Department of Health Sciences is acknowledged for donating the SH-SY5Y cell line.Notes and references
- A. S. DeToma, S. Salamekh, A. Ramamoorthy and M. H. Lim, Chem. Soc. Rev., 2012, 41, 608–621 RSC.
- http://www.alzheimer.ca .
- http://www.alz.org .
- K. P. Kepp, Chem. Rev., 2012, 112, 5193–5239 CrossRef CAS PubMed.
- M. Citron, Nat. Rev. Drug Discovery, 2010, 9, 387–398 CrossRef CAS PubMed.
- Y. Huang and L. Mucke, Cell, 2012, 148, 1204–1222 CrossRef CAS PubMed.
- J. A. Hardy and G. A. Higgins, Science, 1992, 256, 184–185 CAS.
- E. McGowan, F. Pickford, J. Kim, L. Onstead, J. Eriksen, C. Yu, L. Skipper, M. P. Murphy, J. Beard, P. Das, K. Jansen, M. DeLucia, W.-L. Lin, G. Dolios, R. Wang, C. B. Eckman, D. W. Dickson, M. Hutton, J. Hardy and T. Golde, Neuron, 2005, 47, 191–199 CrossRef CAS PubMed.
- I. Kuperstein, K. Broersen, I. Benilova, J. Rozenski, W. Jonckheere, M. Debulpaep, A. Vandersteen, I. Segers-Nolten, K. Van Der Werf, V. Subramaniam, D. Braeken, G. Callewaert, C. Bartic, R. D'Hooge, I. C. Martins, F. Rousseau, J. Schymkowitz and B. De Strooper, EMBO J., 2010, 29, 3408–3420 CrossRef CAS PubMed.
- R. J. O'Brien and P. C. Wong, Annu. Rev. Neurosci., 2011, 34, 185–204 CrossRef PubMed.
- M. P. Mattson, Nature, 2004, 430, 631–639 CrossRef CAS PubMed.
- F. Hane and Z. Leonenko, Biomolecules, 2014, 4, 101–116 CrossRef PubMed.
- C. G. Glabe, J. Biol. Chem., 2008, 283, 29639–29643 CrossRef CAS PubMed.
- J. C. Stroud, C. Liu, P. K. Teng and D. Eisenberg, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7717–7722 CrossRef CAS PubMed.
- R. Kayed, A. Pensalfini, L. Margol, Y. Sokolov, F. Sarsoza, E. Head, J. Hall and C. Glabe, J. Biol. Chem., 2009, 284, 4230–4237 CrossRef CAS PubMed.
- R. Kayed, E. Head, J. L. Thompson, T. M. McIntire, S. C. Milton, C. W. Cotman and C. G. Glabe, Science, 2003, 300, 486–489 CrossRef CAS PubMed.
- C. Haass and D. J. Selkoe, Nat. Rev. Mol. Cell Biol., 2007, 8, 101–112 CrossRef CAS PubMed.
- D. M. Walsh and D. J. Selkoe, J. Neurochem., 2007, 101, 1172–1184 CrossRef CAS PubMed.
- V. Borutaite, R. Morkuniene and G. Valincius, BioMolecular Concepts, 2011, vol. 2, p. 211 Search PubMed.
- P. Giannakopoulos, C. Bouras, F. R. Herrmann, W. L. Klein, E. Kovari, A. Savioz and E. Giacobini, Neurobiol. Aging, 2014, 35, S9 CrossRef PubMed.
- W. L. Klein, J. Alzheimer's Dis., 2013, 33, S49–S65 Search PubMed.
- Q. Nie, X.-g. Du and M.-y. Geng, Acta Pharmacol. Sin., 2011, 32, 545–551 CrossRef CAS PubMed.
- B. Solomon, R. Koppel, D. Frankel and E. Hanan-Aharon, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 4109–4112 CrossRef CAS.
- S. Lee, X. Zheng, J. Krishnamoorthy, M. G. Savelieff, H. M. Park, J. R. Brender, J. H. Kim, J. S. Derrick, A. Kochi, H. J. Lee, C. Kim, A. Ramamoorthy, M. T. Bowers and M. H. Lim, J. Am. Chem. Soc., 2013, 136, 299–310 CrossRef PubMed.
- A. K. Sharma, S. T. Pavlova, J. Kim, D. Finkelstein, N. J. Hawco, N. P. Rath, J. Kim and L. M. Mirica, J. Am. Chem. Soc., 2012, 134, 6625–6636 CrossRef CAS PubMed.
