
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTraumatic brain injury induces elevation of Co in the human brain
Blaine R.
Roberts
a,
Dominic J.
Hare
ab,
Catriona A.
McLean
c,
Alison
Conquest
d,
Monica
Lind
a,
Qiao-Xin
Li
a,
Ashley I.
Bush
 a,
Colin L.
Masters
a,
Maria-Christina
Morganti-Kossmann
d and
Tony
Frugier
*cde
a,
Colin L.
Masters
a,
Maria-Christina
Morganti-Kossmann
d and
Tony
Frugier
*cde
aThe Florey Institute of Neuroscience and Mental Health, The University of Melbourne, Parkville, Victoria, Australia
bElemental Bio-imaging Facility, University of Technology Sydney, Broadway, NSW, Australia
cDepartment of Anatomical Pathology, The Alfred Hospital, Melbourne, Victoria, Australia
dNational Trauma Institute, The Alfred Hospital, Melbourne, Victoria, Australia
eDepartment of Pharmacology and Therapeutics, The University of Melbourne, Parkville Campus, Victoria 3031, Australia. E-mail: tony.frugier@unimelb.edu.au; Fax: +61 3 8344 0241; Tel: +61 3 9035 7662
First published on 14th November 2014
Abstract
Traumatic brain injury (TBI) is the most common cause of death and disability in young adults, yet the molecular mechanisms that follow TBI are poorly understood. We previously reported a perturbation in iron (Fe) levels following TBI. Here we report that the distribution of cobalt (Co) is modulated in post-mortem human brain following injury. We also investigated how the distribution of other biologically relevant elements changes in TBI. Cobalt is increased due to TBI while copper (Cu), magnesium (Mg), manganese (Mn), phosphorus (P), potassium (K), rubidium (Rb), selenium (Se) and zinc (Zn) remain unchanged. The elevated Co has important implications for positron emission tomography neuroimaging. This is the first demonstration of the accumulation of Co in injured tissue explaining the previous utility of 55Co-PET imaging in TBI.
Traumatic brain injury (TBI) is a major health and socioeconomic problem worldwide which according to the World Health Organization projection will become the major cause of death and disability by 2020. Annually there are an estimated 10 million people affected by TBI.1 Repeated or moderate to severe TBI is suspected to be a risk factor for the development of Alzheimer's disease2 and sporadic Parkinson's disease.3 In humans the molecular mechanisms leading to neurodegeneration and poor neurological outcome remain unclear.
Transition elements, although in trace amounts, are vital for biological function and for all facets of life. Approximately half of all enzymes are metalloenzymes.4 Zinc has been implicated as having a positive impact on the outcome of TBI in mouse models,5,6 but there is a lack of information on how metals change as a result of TBI in humans, save for a recent report of iron (Fe) accumulation post-injury.7 In this study we analyze the level of cobalt (Co), copper (Cu), iron (Fe), magnesium (Mg), manganese (Mn), phosphorus (P), potassium (K), rubidium (Rb), selenium (Se) and zinc (Zn) in human brain tissue of patients who died from severe traumatic brain injury.
All procedures were conducted in accordance with the Australian National Health and Medical Research Council's National Statement on Ethical Conduct in Human Research (2007), the Victorian Human Tissue Act (1982), the National Code of Ethical Autopsy Practice (2002) and the Victorian Government policies and practices in relation to post-mortem.
