Thermodynamic and electronic properties of tunable II–VI and IV–VI semiconductor based metal–organic frameworks from computational chemistry
Christopher H.
Hendon
a,
Davide
Tiana
a,
Thomas P.
Vaid
b and
Aron
Walsh
*a
aDepartment of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: a.walsh@bath.ac.uk; Tel: +44 (0)1225 384913
bDepartment of Chemistry, The University of Alabama, Tuscaloosa, Alabama 35487, USA
First published on 24th October 2012
Abstract
Optoelectric control of metal–organic frameworks would open up a new area of applications for hybrid materials. This article reports the calculated thermodynamic and electronic properties of a family of M3(C6X6) metal–organic frameworks (M = Mg, Ca, Zn, Cd, Hg, Ge, Sn, Pb; X = O, S, Se, Te). Herein, we present a systematic approach for studying families of hybrid compounds, and describe extended tunability of their electronic and enthalpic properties through compositional control. It was shown that the formation enthalpy is dictated by the stability of the ligand, and the band gap is tunable depending on both metal and chalcogenide selection. Five compounds were found to be candidate semiconductors as they combine thermodynamic stability with band gaps in the visible range of the electromagnetic spectrum.
1 Introduction
Hybrid metal–organic frameworks (MOFs) are designer materials that bridge the disparity between organic and inorganic structures.1 Whilst most MOF research has been invested in gas adsorption,2,3 hybrid materials have diverse properties, and importantly have potential applications as semiconductors in photovoltaics, photo-emitters and transistors.4,5,6 The multitude of organic ligand–metal combinations denotes the importance of further exploring these interesting compounds.Hybrid framework families are familiar in the literature (e.g. Yaghi's IRMOF-xx series),7 but systematic studies on the effects of chemical changes by metal or ligand coordinating atom substitutions are rare.8 Due to the impracticality of synthetically producing all possible compounds in a family, there is an opportunity to design novel hybrids with desirable properties through computational studies.9 There are many examples of such disparity; a literature search shows that most papers exclude studies of substituting metal composition both experimentally and theoretically.10 This is an artefact of working in a diverse field; it is hard to distinguish what makes a good conductor, and what makes a material insulating.11 The issue is one of volume: we approach this using modern quantum chemical techniques, to systematically screen for potentially interesting electronic and optical properties.
This article expands on the ubiquitous II–VI and IV–VI semiconductors (e.g. CdS, ZnSe, PbTe), and explores the feasibility of their corresponding hybrid organic–inorganic analogues. Taking an archetype 3D framework recently identified for Pb3(C6S6) by Vaid et al.,12,13 we explore both anion and cation modifications: M3(C6X6) (M = Mg, Ca, Zn, Cd, Hg, Ge, Sn, Pb; X = O, S, Se, Te) paying specific attention to formation enthalpy, and potential routes of decomposition.14 These properties were predicted by performing density functional theory (DFT)15,16 calculations on the 32 hybrid frameworks. From the 32 compounds studied, 15 were found to have a negative enthalpy of formation. Of these 15 compounds, 3 had plausible exothermic decomposition routes and hence deemed as thermodynamically unstable materials. Of the remaining 12, 5 candidate semiconducting MOFs had band gaps in the visible region of the electromagnetic spectrum.