- J. Bieschke, M. Herbst, T. Wiglenda, R. P. Friedrich, A. Boeddrich, F. Schiele, D. Kleckers, J. M. Lopez del Amo, B. A. Grüning, Q. Wang, M. R. Schmidt, R. Lurz, R. Anwyl, S. Schnoegl, M. Fändrich, R. F. Frank, B. Reif, S. Günther, D. M. Walsh and E. E. Wanker, Nat. Chem. Biol., 2012, 8, 93–101 CrossRef CAS PubMed.
- P. E. Cramer, J. R. Cirrito, D. W. Wesson, C. Y. D. Lee, J. C. Karlo, A. E. Zinn, B. T. Casali, J. L. Restivo, W. D. Goebel, M. J. James, K. R. Brunden, D. A. Wilson and G. E. Landreth, Science, 2012, 335, 1503–1506 CrossRef CAS PubMed.
- R. A. Cherny, C. S. Atwood, M. E. Xilinas, D. N. Gray, W. D. Jones, C. A. McLean, K. J. Barnham, I. Volitakis, F. W. Fraser, Y.-S. Kim, X. Huang, L. E. Goldstein, R. D. Moir, J. T. Lim, K. Beyreuther, H. Zheng, R. E. Tanzi, C. L. Masters and A. I. Bush, Neuron, 2001, 30, 665–676 CrossRef CAS.
- P. A. Adlard, R. A. Cherny, D. I. Finkelstein, E. Gautier, E. Robb, M. Cortes, I. Volitakis, X. Liu, J. P. Smith, K. Perez, K. Laughton, Q.-X. Li, S. A. Charman, J. A. Nicolazzo, S. Wilkins, K. Deleva, T. Lynch, G. Kok, C. W. Ritchie, R. E. Tanzi, R. Cappai, C. L. Masters, K. J. Barnham and A. I. Bush, Neuron, 2008, 59, 43–55 CrossRef CAS PubMed.
- M. Necula, R. Kayed, S. Milton and C. G. Glabe, J. Biol. Chem., 2007, 282, 10311–10324 CrossRef CAS PubMed.
- J. Bieschke, J. Russ, R. P. Friedrich, D. E. Ehrnhoefer, H. Wobst, K. Neugebauer and E. E. Wanker, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 7710–7715 CrossRef CAS PubMed.
- D. J. Hayne, S. Lim and P. S. Donnelly, Chem. Soc. Rev., 2014, 43, 6701–6715 RSC.
- I. Sasaki, C. Bijani, S. Ladeira, V. Bourdon, P. Faller and C. Hureau, Dalton Trans., 2012, 41, 6404–6407 RSC.
- F. Collin, I. Sasaki, H. Eury, P. Faller and C. Hureau, Chem. Commun., 2013, 49, 2130–2132 RSC.
- C. Hureau and P. Faller, Dalton Trans., 2014, 43, 4233–4237 RSC.
- K. J. Barnham, V. B. Kenche, G. D. Ciccotosto, D. P. Smith, D. J. Tew, X. Liu, K. Perez, G. A. Cranston, T. J. Johanssen, I. Volitakis, A. I. Bush, C. L. Masters, A. R. White, J. P. Smith, R. A. Cherny and R. Cappai, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6813–6818 CrossRef CAS PubMed.
- V. B. Kenche, L. W. Hung, K. Perez, I. Volitakes, G. Ciccotosto, J. Kwok, N. Critch, N. Sherratt, M. Cortes, V. Lal, C. L. Masters, K. Murakami, R. Cappai, P. A. Adlard and K. J. Barnham, Angew. Chem., Int. Ed., 2013, 52, 3374–3378 CrossRef CAS PubMed.
- B. Y.-W. Man, H.-M. Chan, C.-H. Leung, D. S.-H. Chan, L.-P. Bai, Z.-H. Jiang, H.-W. Li and D.-L. Ma, Chem. Sci., 2011, 2, 917–921 RSC.
- D. Valensin, P. Anzini, E. Gaggelli, N. Gaggelli, G. Tamasi, R. Cini, C. Gabbiani, E. Michelucci, L. Messori, H. Kozlowski and G. Valensin, Inorg. Chem., 2010, 49, 4720–4722 CrossRef CAS PubMed.
- D. Valensin, C. Gabbiani and L. Messori, Coord. Chem. Rev., 2012, 256, 2357–2366 CrossRef CAS PubMed.