Trauma brain samples from 27 individuals who died after closed head injury were obtained from the Australian Neurotrauma Tissue and Fluid Bank. Cases were aged between 17 and 78 years (mean = 48 years) and the causes of injury included motor vehicle accident, motorbike accident, nursing home accident, household accident, stair accident and falls. The post-mortem intervals varied between 33 and 129 hours (mean = 81 hours). Patients were divided in 3 groups to compare the acute and delayed times after injury: 10 cases (8 males and 2 females) had survival time of less than 17 minutes, designated ‘acute’ group, when death occurred upon arrivals of the paramedics (cases 1–10); 8 cases (7 males and 1 female) were selected with a survival time between 30 minutes and 3 hours (mean = 1 hour) and designated as ‘early’ group (cases 11–18); and 9 cases (7 males and 2 females) had survival time between 6 and 122 hours (mean = 43 hours) and designated ‘late’ group (cases 19–27). The late cohort also contains tissue from the same area on the non-injured (late-NI) side of the brain. The brain region analyzed was located in proximity of the injured tissue and was identified macroscopically by a neuropathologist (Prof. McLean). Control brain samples of 10 individuals, aged between 16 and 78 (mean = 56 years), without brain injury or other neuropathology were obtained from the National Neural Tissue Resource Centre of Australia (cases 28–37). Clinical information and epidemiological details of all patients are described in Table 1.
| Case | Age | Sex | Cause of injury | PMI (h) | Cause of death | Survival time |
|---|---|---|---|---|---|---|
| a Acute group. b Early group. c Late group. d Control group. | ||||||
| 1a | 51 | M | Motor vehicle accident | 60 | Brain + multiple injuries | <17 min |
| 2a | 63 | M | Household accident | 70 | Brain injury | <17 min |
| 3a | 27 | M | Suicide | 84 | Brain + multiple injuries | <17 min |
| 4a | 41 | M | Suicide | 96 | Brain + multiple injuries | <17 min |
| 5a | 57 | F | Motor vehicle accident | 87 | Brain + multiple injuries | <17 min |
| 6a | 49 | M | Motor vehicle accident | 107 | Brain + multiple injuries | <17 min |
| 7a | 45 | M | Motor vehicle accident | 43 | Brain + multiple injuries | <17 min |
| 8a | 21 | M | Motor vehicle accident | 100 | Brain injury | <17 min |
| 9a | 41.3 | M | Aviation accident | 114 | Brain + multiple injuries | <17 min |
| 10a | 57.6 | F | Motor vehicle accident | 97 | Brain injury | <17 min |
| 11b | 16.8 | M | Motor vehicle accident | 85 | Brain + multiple injuries | <3 h |
| 12b | 78.7 | M | Household accident | 45 | Brain injury | <3 h |
| 13b | 18.3 | M | Motor vehicle accident | 79 | Brain + multiple injuries | <3 h |
| 14b | 34.7 | M | Motorbike accident | 66 | Brain + multiple injuries | <3 h |
| 15b | 22.9 | F | Motor vehicle accident | 108 | Brain + multiple injuries | <3 h |
| 16b | 52.8 | M | Motorbike accident | 65 | Brain + multiple injuries | <3 h |
| 17b | 19.6 | M | Suicide | 33 | Brain + multiple injuries | <3 h |
| 18b | 59.8 | M | Motor vehicle accident | 71 | Brain + multiple injuries | <3 h |
| 19c | 46.0 | M | Fall | 129 | Brain injury | 6 h |
| 20c | 56.3 | M | Motor vehicle accident | 65 | Brain injury | 8 h |
| 21c | 64.6 | M | Fall | 61 | Brain injury | 8 h |
| 22c | 75.9 | M | Staircase fall | 89 | Brain injury | 10 h |
| 23c | 59.6 | F | Motor vehicle accident | 80 | Brain injury | 35 h |
| 24c | 61.7 | M | Fall | 40 | Brain injury | 93 h |
| 25c | 38.9 | F | Staircase fall | 101 | Brain injury | 122 h |
| 26c | 70.9 | M | Motor vehicle accident | 114 | Brain injury | 76 h |
| 27c | 73.7 | M | Fall | 91 | Brain injury | 29 h |
| 28d | 16 | M | — | — | Suicide by hanging | — |