2 Computational details
All electronic and structural calculations were performed within the DFT framework using periodic boundary conditions to approximate the perfect solid. The Vienna ab initio simulation package (VASP),17 a planewave code (with PAW scalar relativistic pseudopotentials),18 was employed for all geometry and electronic calculations. The effect of spin–orbit coupling for selenium and tellurium was not considered due to the high computational cost. However, this approximation is not expected to influence the qualitative results. A constant 3 × 3 × 8 k-point grid was specified for the hexagonal systems, based on it being suitable for the smallest of the hybrid cells, Mg3(C6O6). Starting with the experimentally determined unit cell of Pb3(C6S6), manual elemental substitutions were made for the metal and chalcogen, and the structures were geometrically relaxed using the semi-local Perdew–Burke–Ernzerhof exchange–correlation functional revised for solids (PBEsol).19 For these calculations, a 500 eV plane-wave cutoff basis set was found to be suitable for convergence of the systems, to within 0.01 eV per atom. All systems were relaxed with respect to internal ionic positions and cell parameters, and each compound maintained hexagonal symmetry.In order to obtain quantitative electronic information, in particular for estimations of the band gaps, higher order hybrid-DFT calculations were performed using the HSE06 functional,20 with 25% of the short-range semi-local exchange functional replaced by the exact non-local Hartree–Fock exchange. This functional has been shown to perform consistently with inorganic semiconductors as well as hybrid metal–organic materials.21–24
3 Geometrical parameters
The M3(C6X6) family of hybrids are hexagonal (P6/mmm), with alternating metallic sheets and hexa-substituted benzene analogues. Each metal is cubically coordinated to eight chalcogenide aromatic substituents, Fig. 1. The unit cell a vector increases proportionally to both chalcogen and metal radii and vector c varies approximately 1 Å, monotonically with chalcogen size and metal cationic radius, r(M). Lattice parameters and bond lengths are summarised in Table 1.![Structure of the P6/mmm Ca3(C6O6) hybrid system ([001] – left, truncated [100] – right) with the metal–chalcogenide, carbon–chalcogenide and metal–metal (= c) distances emphasised. M, C and X are depicted in blue, black and red, respectively.](/image/article/2013/TC/c2tc00108j/c2tc00108j-f1.gif) | ||
| Fig. 1 Structure of the P6/mmm Ca3(C6O6) hybrid system ([001] – left, truncated [100] – right) with the metal–chalcogenide, carbon–chalcogenide and metal–metal (= c) distances emphasised. M, C and X are depicted in blue, black and red, respectively. | ||
| System | a | c , r(M–M) | r(M–X) | r(C–X) | System | a | c , r(M–M) | r(M–X) | r(C–X) |
|---|---|---|---|---|---|---|---|---|---|
| MgO | 7.33 | 2.89 | 2.37 | 1.37 | CaO | 7.54 | 3.16 | 2.51 | 1.37 |
| MgS | 8.60 | 3.44 | 2.81 | 1.75 | CaS | 8.80 | 3.72 | 2.95 | 1.76 |
| MgSe | 9.00 | 3.63 | 2.95 | 1.91 | CaSe | 9.19 | 3.88 | 3.08 | 1.93 |
| MgTe | 9.60 | 3.97 | 3.18 | 2.13 | CaTe | 9.81 | 4.17 | 3.30 | 2.15 |
| ZnO | 7.44 | 2.83 | 2.39 | 1.35 | GeO | 7.56 | 3.09 | 2.49 | 1.37 |
| ZnS | 8.53 | 3.32 | 2.76 | 1.72 | GeS | 8.60 | 3.58 | 2.85 | 1.75 |
| ZnSe | 8.91 | 3.51 | 2.89 | 1.89 | GeSe | 8.99 | 3.73 | 2.98 | 1.91 |
| ZnTe | 9.48 | 3.77 | 3.09 | 2.11 | GeTe | 9.56 | 3.98 | 3.17 | 2.13 |
| CdO | 7.68 | 3.04 | 2.51 | 1.36 | SnO | 7.75 | 3.33 | 2.62 | 1.38 |
| CdS | 8.70 | 3.55 | 2.87 | 1.74 | SnS | 8.79 | 3.85 | 2.99 | 1.76 |
| CdSe | 9.07 | 3.73 | 3.00 | 1.90 | SnSe | 9.19 | 3.98 | 3.11 | 1.92 |
