
What do we know about multicomponent reactions? Mechanisms and trends for the Biginelli, Hantzsch, Mannich, Passerini and Ugi MCRs
Haline G. O. Alvim
a,
Eufrânio N. da Silva Júnior
b and
Brenno A. D. Neto
*a
aLaboratory of Medicinal and Technological Chemistry, University of Brasilia (IQ-UnB). Campus Universitário Darcy Ribeiro, P. O. Box 4478, CEP 70904970, Brasilia-DF, Brazil. E-mail: brenno.ipi@gmail.com; Fax: +55 61 32734149; Tel: +55 61 31073867
bLaboratory of Synthetic and Heterocyclic Chemistry, Institute of Exact Sciences, Department of Chemistry, Federal University of Minas Gerais, CEP 31270-901, Belo Horizonte-MG, Brazil. Fax: +55 31 34095700; Tel: +55 31 34095720
First published on 9th October 2014
Abstract
The current manuscript describes the importance, mechanism propositions, evidence and controversies associated with multicomponent reactions (MCRs). The following multicomponent reactions are presented and critically evaluated: the Biginelli, Hantzsch, Mannich, Passerini and Ugi reactions. The aim of this review is to highlight what we already know about the mechanisms associated with these MCRs and the evidence supporting the proposed reaction pathways. Controversies and prospects are also discussed herein.
 Haline G. O. Alvim | Haline G. O. Alvim received her degree in Chemistry from the University of Brasilia (UnB, 2011). In 2013, she completed her Ms.C. in the same University working in the group of Professor B. A. D. Neto and Professor Wender A. da Silva. She is currently working under the supervision of Professor Neto developing new catalytic systems for MCRs and mechanism investigations to complete her Ph.D. |
 Eufrânio N. da Silva Júnior | Eufrânio N. da Silva Júnior received his degree in chemistry from the Catholic University of Brasília (UCB). In 2007, he completed his MSc at the Fluminense Federal University (UFF) and in 2009 he concluded his PhD at the University of Brasilia (UnB). In 2010, he became Professor of Chemistry at the Federal University of Minas Gerais (UFMG). His research interests are focused on click chemistry reactions, mechanism investigation, asymmetric organocatalysis and on the synthesis of heterocyclic and naphthoquinoidal bioactive compounds. Currently, he is also interested in obtaining fluorescent substances for the study of pharmacological and DNA-binding properties. |
 Brenno A. D. Neto | Brenno A. D. Neto was born on December 9th in 1977 in Brazil. He received his degree in Chemistry from the Federal University of Rio Grande do Sul (UFRGS, 2000). In 2006, he completed his Ph.D. in the same University. After a period of postdoctoral work at Tecnopuc (2007) and as a Professor at PUCRS (2008), he became a Professor of Organic and Medicinal Chemistry at the University of Brasília (UnB, since 2009), the University located in the Federal Capital. His research interests include the development of new catalysts and ionic liquids, mechanistic investigation, MCRs and applications of rationally designed selective fluorescent cell markers. His independent career began in the middle of 2006. Until then, he had published 10 articles and, after that (2007–2014), he published more than 50 articles underpinning his research independence. Professor Neto has received some national and international awards, among which can be highlighted: BMOS/RSC Young Investigator Award (2013), Honourable Mention for the Best Brazilian Thesis in Chemistry 2013 (Category: Advisor) and Petrobras Inventor Award for the best-filed patent in 2008. |
1. Introduction
Multicomponent reactions (MCRs) are tools of paramount importance as the central strategy to reach eco-friendly and sustainable transformations in modern chemistry. MCRs have many advantages over stepwise linear synthesis (Scheme 1) such as atom economy, less waste generation, time and energy economy, less human effort, fewer resources required, easy purification issues, and high convergence, among others.1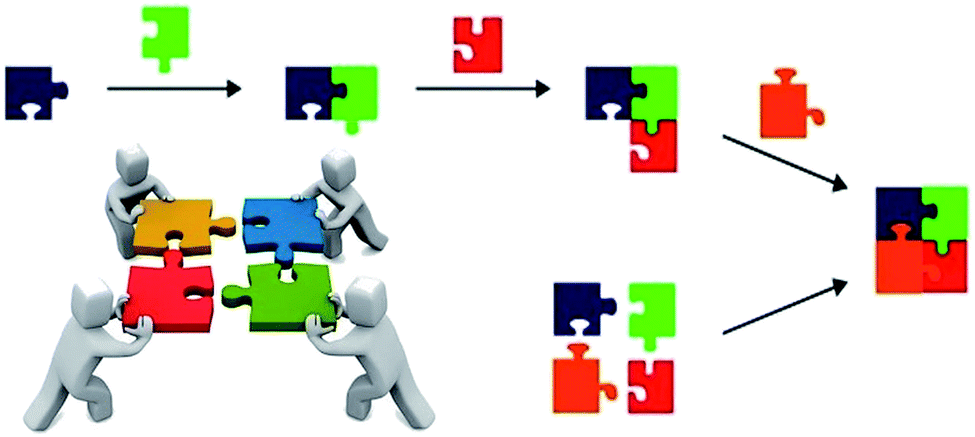 | ||
| Scheme 1 Pictorial view of a stepwise linear synthesis compared to a multicomponent reaction (MCR) approach. The advantages of MCRs are clear-cut in this illustration. | ||
Reactions whereby at least three different components are brought together in a one-pot version, affording a single product, may be summarised under the term ‘multicomponent reactions’. In an ideal MCR, however, the majority of the atoms found in the reagents must be incorporated in the product structure, otherwise this most important feature of MCRs will be broken and the reaction should not be labelled as part of the ‘green’ MCR class. By-products from a MCR are well known to be usually water.
Through MCRs, access to elaborate molecular scaffolds combining both structural diversity and eco-compatible methodologies becomes possible in a single step and in a one-pot version. It is therefore natural to observe MCRs as the key steps for the rapid and efficient synthesis of simple or complex products.
Although the origin of MCRs dates back to the middle of the 19th century, MCRs have experienced an exponential growth in importance and usage especially in recent decades, becoming an unsurpassed synthetic tool in a very prominent position. In the search for multiple-bond-forming efficiency, MCRs may indeed be labelled as the most promising strategy to reach an outstanding combination of efficiency, atom economy and sustainability.
The huge interest in MCRs lies not only in their green and promising characteristics but also in the biological properties commonly observed for the products synthesized straight from multicomponent methodologies. MCRs allow direct and elegant access to bioactive compound libraries and meet the need for never-ending biologically active compound syntheses and discovery. Many MCR adducts have pronounced biological activity even in their racemic mixtures, thus boosting the interest in these compounds and making the process of their obtainment easier and less costly. As one may expect, tests of enantiomerically pure compounds are of huge importance2 because the isomers may have distinct activity and/or potency, but for many MCR derivatives it has been proved that racemic mixtures may be used3 with no harm.
Despite all the promising features and biological importance of MCRs, harsh reaction conditions, reagent excesses, high temperatures, toxic solvents, expensive catalysts, purification issues, low yields, low selectivity and long reaction times are drawbacks still commonly observed for these important synthetic tools. The aforementioned shortcomings diverge, however, with the beneficial features associated with MCRs. Deeper knowledge and comprehension of the mechanisms (under catalysed or non-catalysed conditions) is therefore vital to the development of new catalysts, improved and greener reaction conditions, rational designs, innovative applications and for synthetic predictions.
Intermediates from a MCR are typically not isolated, although sometimes they can be isolated and properly characterized, thus allowing a more precise mechanism proposition for the transformation; and this issue will be better evaluated herein in due course. What is really astonishing about MCR mechanisms is that, independent of the preferred reaction pathway, all mechanisms lead to the same final product, as seen in pictorial Scheme 2. In other words, it can be said that MCRs proceed in an “all roads lead to Rome” fashion i.e. the main product may be formed via various but convergent reaction paths.
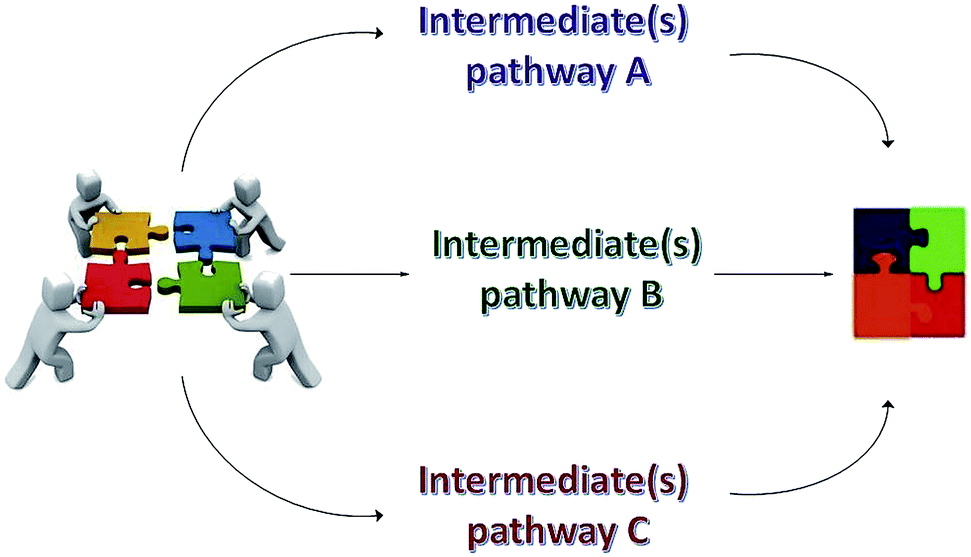 | ||
| Scheme 2 Pictorial view for three possible reaction mechanisms associated with multicomponent transformations. It is noteworthy that even considering two or more reaction pathway possibilities, the final product in a MCR is always the same, independent of the mechanistic pathway. | ||
Among all MCRs already described, special attention must be given to the Biginelli, Hantzsch, Mannich, Passerini and Ugi reactions (Scheme 3). These MCRs are among the most popular, studied and widely used in the synthesis of bioactive compounds, therefore justifying their importance, practicality and promising trends. All these aforementioned MCR types have already been studied by different approaches and spectroscopic/spectrometric techniques which allowed for diverse mechanism propositions depicted from the data generated for all of them.
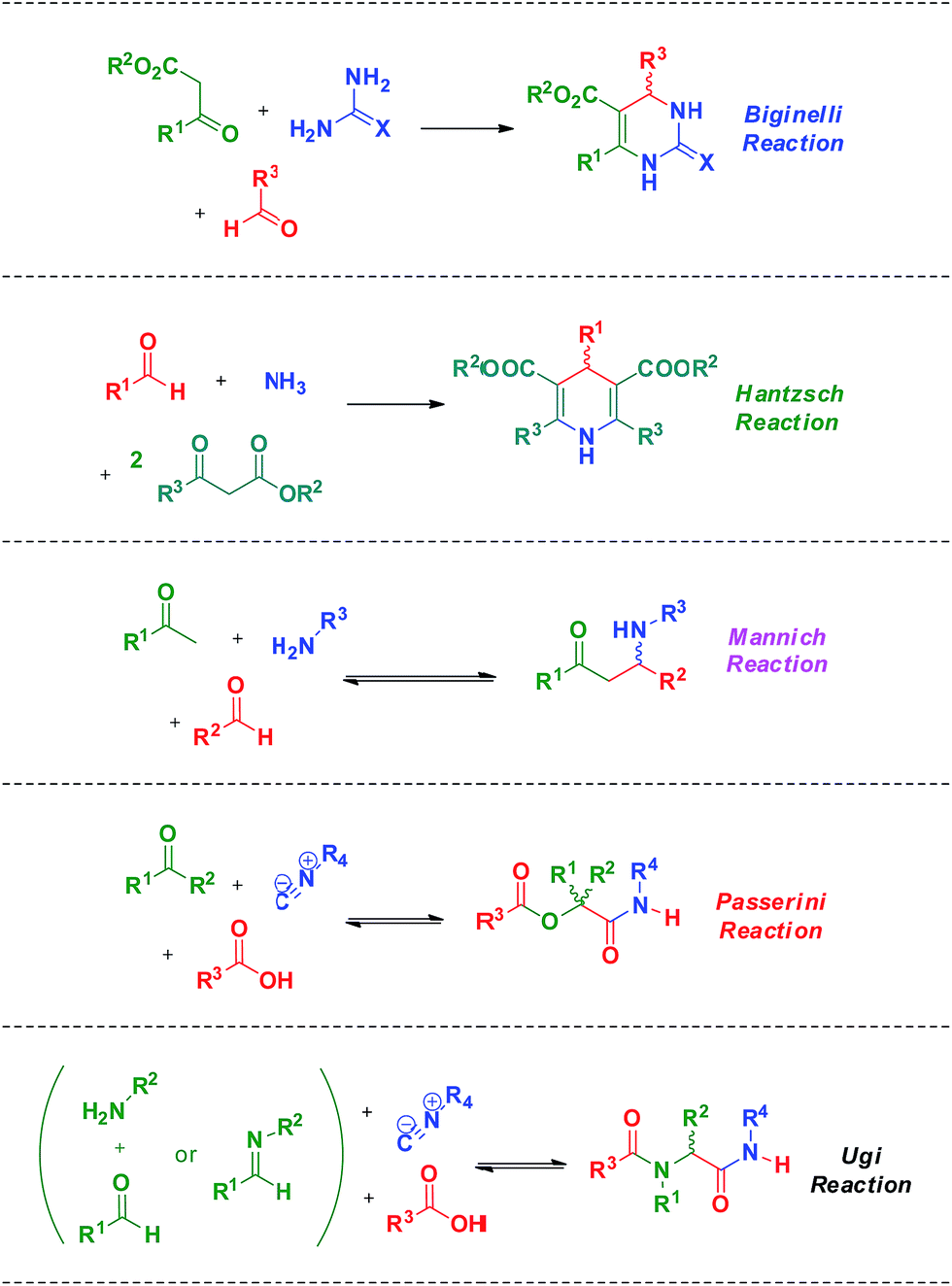 | ||
| Scheme 3 Five examples of MCRs. The Biginelli, Hantzsch, Mannich, Passerini and Ugi reactions are among the most useful, used and studied MCRs. | ||
As expected, all these MCRs have at least two possible reaction pathways. The mechanism disclosed is therefore essential towards rational design of catalysts, ligands, reagents, stereo- and electronic controls and, most importantly, for the prediction of new molecules which may be useful as bioactive compounds or synthetic intermediates in total syntheses and others. MCR mechanisms generally evoke a cascade of successive bimolecular reactions, although sometimes termolecular steps, requiring the participation of at least three chemical entities in the transition state, are also evoked to explain a specific transformation to afford a particular intermediate or final product during a MCR.
At this point it is important to limit the scope of this tutorial review. In the present manuscript, it is intended that the focus will be the mechanism propositions, the evidence and results to support these propositions, the controversies and trends associated with these five widely used MCRs i.e. the Biginelli, Hantzsch, Mannich, Passerini and Ugi reactions. In this manuscript, only those articles with some contribution to the comprehension of the evoked mechanisms for these five important MCRs are cited, independently of whether theoretical or experimental data (or both) are presented. There are several manuscripts available in the scientific literature with a simple mechanistic description (or proposition or plausible mechanism) without any supportive data. This kind of article will only be cited as an exception and, in a general way, these publications are not the object of this review. A critical evaluation of the evidence and propositions for each of those MCRs is to be given and, finally, some trends are suggested, regarding the mechanism investigation of the MCRs in question.
2. The Biginelli multicomponent reaction
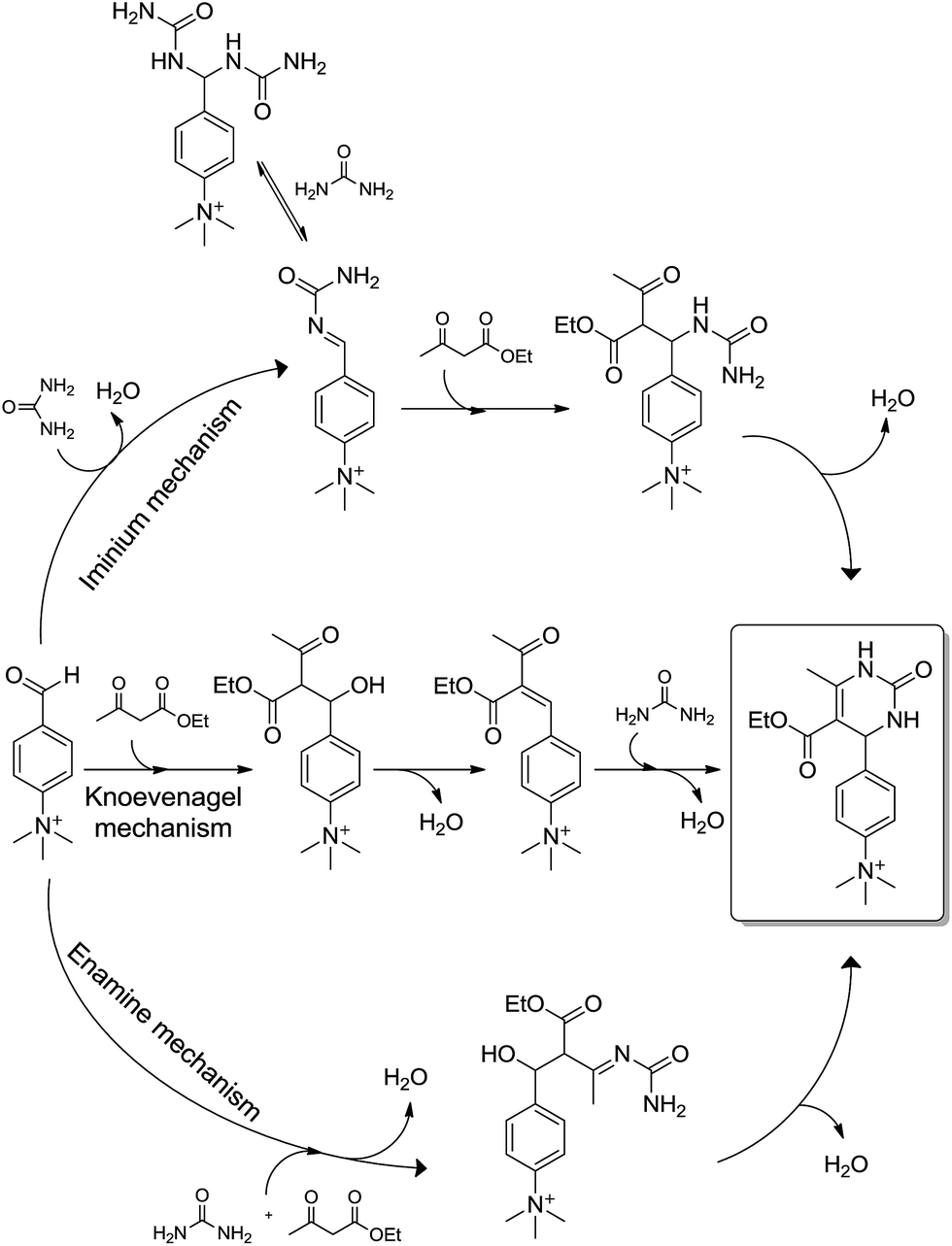
The Biginelli MCR (Scheme 3) is a three-component reaction (3CR) discovered in 1891 by Pietro Biginelli.4 This very elegant and useful 3CR is widely used in the synthesis of 3,4-dihydropyrimidin-2(1H)-ones (or -thiones), which are also referred to as DHPMs. DHPMs are especially important because of their commonly observed biological activities as calcium channel modulators, mitotic Kinesin inhibitors, adrenergic receptor antagonists, antibacterials, antivirals, and others, as reviewed elsewhere.5 Interestingly, many DHPMs are successfully tested in their racemic mixtures with impressive results.6 Today, there are three (hotly) debated mechanism propositions accepted for this MCR i.e. iminium-, enamine- or Knoevenagel mechanisms, which will be analysed individually. For those readers interested in a more detailed description of the evolution of the Biginelli reaction mechanism, some helpful reviews are available.7,8 Interestingly, this elegant MCR was born to be controversial, especially if one takes into account that the original structure suggested for the Biginelli adduct was not a heterocycle, but an open chain structure (an uramidocrotonate derivative as shown in Scheme 4) from the reaction of a mixture of urea, salicylaldehyde, and ethyl acetoacetate at reflux in absolute ethanol. The structure initially assigned had to be later revisited by Pietro Biginelli himself in 1893, the year the full account of the Biginelli reaction was published.4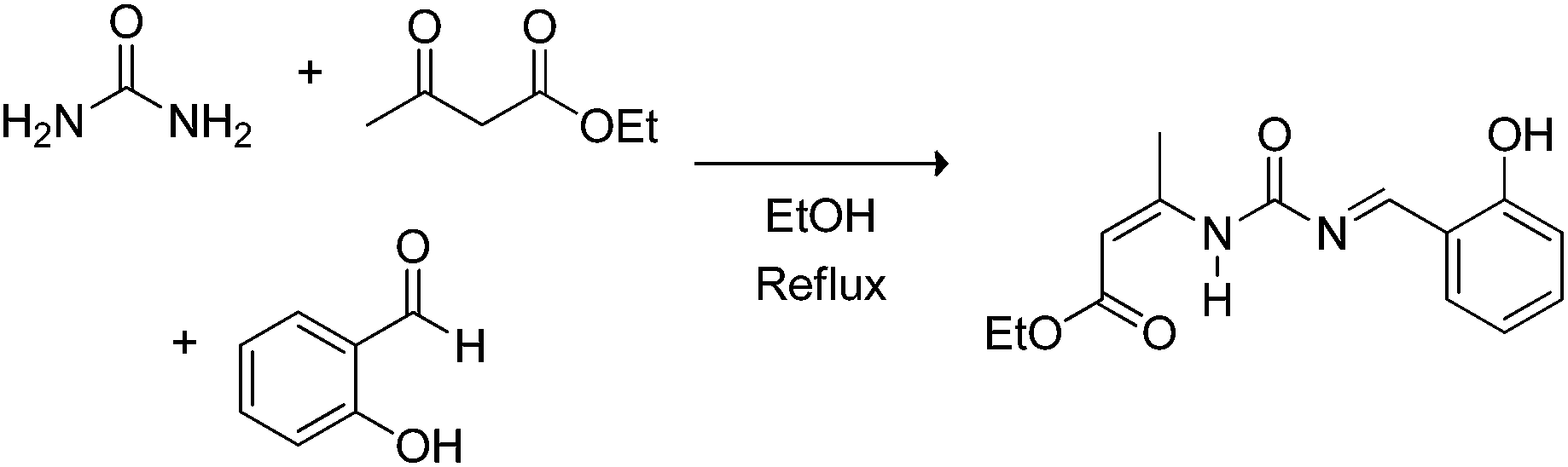 | ||
| Scheme 4 The Biginelli MCR as originally reported by Pietro Biginelli in 1891.4 Two years later (1893), Biginelli published full accounts of the reaction revisiting the original structure.4 | ||
What today has become known as the Knoevenagel mechanism (Scheme 5) is based on the findings of Sweet and Fissekis.9 Although the Knoevenagel-based mechanism is still accepted as a possible reaction pathway for the three-component Biginelli reaction, it is unlikely, since further evidence indicates other preferred reaction pathways, as will be shown in this section.