| 29d | 48.7 | M | — | 50 | Cardiac failure | — |
| 30d | 51.6 | M | — | 64 | Asthma | — |
| 31d | 52.3 | M | — | 52 | Cardiomyopathy | — |
| 32d | 59.6 | M | — | 43 | Pulmonary embolism | — |
| 33d | 64.1 | M | — | 24 | Ischaemic heart disease | — |
| 34d | 66.9 | M | — | 10 | Pneumonia | — |
| 35d | 64.4 | M | — | 24 | Pulmonary embolism | — |
| 36d | 77.5 | M | — | 53 | Myocardial infarction | — |
| 37d | 60 | F | — | 48 | Myocardial infarction | — |
Approximately 0.25 g of frozen tissue was allowed to thaw from −80 °C on ice and then homogenized using a BioMasher (Omni International). Tissue was placed in the BioMasher, the plunger was inserted and then the apparatus was centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm with a benchtop centrifuge. After centrifugation Tris buffer saline (TBS, 50 mM Tris pH 8.0, 150 mM NaCl) containing EDTA free protease inhibitors (Roche) was added at a ratio of 1:4 (w/v). The sample was centrifuged at 175000g for 30 minutes at 4 °C. The TBS supernatant was collected and stored at −80 °C before analysis. The resulting pellet was washed with a volume of TBS equal to the amount used for homogenization and centrifuged for 15 minutes at 175000g at 4 °C. The TBS wash supernatant was removed and the pellet was resuspended in 7 M urea 2 M thiourea 4% CHAPS 30 mM bicine pH 8.5 and centrifuged as before. The resulting pellet was then incubated with 70% formic acid for 16 hours at room temperature before being centrifuged at 16000g for 30 minutes.
000 rpm with a benchtop centrifuge. After centrifugation Tris buffer saline (TBS, 50 mM Tris pH 8.0, 150 mM NaCl) containing EDTA free protease inhibitors (Roche) was added at a ratio of 1:4 (w/v). The sample was centrifuged at 175000g for 30 minutes at 4 °C. The TBS supernatant was collected and stored at −80 °C before analysis. The resulting pellet was washed with a volume of TBS equal to the amount used for homogenization and centrifuged for 15 minutes at 175000g at 4 °C. The TBS wash supernatant was removed and the pellet was resuspended in 7 M urea 2 M thiourea 4% CHAPS 30 mM bicine pH 8.5 and centrifuged as before. The resulting pellet was then incubated with 70% formic acid for 16 hours at room temperature before being centrifuged at 16000g for 30 minutes.
Inductively coupled plasma mass spectrometry (ICP-MS) was used to determine the quantity of Co, Cu, Fe, Mg, Mn, P, K, Rb, Se and Zn in the TBS soluble, membrane and formic acid extracted homogenates. Supernatants were diluted 1:15 with 1% nitric acid (Suparpur, Merck). Measurements were made using an Agilent 7700 series ICP-MS instrument under routine multi-element operating conditions using a helium collision gas cell. The instrument was calibrated using 0, 5, 10, 50 and 100 ppb of certified multi-element ICP-MS standard calibration solutions (ICP-MS-CAL2-1, ICP-MS-CAL-3 and ICP-MS-CAL-4, Accustandard) for a range of elements. We used a certified internal standard solution containing 200 ppb of yttrium (89Y) as an internal control (ICP-MS-IS-MIX1-1, Accustandard). The sample was introduced via the automated liquid sampler (Agilent) using a peristaltic pump at a flow rate of 0.4 mL min−1.
Statistical analysis was performed using SigmaStat (SysStat, San Jose, CA), Prism 5.0 (GraphPad, La Jolla, CA) and SPSS software (SPSS Inc., Chicago, IL). Kolmogorov–Smirnov test (with Lilliefors' correction) was used to test data for normality within each group and values were transformed by natural logarithm calculation if required. One-way ANOVA was followed by multiple comparisons using the Tukey's post-hoc test to identify significant difference between trauma and control groups. Statistical significance was considered at the 5% level (p < 0.05).