| CdTe | 9.58 | 4.09 | 3.21 | 2.12 | SnTe | 9.75 | 4.24 | 3.30 | 2.14 |
| HgO | 8.27 | 2.99 | 2.68 | 1.30 | PbO | 7.81 | 3.38 | 2.65 | 1.38 |
| HgS | 8.74 | 3.63 | 2.91 | 1.72 | PbS | 8.92 | 3.93 | 3.05 | 1.76 |
| HgSe | 9.08 | 3.84 | 3.04 | 1.89 | PbSe | 9.29 | 4.06 | 3.16 | 1.92 |
| HgTe | 9.67 | 4.09 | 3.24 | 2.09 | PbTe | 9.86 | 4.31 | 3.35 | 2.15 |
Zn, Cd, Hg, Mg, Sn and Pb were selected because they are well documented to have desirable semiconducting properties when bound to chalcogenides.25–27 Whilst less studied as inorganic semiconductors, Ge and Ca were also screened to extend this study to other relevant metals in those respective groups. Of the compounds examined in this article, only Pb3(C6S6) has been synthesised. The other metal structures were fixed with the same space group. Each system was then optimised allowing cell volume, lattice parameters and atomic positions to vary. A further comment must be made about the ligand selection itself. The precursor to the oxide ligand, benzenehexol, is commercially available. Benzenehexathiol may be prepared by the synthetic procedure reported by Harnisch and Angelici.28 The other two compounds, benzenehexaselenide and benzenehexatelluride, have not yet been synthesised. Mono-selenating/tellurating aromatics is a synthetic challenge in itself; to our knowledge, 1,2-phenyl ditelluride hybrids are the most extreme case thus far, forming an unusual complex with titanium.29 This makes the task of isolating the hexa-substituted ligand a major challenge. They are still included in this study as they are exciting from a chemical perspective. Since all systems studied result in local stability, the addition of the metal in the cubic coordination environment may in fact allow future formation of as yet unobserved ligands.
Progressive changes in chalcogenide (O → Te) are expected to dictate the semiconducting properties. Specifically, the anion radius, r(X2–), increases and the material band gap decreases through a decrease in the ionisation potential of the C6X6 group (following the binding energy of the anion p states). In addition, the metal–metal distance (r(M–M)), which is equivalent to the distance between aromatic moieties (r(π–π)), and c vector, increases with the size of aromatic substituent. There may be an equilibrium between chalcogen and metal selection such that π stacking and metal–metal interactions (if any) are ideally paired. These distances are illustrated in Fig. 1.
II–VI and IV–VI metal chalcogenides occupy a variety of geometries, with wurtzite, zinc blende and rocksalt being the most familiar. HgO (and α-HgS) has an unusual linear coordination, whilst the subsequent monotonic chalcogen analogues (and β-HgS) are all 4-coordinate tetrahedrally orientated. The change in coordination environment is attributed to the progressive ‘softening’ of the chalcogenide and the decreased binding energy of the valence p orbitals.30 This geometric change in the binary compounds is not accounted for in this study, as we impose the initial hexagonal structure upon each system.
Table 1 shows the unit cell dimensions increase monotonically with the chalcogen radii for a fixed metal: all systems demonstrate ∼2 Å increase transitioning from r(O2−) to r(Te2−). The cation radius, r(M2+), should also be considered, as it contributes to the cell parameters, e.g. the compounds formed from Ca and Cd (r(Ca2+) = 1.00 Å, r(Cd2+) = 0.95 Å)31 have similar cell parameters. The cation radii do not appear to correlate with the band gap, suggesting metal–metal interactions are negligible in these systems.