- L. Messori, M. Camarri, T. Ferraro, C. Gabbiani and D. Franceschini, ACS Med. Chem. Lett., 2013, 4, 329–332 CrossRef CAS PubMed.
- A. Kumar, L. Moody, J. F. Olaivar, N. A. Lewis, R. L. Khade, A. A. Holder, Y. Zhang and V. Rangachari, ACS Chem. Neurosci., 2010, 1, 691–701 CrossRef CAS PubMed.
- J. Reedijk, Platinum Met. Rev., 2008, 52, 2–11 CrossRef CAS.
- M. J. Clarke, Coord. Chem. Rev., 2002, 232, 69–93 CrossRef CAS.
- A. Bergamo, C. Gaiddon, J. H. M. Schellens, J. H. Beijnen and G. Sava, J. Inorg. Biochem., 2012, 106, 90–99 CrossRef CAS PubMed.
- B. K. Keppler, M. Henn, U. M. Juhl, M. R. Berger, R. Niebl and F. E. Wagner, Prog. Clin. Biochem. Med., 1989, 10, 41–69 CAS.
- H. Depenbrock, S. Schmelcher, R. Peter, B. K. Keppler, G. Weirich, T. Block, J. Rastetter and A. R. Hanauske, Eur. J. Cancer, 1997, 33, 2404–2410 CrossRef CAS.
- A. Galeano, M. R. Berger and B. K. Keppler, Arzneim.-Forsch./Drug Res., 1992, 42, 821–824 CAS.
- P. Heffeter, M. Pongratz, E. Steiner, P. Chiba, M. A. Jakupec, L. Elbling, B. Marian, W. Körner, F. Sevelda, M. Micksche, B. K. Keppler and W. Berger, J. Pharmacol. Exp. Ther., 2005, 312, 281–289 CrossRef CAS PubMed.
- S. Kapitza, M. A. Jakupec, M. Uhl, B. K. Keppler and B. Marian, Cancer Lett., 2005, 226, 115–121 CrossRef CAS PubMed.
- S. Kapitza, M. Pongratz, M. A. Jakupec, P. Heffeter, W. Berger, L. Lackinger, B. K. Keppler and B. Marian, J. Cancer Res. Clin. Oncol., 2005, 131, 101–110 CrossRef CAS PubMed.
- E. D. Kreuser, B. K. Keppler, W. E. Berdel, A. Piest and E. Thiel, Semin. Oncol., 1992, 19, 73–81 CAS.
- C. G. Hartinger, S. Zorbas-Seifried, M. A. Jakupec, B. Kynast, H. Zorbas and B. K. Keppler, J. Inorg. Biochem., 2006, 100, 891–904 CrossRef CAS PubMed.
- C. G. Hartinger, M. A. Jakupec, S. Zorbas-Seifried, M. Groessl, A. Egger, W. Berger, H. Zorbas, P. J. Dyson and B. K. Keppler, Chem. Biodiversity, 2008, 5, 2140–2155 CAS.
- F. Lentz, A. Drescher, A. Lindauer, M. Henke, R. A. Hilger, C. G. Hartinger, M. E. Scheulen, C. Dittrich, B. K. Keppler and U. Jaehde, Anti-Cancer Drugs, 2009, 20, 97–103 CrossRef CAS PubMed.
- C. G. Hartinger, S. Hann, G. Koellensperger, M. Sulyok, M. Groessl, A. R. Timerbaev, A. V. Rudnev, G. Stingeder and B. K. Keppler, Int. J. Clin. Pharmacol. Ther., 2005, 43, 583–585 CrossRef CAS.
- A. R. Timerbaev, A. V. Rudnev, O. Semenova, C. G. Hartinger and B. K. Keppler, Anal. Biochem., 2005, 341, 326–333 CrossRef CAS PubMed.
- N. Cetinbas, M. Webb, J. Dubland and C. Walsby, JBIC, J. Biol. Inorg. Chem., 2010, 15, 131–145 CrossRef CAS PubMed.
- P. Heffeter, K. Bock, B. Atil, M. A. R. Hoda, W. Korner, C. Bartel, U. Jungwirth, B. K. Keppler, M. Micksche, W. Berger and G. Koellensperger, J. Biol. Inorg. Chem., 2010, 15, 737–748 CrossRef CAS PubMed.