Quantitative measurement of Co, Cu, Fe, Mg, Mn, P, K, Rb, Se and Zn was performed on the three extracted pools of material: soluble, membrane and formic acid (Table 2). The total level of each element measured were consistent with the total level per gram of wet tissue for Co, Cu, Fe, Mg, Mn, P, K, Rb, Se and Zn levels previously reported for normal human brain tissue.8–10 Cobalt and Fe (previously reported by Ayton et al.7) were the only elements that changed significantly in response to TBI. Co was significantly elevated in the acute TBI patients (Fig. 1a). The elevation of Co was restricted to the soluble fraction with no evidence of a change in Co levels in the membrane or FA fractions (Table 2). Although the elevation in Co remained significantly elevated in the total (Fig. 1b, [total] = [soluble + membrane + FA]) the signal to noise was much greater due to the addition of the membrane and FA fractions. The elevation of Co returned to baseline 3 hours post-injury.
| Co (ng g−1) | Cu (μg g−1) | Fe (μg g−1) | Mg (μg g−1) | Mn (ng g−1) | P (mg g−1) | K (mg g−1) | Rb (μg g−1) | Se (ng g−1) | Zn (μg g−1) | |
|---|---|---|---|---|---|---|---|---|---|---|
| a Excludes values from the ‘acute’ cohort (n = 25). b Excludes values from the ‘late’ cohort (n = 25; reported in Ayton et al.7). c Total = soluble + membrane + formic acid. | ||||||||||
| Soluble | 1.0 ± 0.7 (0.3–2.7)b | 2.2 ± 0.9 (0.8–4.4) | 8.7 ± 4.2 (2.5–25.3)a | 58.7 ± 28 (21–189) | 51 ± 24 (19–145) | 0.5 ± 0.3 (0.2–1.6) | 1.5 ± 0.7 (0.4–4.4) | 2.2 ± 1.0 (0.6–5.3) | 57 ± 26 (19–152) | 2.8 ± 1.1 (1.3–7.6) |
| Membrane | 1.4 ± 0.6 (0.4–3.3) | 1.3 ± 0.4 (0.6–2.3) | 36.9 ± 10 (19–63) | 34.5 ± 10 (19–73) | 118 ± 34 (56–206) | 1.0 ± 0.4 (0.3–1.9) | 0.4 ± 0.1 (0.2–1.0) | 0.6 ± 0.2 (0.3–1.4) | 79 ± 35 (0.5–150) | 5.1 ± 2.6 (1.9–18) |