4 Thermodynamic properties
A geometric examination of these highly symmetric systems sheds little insight on their stability. Short of a synthetic approach, stability can be deduced by the enthalpy of formation (ΔHf). This is defined as the enthalpy difference between the compounds and their constituent elements in their standard states (described by eqn (1)). These energies are summarised in Table 2. Notably, ΔHf increases proportionally to chalcogen size, progressively becoming less stable both theoretically and synthetically.| 3M + 6C + 6X → M3(C6X6) | (1) |
| M3(C6X6) → 3C + 3CX + 3MX | (2) |
| M3(C6X6) → (C6X3)n + 3MX | (3) |
| System | ΔHf | ΔHcx | ΔHpoly | System | ΔHf | ΔHcx | ΔHpoly |
|---|---|---|---|---|---|---|---|
| MgO | −17.55 | −0.57 | 21.71 | CaO | −23.95 | 4.27 | 26.55 |
| MgS | −4.61 | 36.54 | 20.56 | CaS | −10.83 | 38.28 | 22.30 |
| MgSe | −1.83 | 33.10 | — | CaSe | −8.18 | 34.53 | — |
| MgTe | 2.83 | — | — | CaTe | −3.44 | — | — |
| ZnO | −9.09 | −1.03 | 21.25 | GeO | −8.80 | −8.01 | 14.27 |
| ZnS | 1.50 | 34.44 | 18.46 | GeS | 0.80 | 37.42 | 21.44 |
| ZnSe | 3.65 | 31.16 | — | GeSe | 2.57 | 33.80 | — |
| ZnTe | 6.87 | — | — | GeTe | 5.73 | — | — |
| CdO | −9.99 | 2.29 | 24.58 | SnO | −9.89 | 0.35 | 22.63 |
| CdS | 0.30 | 36.80 | 20.82 | SnS | −0.02 | 37.83 | 21.85 |
| CdSe | 2.31 | 33.15 | — | SnSe | 1.83 | 34.60 | — |
| CdTe | 5.65 | — | — | SnTe | 5.12 | — | — |
| HgO | −6.33 | 4.22 | 26.50 | PbO | −11.35 | 2.90 | 25.18 |
| HgS | 2.83 | 36.92 | 20.94 | PbS | −0.62 | 38.00 | 22.02 |
| HgSe | 4.21 | 33.40 | — | PbSe | 0.83 | 34.62 | — |
| HgTe | 6.12 | — | — | PbTe | 4.26 | — | — |
One of the major issues with hybrid frameworks is that generally they decompose upon heating. Some creativity can be applied when examining decomposition products, as characterisation of such species is in its infancy. The two examples, the carbon monochalcogenide (ΔHcx, eqn (2)) and the ill-defined polymer (ΔHpoly, eqn (3), Fig. 2) products are outlined in this paper. They exemplify trends and potential, and we encourage the reader to envisage other decomposition pathways. The products of eqn (2) and (3), appear to be in relatively high energy states, but do show an inverse relationship with the stability of ΔHf. A third, inferred, decomposition pathway is the direct reverse reaction of formation (ΔHf, eqn (1)). Whilst complete dissociation to the standard states is often far-fetched (for example, systems with values of ΔHf close to 0, e.g. the GeS, PbSe, CdS hybrids), it is possible to imagine other decomposition products that may occur. Studies into the structures of experimentally determined decomposed products is an area that will become progressively more important once a family of MOFs has been described for potential applications.
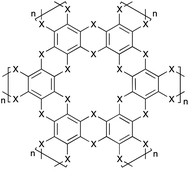
Mg3(C6S6), Ca3(C6S6), Sn3(C6S6), and Pb3(C6S6) all have negative (exothermic) enthalpies of formation. Pb3(C6S6) is unsurprising as this result coincides with experiment. Sn3(C6S6) is an attractive option as there is currently interest in binary chalcogenides for application in earth-abundant solar cells;32 perhaps this hybrid material may be of interest. From Bader partial charge analysis33 of the group 2 hybrids, the products form salt-like bonding between the metals and ligands. Both Mg and Ca form a thermodynamically stable network with the hexaselenide compounds. We propose this to be due to the small ionic radii of Mg/Ca, allowing for both selenide orbital overlap between sheets (as shown in Fig. 3), and because Mg/Ca form highly ionic bonds with the ligands. This poses an interesting problem: selenium is attractive as a semiconductor component, but C6(SeH)6 has not been synthesised itself, let alone the hybrid Mg3(C6Se6). Perhaps the products, Mg3(C6Se6) and Ca3(C6Se6), will not be ideal semiconductors, but they exemplify a family of seleno-hybrids thus-far unexplored. It may be possible to construct these systems by a different synthetic procedure, but this exemplifies the problematic nature of selenide and telluride chemistry.