- F. Piccioli, S. Sabatini, L. Messori, P. Orioli, C. G. Hartinger and B. K. Keppler, J. Inorg. Biochem., 2004, 98, 1135–1142 CrossRef CAS PubMed.
- J. Hardy and D. J. Selkoe, Science, 2002, 297, 353–356 CrossRef CAS PubMed.
- D. J. Selkoe, Nat. Med., 2011, 17, 1060–1065 CrossRef CAS PubMed.
- L. S. Wolfe, M. F. Calabrese, A. Nath, D. V. Blaho, A. D. Miranker and Y. Xiong, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 16863–16868 CrossRef CAS PubMed.
- A. M. Klein, N. W. Kowall and R. J. Ferrante, Ann. N. Y. Acad. Sci., 1999, 893, 314–320 CrossRef CAS PubMed.
- A. K. Sharma, S. T. Pavlova, J. Kim, J. Kim and L. M. Mirica, Metallomics, 2013, 5, 1529–1536 RSC.
- M. I. Webb and C. J. Walsby, Metallomics, 2013, 5, 1624–1633 RSC.
- M. I. Webb, B. Wu, T. Jang, R. A. Chard, E. W. Y. Wong, M. Q. Wong, D. T. T. Yapp and C. J. Walsby, Chem. – Eur. J., 2013, 19, 17031–17042 CrossRef CAS PubMed.
- P. Faller, C. Hureau and O. Berthoumieu, Inorg. Chem., 2013, 52, 12193–12206 CrossRef CAS PubMed.
- B. Cebrián-Losantos, E. Reisner, C. R. Kowol, A. Roller, S. Shova, V. B. Arion and B. K. Keppler, Inorg. Chem., 2008, 47, 6513–6523 CrossRef PubMed.
- K. S. Kim, Neurosci. Res. Commun., 1988, 2, 121–130 CAS.
- H. W. Querfurth and F. M. LaFerla, N. Engl. J. Med., 2010, 362, 329–344 CrossRef CAS PubMed.
- M. Bucciantini, E. Giannoni, F. Chiti, F. Baroni, L. Formigli, J. Zurdo, N. Taddei, G. Ramponi, C. M. Dobson and M. Stefani, Nature, 2002, 416, 507–511 CrossRef CAS PubMed.
- F. M. LaFerla, Nat. Rev. Neurosci., 2002, 3, 862–872 CrossRef CAS PubMed.
- H. Jang, F. T. Arce, S. Ramachandran, R. Capone, R. Azimova, B. L. Kagan, R. Nussinov and R. Lal, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6538–6543 CrossRef CAS PubMed.
- K. Ono, M. Condron and D. Teplow, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14745–14750 CrossRef CAS PubMed.
- C. Barucker, A. Harmeier, J. Weiske, B. Fauler, K. F. Albring, S. Prokop, P. Hildebrand, R. Lurz, F. L. Heppner, O. Huber and G. Multhaup, J. Biol. Chem., 2014, 289, 20182–20191 CrossRef CAS PubMed.
- F. Zhou, Y. Xu and X.-Y. Hou, J. Neurosci. Res., 2014, 92, 808–817 CrossRef CAS PubMed.
- J. Luo, I. Mohammed, S. K. T. S. Wärmländer, Y. Hiruma, A. Gräslund and J. P. Abrahams, Biomacromolecules, 2014, 15, 1985–1991 CrossRef CAS PubMed.
- E. Hayden and D. Teplow, Alzheimer's Res. Ther., 2013, 5, 60 CrossRef PubMed.
- P. Prangkio, E. Yusko, D. Sept, J. Yang and M. Mayer, PLoS One, 2012, 7, e47261 CAS.
- B. Kagan and J. Thundimadathil, in Proteins Membrane Binding and Pore Formation, ed. G. Anderluh and J. Lakey, Springer, New York, 2010, vol. 677, pp. 150–167 Search PubMed.
- Q. Wang, X. Yu, K. Patal, R. Hu, S. Chuang, G. Zhang and J. Zheng, ACS Chem. Neurosci., 2013, 4, 1004–1015 CrossRef CAS PubMed.
- R. Liu, H. Barkhordarian, S. Emadi, C. B. Park and M. R. Sierks, Neurobiol. Dis., 2005, 20, 74–81 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, dot blot assay, KP1019 and Aβ1–42 toxicity in the SH-SY5Y cell line. See DOI: 10.1039/c4mt00252k |
| This journal is © The Royal Society of Chemistry 2015 |