| Formic Acid | 3.5 ± 1.3 (2–9) | 1.3 ± 0.3 (0.8–2.1) | 32.3 ± 12 (10–55) | 42.0 ± 14 (17–78) | 119 ± 31 (66–210) | 1.2 ± 0.3 (0.7–2.1) | 0.3 ± 0.1 (0.1–0.6) | 0.4 ± 0.2 (0.2–0.9) | 134 ± 35 (70–260) | 6.3 ± 1.6 (4–11) |
| Totalc | 5.8 ± 2 (4 + 10)a | 4.8 ± 1.2 (2.5–7.5) | 76.0 ± 17 (39–107)b | 136.2 ± 36 (63–255) | 291 ± 60 (198–425) | 2.7 ± 0.6 (1.7–4.2) | 2.2 ± 0.7 (0.8–5.0) | 3.2 ± 1.0 (1.2–6.0) | 270 ± 60 (123–400) | 14.3 ± 4.4 (8.6–29.5) |
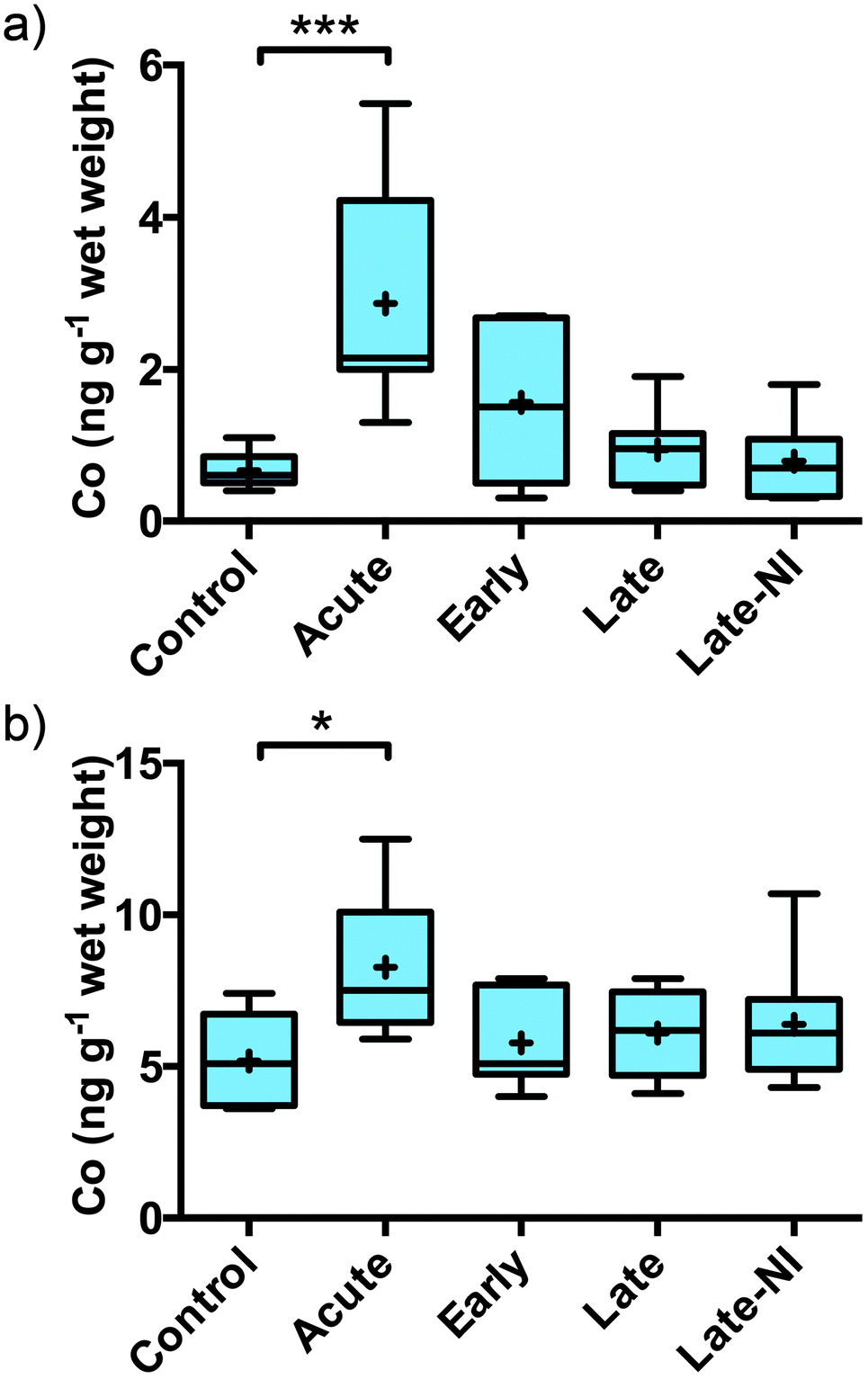 | ||
| Fig. 1 Elevated levels of Co in the human brain following TBI as measured by ICP-MS. (a) Co is significantly elevated in the acute TBI cases in the TBS soluble extracted material compared to all other groups (***Tukey post-hoc test p < 0.001) and (b) in the total Co levels calculated as the sum of soluble, membrane and formic acid extracted Co is also significantly elevated but to a lesser extent (*Tukey post-hoc test p < 0.05). Boxes represent interquartile range; error bars represent minimum and maximum values; + represents mean and line represents median. | ||
Finally, the fractionation protocol allowed a more detail analysis about how the elements are distributed in the tissue (Fig. 2). Analyzing the proportion of an element in each fraction showed that elements K and Rb are greater than 70% associated with the soluble extracted material consistent with these elements being free ions.11 Alternatively, elements largely associated to membrane bound proteins, such as Fe, had greater than 80% of the element associated with the membranous pellet (membrane plus FA fraction) consistent with Fe being a cofactor for membrane associated proteins involved in the electron transport chain, for example.