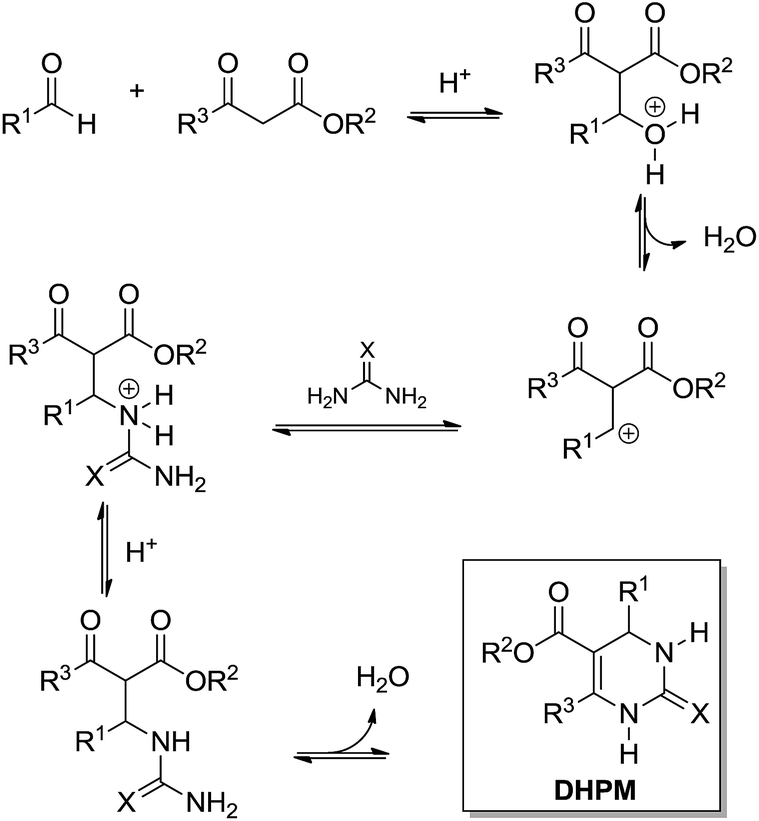 | ||
| Scheme 5 The Knoevenagel mechanism for the Biginelli reaction. | ||
The enamine mechanism (Scheme 6) has its origin in the mechanistic investigation of Folkers and Johnson in 1933,10 which was indeed the first attempt at a mechanistic rationale of the Biginelli reaction transformation. In their work10 it was suggested that this 3CR may proceed via one of the three possible primary bimolecular reaction intermediates (Scheme 7) afforded by a combination of urea, benzaldehyde and ethyl acetoacetate (usually referred to as the model Biginelli reaction).
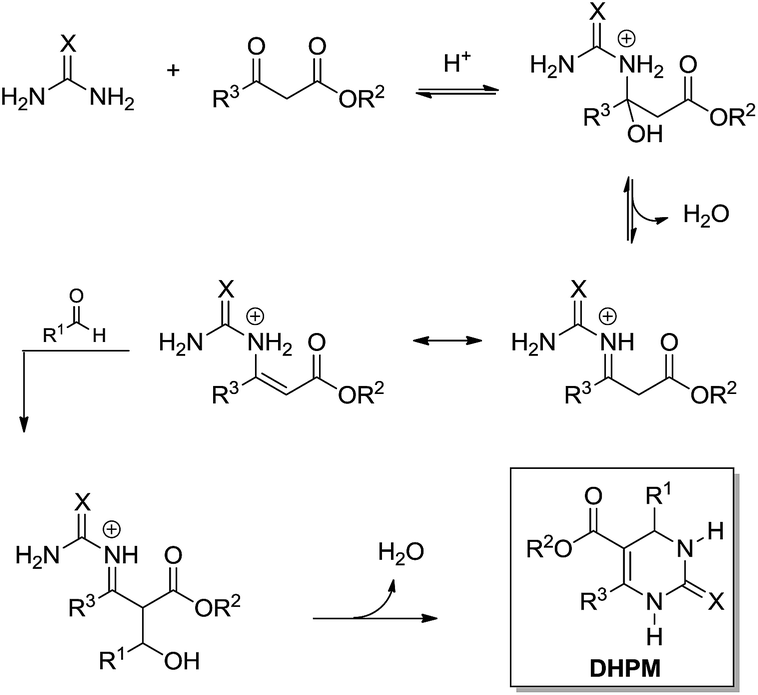 | ||
| Scheme 6 The enamine-based mechanism for the Biginelli reaction. | ||
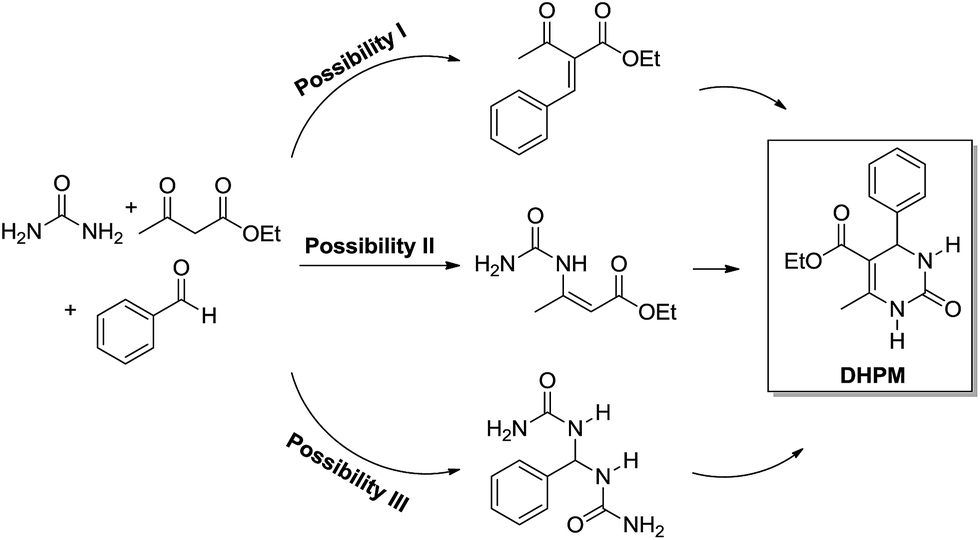 | ||
| Scheme 7 Three possible primary bimolecular reaction intermediates which may afford the Biginelli adduct. Scheme based on the original work of Folkers and Johnson.10 Note that the bisureide derivative (Possibility III) is evoked as the intermediate and not the iminium ion (from the first urea addition to the aldehyde). Two urea additions to the aldehyde yield the bisureide intermediate. | ||
Interestingly, in 2007 Cepanec and coworkers11 presented a series of experiments showing the Biginelli 3CR goes through the enamine mechanism when catalysed by SbCl3 in anhydrous MeCN. In the report, bimolecular reactions catalysed by SbCl3 are described to depict the preferred reaction pathway (Scheme 8). The mixture of ethyl acetoacetate and urea afforded the ureidocrotonate intermediate which could be isolated in 9% yield after a preparative chromatography. When treated with benzaldehyde (even at room temperature) the ureidocrotonate intermediate afforded the expected DHPM in almost quantitative yields. The other two possible combinations (ethyl acetoacetate + benzaldehyde or urea + benzaldehyde) afforded no product under the tested conditions (Scheme 8).
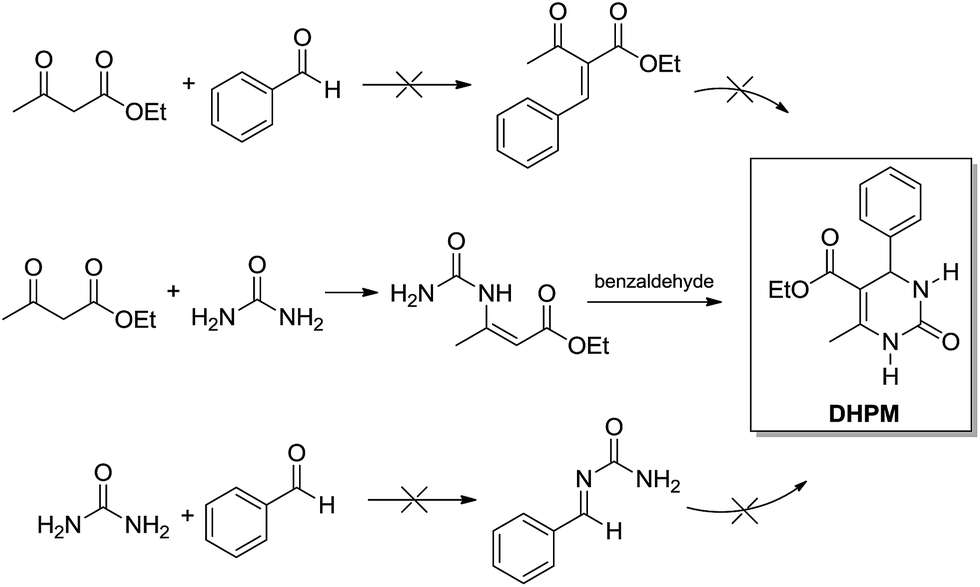 | ||
| Scheme 8 SbCl3 (20–100 mol%) as the promoter of the reaction in anhydrous MeCN (room temperature to reflux). The experiments allowed the determination of the enamine mechanism as the preferred reaction pathway for the Biginelli reaction.11 | ||
Cepanec and coworkers11 also suggested that many described Biginelli reactions catalysed by Lewis acids in aprotic solvents probably follow this reaction pathway; and that many reports have only assumed a plausible mechanism according to Folkers,10 Johnson10 and Kappe12 without any real proof. Further work of Litvic and coworkers13 using [Al(H2O)6](BF4)3 as the catalyst returned similar results, in accordance with Cepanec's description.11
In 1997, Kappe12 reported a re-examination of the Biginelli mechanism based on NMR experiments, providing therefore strong evidence for the iminium mechanism (Scheme 9). Mixtures of urea (or N-methylurea), benzaldehyde and ethyl acetoacetate were monitored by 1H and 13C NMR (in CD3OH in the presence of catalytic amounts of HCl). All evidence pointed firmly to the iminium mechanism and discarded the Knoevenagel and enamine pathways. The results also indicated the first urea addition to the aldehyde as the rate-determining step (slow). Interestingly, it is suggested that in the presence of ethyl acetoacetate, the iminium ion undergoes an additional reaction towards the Biginelli adduct formation, whereas in the absence of the 1,3-dicarbonyl compound the second urea addition takes place furnishing the bisureide derivative (Scheme 10). Arguing that the equilibrium for the formation of the intermediates from the Knoevenagel and enamine pathways lies far to the side of the reactants, and based on the experimental evidence, these two reaction pathways were discarded.
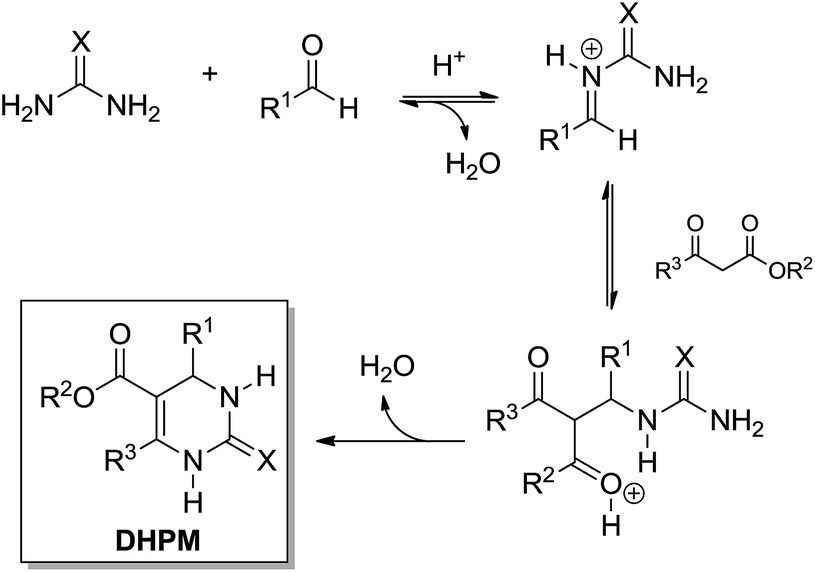 | ||
| Scheme 9 The iminium mechanism for the Biginelli reaction. | ||
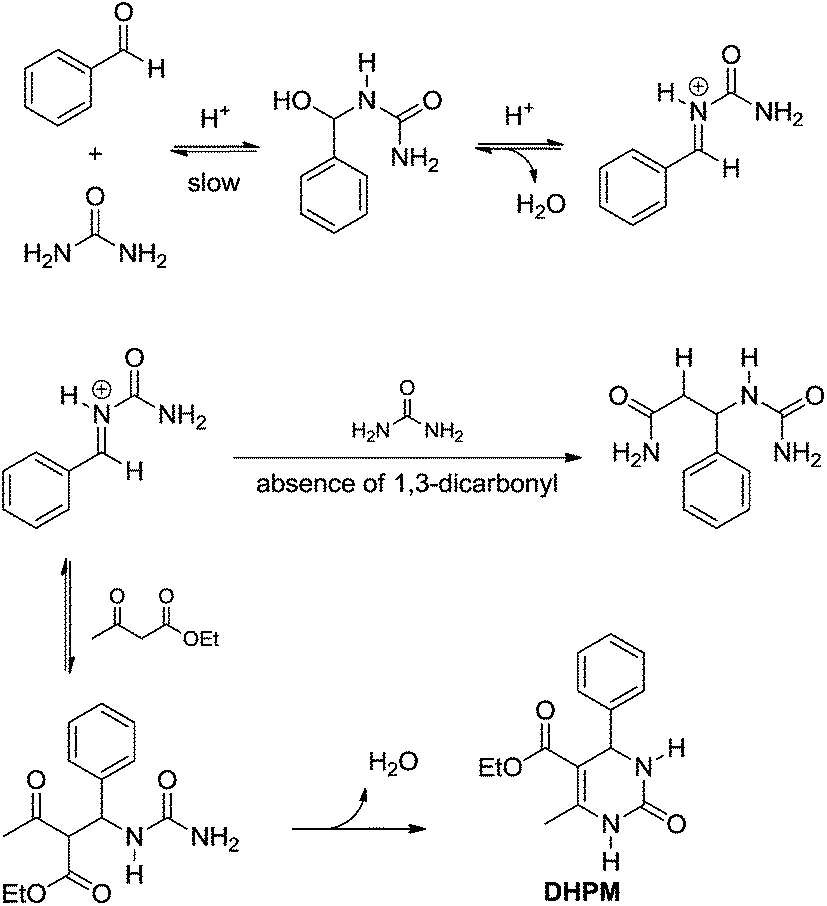 | ||
| Scheme 10 The iminium-based mechanism depicted from the reexamination of the Biginelli mechanism based on 1H and 13C NMR.12 The use of N-methylurea instead of urea afforded similar results. Note the highly reactive N-acyliminium is the key intermediate instead of the bisureide derivative. | ||
Lately, the mechanism of the Biginelli reaction was investigated using Bronsted acid catalysis (formic acid) under the light of both electrospray (tandem) mass spectrometry – ESI-MS(/MS) – and DFT calculations.14 The online reaction monitoring allowed interesting conclusions. In the absence of ethyl acetoacetate, the authors were capable of detecting and characterizing not only the iminium intermediate (m/z 149) but also the protonated bisureide derivative (m/z 209) as well. Protonated hemiaminal derivative (m/z 167) could also be intercepted and characterized prior to its dehydration, affording the N-acyliminium derivative (m/z 149). The online monitoring of the 3CR (1 mmol of each reagent of the model reaction in aqueous methanol solution (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) and 0.1% of formic acid) returned very elucidative results. By means of ESI-MS(/MS) it was possible to follow the reaction for more than 24 h. After 5 min, however, the protonated Biginelli adducted (m/z 261) could already be noted in the spectrum and subjected to structural characterization by ESI-MS/MS. The addition of urea to the 1,3-dicabonyl (Scheme 11) afforded a postulated dormant intermediate that reverts to reagents during the course of the reaction. A mixture of ethyl acetoacetate and urea in the absence of the aldehyde pointed to the same conclusions from the 3CR (Scheme 11) and no protonated enamine (m/z 173) could be detected.
:1 v/v) and 0.1% of formic acid) returned very elucidative results. By means of ESI-MS(/MS) it was possible to follow the reaction for more than 24 h. After 5 min, however, the protonated Biginelli adducted (m/z 261) could already be noted in the spectrum and subjected to structural characterization by ESI-MS/MS. The addition of urea to the 1,3-dicabonyl (Scheme 11) afforded a postulated dormant intermediate that reverts to reagents during the course of the reaction. A mixture of ethyl acetoacetate and urea in the absence of the aldehyde pointed to the same conclusions from the 3CR (Scheme 11) and no protonated enamine (m/z 173) could be detected.