 | ||
| Fig. 3 Charge density maps of Mg3(C6O6), Pb3(C6S6) and Zn3(C6Se6). Valence and conduction bands were calculated with HSE06, using 3 × 3 × 8 k-points. Mg3(C6O6) shows π stacking interactions in both the valence and conduction bands accounting for the low band gap in this ionic solid. Pb3(C6S6) shows a π → pσ transition, whilst Zn3(C6Se6) shows chalcogenide centered excitation. | ||
Table 2 summarises the enthalpy of formation, and decomposition via two proposed pathways. A negative formation enthalpy may suggest that there is some thermodynamic motivation for the compounds to remain in their hexagonal hybrid state. This is unlikely for the d-block metals, which rarely form cubic coordination environments, but we continue studying the systems that have negative formation enthalpies, since they are in a stable local minimum. Positive formation energies suggest either a kinetic or meta-stable product, as all systems are locally stable (i.e. no negative phonon modes). There was a predictable (and observed) increase in formation energy as chalcogen is substituted from O → Te. As discussed previously, oxides should be considered as an extremity as the c vector is small, and hence the structures often act metallic. Oxygen forms highly ionic bonds with lighter metals, making systems like Ca3(C6O6) act more like a dielectric; the high formation enthalpies confirm such sentiments. This phenomenon is discussed in more depth with reference to band gaps in Section 5.
Similarly, the other extremity, tellurides, exemplify meta-stability in the these calculated systems. They are expected to also act metallic as the 5p valence orbitals of tellurium are extremely diffuse. Bearing in mind that C6(TeH)6 cannot be isolated experimentally, it is unsurprising that the formation enthalpy for the tellurides is frequently positive. There is an exception; Ca3(C6Te6). Indeed, its formation enthalpy is negative, suggesting that there may be a happy medium between the diffuse tellurium orbitals and the electropositive Ca ion. The other tellurium hybrids demonstrate plausible structures with positive formation enthalpies, suggesting the compounds may be likely to dissociate. It is unsurprising that CS and CSe decomposition products give large (endothermic) enthalpies of formation; both are highly unstable themselves. Instability of carbon monotelluride (a hypothetical decomposition product) and the (C6Te3)n and (C6Se3)n ill-defined polymer prevented completion of Table 2 for all routes of decomposition.
5 Electronic properties
The electronic properties of hybrid frameworks are poorly understood, however, they are well documented for the parent II–VI and IV–VI compounds. For example, SnS, SnSe and SnTe have band gaps of 1.42, 0.9 and 0.36 eV, respectively.34 They show a trend to decreasing band gap with increasing r(X2−). There are exceptions to this general rule: oxides and sulfide band gaps can behave in opposite ways to expected, e.g. the band gaps of ZnS and CdS are significantly larger than ZnO and CdO; the origin of these anomalies remains poorly understood. A prototypical series is the Cd chalcogenides: CdO, CdS, CdSe and CdTe have band gaps of 1.09, 2.48, 1.73 and 1.48 eV, respectively.34 The family of MOFs described here should show similar trends. Quantitative band gap calculations were performed on the structures discussed in Table 1, with the results summarised in Fig. 5.The group 2 (IIA) metals show periodic trends, with band gaps decreasing rapidly with increasing metal size, converging to the telluride variants. They have unusually small band gaps for classical salts. The origin of these small band gaps is best visualised in Fig. 3, when c is small, π orbital overlap becomes significant. The depicted electron density in the upper valence and lower conduction bands, show the segregation of carbon p-orbitals as r(X2−) increases. Using a single k-point, a similar charge density map has been previously described by Walsh.35 It is due to this stacking that the band gaps for the salt-like systems are smaller than expected. In fact, π stacking can be an important characteristic in metal–organic frameworks, and one of the tunable properties.36 A classic example of these interactions is the benzene–benzene π stacking (distance between aromatics is 3.7 Å (synonymous to ‘r(M–M)’),37 which is near the upper limit of r(M–M) in these hybrids. This suggests that π stacking is significant in these MOFs. It is notable that the ligand-based π → π* excitation represents the band gap in the salt-like MOFs. Mg therefore has a lower band gap than Ca in all instances, because the Mg3(C6X6) c vector is smaller than the corresponding Ca3(C6X6) c vector. For the other systems, as r(X2−) increases, the highest occupied molecular orbital transitions from being carbon/sulfur π centred to chalcogenide centred, reducing the significance of the π stacking interaction. The valence and conduction band densities of Pb3(C6S6) are described in Fig. 3, and the HSE06 band structure is shown in Fig. 4.
 | ||
| Fig. 4 The HSE06 band structure calculated for Pb3(C6S6). The Fermi energy has been adjusted to 0 eV to clearly depict the band gap (1.70 eV) described in Fig. 5. | ||
The group 12 (IIB) cations (Zn, Cd and Hg) show significantly smaller band gap values for their oxide and sulfide derivatives than the other non-transition metals examined. The Cd family demonstrates the same chemical trends as inorganic CdX; the oxide has a lower band gap than the sulfide. This trend does not extend to the Zn family as it does in the inorganic ZnX series. Both Zn and Hg show predictable decreases in band gap with increasing r(X2−), followed by an increase in band gap for their subsequent telluride analogues. This sudden increase is unusual, and may be an artefact of the benzene hexatelluride. Irrespective of metal, these systems have relatively small band gaps in all cases. Indeed, inorganic HgTe exhibits valence and conduction band crossing; in essence forming a metallic system. Hg3(C6Te6) results in a separation of this intersection, to ∼0.35 eV, Fig. 5. Of the systems examined in this study, group 12 metals appear to be the least likely to ascertain this geometry in reality. Zn and Cd infrequently adopt cubic coordination, so this is not surprising. Attempted synthesis of these compounds may yield variable conformations, perhaps resulting in favourable materials. A similar π stacking argument to the group 2 metals can be made for these systems, as they have some of the smallest c vectors in this study. Analysis of the valence and conduction band wavefunctions reveals the same trends as discussed for the group 2 metals, with the added tendency toward metallic behaviour.
 | ||
| Fig. 5 Band gap (eV) calculated using the HSE06 functional for the 32 hybrid systems (M3(C6X6)), subdivided into 4 groups (ligand depicted, top) and further subdivided into group 2, 12 and 14 metals, denoted by colour. | ||
Hybrids formed from the group 14 metals (in their lower divalent oxidation states) show the familiar trend towards the lowest band gap for heavier chalcogens. Hybrid-Ge compounds are interesting as the s and p electrons of atomic Ge are close in energy. Inorganic GeO has never been crystallised; the stable oxide of Ge is GeO2. Hence, the decomposition pathway of Ge3(C6O6) was calculated making the approximation that E(GeO) is defined as GeO2 − 1/2O2. Ge3(C6X6) forces Ge into the 2+ oxidation state, s2p0, resulting in Ge3(C6O6) showing a near metallic band gap. Pb3(C6S6) has one of the highest band gaps in this study, and both Ge3(C6X6) and Pb3(C6X6) show the previously discussed trend inversion when examining oxide and sulfide hybrids. A note should be made that π stacking is not significant in these systems because the larger metal radii force the sheets too far apart, even for the oxide case.
6 Conclusions
The initial family of 32 hybrids were screened for enthalpic stability. After local geometry optimisation, all systems converged to the same conformation. Systems with positive enthalpy of formation were eliminated as candidates for synthesis. Of the remaining 15 hybrids, a further 3 were eliminated due a predicted exothermic decomposition product. Of the remaining 12 only 5 hybrids (Ca3(C6O6), Ca3(C6S6), Sn3(C6O6), Pb3(C6O6) and Pb3(C6S6)) were of interest as they fulfilled the negative formation enthalpy requirement, have band gaps predicted to be in the visible light spectrum, and result in promising compounds for applications as photo-active materials. Pb3(C6Se6) also showed desirable electronic properties, but had a slightly positive formation enthalpy. The exemplifies the importance of synthetic plausibility; the benzene hexaselenide/hexatelluride poses a synthetic challenge. Ca3(C6S6) and Pb3(C6S6) have similar electronic properties which is unusual, and the application of Ca in semiconductors is a rarity due to its high ionic character.38 Sn3(C6O6) and Sn3(C6S6) both appear to be interesting low band gap semiconductors, and are the likely synthetic extension of this work.After careful examination of both the electronic and geometric data it is apparent that for control of band gap and thermodynamic stability, three parameters must be considered:
• Distance between both metal and aromatic layers; here represented by the lattice c vector. For this family of hybrid materials, it is not metal–metal interactions that influence the band gap, but rather the distance between aromatic moieties, allowing for pz-orbital overlap.
• Chalcogen selection, X; the selection of chalcogen influences both thermodynamic stability and band gap. Heavier chalcogens reduce the band gap as their ionisation potentials decrease.
• Metal selection, M; not only the ‘size’ of the cation, but also the magnitude of localised charge (electronegativity). Ca and Mg form salts (usually indicating large band gaps), whilst the group 14 metals are more covalent, with smaller band gaps. It was also observed that r(M2+) dictates the cell parameters, influencing the magnitude of π stacking in the X = O, S systems.
Systematic studies of this nature are indicative as to the relationship between the nanoarchitecture of novel MOF materials and their properties. The obvious drawback of screening families of hybrids computationally is that the 3D structure is imposed on the compounds. Thus, there are many facets of extended work for this class of hybrid: finding global structural minima for the systems that were energetically unfavourable, but synthetically plausible (e.g. Zn3(C6S6), Cd3(C6S6); studying doping of systems through both cation and ligand exchange; and also predicting carrier transport phenomena.
Acknowledgements
A.W. is supported a Royal Society University Research Fellowship, while C.H.H. and D.T. are funded under an ERC Starting Grant. The work benefited from the University of Bath's High Performance Computing Facility, and access to the HECToR supercomputer through membership of the UKs HPC Materials Chemistry Consortium, which is funded by EPSRC (Grant no. EP/F067496).References
- F. X. Llabres i Xamena, A. Corma and H. Garcia, J. Phys. Chem. C, 2007, 111, 80–85 CAS.
- K. Sillar, A. Hofmann and J. Sauer, J. Am. Chem. Soc., 2009, 131, 4143–4150 CrossRef CAS.
- H. Kim, C. Rey, M. J. Glimcher, J. B. M. Res, N. L. Rosi, J. Eckert, M. Eddaoudi, T. Vodak, J. Kim, M. O. Keeffe and O. M. Yaghi, Science, 2003, 300, 1127–1129 CrossRef.
- O. M. Yaghi, C. E. Davis, G. Li and H. Li, J. Am. Chem. Soc., 1997, 119, 2861–2868 CrossRef CAS.
- O. M. Yaghi, R. Jernigan, H. Li, C. E. Davis and T. L. Groy, Dalton Trans., 1997, 2383–2384 RSC.
- M. Alvaro, E. Carbonell, B. Ferrer, F. X. Llabrés i Xamena and H. Garcia, Chem.–Eur. J., 2007, 13, 5106–5112 CrossRef CAS.
- D. Tranchemontagne, J. Hunt and O. Yaghi, Tetrahedron, 2008, 64, 8553–8557 CrossRef CAS.
- H. Lin and P. A. Maggard, Cryst. Growth Des., 2010, 10, 1323–1331 CAS.
- J. Li, W. Bi, W. Ki, X. Huang and S. Reddy, J. Am. Chem. Soc., 2007, 129, 14140–14141 CrossRef CAS.
- C. H. Hendon, D. Tiana and A. Walsh, Phys. Chem. Chem. Phys., 2012, 14, 13120 RSC.
- A. K. Cheetham and C. N. R. Rao, Science, 2007, 318, 58–59 CrossRef CAS.
- D. L. Turner, T. P. Vaid, P. W. Stephens, K. H. Stone, A. G. DiPasquale and A. L. Rheingold, J. Am. Chem. Soc., 2008, 130, 14–15 CrossRef CAS.
- A. Stott, T. Vaid, E. Bylaska and D. Dixon, J. Phys. Chem. C, 2012, 3, 8370–8378 Search PubMed.
- T. Dunning Jr, J. Phys. Chem. A, 2000, 104, 9062–9080 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, 1133–1138 CrossRef.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 155, 864–871 CrossRef.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- P. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- J. Perdew, A. Ruzsinszky, G. Csonka and O. Vydrov, Phys. Rev. Lett., 2008, 2–6 Search PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207 CrossRef CAS.
- A. Walsh, J. Phys. Chem. Lett., 2010, 1, 1284–1287 CrossRef CAS.
- K. Hummer, J. Harl and G. Kresse, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 5205 CrossRef.
- Y.-S. Kim, K. Hummer and G. Kresse, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 1–9 Search PubMed.
- J. Wróbel, K. Kurzydowski, K. Hummer, G. Kresse and J. Piechota, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 1–8 CrossRef.
- W.-T. Yao and S.-H. Yu, Adv. Funct. Mater., 2008, 18, 3357–3366 CrossRef CAS.
- S. Wang, D. B. Mitzi, C. A. Feild and A. Guloys, J. Am. Chem. Soc., 1995, 5297–5302 CrossRef CAS.
- A. Walsh, D. J. Payne, R. G. Egdell and G. W. Watson, Chem. Soc. Rev., 2011, 40, 4455–4463 RSC.
- J. A. Harnisch and R. J. Angelici, Inorg. Chim. Acta, 2000, 300–302, 273–279 CrossRef CAS.
- H. Kopf and T. Klapotke, J. Chem. Soc., 1986, 1192–1193 Search PubMed.
- P. Glans, T. Learmonth, C. McGuinness, K. Smith, J. Guo, A. Walsh, G. Watson and R. Egdell, Chem. Phys. Lett., 2004, 399, 98–101 CrossRef CAS.
- R. Shannon, Acta Crystallogr., Sect. A: Found. Crystallogr., 1976, 751–767 CrossRef CAS.
- S. Bashkirov, V. Gremenok, V. Ivanov, V. Lazenka and K. Bente, Thin Solid Films, 2012, 520, 5807–5810 CrossRef CAS.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS.
- O. Madelung, Semiconductors: Data Handbook, Springer, 3rd edn, 2004, p. 691 Search PubMed.
- A. Walsh, Proc. R. Soc. A, 2011, 467, 1970–1985 CrossRef CAS.
- T. Narayan and T. Miyakai, J. Am. Chem. Soc., 2012, 138, 4–7 Search PubMed.
- M. O. Sinnokrot, E. F. Valeev and C. D. Sherrill, J. Am. Chem. Soc., 2002, 124, 10887–10893 CrossRef CAS.
- O. Compton, E. Carroll, J. Kim, D. Larsen and F. Osterloh, J. Phys. Chem. C, 2007, 111, 14589–14592 CAS.
| This journal is © The Royal Society of Chemistry 2013 |