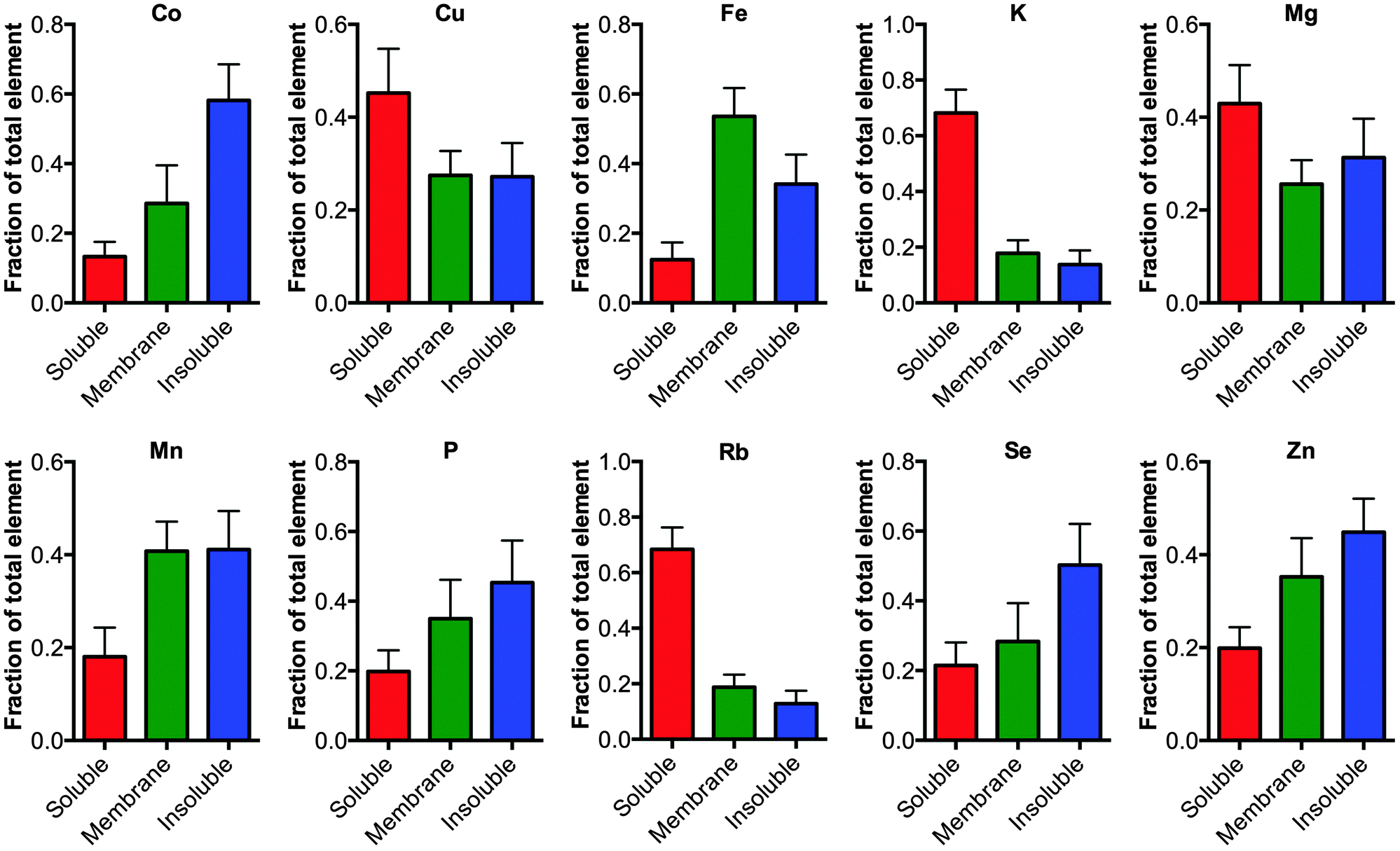 | ||
| Fig. 2 Distribution of elements in the cellular fractions. The elements Co, Cu, Fe, Mg, Mn, P, K, Rb, Se and Zn were measured in the soluble, membrane and formic acid fractions using ICP-MS, no significant changes were observed between the tissue from TBI and controls (one-way ANOVA) except for Co as noted in Fig. 1. The fraction of each element in the respective pool is graphed (mean ± 1 standard deviation). As expected elements that are truly fee ions such as Rb and K are mostly distributed in the soluble phase. | ||
Calcium (Ca) overload and inflammatory process are both attributed to the resulting neuronal death. Co is used as a surrogate marker of Ca accumulation in degenerating neurons.12 Radioisotopes of Co have been successfully used to detect ischemic damage, extravasation or inflammation in several neurodegenerative pathologies.13–15 This is the first study to demonstrate that TBI induces the specific uptake of in situ Co into brain tissue. Cobalt is predominantly found as cobalamin (vitamin B12), which is used as a cofactor for methyl transfer reactions that are vital for DNA synthesis and fatty acid synthesis. Alternatively, Co is also utilized in a vitamin B12 independent fashion in the enzyme methionine aminopeptidase 2,16 which removes the N-terminal methionine of newly synthesized proteins and plays a vital role in the angiogenesis of blood vessels and as such is a target of anticancer angiogenesis compounds. Long-term hypoxia in mice has been shown to both elevate brain Co levels and increase activity of vitamin B12.17 Previous positron emission