
Synthesis and encapsulation of an amphiphilic thermoresponsive star polymer with β-cyclodextrin and hyperbranched poly(oligo(ethylene glycol)methacrylate) as building blocks†
Abstract
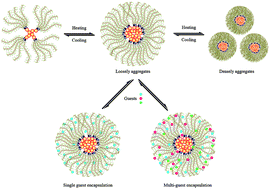
Novel macromolecular star polymers with triazole and cyclodextrin (CD) segments as branch points and poly(oligo(ethylene glycol)methacrylate) (POEGMAs) as dense hydrophilic branches were synthesized via a combination of azide–alkyne click chemistry and atom transfer radical polymerization (ATRP). Firstly, a tetrafunctional linking agent (PETP) was prepared by the reaction of pentaerythritol (PE) and propargyl bromide, and then a four arm β-CD terminated star polymer (PE-CD) was obtained through the click chemistry reaction. Finally, thermally-responsive star polymers (PE-CD–POEGMAs) with PE as the central core, triazole and CD segments as branch points, and POEGMAs as side branches were synthesized by the ATRP of MEO2MA and OEGMA using PE-CD terminated with bromines as macroinitiators. A study on the thermoresponsivity and morphology of PE-CD–POEGMAs indicated that polymeric nano-aggregates existed as multimolecular micelles and behaved with tunable thermosensitivity, which were driven by the strong hydrophobic–hydrophilic interactions in the inner core and outer shell. The encapsulation capacities towards multi-guest molecules were investigated and the results indicated that water soluble guests could be encapsulated by PE-CD–POEGMAs, and the guest encapsulation capacities were derived from the special star molecular structure properties of PE-CD–POEGMAs and the synergistic encapsulation phenomenon of different guest molecules. This unique amphiphilic star polymer illustrated the potential applications in supramolecular science, drug delivery and other nanotechnology applications.
 Please wait while we load your content...
Please wait while we load your content...