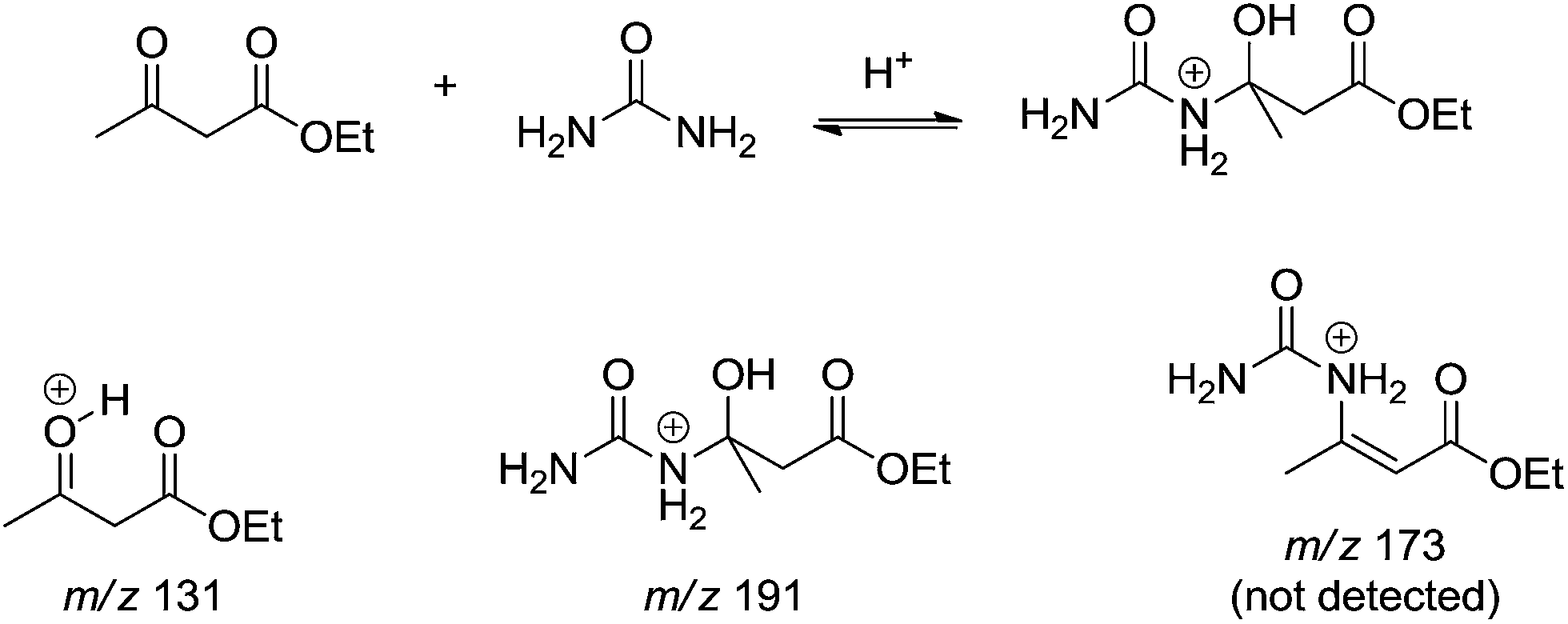 | ||
| Scheme 11 A postulated dormant intermediate formation from the enamine reaction pathway detected and characterized by ESI-MS(/MS).14 Those intermediates were detected in the three-component reaction or in the bimolecular reaction between urea and ethyl acetoacetate in the absence of benzaldehyde. | ||
The reaction of ethyl acetoacetate and benzaldehyde in the absence of urea was conducted as well. After 24 h some intermediates postulated from the Knoevenagel mechanism were detected and characterized (Scheme 12). Except for the intermediate of m/z 237 no other intermediate could be detected during the monitoring of the 3CR version. In the bimolecular version, however, the protonated product from the condensation reaction (m/z 219), and both the protonated benzaldehyde (m/z 107) and ethyl acetoacetate (m/z 131) could be noted. The MS data allowed the authors to suggest that the iminium mechanism is highly favoured under Bronsted acid catalysis.
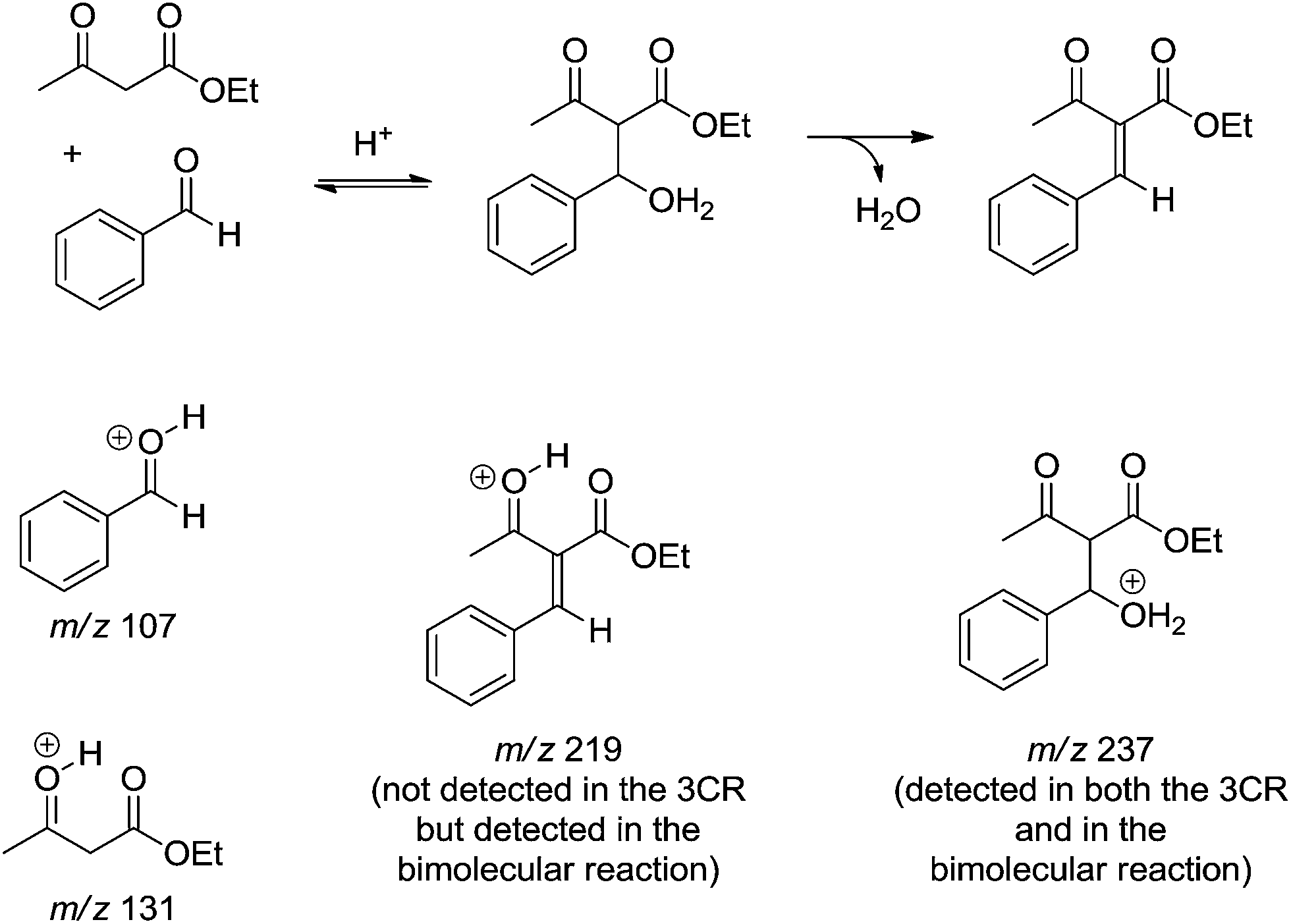 | ||
| Scheme 12 A postulated dormant intermediate from the Knoevenagel reaction pathway detected and characterized by ESI-MS(/MS).14 The intermediate of m/z 237 could be detected in the three-component reaction, but the other key intermediate (m/z 219) could only be detected after 24 h of reaction from a mixture of ethyl acetoacetate and benzaldehyde in the absence of urea. | ||
DFT calculations (including solvent effects) have also indicated that the iminium mechanism is the kinetically and thermodynamically favoured one. Curiously, DFT calculations for the Knoevenagel mechanism revealed the highest energy barrier in accordance with MS data. Based on the data obtained by this landmark work,14 the authors could suggest that the iminium pathway (as proposed by Kappe12) is highly favoured and the enamine and Knoevenagel pathways could be kinetically discarded.
A recent report questioned the conclusions that Lewis acids may prefer the enamine-like reaction pathway.15 The mechanism of the Lewis acid-catalysed Biginelli reaction has been shown to be far more complex than Bronsted-catalysed versions of the reaction. The report re-examined the mechanism of the reaction catalysed by CuCl2 in the ionic liquid (IL) 1-butyl-3-methylimidazolium hexafluorophosphate (BMI·PF6). Based on 1H and 13C NMR, ESI-MS(/MS) and theoretical calculations (DFT), the authors showed the role of the reagents in the formation and stabilization of the reactive intermediates, demonstrating, therefore, important features for the Lewis acid-catalysed 3CR (Scheme 13).
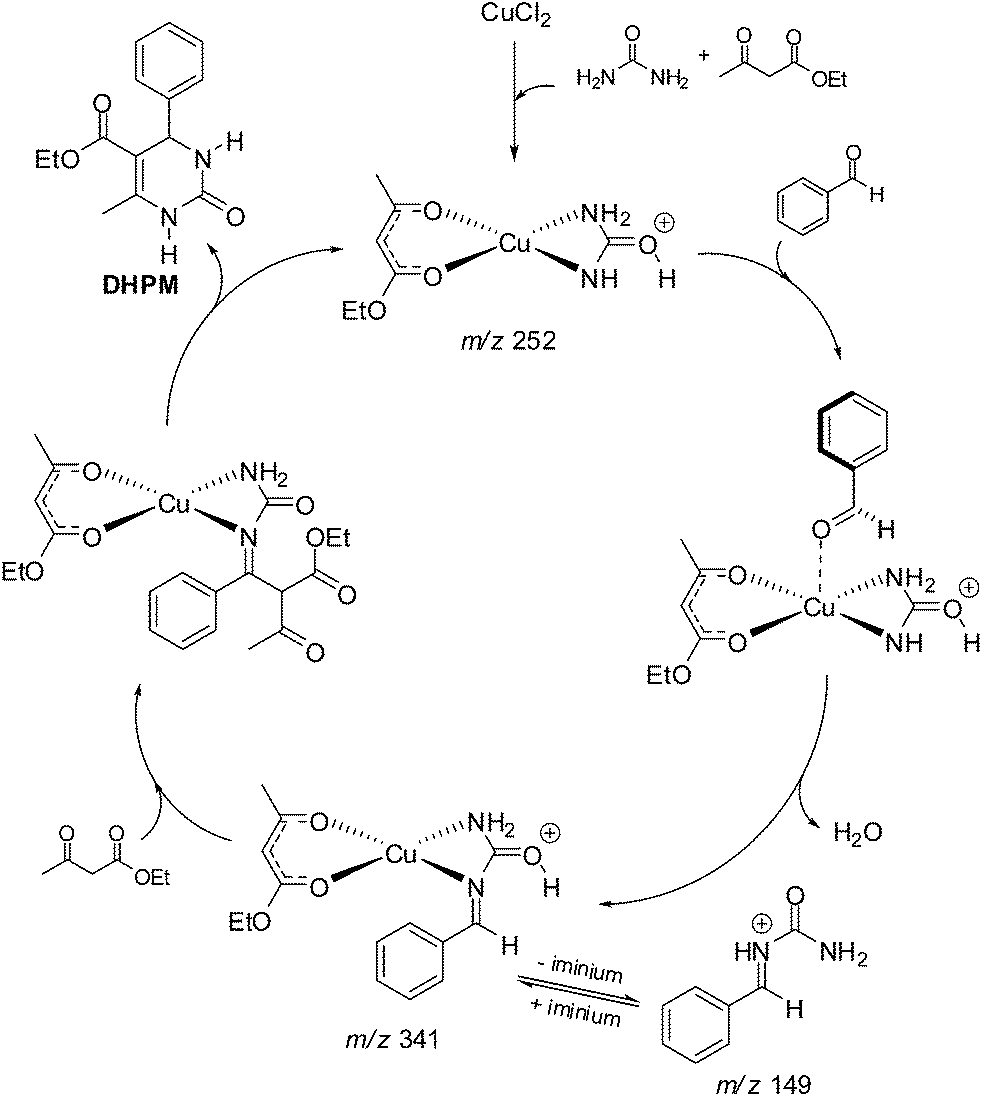 | ||
| Scheme 13 Catalytic cycle for the Lewis acid-catalysed Biginelli reaction (model reaction) based on the detected (and characterized) intermediates monitored by ESI(+)-MS(/MS).15 The detected intermediates are indicated by their respective m/z. Note the reagents play a role in the formation and stabilization of the copper complex derivatives and are thus essential to further the reaction. | ||
Remarkably, the conclusion was that even through a more complex mechanism, the authors could claim the preferred reaction pathway is based on the iminium mechanism and indicated that the conclusions drawn by Kappe12 were essentially correct even considering a Lewis acid-catalysed version of the Biginelli reaction. The NMR data proved to be in accordance with the observed intermediates detected and characterized by ESI-MS(/MS). DFT calculations helped in the comprehension of the anion effect ([PF6]−) (from BMI·PF6) for the formation and stabilization of the charged and polar intermediates. Indeed, the IL effect was seen to play a fundamental role in the success of the catalytic system (Lewis acid catalysis in ILs). The main IL effects depicted by DFT calculations was the formation of ion pairs (and larger supramolecular aggregates) and the orientation of the reagents in the proposed catalytic cycle. The authors made a comment that the actual catalytic cycle is, indeed, more complex than the usually presented “textbook” mechanism in which a Lewis acid is shown interacting with the aldehyde to activate its carbonyl towards a nucleophilic addition. NMR experiments were found in accordance with DFT calculations and a deshielding effect of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (of the aldehyde) was noted in the presence of the Lewis acid and the IL.
O (of the aldehyde) was noted in the presence of the Lewis acid and the IL.
An investigation into the mechanism of the Biginelli reaction (model reaction) catalysed by a Bronsted acid-containing task-specific ionic liquid (TSIL) incorporating anionic heteropolyacid ([PW12O40]3−), and carried out in the IL 3-methylimidazolium (bis(trifluoromethylsulfonyl)imide) (BMI·NTf2), by means of ESI-MS(/MS) and DFT calculations, indicated once more that the iminium pathway was preferred over the enamine or Knoevenagel mechanisms.16 It is worth noting that, just like De Souza et al.,14 the authors noted the presence of a dormant intermediate of m/z 237 (see the structure in Scheme 12) which is in accordance with the Knoevenagel reaction pathway. However, all other detected and characterized intermediates were in accordance with the N-acyliminium mechanism re-examined by Kappe;12 and no other Knoevenagel-based intermediate could be detected during the reaction time monitoring. Initial addition of ethyl acetoacetate to benzaldehyde was reversible, and a different reaction pathway rather than the iminium mechanism would be very unlikely.
At this point it is important to highlight to readers that until recently, no actual quantitative kinetic study was available for the Biginelli reaction. If one considers the three proposed reaction mechanisms and the number of possible intermediates, it can be appreciated how difficult the task of complete kinetic investigation of the Biginelli reaction is. However, to overcome this major problem, a global kinetic approach varying the concentration of the three substrates was performed by Neto and coworkers.17 This work was indeed the first to show a kinetic study on the Biginelli reaction supporting the proposed mechanism. In the study the reaction was catalysed by a dual activation mode TSIL as the catalyst, i.e. a TSIL with a Bronsted acid in the cation and a Lewis acid in the anion. The study was also based on the conclusions supported by ESI-MS(/MS), NMR (1H and 13C) and DFT calculations. Based on all data obtained from the kinetics and spectroscopic/spectrometric analyses, the authors could prove a clear-cut preference for the iminium mechanism and could (undoubtedly) discard the possibility of the enamine or Knoevenagel pathways. The preferred mechanism, promoted by the catalyst with dual activation mode, basically followed the N-acyliminium pathway (Scheme 14) with a specific role for the cation (Bronsted acid) and for the anion (Lewis acid).
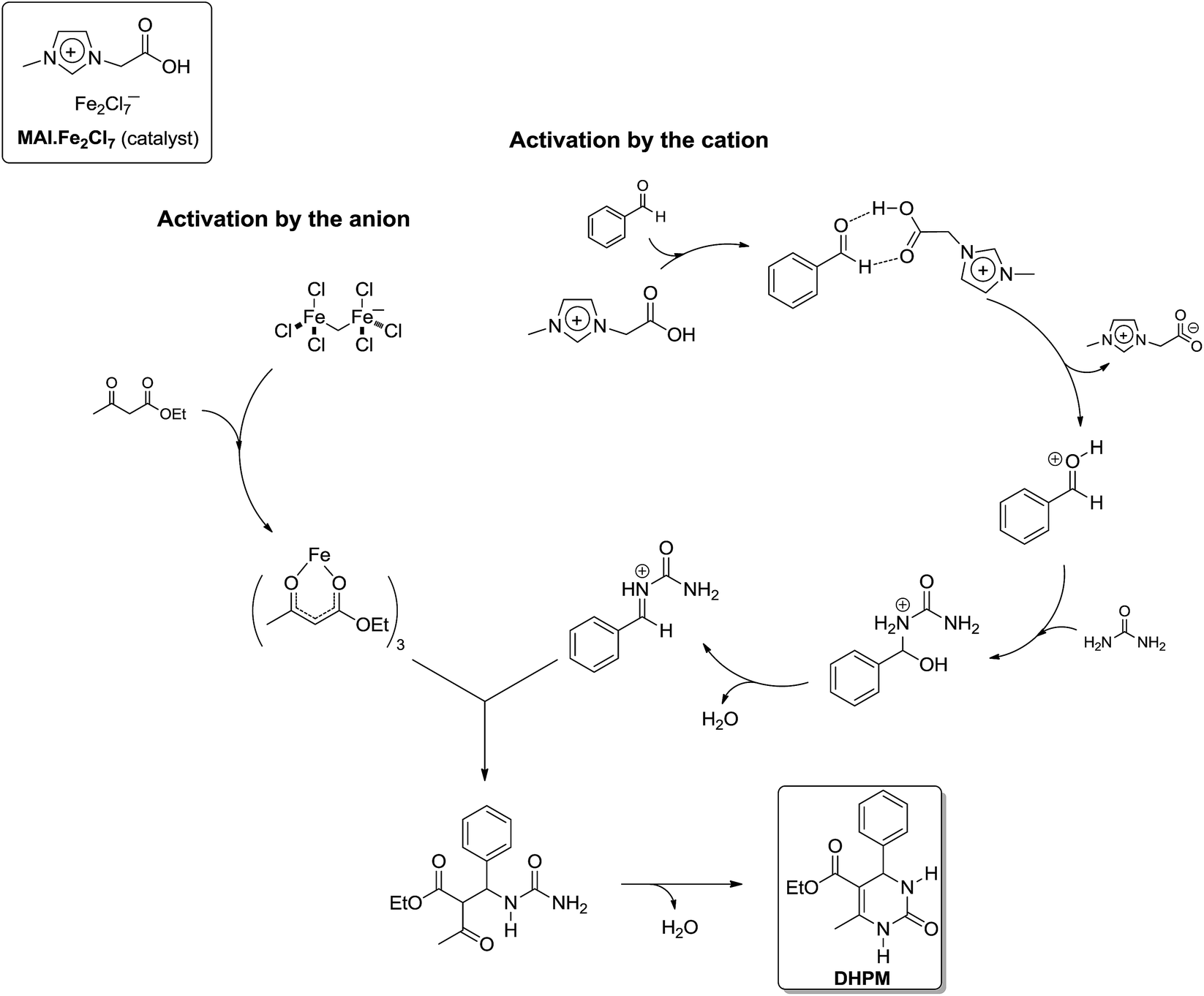 | ||
| Scheme 14 Proposed catalytic mechanism promoted by a task-specific ionic liquid with dual activation mode, in which both the anion (iron complex) and the cation (imidazolium) moieties play a role in promoting the reaction.17 Note that this proposed mechanism is compatible with the iminium mechanism for the Biginelli reaction. | ||
The mechanism, which is based on experimental data, is the first describing the cooperative catalytic effect of a Bronsted and Lewis acid in the synthesis of DHPM. The IL effect was disclosed for the reactions in the IL 1-butyl-3-methylimidazolium tetrafluoroborate (BMI·BF4), showing the anion had a role not only in the stabilization of charge and polar intermediates through ion-pairing effect, but also in the activation of the protonated aldehyde, as shown in Scheme 15. Once the aldehyde is activated, the first urea addition is observed and, once more, this reaction step is in accordance with the iminium mechanism for the Biginelli reaction. A comment on the kinetic experiments is also necessary. Although a global kinetic analysis was performed, the kinetic constants returned interesting results. The analyses revealed the only plausible mechanism is the iminium mechanism and the other two could be discarded. Usually, urea excess is used in most of the reported articles describing the Biginelli synthesis. However, the authors proved that, when the iminium mechanism is the preferential reaction pathway, the only reagent which should not be used in excess is urea.
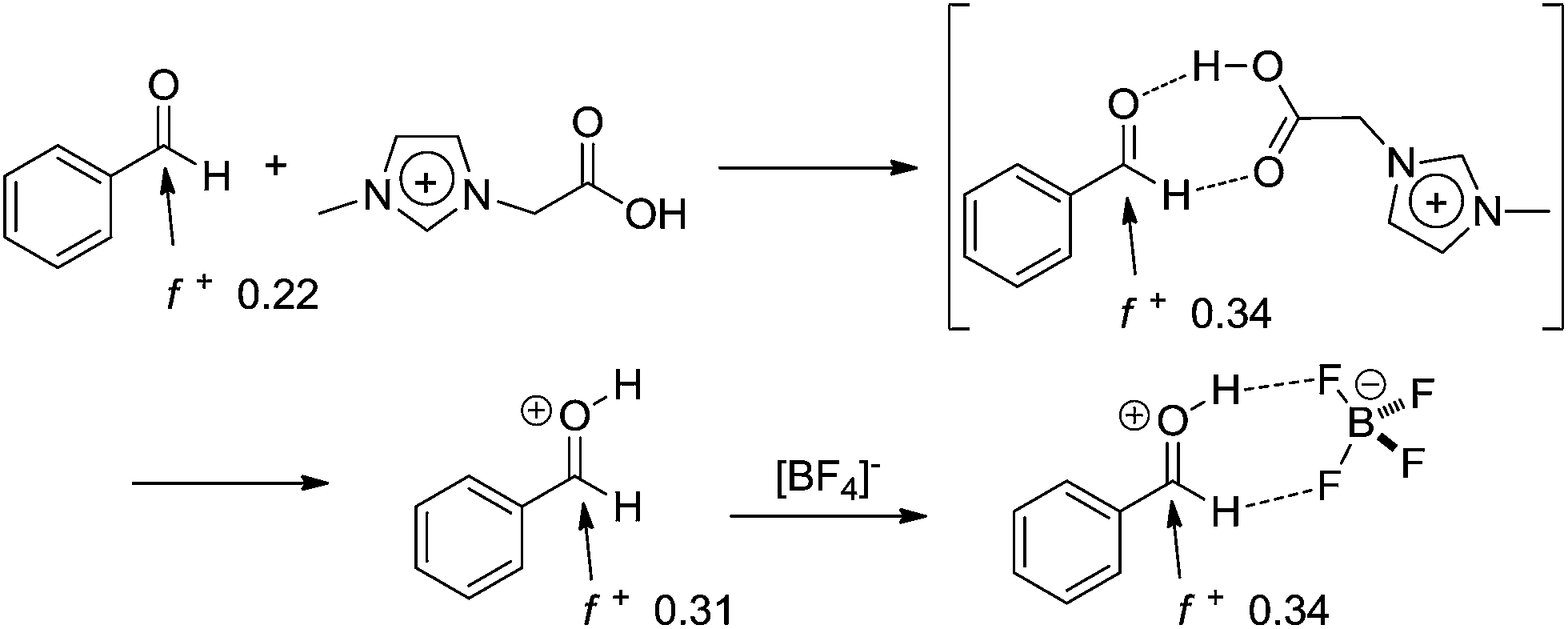 | ||
| Scheme 15 Aldehyde activation by the cation of the task-specific ionic liquid and ionic liquid effect (ion-pairing formation) in BMI·BF4.17 The calculated Fukui functions of the indicated atoms are also shown. Note that once the aldehyde is activated, the urea addition is more prone to take place and the reaction will continue through the iminium mechanism. | ||
Solvent effects are usually neglected issues with regard to the Biginelli 3CR. Recently, Clark and coworkers18 described a landmark investigation into catalytic and solvent effects, demonstrating their combined role. A vital contribution of the article is the reactivity analysis of the 1,3-dicarbonyl compound and how the solvent choice affects its reactivity and the yields of the reaction. The authors concluded that the solvent has a huge influence over the reaction, but they also demonstrated that the quantity of available enol tautomer in the reaction mixture is fundamental to further the reaction. As one can expect, the quantity of the available enol tautomer is directly associated with the solvent choice. It is also important to highlight that authors tested the Biginelli mechanism using a classical Bronsted acid (HCl) and a Lewis acid (ZnCl2). Based on their analyses, it was possible to conclude that the mechanism indicated by Kappe12 seems to be correct even with Lewis acids. Conclusions proved to be also in agreement with those reported by Neto15 for the reactions conducted in ILs, and in opposition to those of Cepanec11 and Litvic.13
Despite the fact that the Biginelli reaction is typically catalysed by a Bronsted or a Lewis acid, it is also possible to find an interesting mechanistic investigation by a Bronsted base-catalysed version of the reaction.19 It has been shown that the reaction pathway is mostly dependent on the use of urea or thiourea. The authors' conclusions were based on reactions of pre-formed compounds known as possible intermediates of the Biginelli-like reaction (Scheme 16) with urea or thiourea.
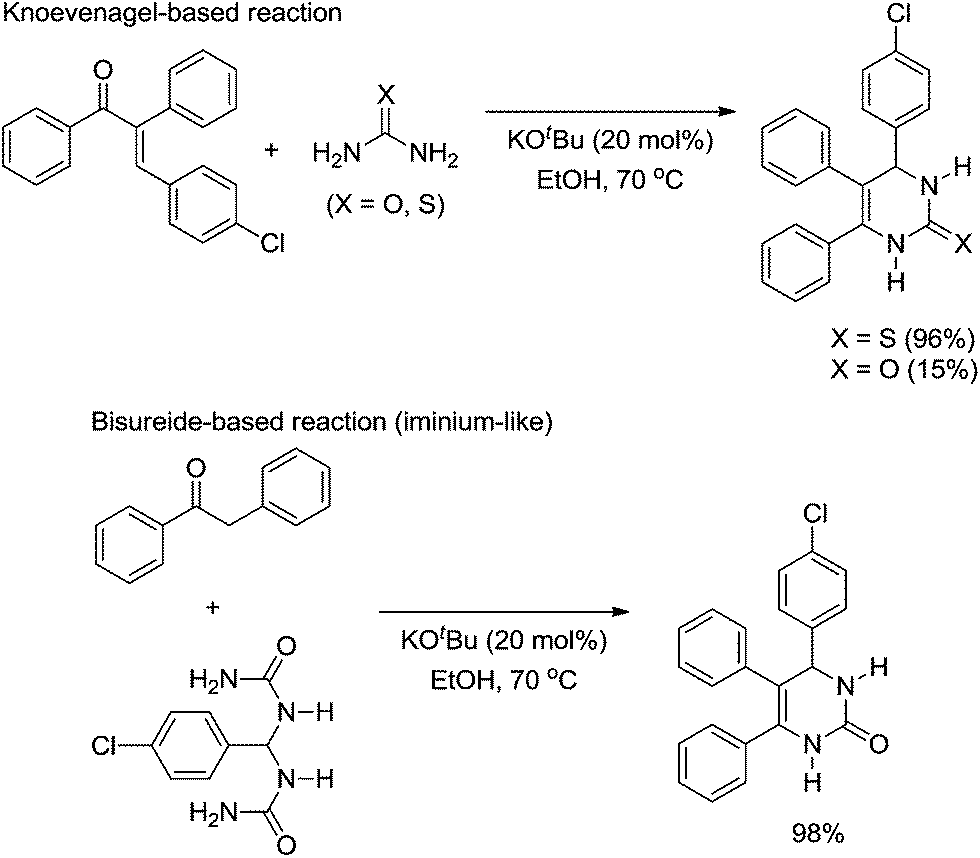 | ||
| Scheme 16 Reaction of urea and thiourea with pre-formed intermediates.19 | ||
In the original publication,19 the authors did not name the two reaction pathways (shown in Scheme 16) as Knoevenagel- and bisureide-based mechanisms; however, we would like to do so now. In the Knoevenagel-based pathway it is noted that the reaction proceeds almost quantitatively with thiourea, whereas in the reaction with urea as the reagent, the observed yield of the Biginelli adduct was very low. In the meantime, the reaction of the bisureide intermediate with urea afforded the Biginelli adduct in almost quantitative yields. Based on the experimental observations, the authors were able to propose a mechanistic rationale for the Biginelli-like reaction under basic conditions (Scheme 17).
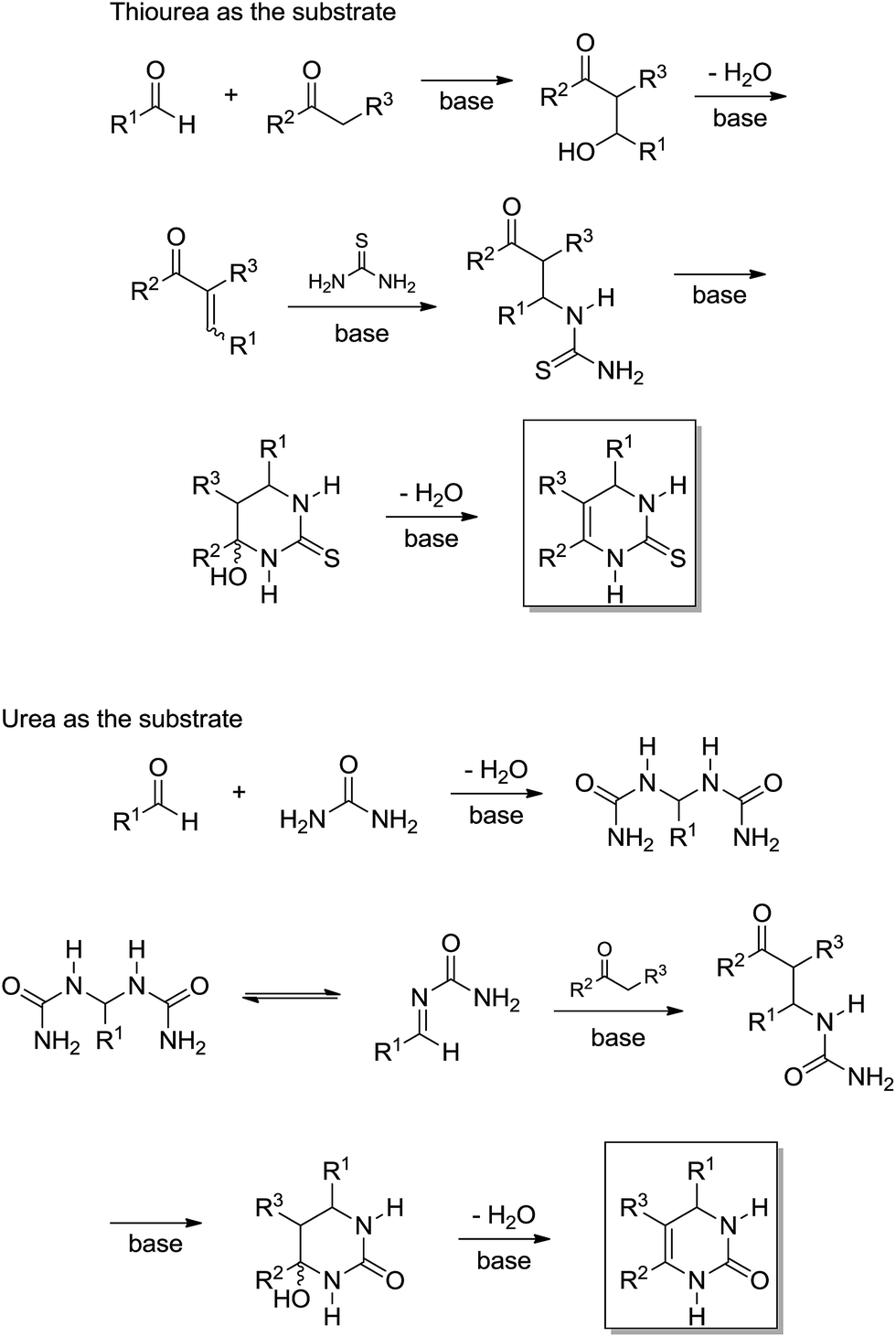 | ||
| Scheme 17 Mechanism rationale for the Biginelli reaction under basic conditions.19 Note that, for the first time, a Biginelli-type reaction had two totally different mechanisms based on a reagent selection (urea or thiourea). | ||
Later, based on ESI-MS(/MS) analysis, a different mechanistic proposition for a base-catalysed Biginelli reaction showed a preferred reaction pathway favouring the enamine-like mechanism (Scheme 18).20
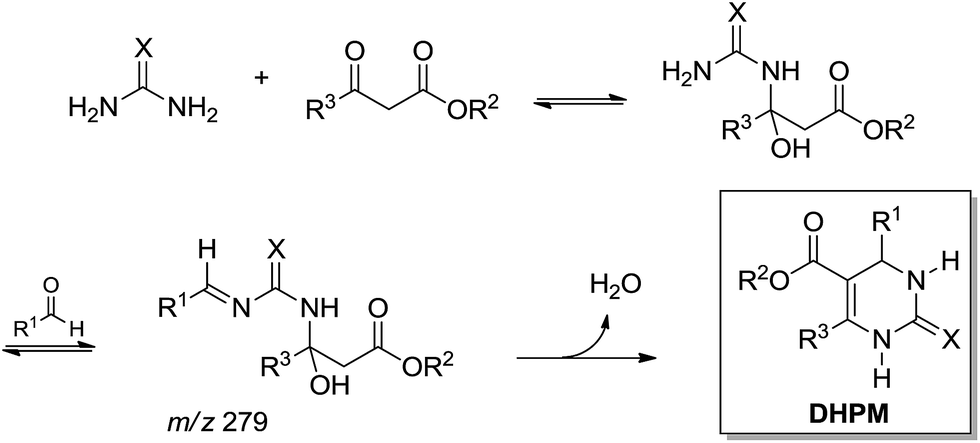 | ||
| Scheme 18 Base-catalysed Biginelli reaction proposition based on ESI-MS(/MS) analyses.20 Note that the intermediate of m/z 279 was detected and characterized by ESI(+)-MS/MS, therefore in its protonated form (the proton has been omitted for clarity). | ||
Curiously, the intermediate of m/z 279 was detected and characterized in the positive ion mode, therefore in its protonated form, despite the fact that the reaction was base-catalysed.
One last and important feature of the Biginelli reaction is the so-called ‘catalyst-free’ version. Very recently, it has been demonstrated that reactions conducted without any catalyst, and in a solvent-free versions, have a clear competition between the three possible mechanisms.21 The conclusions were based on NMR, DFT calculations and, mainly, on kinetics and high resolution quantitative ESI-MS(/MS) analyses using a charge-tagged aldehyde, which in turn, allowed an online monitoring of the formation and consumption of the reagents, intermediates and the final Biginelli adduct (Scheme 19) without the need of protonation or deprotonation (the charge tag strategy22 for MS). The strategy of using charge-tagged reagents proved to be essential to a precise mechanistic analysis without any catalyst, especially because protonation (or deprotonation) or coordination with any metal (to follow a residual charge from complex formation) would result in a considerable change in the reactivity of the reagents, therefore not representing the real mechanism of a catalyst-free version of the reaction.
 | ||
| Scheme 19 The proposed mechanistic competition for the catalyst-free (and solvent-free) Biginelli reaction.21 The charge-tagged intermediates have been detected and characterized by ESI(+)-MS and ESI(+)-MS/MS. Note the strategy allowed the online monitoring without protonation or deprotonation of the intermediates. | ||
A very important conclusion from the study was that the role of a catalyst is not only to improve yields and shorten reaction times, but the promoter also plays a fundamental role in improving the selectivity towards one reaction pathway, which was severely compromised without any catalyst. This knowledge is fundamental to the achievement of more efficient catalysts and greener reaction conditions, especially for the asymmetric versions of the Biginelli MCR.
3. The Hantzsch multicomponent reaction
The Hantzsch synthesis is a 3CR announced in the 19th century by Arthur Hantzsch (1882) and widely used for direct synthesis of 1,4-dihydropyridine (DHPs) derivatives. Many DHPs are commonly known for their biological activity as calcium channel blockers, as well as many other pharmacological activities such as antitumoral, bronchodilating, antidiabetic, neurotropic, HIV protease inhibitors described for several DHPs.6 Interestingly, because of DHPs' similarity with the natural product enzyme co-factors NAD(P)H and their oxidized forms NAD(P), the Hantzsch adducts (also referred to as Hantzsch esters) proved to have a great potential as hydride (or hydrogen) transfer reagents.6 These reactions include several asymmetric versions of the H-transfer process with excellent levels of enantioselectivities.6 The possibility of biological and chemical application of DHPs has fostered interest in these derivatives. As a consequence, many reports describing new methodologies and conditions for the synthesis of these important compounds have been described in the scientific literature.6 Despite the large number of available methodologies for the synthesis of DHPs, the mechanism of the Hantzsch 3CR is still the object of scientific debates.The Hantzsch 3CR has, perhaps, one of the most complexes mechanisms among all MCRs. In a general way, the mechanism of the Hantzsch reaction is not even appropriately discussed in review articles. Depending on the reaction conditions and substrates selected for the transformation, side reactions may take place in competition with the Hantzsch ester formation. Today, five reaction pathways may be summarized for the Hantzsch 3CR, as seen in Scheme 20. Once more, we do not intend to describe the evolution of the mechanism propositions, rather preferring to focus on the evidence published to support these five possible mechanistic paths. Therefore, although this topic is usually overviewed, the readers are urged to peruse comprehensive surveys on the subject to understand the history behind these propositions. Overall, these propositions are logical (Scheme 20) and have been evoked based on the general knowledge of reaction mechanisms and in the pioneering works dating back to the 19th century. With the benefit of hindsight, it now seems that a better critical evaluation of these mechanisms is possible. Not surprisingly, only a few reports describe a critical analysis of the Hantzsch mechanism and suggest a preferred reaction pathway based on solid evidences. A few reports also describe the mechanism for side reactions, which in some cases, depending on the interest, the sideproduct may be of note as well.
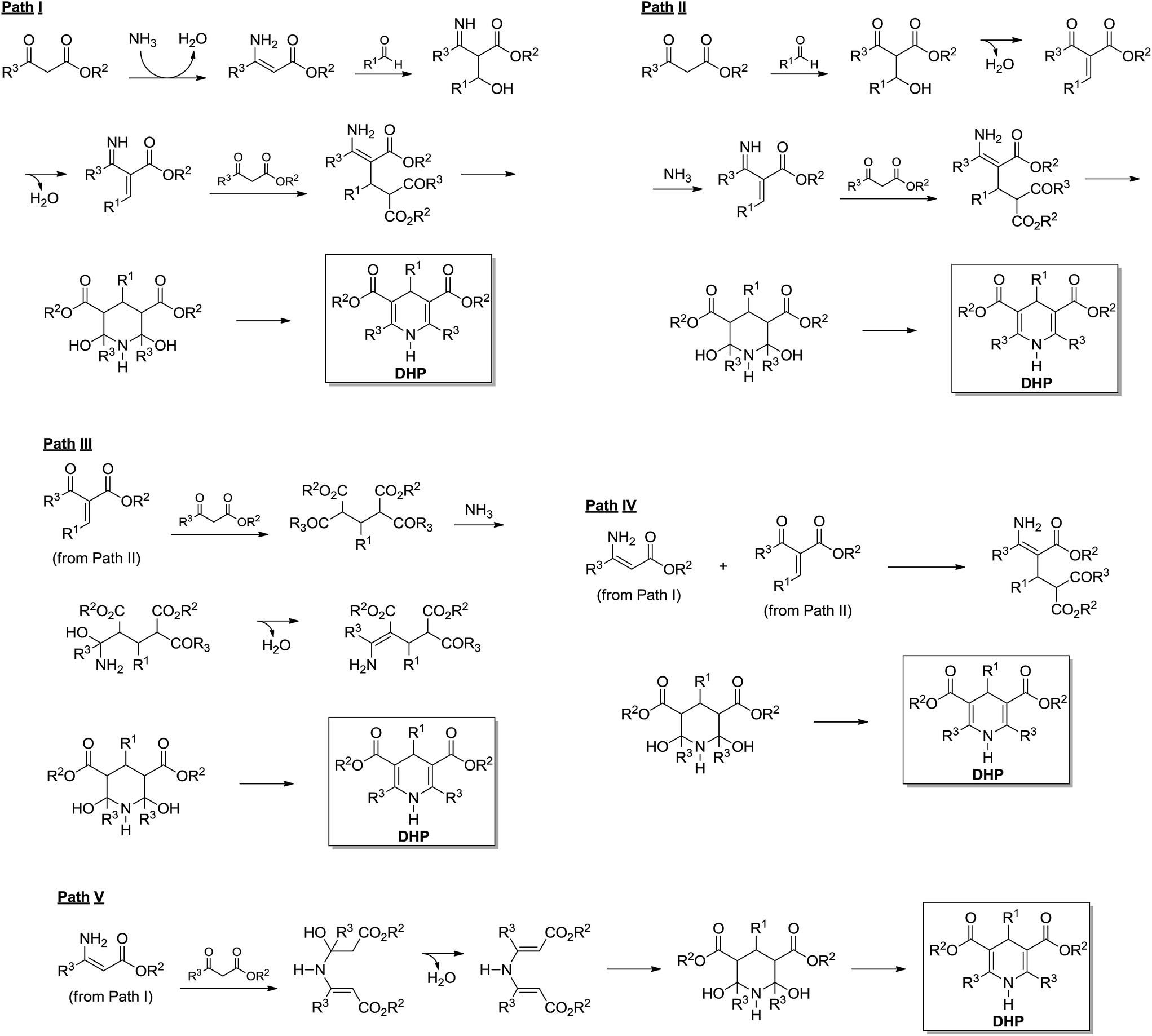 | ||
| Scheme 20 Possible mechanistic paths currently evoked for the Hantzsch MCR. | ||
Based on 15N and 13C NMR spectroscopy, a study by Katritzky and coworkers23 reported one of the most solid contributions towards the understanding of this truncated mechanism. The report also describes previously published evidence for these five propositions. Before this ground-breaking work,23 the mechanistic evidences were mostly based on pH effects on substituted benzaldehyde derivatives and on the basis of product composition, and are therefore not of interest to this review. In the NMR study, it was observed that two intermediates are always formed i.e. the enamine (Scheme 20, Path I) and the chalcone analogue (Scheme 20, Path II), which is formed after the first water elimination. The dienamine intermediate (Scheme 20, Path V) could be detected in the early stages of the reaction, therefore indicating this compound was a metastable sideproduct instead of an actual intermediate in the reaction. Interestingly, depending on the nature of the 1,3-dicarbonyl reagent, the dienamine was not observed. Preformed enamine and chalcone (Scheme 20, Paths I and II) intermediates have been used in the mechanism investigation and pointed to a preferred reaction pathway such as Path IV shown in Scheme 20. This result was found in accordance with the NMR monitoring of the 3CR. The authors tested several 1,3-dicarbonyl derivatives with similar results and the rate limiting step of the Hantzsch reaction has also been suggested as the chalcone analogue formation.
The Hantzsch MCR is highly sensitive to the reaction conditions i.e. presence or absence of a solvent, catalysed and non-catalysed versions, substituent effects on the reagents, reaction time, temperature, presence of a base or an acid and etc. For this reason, side reactions have been noted and described with improved conditions to select the formation of other products rather than the Hantzsch ester. All these aforementioned variants usually have pronounced effects on the Hantzsch 3CR. Considering however some of the side-products observed during the reaction are of synthetic interest, and that the competitive mechanisms are also important to understand the mechanism of the Hantzsch reaction, we believe it is worth looking closely at reports with a mechanistic contribution to understand these side reactions.
Aromatized compounds from the oxidation of the Hantzsch ester, 2-arylpyridine and 1,2-dihydropyridine derivatives have been described to form competitively from the Hantzsch reaction (Fig. 1)24 instead of the expected 1,4-dihydropyridine. Improved conditions and the competitive mechanism to select the formation of DHPs or 2-arylpyridine derivatives have been summarized in the study and a mechanism was evaluated (Scheme 21).24 Several parameters were tested to select one of the possible products. Temperature, solvents, base presence (or absence) and reaction time were among the investigated parameters. The results showed that all of these parameters had an important role for selecting the synthesis of one of these products (Fig. 1). Remarkably, good selectivities could be observed for the formation of the Hantzsch ester, the 1,2-dihydropyridine and for the 2-arylpyridines. The product selection, as one can expect, was closely associated with good control of all described reaction conditions.
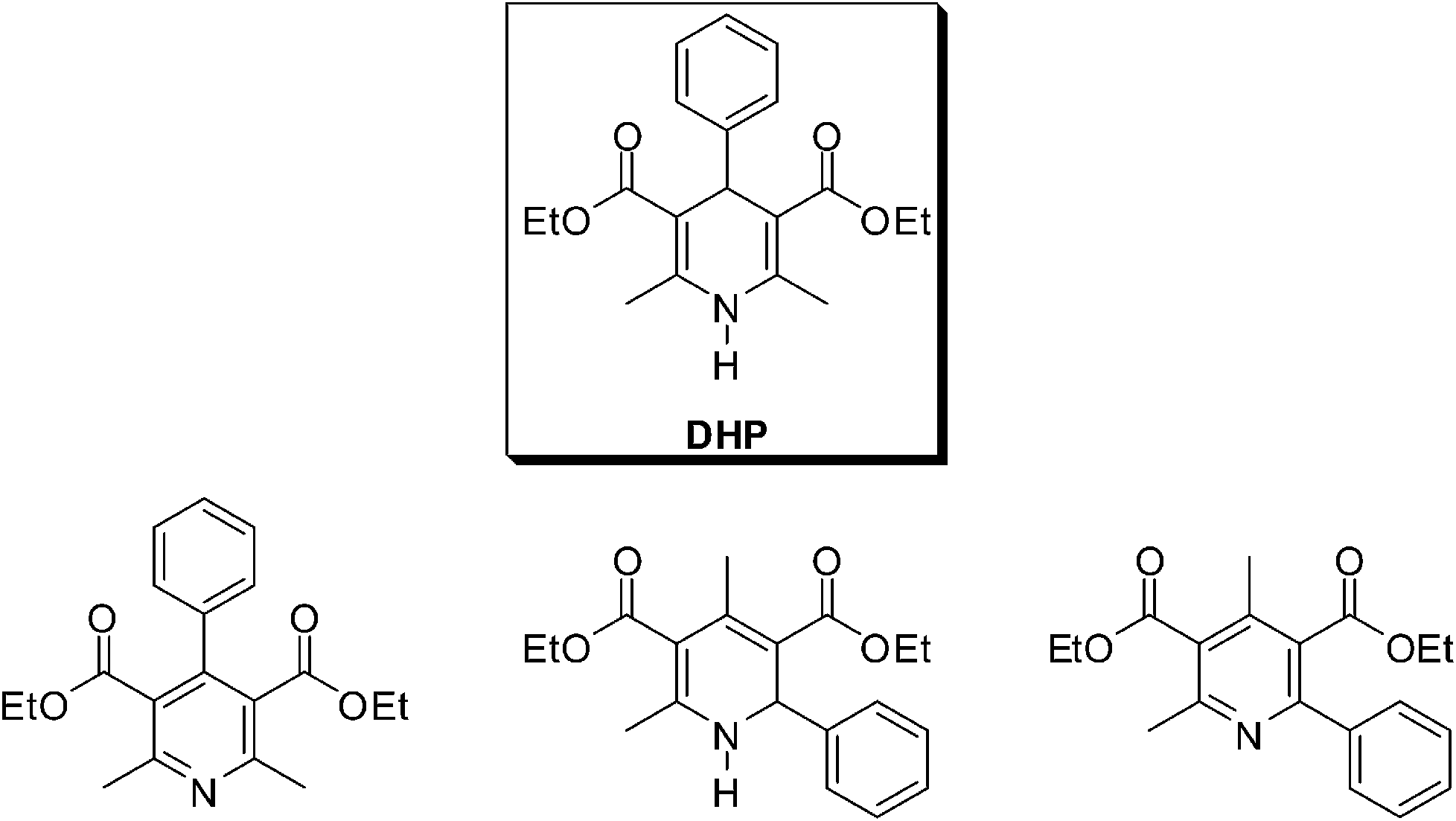 | ||
| Fig. 1 Products that could be observed in the Hantzsch reaction. (Top) 1,4-Dihydropyridine (Hantzsch ester, DHP). (Bottom) From left to right: the aromatized compound, 2-arylpyridine and 1,2-dihydropyridine derivatives. The formation of such compounds is highly dependent on the reaction conditions and their formation can be tuned by using the appropriate reactional condition.24 | ||
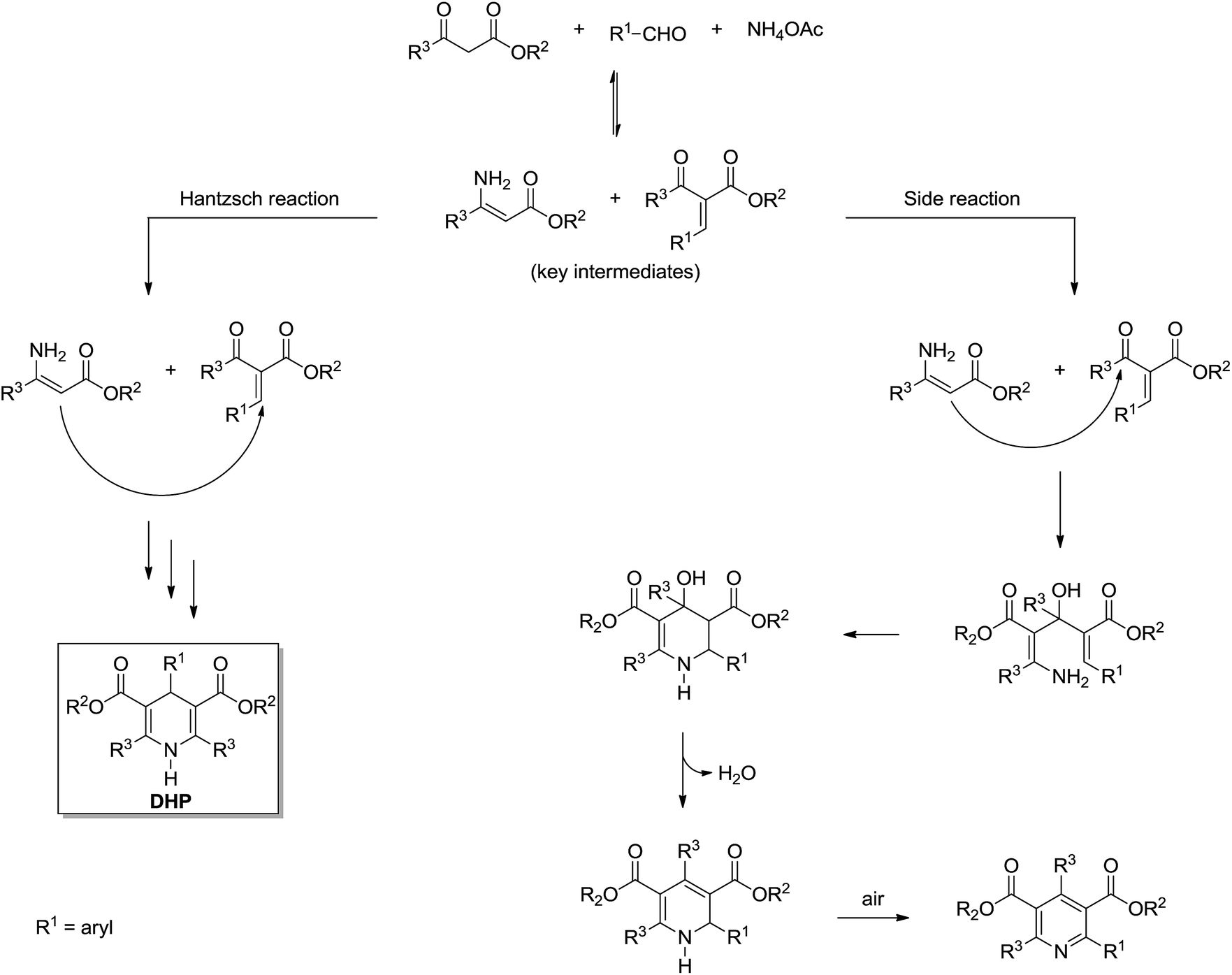 | ||
| Scheme 21 Formation of 2-arylpyridine derivatives in competition with the Hantzsch reaction expected pathway.24 Note that 1,2-dihydropyridines are formed prior to the formation of 2-arylpyridines and, depending on the reaction conditions used, this could be the major product isolated. | ||
A compelling study demonstrated three competitive reaction pathways for the condensation of 5-aminopyrazoles, aldehydes, and 1,3-cyclic diketones (Scheme 22).25 The reaction could afford the Hantzsch-type ester (Scheme 22, Path A), the Biginelli-type adduct (Scheme 22, Path B) or pyrazolo[4,3-c]quinolizin-9-one products (Scheme 22, Path C). Temperature and the nature of the catalyst also played a fundamental role in the mechanism selection. At ambient temperature and under neutral conditions the reaction preferentially proceeds via the formation of a kinetically controlled intermediate, thus yielding a Biginelli-type adduct, as shown in Scheme 23.
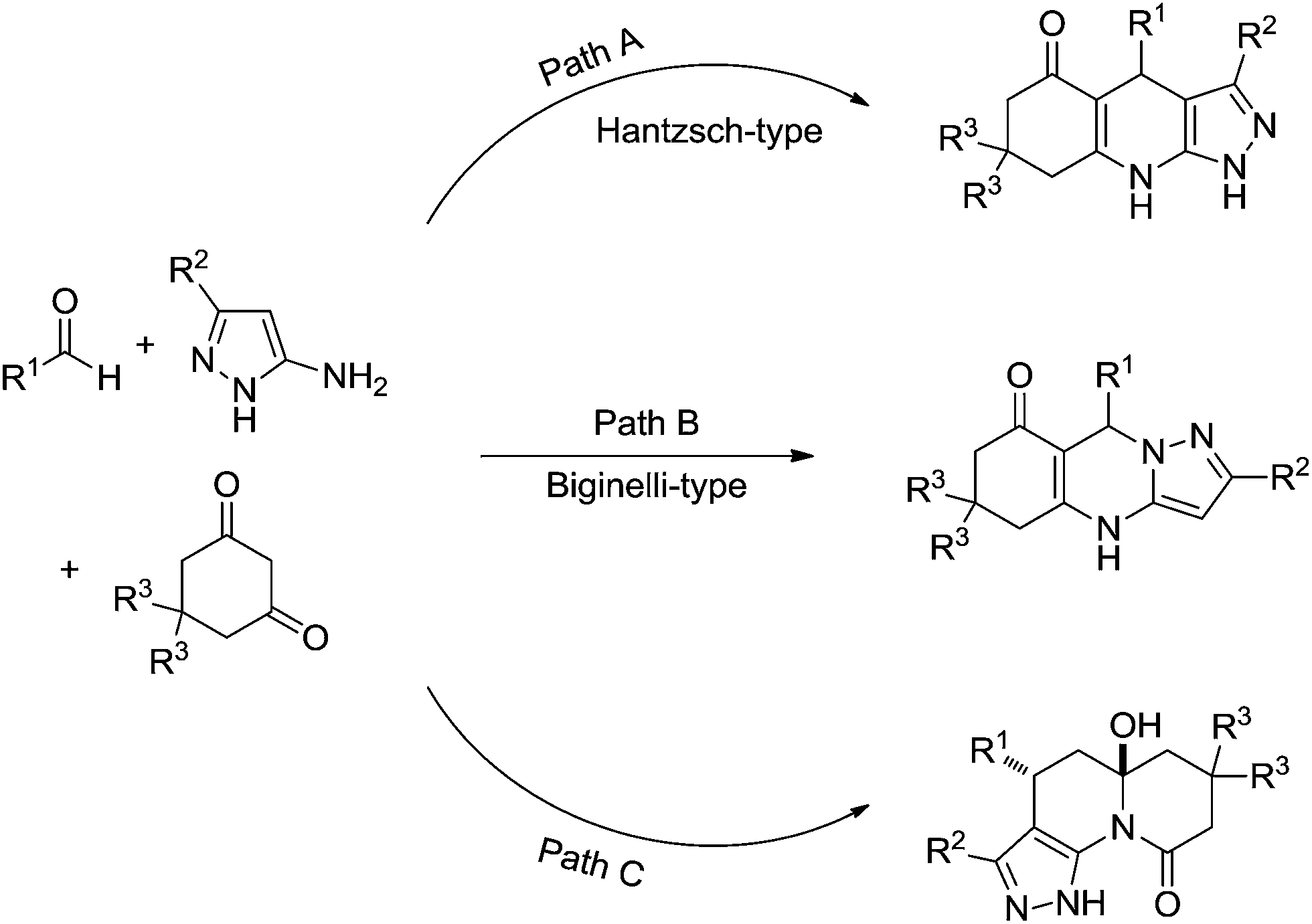 | ||
| Scheme 22 Three possible reaction pathways for the condensation of 5-aminopyrazoles, aldehydes, and 1,3-cyclic diketones.25 | ||
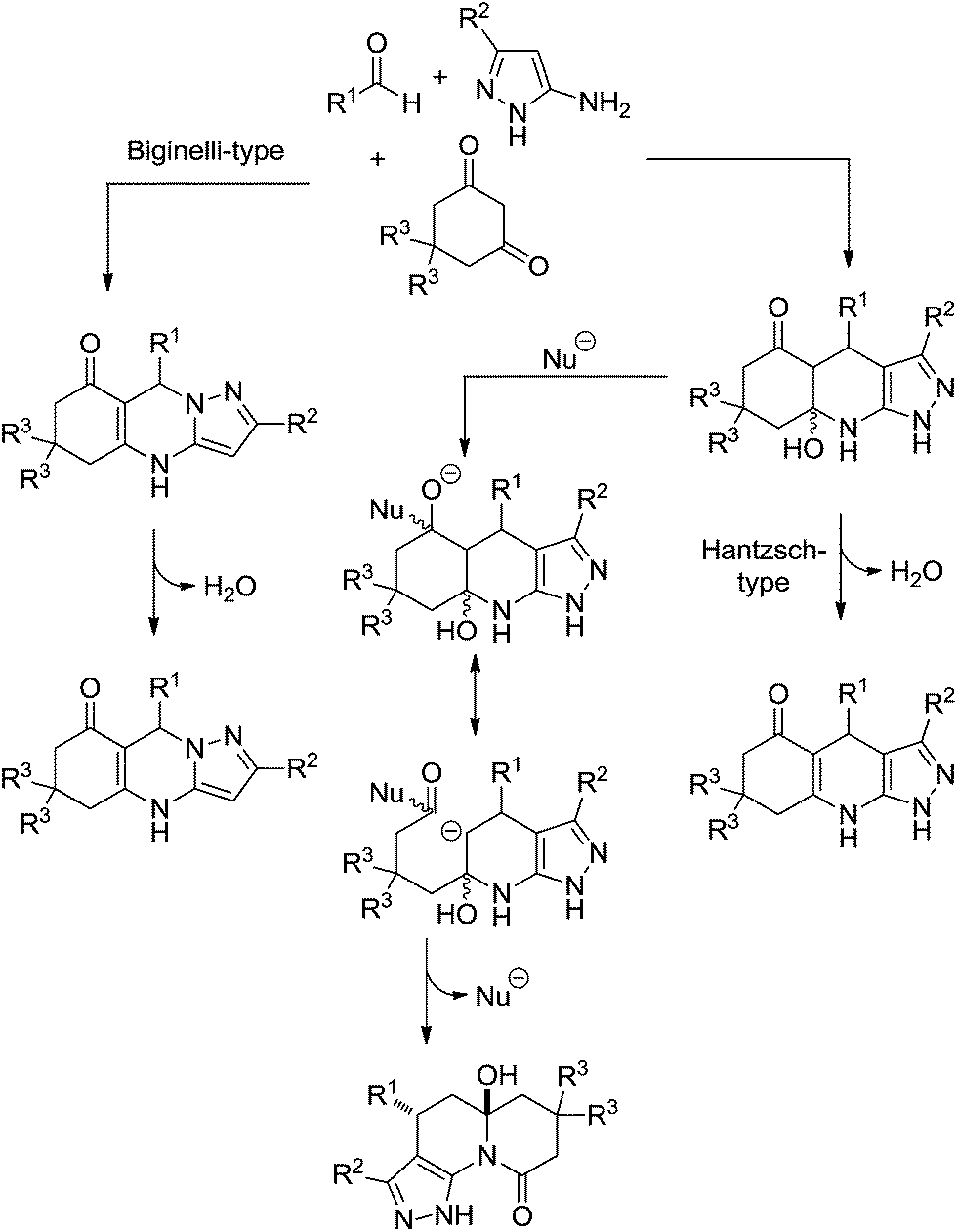 | ||
| Scheme 23 The proposed mechanism for the condensation of 5-aminopyrazoles, aldehydes, and 1,3-cyclic diketones based on experimental evidence.25 | ||
Upon increasing the reaction temperature and by adding triethylamine to the mixture it was possible to demonstrate the condensation undergone preferentially through a tricycle compound, which is a common intermediate in the competition between the Hantzsch-type reaction and the formation of pyrazoloquinolinone products (Scheme 23).
To select between the Hantzsch-type reaction and the formation of pyrazoloquinolinone, not only does temperature play a role, but the nature of the amine (catalyst) also had a crucial role to further the transformation of the common tricyclic intermediate. It was shown that tertiary bases (triethylamine or N-methylmorpholine) favoured the Hantzsch-like pathway, whereas strong bases of relatively small size (e.g. hydroxide, or methoxide) afforded either mixtures or exclusively pyrazoloquinolinones (depending on the reaction conditions) because such bases could act as nucleophiles.
Later, two other important works26,27 described a side-reaction from the Hantzsch(-like) reaction, and the mechanisms were based on these already shown in Schemes 21 and 23.
Recently, a breakthrough was published by Garden, Eberlin and coworkers28 describing a mechanistic investigation of the Hantzsch reaction using charge-tagged reagents (the charge tag strategy22) for ESI-MS(/MS) online monitoring of the reaction. The study allowed the proposition of a comprehensive mechanism and the equilibriums involved (Scheme 24). Moreover, the results obtained by ESI-MS(/MS) were found to be in accordance with some conclusions depicted by NMR analyses23 such as that the Knoevenagel-like intermediate seemed indeed to participate in the rate limiting step of the Hantzsch reaction.
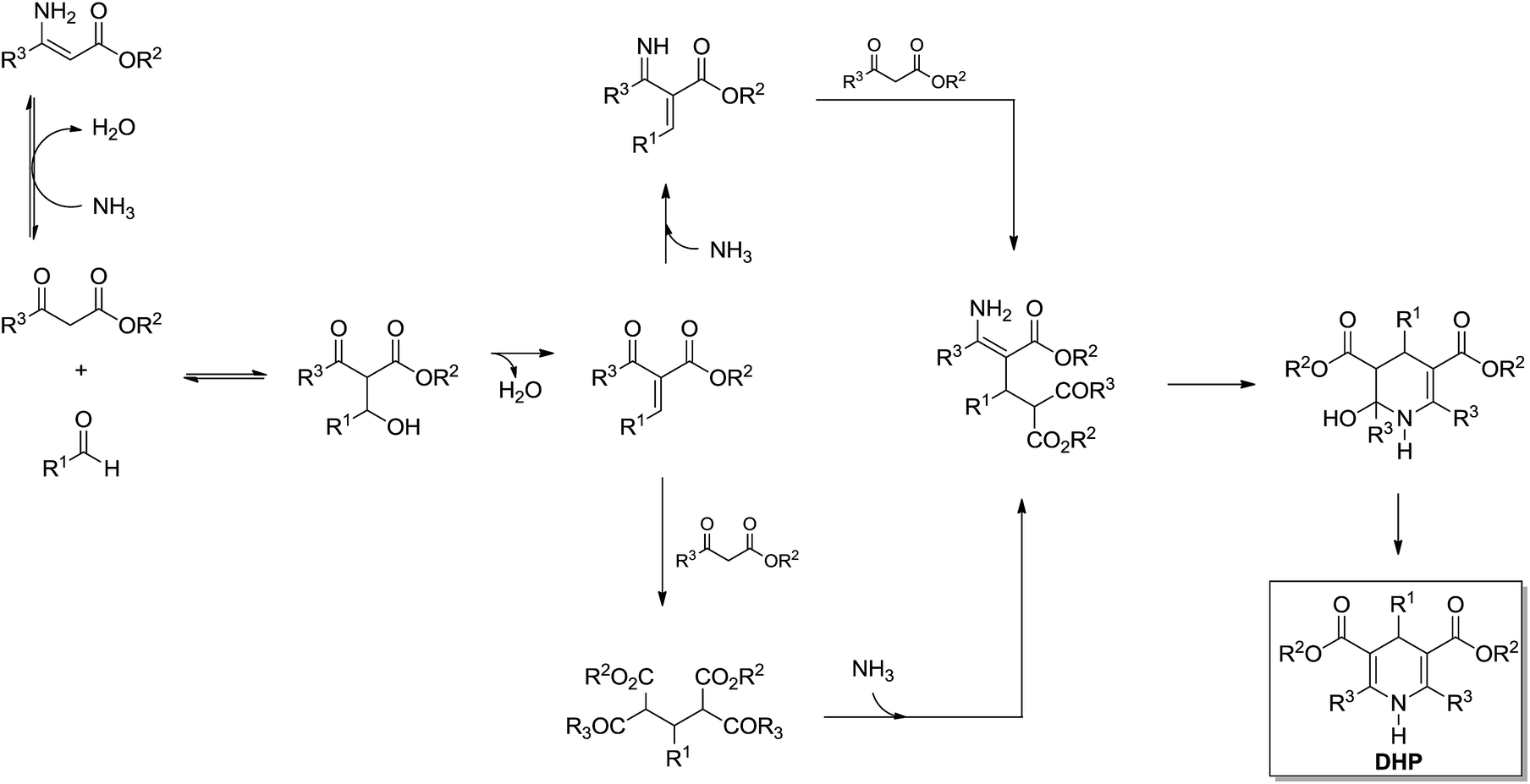 | ||
| Scheme 24 The Hantzsch 3CR mechanism proposition based on ESI-MS(/MS) analyses using the charge tag strategy for MS online monitoring.28 | ||
4. The Mannich multicomponent reaction
The Mannich 3CR (discovered in 1912) is a very useful synthetic tool applied in the synthesis of β-amino carbonyl compounds (BAC). Among all MCRs, the Mannich 3CR has the least controversial mechanism and its reaction pathway is almost universally accepted. Interestingly, there are many asymmetric catalysed versions of the Mannich MCR. The Mannich MCR is also called ‘direct Mannich’ reaction. When preformed enolates (or modified enolates) are used, the Mannich reaction is referred to as the indirect Mannich reaction,29 and this version is also widely used. The most controversial issue related to the Mannich MCR is not the mechanism itself, but the chiral induction step involved in this transformation. This is especially true for organocatalysed versions of the Mannich MCR because the observed stereocontrol is not yet well understood, but this issue has already been reviewed for the Mannich reaction.30 As one can expect, any new chiral system has unique transition states and stereocontrol associated with the transformation. Even though the basic Mannich reaction mechanism has the same sequence, that is, imine (or iminium) formation followed by trapping the enol (or enolate), affording the β-amino carbonyl compound. The imine (or iminium) intermediate is formed from the condensation reaction between the aldehyde and the amine (primary or secondary) and it is the first step of the reaction.The first plausible attempt to understand the Mannich 3CR mechanism was based on a kinetic approach using ethylmalonic acid with formaldehyde and dimethylamine as the model reaction.31 It is interesting to highlight that, based on their results, the authors could propose a very accurate mechanism for the transformation (Scheme 25) in 1949.
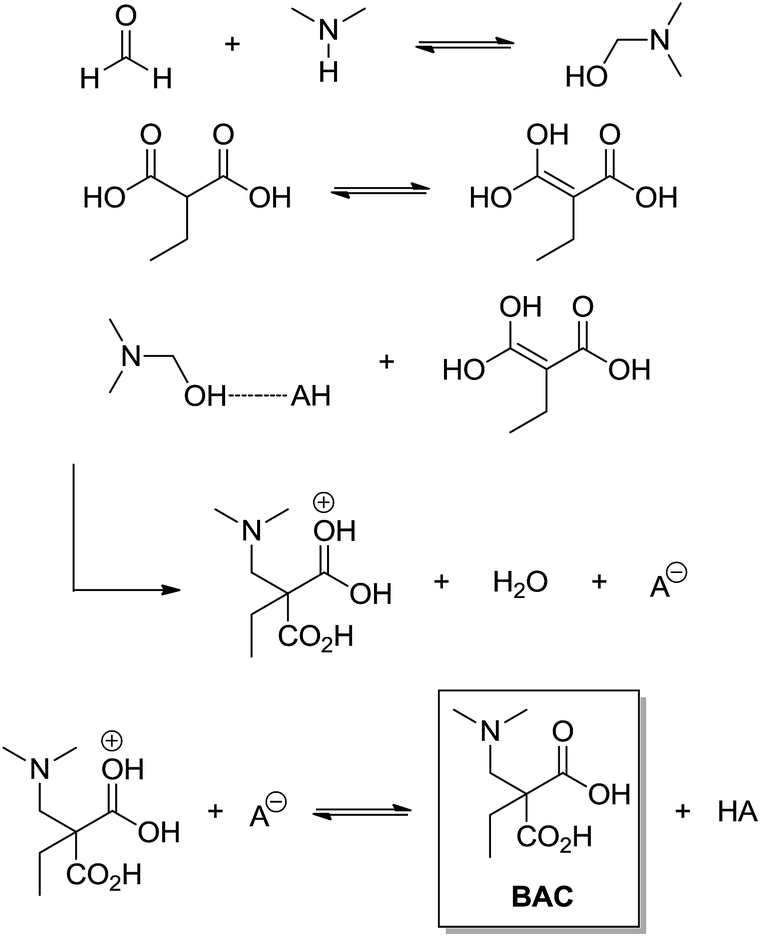 | ||
| Scheme 25 The three-component Mannich reaction mechanism as proposed by Alexander and Underhill.31 Note it is an acid-catalysed version and no imine (or iminium) is noted in the reaction mechanism. | ||
The study proposed that the Mannich 3CR follows third order kinetics and, therefore, any further mechanistic consideration should take this fact into account. Alternative routes were also proposed32 and refuted,33 but with no significant differences from that shown in Scheme 25, that is, the basis for the mechanism was that proposed by Alexander and Underhill in 1949.31 Since then, some new approaches have been used in the investigation of the direct Mannich mechanism, such as isotopic labelling,34 infrared35 (also using organozinc reactants), NMR36 (for a Mannich-type reaction) and ESI-MS(/MS) for the Mannich-type α-methylenation of ketoesters.37 These contributions, like the studies of the indirect Mannich version, allowed the consolidation of the iminium formation (from the aldehyde and amine condensation) as the key intermediate for the Mannich MCR. Interestingly, this is the main difference observed in the current accepted mechanism for the direct Mannich reaction when compared to the original proposition of Alexander and Underhill.31
Except for asymmetric versions, which means that the catalytic system has unique characteristics and requires specific investigations (especially for the understanding of chiral induction), a direct investigation of the Mannich 3CR has only very recently been reported and the study was based on ESI-MS(/MS) and DFT calculation,38 which also revealed the role of ion-pairing effects (ionic liquid effect) for reaction success (Scheme 26).
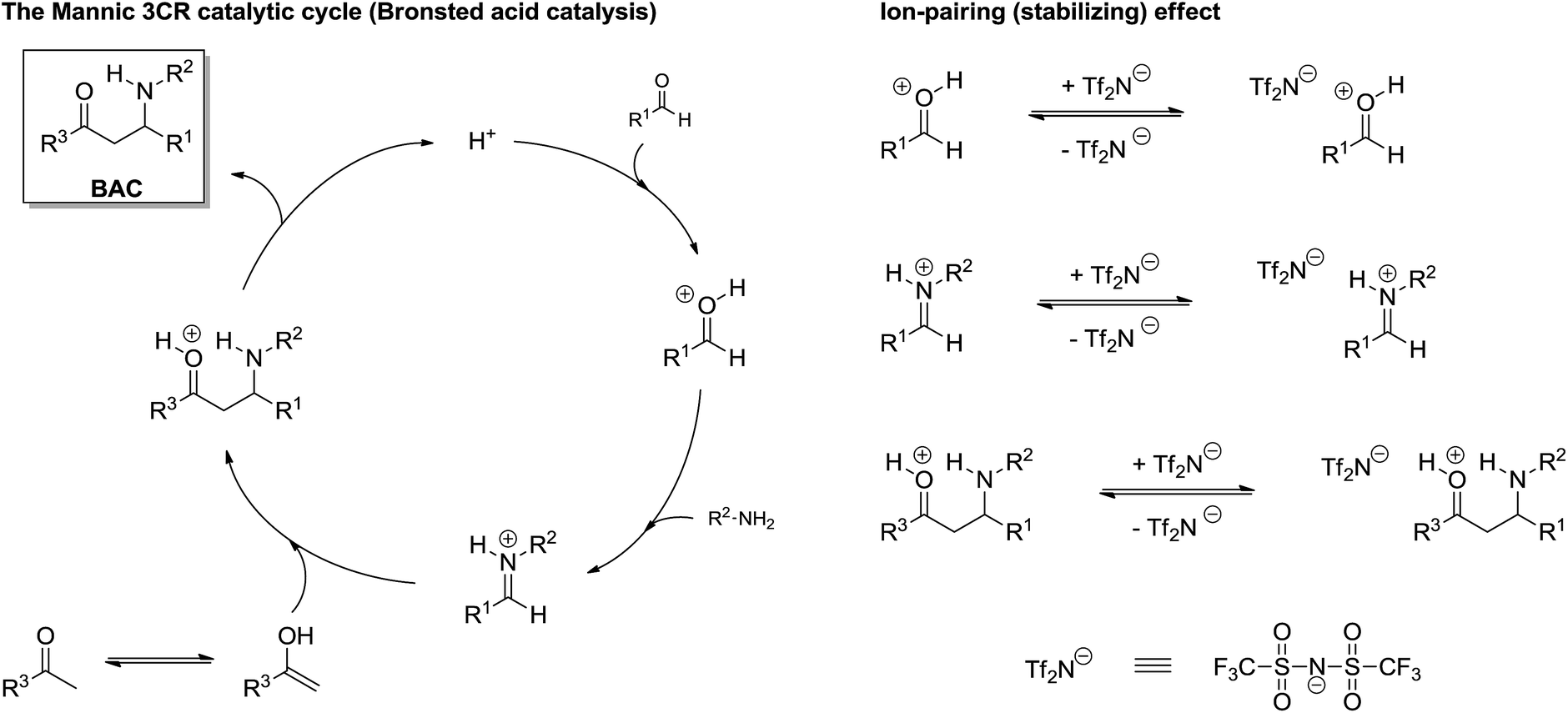 | ||
| Scheme 26 Mannich three-component catalytic cycle with ionic liquid effect under Bronsted acid catalysis.38 Note the ion-pairing effect (right) stabilizes the charged intermediates from the catalytic cycle (left). Also note the iminium ion formation from the condensation of the aldehyde treated with the amine. | ||
Despite the direct observation of the charged intermediates and the contribution to the comprehension of the positive intervention of ionic liquids, once again, the mechanism and the data supporting it pointed to the accuracy of the original proposition.
5. The Passerini and Ugi multicomponent reactions
The Passerini (announced in 1921) 3CR and Ugi 4CR (described in 1959) reactions are part of a class of MCRs called isocyanide-based MCRs (also referred to as IMCRs). These two IMCRs are intimately bonded and their mechanisms have many common features. We therefore decided to discuss their mechanisms in a single section. Variations for the Ugi and Passerini IMCRs and the contributions from these variations to the understanding of the mechanism involved with these two MCRs have been already reviewed.39 The chemistry of isocyanide derivatives plays a role as well, and the unique properties and reactivity of such compounds have already been reviewed.40Despite their many similarities, the differences are also important. The Passerini reaction, for instance, is typically accelerated in aprotic solvents, indicating a non-ionic mechanism and thus contrasting with the Ugi reaction, as reviewed elsewhere.40 A plausible mechanism for the Passerini 3CR is shown in Scheme 27. The Passerini 3CR mechanism is based on the isocyanide insertion in the loosely hydrogen-bonded adduct afforded from the acid and aldehyde interaction. The intermediate with the three components has not been isolated and immediately undergoes a rearrangement, affording the Passerini adduct.
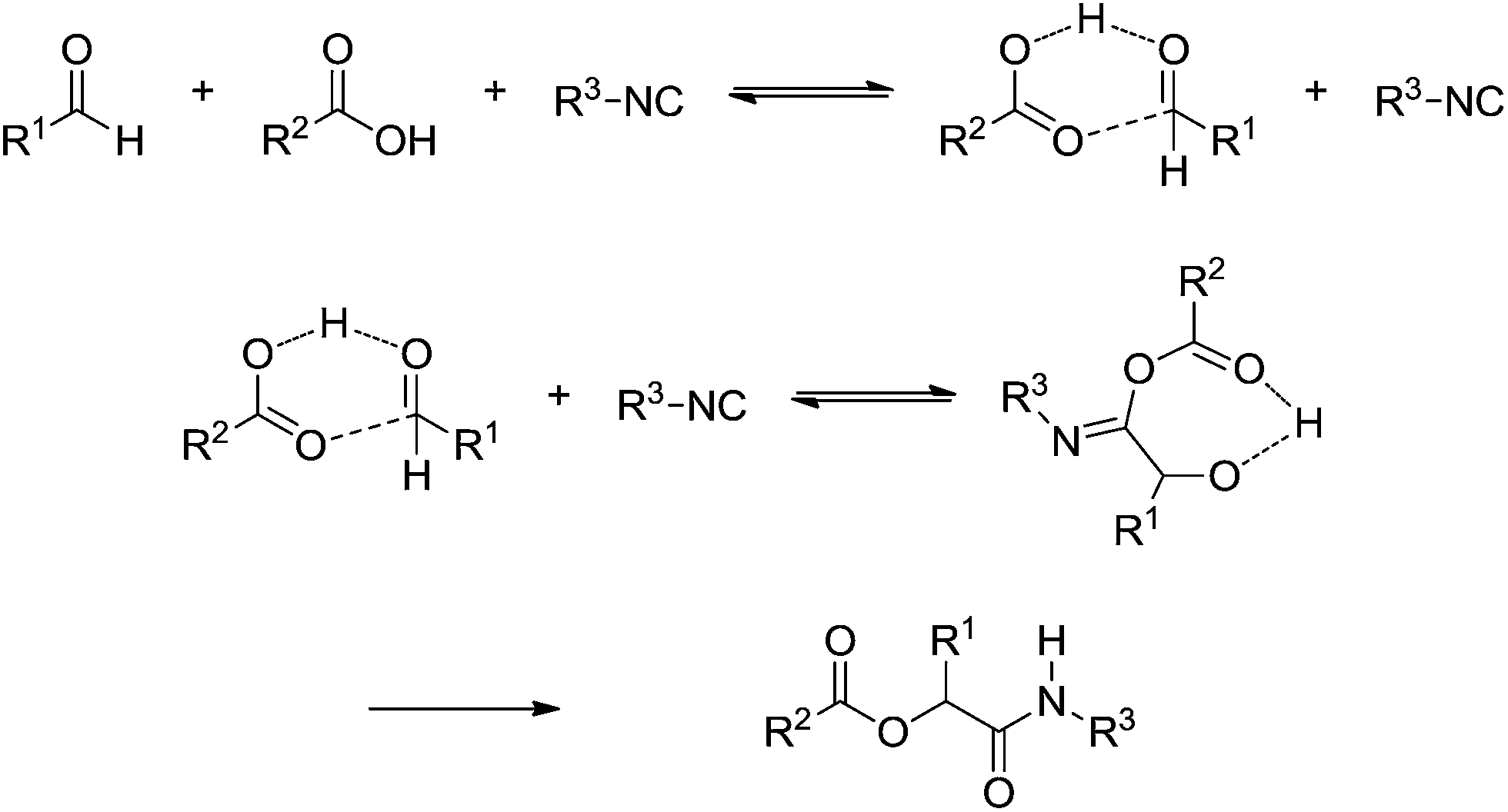 | ||
| Scheme 27 The proposed mechanism for the Passerini three-component reaction based on the literature evidence.40 | ||
The importance of the hydrogen-bonded structure has been evidenced by the work of Lamberth and coworkers41 of a highly stereoselective Passerini reaction. Curiously, in 1951 a third-degree reaction order was proposed based on kinetic data.42 The proposition already shown in Scheme 27 fulfils this hypothesis and was found to be in accordance with much evidence.43
Breakthrough work was later published by Floriani and coworkers44 describing the role of TiCl4 as a Lewis acid promoter of the Passerini reaction (Scheme 28).
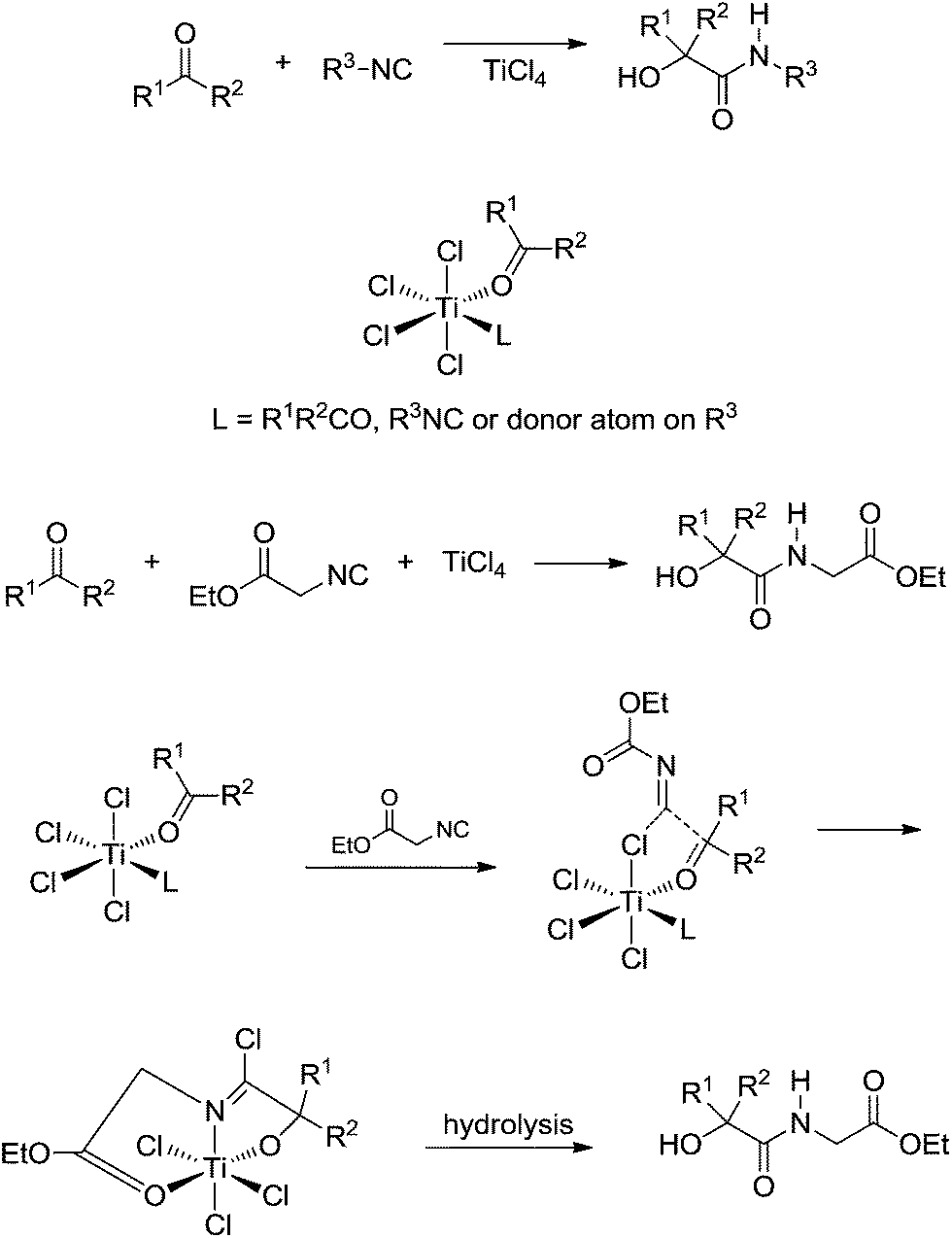 | ||
| Scheme 28 The proposed mechanism for the Passerini reaction catalysed by TiCl4.44 | ||
The study pointed to a major difference from Bronsted- and Lewis-catalysed Passerini, which is, using titanium as the metal, self-assembling properties are observed for the reagents around the metal centre. In general, this may be the whole idea for metal-catalysed Passerini reaction, especially for the d (transition metals) and f (lanthanide and actinide) metals. Some asymmetric versions of the Passerini reaction have already been described using chiral transition metal complexes45 and, in this sense, the role of the reagents for the reactive intermediates must also play a role during the chiral induction step.
A recent theoretical study46 provided a seminal contribution based exclusively on theoretical calculations that helped understand the Passerini reaction mechanism more fully. Transition states with four-components were calculated, as shown in Scheme 29. The role of an extra acidic component (the fourth component) to facilitate the observed rearrangements before the final adduct formation was demonstrated. Based on the results, the authors suggested that the Passerini reaction undergoes a pseudo-four-component reaction pathway to afford the desired product.
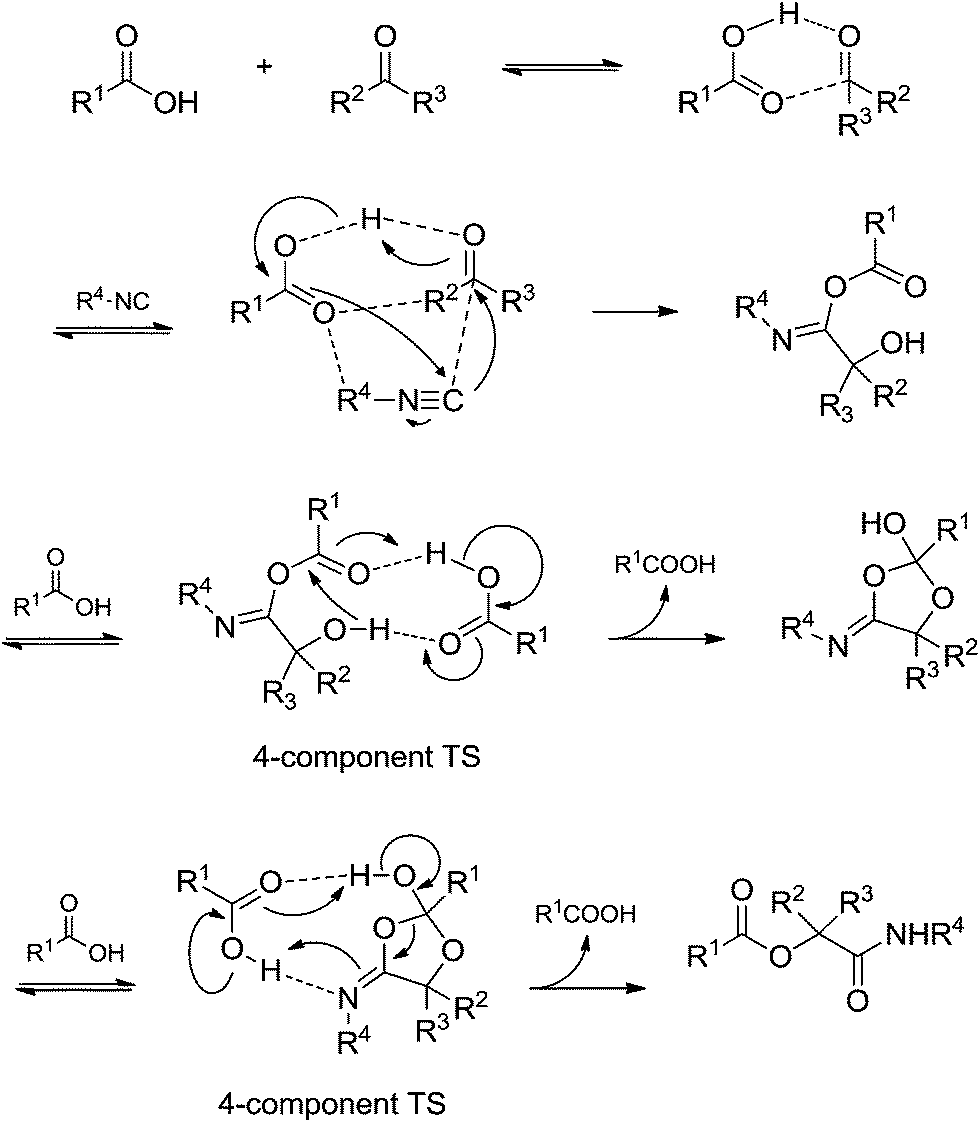 | ||
| Scheme 29 Passerini reaction mechanism based on theoretical calculations. Note the occurrence of transition states with four components.46 | ||
The Ugi four-component reaction is one of the most important IMCRs to access peptide-like structures. The transformation was first reported in 1959 by Ivar Ugi,47 who also brought to light many important features for understanding the mechanism of his eponymous reaction. The Ugi reaction is likely favoured in polar protic solvents, in contrast to the Passerini reaction, but there are also many available examples describing success with the reaction using polar aprotic solvents.43 The basic mechanism of the Ugi reaction (Scheme 30) was widely accepted with a few controversies, as will be discussed.
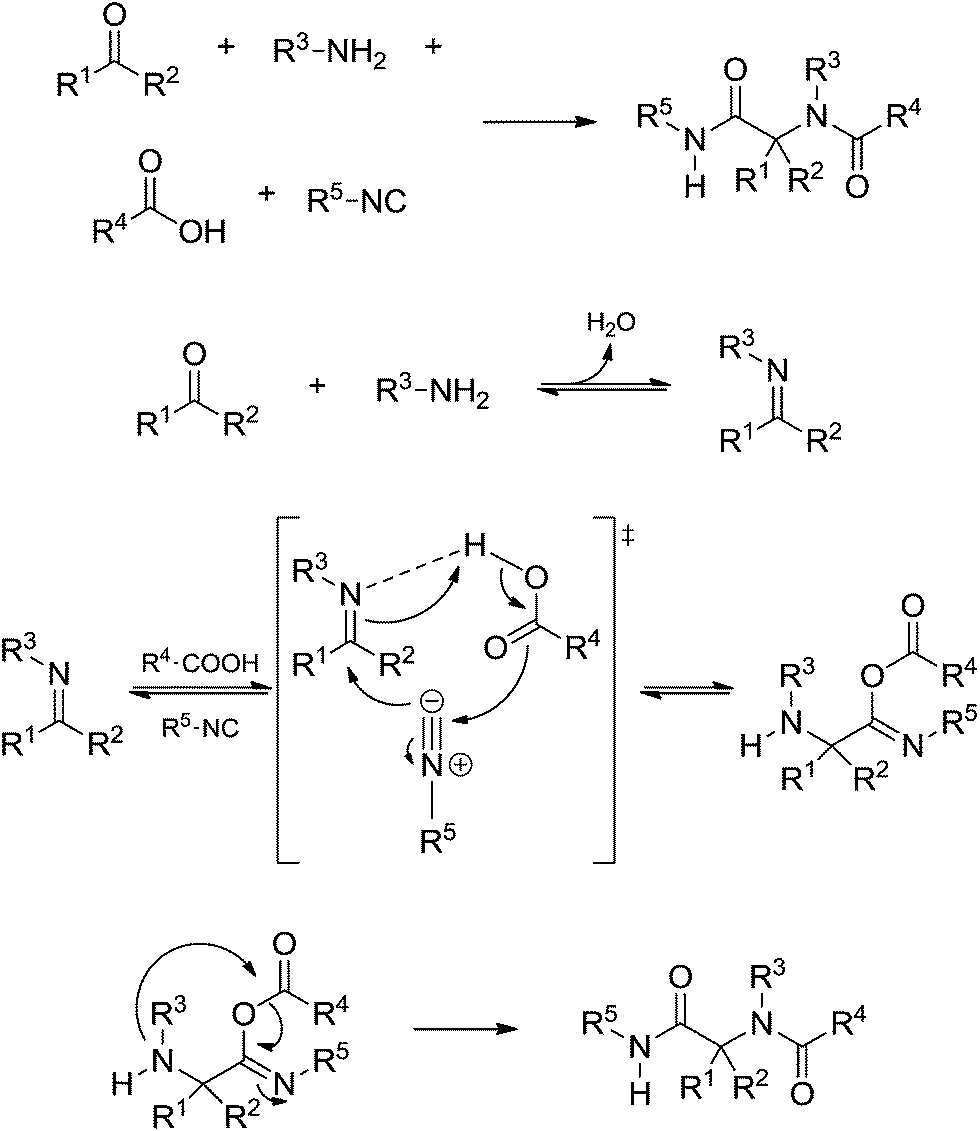 | ||
| Scheme 30 The general (and accepted) Ugi four-component reaction mechanism. | ||
The Ugi reaction begins with the in situ imine (or iminium) formation followed by a three-component transition state. The last step is the Mumm rearrangement (or Smile rearrangement, depending on the substrate) leading to the Ugi adduct. The general proposition shows a transition state bearing the imine (or iminium), the acid and the isocyanide. The presence of three components is the first issue discussed regarding the Ugi reaction mechanism. Instead, the iminium (or imine) would be trapped by the isocyanide followed by addition of the acid (Scheme 31).
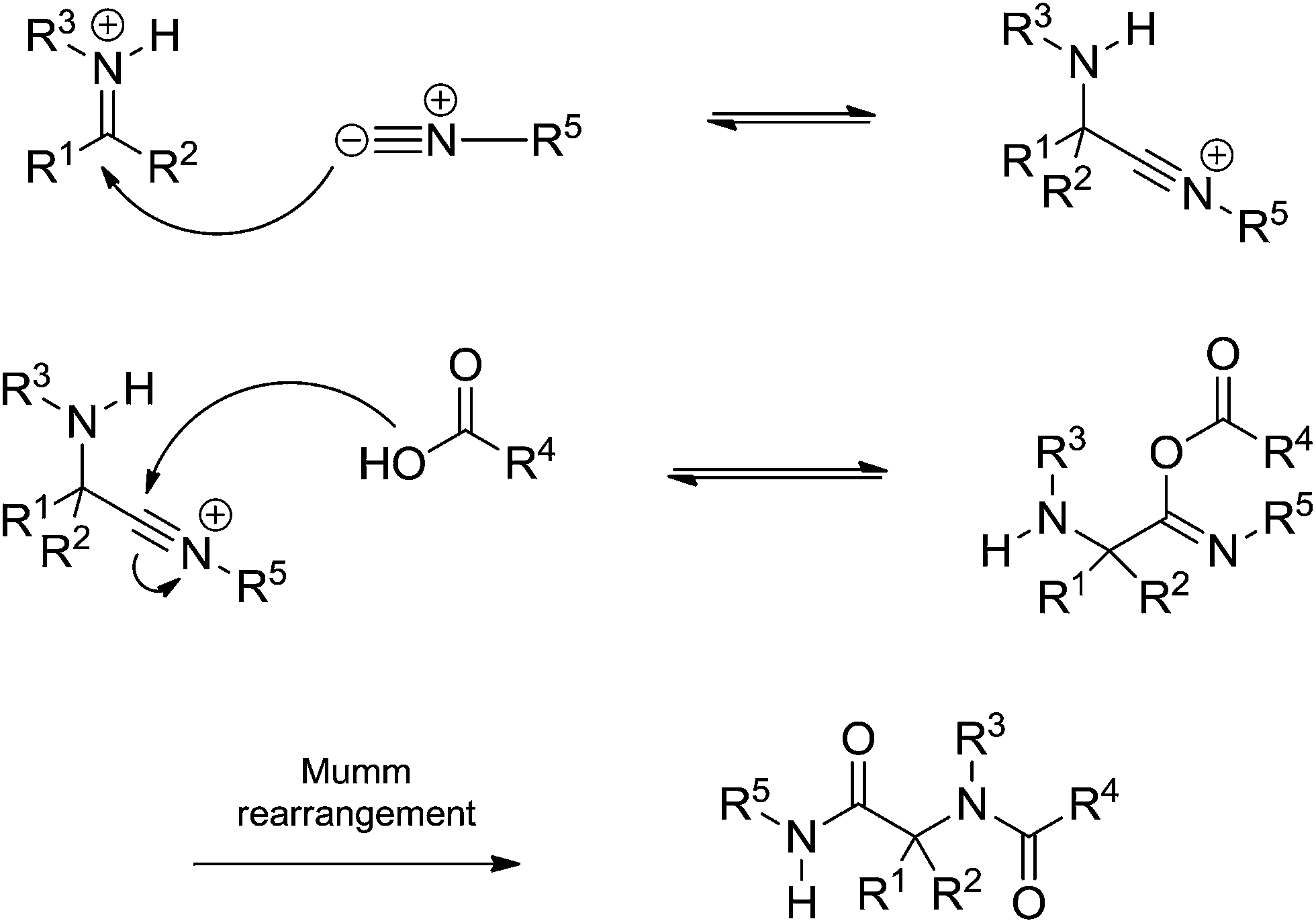 | ||
| Scheme 31 Isocyanide addition to the iminium (or imine) intermediate followed by acid addition before the Mumm rearrangement. | ||
Most synthetic development of the Ugi reaction relied on the mechanistic proposition, with all its associated assumptions, described in Scheme 31. Fleurat-Lessard and coworkers,48 however, have recently challenged fifty years of established views on the Ugi reaction. In the seminal work based on theoretical calculations, the possibility of the acid being added to the iminium intermediate followed by isocyanide insertion was described (Scheme 32).
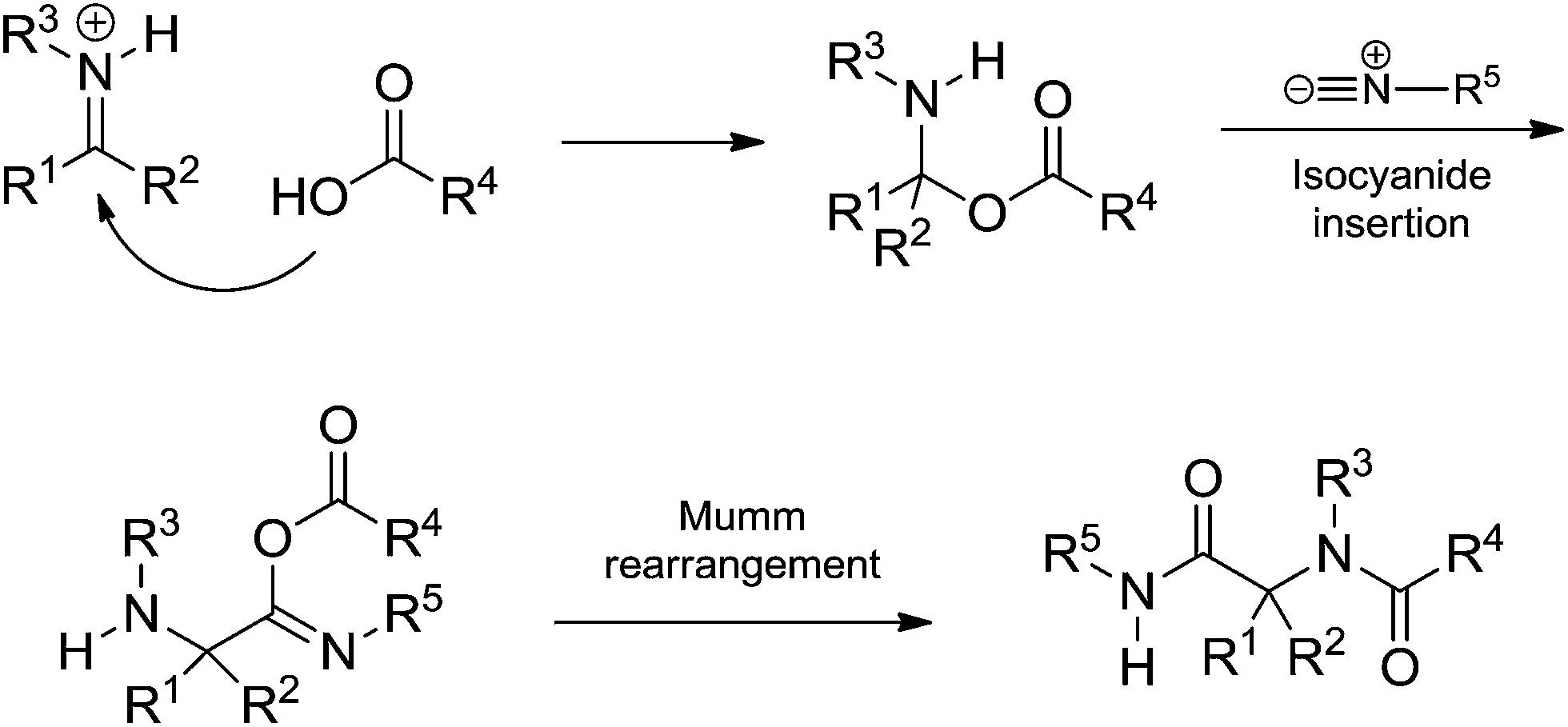 | ||
| Scheme 32 Alternative mechanism by acid addition followed by isocyanide insertion before the Mumm rearrangement. The proposition was mostly based on theoretical calculations.48 | ||
The detailed study showed the transition states associated with each step for the Ugi-Mumm and Ugi-Smile rearrangements and also the details for the possible isocyanide insertion after the acid addition.
A mechanistic study on the Ugi four-component reaction based on the charge tag strategy22 using ESI-MS(/MS) and DFT calculations was recently described.49 Interesting intermediates could be efficiently detected and characterized by collision-induced dissociation experiments (Fig. 2).
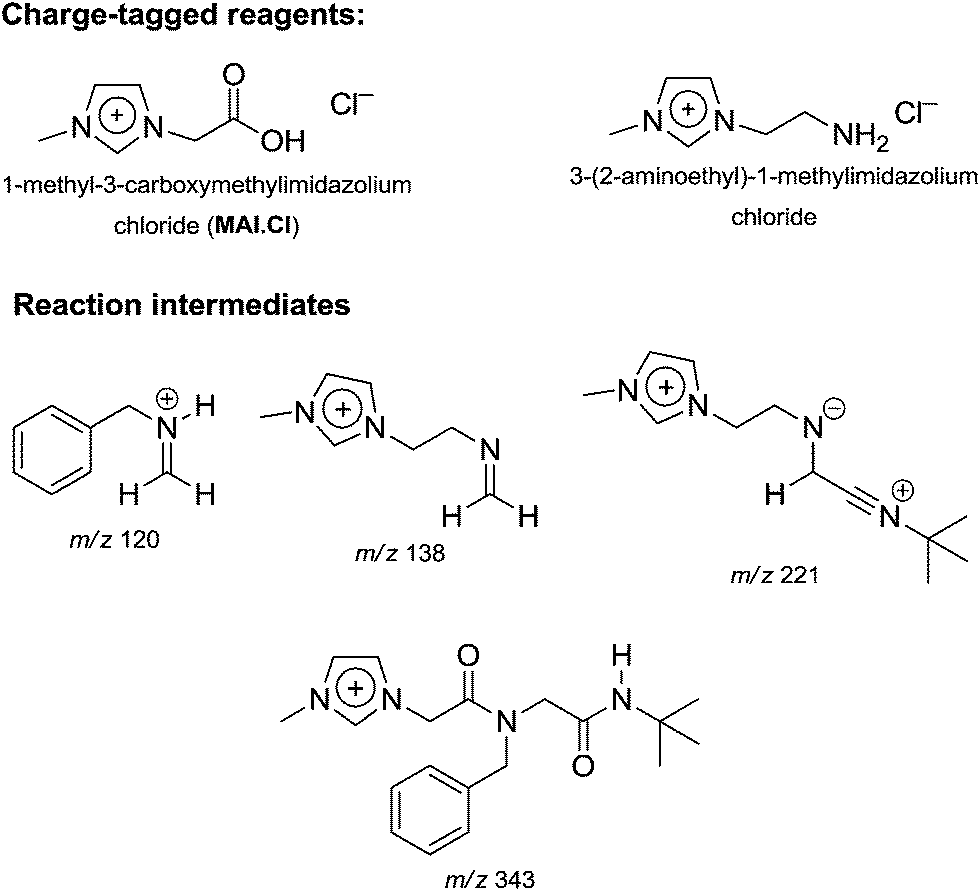 | ||
| Fig. 2 Charge-tagged reagents and intermediates from the Ugi reaction detected and characterized by ESI-MS(/MS) during online monitoring.49 Note the intermediates pointed to the accuracy of the classical mechanistic view on the Ugi mechanism. | ||
The experiments pointed to the accuracy of the classical view of the Ugi reaction mechanism described in Scheme 31 and, curiously, no protonated isocyanide could be detected. Some by-products from parallel reactions were also noted during online monitoring of the reaction (Scheme 33) but these reactions were only noted due to the high sensitivity of MS technique and are indeed unlikely. The possibility of the alternative route shown in Scheme 32, however, could not be ruled out, as pointed out in the article.49 DFT calculations were found to be in accordance with the importance of the solvent effect for the Mumm (or Smile) rearrangement.49,50
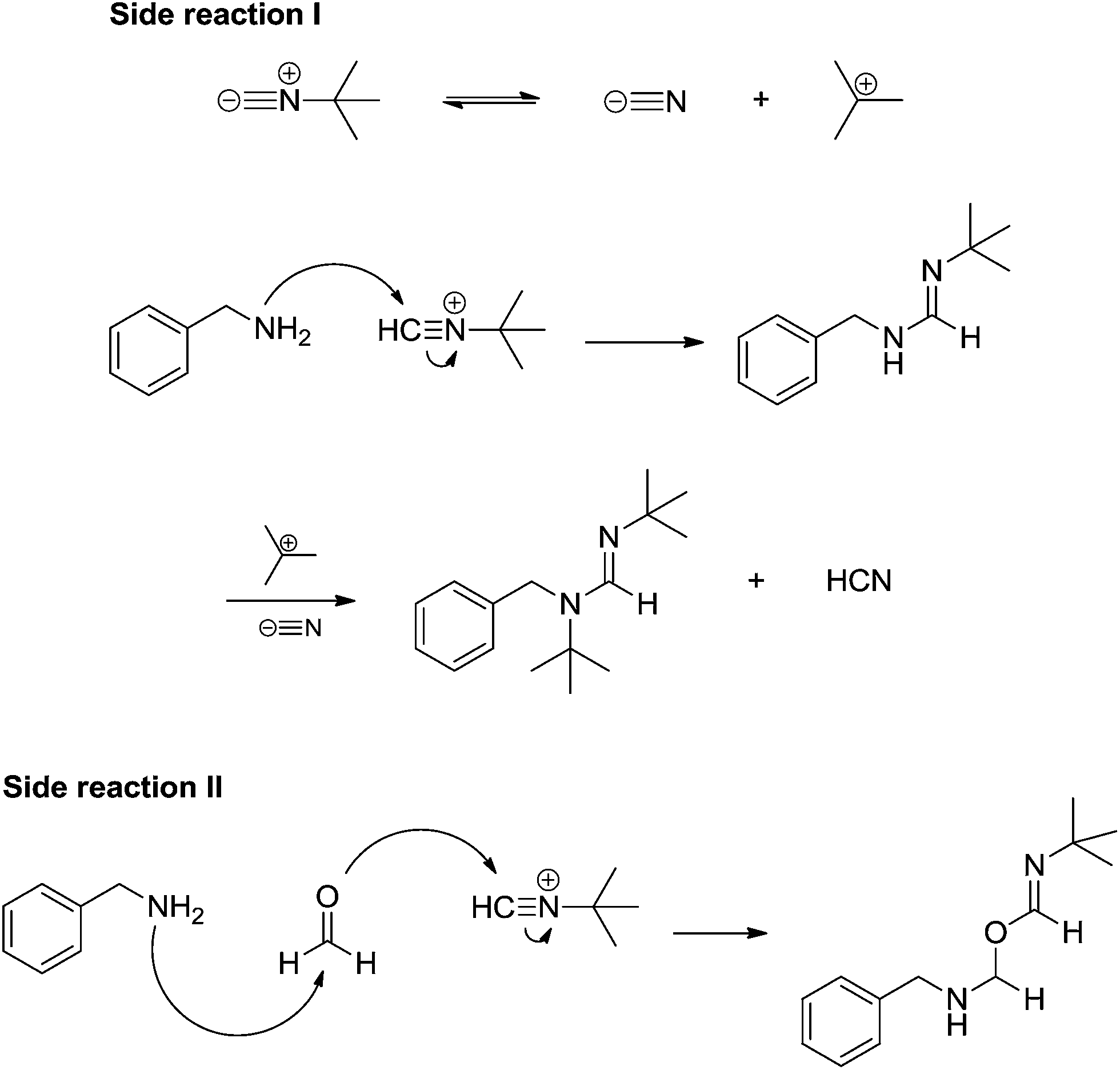 | ||
| Scheme 33 Side reactions noted during ESI-MS(/MS) online monitoring of the Ugi four-component reactions with charge-tagged reagents.49 | ||
6. Summary and outlook
MCRs are, without doubt, a paramount and useful tool incorporated in the arsenal available in the modern synthetic toolbox used by chemists around the world. Modern diversity-oriented synthesis is somehow inwardly associated with MCRs. Regarding green chemistry requirements, MCRs proved to have, at least in theory, all the features needed for sustainable syntheses. Unfortunately, the majority of the available reports only use these principles as catchwords. Use of ‘buzzwords’ such as eco-friendly, sustainable and green should be strongly avoided, unless the report brings real improvements for MCRs as green tools. It is imperative to start an era of more complicated and rewarding studies on MCRs and not to produce more seemingly random catalyst screenings. Only then will MCRs truly occupy the prominent position envisaged for this kind of reaction. MCRs do indeed have much more to offer on this subject.Real improvements certainly require deep knowledge on the mechanism of the MCR transformation and, in general, fine details of MCR mechanisms are only now starting to emerge. Theoretical approaches have, therefore, much to offer, and many real developments are due to some exceptional theoretical contributions. Only a few kinetic data are available in the MCR literature; and this is mostly because of the high level of complication associated with real-time monitoring (and quantification) of all possibilities of intermediates and reaction pathways involved in multicomponent transformations. NMR, IR and MS have proved to be the most effective techniques applied so far in elucidating MCR mechanisms, with special prominence of ESI-MS(/MS).
The Biginelli MCR is perhaps the most popular 3CR transformation. This important transformation, however, lacks actual improvements in its experimental condition. Reagent excesses, high temperatures, large amounts of catalysts and only a few effective asymmetric versions are severe limitations to further the syntheses and applications of DHPMs. It is still not clear how to select a reaction pathway for the Biginelli reaction and, to this end, much effort is required to gain a deep understanding of the key parameters for an effective transformation. Even so, the iminium-based mechanism is favoured over the other possibilities for most of the reaction conditions reported so far.
The Hantzsch 3CR is experiencing similar drawbacks associated with the Biginelli reaction. The possibility of five different reaction pathways is an additional complicating factor. Much is still needed to understand how to select a mechanistic pathway for the synthesis of DHPs using the Hantzsch MCR, especially for asymmetric versions applied in the synthesis of bioactive DHPs.
The Mannich MCR is one of the most ‘domesticated’ 3CRs and there is not much doubt about the reaction mechanism. In general, asymmetric versions and the role of chiral inductors are discussed. The specific step of chiral induction and the conditions for an effective stereocontrol are the most important issues under discussion for this transformation. Overall, the Mannich MCR mechanism goes through an iminium intermediate followed by the addition of the other reaction partner.
The Passerini and Ugi IMCRs still have many issues to solve in their respective mechanisms of transformations. Asymmetric versions are highly controversial, especially because real stereocontrol falls short. Chiral induction steps are still unclear. The role of solvents and reagents for these IMCRs are hotly debated and new evidence for alternative mechanisms has only recent been reported and discussed in the scientific literature. Despite the high atom economy associated with these two reactions, preparative methods for isocyanides are imperative for furthering the green issues associated with the Passerini and Ugi IMCRs.
Overall, little is known about the mechanisms and chiral induction of these five most popular and important MCRs. Considering that fine details have started to emerge only in recent years, the possibility for innovation and creative applications is almost unlimited. The wide avenue for new findings regarding mechanisms and applications of MCRs is just waiting to be travelled.
Acknowledgements
This work has been supported by CAPES, CNPq, FINEP-MCT, FINATEC, FAPEMIG, FAPDF, INCT-Catalysis and DPP-UnB.Notes and references
- R. C. Cioc, E. Ruijter and R. V. A. Orru, Green Chem., 2014, 16, 2958–2975 RSC.
- L. Banfi, A. Basso, L. Moni and R. Riva, Eur. J. Org. Chem., 2014, 2014, 2005–2015 CrossRef CAS.
- P. Slobbe, E. Ruijter and R. V. A. Orru, Med. Chem. Commun., 2012, 3, 1189–1218 RSC.
- G. C. Tron, A. Minassi and G. Appendino, Eur. J. Org. Chem., 2011, 5541–5550 CrossRef CAS.
- C. O. Kappe, Eur. J. Med. Chem., 2000, 35, 1043–1052 CrossRef CAS.
- A. Domling, W. Wang and K. Wang, Chem. Rev., 2012, 112, 3083–3135 CrossRef CAS PubMed.
- C. O. Kappe, Tetrahedron, 1993, 49, 6937–6963 CrossRef CAS.
- Suresh and J. S. Sandhu, ARKIVOC, 2012, 66–133 CAS.
- F. Sweet and J. D. Fissekis, J. Am. Chem. Soc., 1973, 95, 8741–8749 CrossRef CAS.
- K. Folkers and T. B. Johnson, J. Am. Chem. Soc., 1933, 55, 3784–3791 CrossRef CAS.
- I. Cepanec, M. Litvic, M. Filipan-Litvic and I. Grungold, Tetrahedron, 2007, 63, 11822–11827 CrossRef CAS PubMed.
- C. O. Kappe, J. Org. Chem., 1997, 62, 7201–7204 CrossRef CAS PubMed.
- M. Litvic, I. Vecenaj, Z. M. Ladisic, M. Lovric, V. Vinkovic and M. Filipan-Litvic, Tetrahedron, 2010, 66, 3463–3471 CrossRef CAS PubMed.
- R. De Souza, E. T. da Penha, H. M. S. Milagre, S. J. Garden, P. M. Esteves, M. N. Eberlin and O. A. C. Antunes, Chem.–Eur. J., 2009, 15, 9799–9804 CrossRef CAS PubMed.
- L. M. Ramos, A. Tobio, M. R. dos Santos, H. C. B. de Oliveira, A. F. Gomes, F. C. Gozzo, A. L. de Oliveira and B. A. D. Neto, J. Org. Chem., 2012, 77, 10184–10193 CrossRef CAS PubMed.
- H. G. O. Alvim, T. B. de Lima, H. C. B. de Oliveira, F. C. Gozzo, J. L. de Macedo, P. V. Abdelnur, W. A. Silva and B. A. D. Neto, ACS Catal., 2013, 3, 1420–1430 CrossRef CAS.
- L. M. Ramos, B. C. Guido, C. C. Nobrega, J. R. Corrêa, R. G. Silva, H. C. B. de Oliveira, A. F. Gomes, F. C. Gozzo and B. A. D. Neto, Chem.–Eur. J., 2013, 19, 4156–4168 CrossRef CAS PubMed.
- J. H. Clark, D. J. Macquarrie and J. Sherwood, Chem.–Eur. J., 2013, 19, 5174–5182 CrossRef CAS PubMed.
- Z. L. Shen, X. P. Xu and S. J. Ji, J. Org. Chem., 2010, 75, 1162–1167 CrossRef CAS PubMed.
- M. K. Raj, H. S. P. Rao, S. G. Manjunatha, R. Sridharan, S. Nambiar, J. Keshwan, J. Rappai, S. Bhagat, B. S. Shwetha, D. Hegde and U. Santhosh, Tetrahedron Lett., 2011, 52, 3605–3609 CrossRef PubMed.
- H. G. O. Alvim, T. B. Lima, A. L. de Oliveira, H. C. B. de Oliveira, F. M. Silva, F. C. Gozzo, R. Y. Souza, W. A. da Silva and B. A. D. Neto, J. Org. Chem., 2014, 79, 3383–3397 CrossRef CAS PubMed.
- J. Limberger, B. C. Leal, A. L. Monteiro and J. Dupont, Chem. Sci., 2014 10.1039/c1034sc02151g.
- A. R. Katritzky, D. L. Ostercamp and T. I. Yousaf, Tetrahedron, 1986, 42, 5729–5738 CrossRef CAS.
- L. Shen, S. Cao, J. J. Wu, J. Zhang, H. Li, N. J. Liu and X. H. Qian, Green Chem., 2009, 11, 1414–1420 RSC.
- V. A. Chebanov, V. E. Saraev, S. M. Desenko, V. N. Chernenko, I. V. Knyazeva, U. Groth, T. N. Glasnov and C. O. Kappe, J. Org. Chem., 2008, 73, 5110–5118 CrossRef CAS PubMed.
- K. A. Undale, Y. Park, K. Park, D. H. Dagade and D. M. Pore, Synlett, 2011, 791–796 CAS.
- C. Allais, F. Lieby-Muller, J. Rodriguez and T. Constantieux, Eur. J. Org. Chem., 2013, 4131–4145 CrossRef CAS.
- V. G. Santos, M. N. Godoi, T. Regiani, F. H. S. Gama, M. B. Coelho, R. O. M. A. de Souza, M. N. Eberlin and S. J. Garden, Chem.–Eur. J., 2014, 20, 12808–12816 CrossRef CAS PubMed.
- B. List, J. Am. Chem. Soc., 2000, 122, 9336–9337 CrossRef CAS.
- R. Gomez Arrayas and J. C. Carretero, Chem. Soc. Rev., 2009, 38, 1940–1948 RSC.
- E. R. Alexander and E. J. Underhill, J. Am. Chem. Soc., 1949, 71, 4014–4019 CrossRef CAS.
- S. V. Lieberman and E. C. Wagner, J. Org. Chem., 1949, 14, 1001–1012 CrossRef CAS.
- T. F. Cummings and J. R. Shelton, J. Org. Chem., 1960, 25, 419–423 CrossRef CAS.
- A. V. Logan, J. L. Huston and D. L. Dorward, J. Am. Chem. Soc., 1951, 73, 2952–2953 CrossRef CAS.
- E. Le Gall, S. Sengmany, C. Haurena, E. Leonel and T. Martens, J. Organomet. Chem., 2013, 736, 27–35 CrossRef CAS PubMed.
- M. B. Schmid, K. Zeitler and R. M. Gschwind, J. Org. Chem., 2011, 76, 3005–3015 CrossRef CAS PubMed.
- C. D. F. Milagre, H. M. S. Milagre, L. S. Santos, M. L. A. Lopes, P. J. S. Moran, M. N. Eberlin and J. A. R. Rodrigues, J. Mass Spectrom., 2007, 42, 1287–1293 CrossRef CAS PubMed.
- H. G. O. Alvim, G. A. Bataglion, L. M. Ramos, A. L. de Oliveira, H. C. B. de Oliveira, M. N. Eberlin, J. L. de Macedo, W. A. da Silva and B. A. D. Neto, Tetrahedron, 2014, 70, 3306–3313 CrossRef CAS PubMed.
- A. Domling, Chem. Rev., 2006, 106, 17–89 CrossRef PubMed.
- A. Domling and I. Ugi, Angew. Chem., Int. Ed., 2000, 39, 3169–3210 CrossRef.
- R. Frey, S. G. Galbraith, S. Guelfi, C. Lamberth and M. Zeller, Synlett, 2003, 1536–1538 CAS.
- R. H. Baker and D. Stanonis, J. Am. Chem. Soc., 1951, 73, 699–702 CrossRef CAS.
- G. Koopmanschap, E. Ruijter and R. V. A. Orru, Beilstein J. Org. Chem., 2014, 10, 544–598 CrossRef PubMed.
- T. Carofiglio, P. G. Cozzi, C. Floriani, A. Chiesi-Villa and C. Rizzoli, Organometallics, 1993, 12, 2726–2736 CrossRef CAS.
- U. Kusebauch, B. Beck, K. Messer, E. Herdtweck and A. Domling, Org. Lett., 2003, 5, 4021–4024 CrossRef CAS PubMed.
- S. Maeda, S. Komagawa, M. Uchiyama and K. Morokuma, Angew. Chem., Int. Ed., 2011, 50, 644–649 CrossRef CAS PubMed.
- I. Ugi, Angew. Chem., Int. Ed., 1959, 71, 386 Search PubMed.
- N. Cheron, R. Ramozzi, L. El Kaim, L. Grimaud and P. Fleurat-Lessard, J. Org. Chem., 2012, 77, 1361–1366 CrossRef CAS PubMed.
- G. A. Medeiros, W. A. da Silva, G. A. Bataglion, D. A. C. Ferreira, H. C. B. de Oliveira, M. N. Eberlin and B. A. D. Neto, Chem. Commun., 2014, 50, 338–340 RSC.
- R. Ramozzi, N. Chéron, L. El Kaïm, L. Grima`ud and P. Fleurat-Lessard, Chem.–Eur. J., 2014, 20, 9094–9099 CAS.
| This journal is © The Royal Society of Chemistry 2014 |