tomography studies have used 55CoCl2 to detect areas of ischemic damage, suggesting that the observed increase in Co in acute patients may also be due to an influx of non-vitamin B12 Co. Additionally, the cellular fractionation of the tissue into soluble and membrane pools demonstrate that the increased Co is due to an actual accumulation of Co, likely from the blood. There is an emerging role for cobalamin in regulating the production of growth factors and cytokines in the CNS18 and this may be related to the increased uptake of this element in response to TBI. Vitamin B12 and its cobalamin products,19 specifically the thiolato-derivatives20 have also been shown to possess potent antioxidant properties in vitro, which may explain the rapid influx of Co to brain regions under increased oxidative stress, as is the case following acute TBI.21
Acknowledgements
This study was supported by the Victorian Neurotrauma Initiative. Brain tissues were obtained from the Victorian Brain Bank Network, supported by The National Trauma Research Institute, The University of Melbourne, The Florey Institute of Neuroscience and Mental Health, The Victorian Institute of Forensic Medicine and funded by the Victorian Neurotrauma Initiative, Neurosciences Australia, the National Health & Medical Research Council of Australia and the Victorian Government's Operational Infrastructure Support Program.Notes and references
- A. A. Hyder, C. A. Wunderlich, P. Puvanachandra, G. Gururaj and O. C. Kobusingye, NeuroRehabilitation, 2007, 22, 341–353 Search PubMed.
- V. E. Johnson, W. Stewart and D. H. Smith, Nat. Rev. Neurosci., 2010, 11, 361–370 CAS.
- S. M. Goldman, C. M. Tanner, D. Oakes, G. S. Bhudhikanok, A. Gupta and J. W. Langston, Ann. Neurol., 2006, 60, 65–72 CrossRef PubMed.
- K. J. Waldron, J. C. Rutherford, D. Ford and N. J. Robinson, Nature, 2009, 460, 823–830 CrossRef CAS PubMed.
- B. Young, L. Ott, E. Kasarskis, R. Rapp, K. Moles, R. J. Dempsey, P. A. Tibbs, R. Kryscio and C. McClain, J. Neurotrauma, 1996, 13, 25–34 CrossRef CAS.
- P. Doering, M. Stoltenberg, M. Penkowa, J. Rungby, A. Larsen and G. Danscher, PLoS One, 2010, 5, e10131 Search PubMed.
- S. Ayton, M. Zhang, B. R. Roberts, L. Q. Lam, M. Lind, C. McLean, A. I. Bush, T. Frugier, P. J. Crack and J. A. Duce, Free Radical Biol. Med., 2014, 69, 331–337 CrossRef CAS PubMed.
- R. M. Parr and D. M. Taylor, Biochem. J., 1964, 91, 424–431 CAS.
- M. T. Rajan, K. S. Jagannatha Rao, B. M. Mamatha, R. V. Rao, P. Shanmugavelu, R. B. Menon and M. V. Pavithran, J. Neurol. Sci., 1997, 146, 153–166 CrossRef CAS.
- N. A. Larsen, H. Pakkenberg, E. Damsgaard and K. Heydorn, J. Neurol. Sci., 1979, 42, 407–416 CrossRef CAS.
- A. S. Relman, Yale J. Biol. Med., 1956, 29, 248–262 CAS.
- N. E. Andersen, J. Gyring, A. J. Hansen, H. Laursen and B. K. Siesjö, J. Cereb. Blood Flow Metab., 1989, 9, 381–387 CrossRef CAS PubMed.
- H. M. Jansen, J. van der Naalt, A. H. van Zomeren, A. M. Paans, L. Veenma-van der Duin, J. M. Hew, J. Pruim, J. M. Minderhoud and J. Korf, J. Neurol., Neurosurg. Psychiatry, 1996, 60, 221–224 CrossRef CAS.
- J. De Reuck, P. Santens, K. Strijckmans and I. Lemahieu, J. Neurol. Sci., 2001, 193, 1–6 CrossRef CAS.
- H. M. Jansen, J. Pruim, A. M. vd Vliet, A. M. Paans, J. M. Hew, E. J. Franssen, B. M. de Jong, J. G. Kosterink, R. Haaxma and J. Korf, J. Nucl. Med., 1994, 35, 456–460 CAS.
- Y. Hu, F. Vanhaecke, L. Moens, R. Dams, P. del Castilho and J. Japenga, Anal. Chim. Acta, 1998, 373, 95–105 CrossRef CAS.
- S. C. Veasey, J. Lear, Y. Zhu, J. B. Grinspan, D. J. Hare, S. Wang, D. Bunch, P. A. Doble and S. R. Robinson, Sleep, 2013, 36, 1471–1481 Search PubMed.
- G. Scalabrino, J. Neurochem., 2009, 111, 1309–1326 CrossRef CAS PubMed.
- E. S. Moreira, N. E. Brasch and J. Yun, Free Radical Biol. Med., 2011, 51, 876–883 CrossRef CAS PubMed.
- C. S. Birch, N. E. Brasch, A. McCaddon and J. H. Williams, Free Radical Biol. Med., 2009, 47, 184–188 CrossRef CAS PubMed.
- V. A. Tyurin, Y. Y. Tyurina, G. G. Borisenko, T. V. Sokolova, V. B. Ritov, P. J. Quinn, M. Rose, P. Kochanek, S. H. Graham and V. E. Kagan, J. Neurochem., 2000, 75, 2178–2189 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |