
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Characterization and quantification of microplastics and organic pollutants in mussels by microwave-assisted sample preparation and analytical pyrolysis†
Greta
Biale
a,
Jacopo
La Nasa‡
ab,
Lorenzo
Fiorentini
a,
Alessio
Ceccarini
ab,
Diego
Carnaroglio
c,
Marco
Mattonai
 *a and
Francesca
Modugno
ab
*a and
Francesca
Modugno
ab
aDepartment of Chemistry and Industrial Chemistry, Via Giuseppe Moruzzi 13, 56124, Pisa, Italy. E-mail: marco.mattonai@dcci.unipi.it
bCISUP Centre for Instrument Sharing, University of Pisa, Pisa, Italy
cMilestone Srl, Sorisole, Bergamo, Italy
First published on 21st November 2023
Abstract
Sampling, separation, detection, and characterization of micro- and nanoplastic pollutants is a critical goal to assess their amount, fate, and the related hazards for ecosystems. There is still a major lack of understanding of the most relevant mechanisms of interaction and exchange of this class of pollutants with the environment and with organisms. In the last few years a number of studies highlighted the importance of the evaluation of the chemical species associated with the presence of microplastics in the environment, such as plasticizers, low-molecular weight degradation products, and different kinds of organic contaminants. In this work we combined microwave-assisted extraction and digestion, together with analytical pyrolysis coupled with gas chromatography and mass spectrometry (Py-GC-MS), to quantify microplastics together with different classes of associated pollutants. This method was developed using mussels as a matrix and it can be potentially applied to characterize and quantify, together with microplastics, polymer additives (phthalate plasticizers, UV stabilizers, etc.), polycyclic aromatic hydrocarbons (PAHs), polychlorinated biphenyls (PCBs), and emerging contaminants like anti-inflammatory drugs. This method allowed the quantification of more than 40 different contaminants in a single chromatographic run, with recoveries higher that 87% in most cases and limits of detection/quantitation in the nanogram range. The method was also tested on a standard microplastic calibration mixture containing 11 different polymers, and recoveries higher than 84% were obtained in most cases.
Environmental significanceIn recent years, there has been a noticeable increase in studies evaluating interactions between chemical species and microplastics in the environment. While several established analytical approaches exist for characterizing and quantifying microplastics in environmental samples, the accurate quantification of both microplastics and associated chemical species, such as plasticizers, adsorbed persistent organic pollutants, and aliphatic aromatic hydrocarbons, remains a challenge. This study introduces a new analytical approach, employing microwave-assisted sample pretreatments and analytical pyrolysis, to characterize and quantify microplastics and various classes of pollutants. Implementing this method could offer new insights into pollution studies, providing a comprehensive understanding of the behavior of contaminants in the environment. |
1. Introduction
Microplastic (MP) pollution is, nowadays, one of the most discussed environmental problems, being acknowledged at the same time as a social, a health,1 and an ecological issue.2,3 Researchers have confirmed that MPs need to be considered reactive systems,4 being plastic degraded by various biological and atmospheric agents,5 releasing degradation products and additives in the environment,6,7 and interacting with different kinds of pollutants.8 In the last years different analytical approaches have been optimized and applied for MPs research.9 In fact, prior the actual analysis, the samples need to be pretreated. Pretreatment is commonly considered the most crucial, and time-consuming step in MP analysis protocols, and it usually consists of several steps, that vary on the basis of the complexity of the matrix.10When biota samples such as shellfish are taken into account for MPs analysis, the sample pretreatments are usually aimed at eliminating the animal tissue from the sample before MP determination,11–13 and the most used ones for this purpose are digestion promoted by alkali solutions,14 and oxidation.15 Less common approaches use acid solutions,16 and enzymes treatment17,18 to remove all the proteinaceous fraction. Sometimes digestion is followed by a density separation step.19
Pyrolysis-based techniques, and in particular pyrolysis-gas chromatography-mass spectrometry (Py-GC-MS), proved to be very powerful tools for the detection and quantification of MPs in different kind of matrices,20 from marine biota, to sediments and even spider webs.14,21
In this study we propose an innovative approach based on microwave-assisted pretreatments and Py-GC-MS, to quantify in mussels not only MPs, but also different kind of organic contaminants frequently found in the marine environment like polychlorinated biphenyls (PCBs),22–24 polycyclic aromatic hydrocarbons (PAHs),24–26 phthalates (phthalic acid esters, PAEs),27–29 and a series of compounds commonly defined as contaminants of emerging concerns (CECs). CECs are compounds, such as some pharmaceuticals, pesticides, and UV filters, found in waterbodies that may have a negative impact on the marine ecosystem and on the biota, and are currently under no regulations.30 The efficiency of the proposed method was tested using commercial flour deriving from lyophilized mussels (Perna Canaliculus) as matrix to avoid, in this pilot study, the lyophilization step that may be necessary when approaching analysis of mussels.
Mussels have been frequently studied because they are filter-feeding organisms, capable of processing large quantities of water at a rate of one and a half liters per hour (quantity related to a single mussel).31–33 This along with their widespread presence in the marine environments in a wide range of latitudes, makes them suitable for monitoring studies in which mussels are employed as water pollution sentinels.33 Moreover, in a recent study, different species of mussels were tested as active agents to reduce waterborne microplastics, and it has been observed that, under controlled laboratories conditions, 1 kg of mussels is capable of filter out 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 146 MPs per hour.34
146 MPs per hour.34
In this work we take a step forward in the use of microwave-assisted pretreatment,27,35 combining its extraction power with the potential of Py-GC-MS in order to reduce the time required for the sample preparation and enhance the sensitivity of the method.
The samples underwent two separate and parallel steps: a microwave-assisted hydrolysis for the digestion of the matrix before MPs quantification, and a microwave-assisted solvent extraction for the quantification of the organic contaminants.
The samples spiked with solutions of standard reference contaminants underwent microwave-assisted extraction and the resulting extracts were subjected to thermal desorption at 350 °C. Samples spiked with MPs underwent microwave-assisted digestion and subsequently analyzed by Py-GC-MS at 600 °C. EGA-MS analysis was used to optimize the temperature to be used in the thermal desorption step.
2. Materials and methods
2.1 Chemicals
The method validation for the PCBs was performed using the PCB standard solution 7 (Analytical grade, Merck, US) containing 2,4,4′-trichlorobiphenyl (PCB28), 2,2′,5,5′-tetrachlorobiphenyl (PCB52), 2,2′,4,5,5′-pentachlorobiphenyl (PCB101), 2,2′,3,4,4′,5′-hexachlorobiphenyl (PCB138), 2,2′,4,4′,5,5′-hexachlorobiphenyl (PCB153), 2,2′,3,4,4′,5,5′-heptachlorobiphenyl (PCB180), and decachlorobiphenyl (PCB209). The optimization for the PAHs was performed using the PAH Calibration Mix (certificate reference material, Merck) containing naphthalene, acenaphthylene, acenaphthene, fluorene, phenanthrene, anthracene, fluoranthene, pyrene, benz(a)anthracene, chrysene, benzo(b)fluoranthene, benzo(k)fluoranthene, benzo(a)pyrene, dibenz[a,h]anthracene, benzo[g,h,i]perylene, and indeno[1,2,3-C,D]pyrene. The CECs used in the study were (analytical grade, Merck) methiocarb, diclofenac, 3-tert-butyl-4-hydroxyanisole, and 2-ethylhexyl 4-methoxycinnamate, while the PAEs were (analytical grade, Merck) dimethyl phthalate, dibutyl phthalate, bis(8-methylnonyl) phthalate, bis(7-methyloctyl) phthalate, benzyl butyl phthalate, and bis(2-ethylhexyl) phthalate. Hexamethyldisilazane (HMDS, analytical grade, Merck) was used as derivatizing agent in the pyrolysis analysis.To optimize the microwave-assisted digestion for the quantification of MPs, and to build the calibration curves for the determination of the polymers, a microplastic calibration standard (MPs-SiO2, Frontier Laboratories, Japan) was used, containing the following polymers: PE, PP, PS, ABS, SBR, PMMA, PC, PVC, PET, N6, and N66 diluted in silicon dioxide (SiO2). Monopalmitin, dipalmitin, and tripalmitin used as references for the EGA-MS analysis were purchased from Merck (purity > 99%).
2.2 Microwave-assisted approaches
The recovery of the MPs by filtration after the digestion was performed using Whatman® QM-H quartz fiber membrane filters (penetration 0.3 μm at 15 cm s−1).
2.3 Evolved gas analysis-mass spectrometry
EGA-MS experiments were performed using an EGA/PY-3030D micro-furnace pyrolyzer (Frontier Laboratories) coupled to a 6890 gas chromatograph and a 5973 mass spectrometric detector (Agilent Technologies). The temperature ramp of the pyrolysis furnace began at 50 °C and increased steadily at a rate of 10 °C per minute until it reached 700 °C. Simultaneously, the temperature of the interface connecting the pyrolysis furnace to the GC-MS system was automatically maintained 100 °C above the furnace temperature, with a maximum limit of 300 °C. For the GC injector, it operated in split mode at a fixed temperature of 280 °C, using a 20:1 split ratio.
The evolved pyrolysis products were sent to the mass spectrometer detector through an UADTM-2.5N deactivated stainless-steel capillary tube (3 m × 0.15 mm, Frontier Laboratories) held at 300 °C. The gas carrier was helium (1 mL min−1, 99.9995% purity). The temperature of the transfer line to the mass spectrometer was set at 280 °C. The mass spectrometer was operated in EI positive mode (70 eV, m/z range 35–700). The temperature of the ion source and quadrupole analyzer was 230 °C and 150 °C, respectively.
2.4 Analytical pyrolysis-gas chromatography-mass spectrometry
The analyses were performed using a multi-shot pyrolyzer EGA/PY-3030D coupled with an AS-1020E autosampler (Frontier Laboratories). The system was interfaced with an Agilent Technologies 8890 gas chromatograph, which was combined with a 5977B mass selective single quadrupole mass spectrometer detector (Agilent Technologies, USA). The chromatographic separation for both the analysis was performed using a HP-5MS capillary column (30 m × 0.25 mm, film thickness 0.25 μm, Agilent Technology).:1 ratio. The chromatographic conditions for the analysis were 35 °C held for 6 min, 20 °C min−1 to 310 °C held for 40 min. The helium (99.9995% purity) gas flow was set in constant flow mode at 1.0 mL min−1. The mass spectrometer was operated in EI positive mode both in scan and selected ion monitoring (SIM) acquisition.
Quantitation was performed by monitoring sets of m/z signals characteristic of each compound, and integrating the chromatographic peaks in the ion profiles. The list of the m/z ions used for the SIM acquisition is reported in Table S.1 the ESI.†
:1 ratio.
The GC temperature program was 40 °C for 5 min, followed by a 10 °C min−1 ramp to 310 °C kept for 20 min. The helium gas (99.9995% purity) flow was set to 1.2 mL min−1. The mass spectrometer operated in EI positive mode (70 eV) in the mass range m/z 35–700. The mass spectrometric identification of the pyrolysis products was performed using the NIST20 library and the F-Search library (Frontier Laboratories).
Quantitation of the polymers was performed by selecting a characteristic pyrolysis product for each polymer as molecular marker for that polymer and a characteristic m/z signal in its mass spectrum, and integrating the corresponding chromatographic peak in the ion profile. The markers used for the quantification of the polymers and the corresponding m/z signals are reported in Table S.2 in the ESI.† The choice of polymeric marker compounds and corresponding m/z signals was based on available literature references.36
2.5 QA/QC
:1 ratio. The chromatographic conditions were 60 °C for 1 min, followed by a 20 °C min−1 ramp to 310 °C kept for 10 min. The helium gas (99.9995% purity) flow was set to 1.2 mL min−1. The first run was performed using an empty pyrolysis cup added with 5 μL of HMDS to remove the most polar compounds, while the second analysis was performed without the derivatizing agent to remove possible excesses of derivatizing agent in the Py-GC-MS system.
3. Results and discussion
3.1 Optimization of the desorption temperature
Evolved gas analysis was used to define the optimal temperatures to perform the thermal desorption of the pollutants and the pyrolysis of the polymers. Fig. 1 reports as examples the thermograms obtained for a selection of PAHs, PCBs, phthalates, and CECs, together with the thermal profile of the microplastic calibration standard containing 11 polymers, and acylglycerols.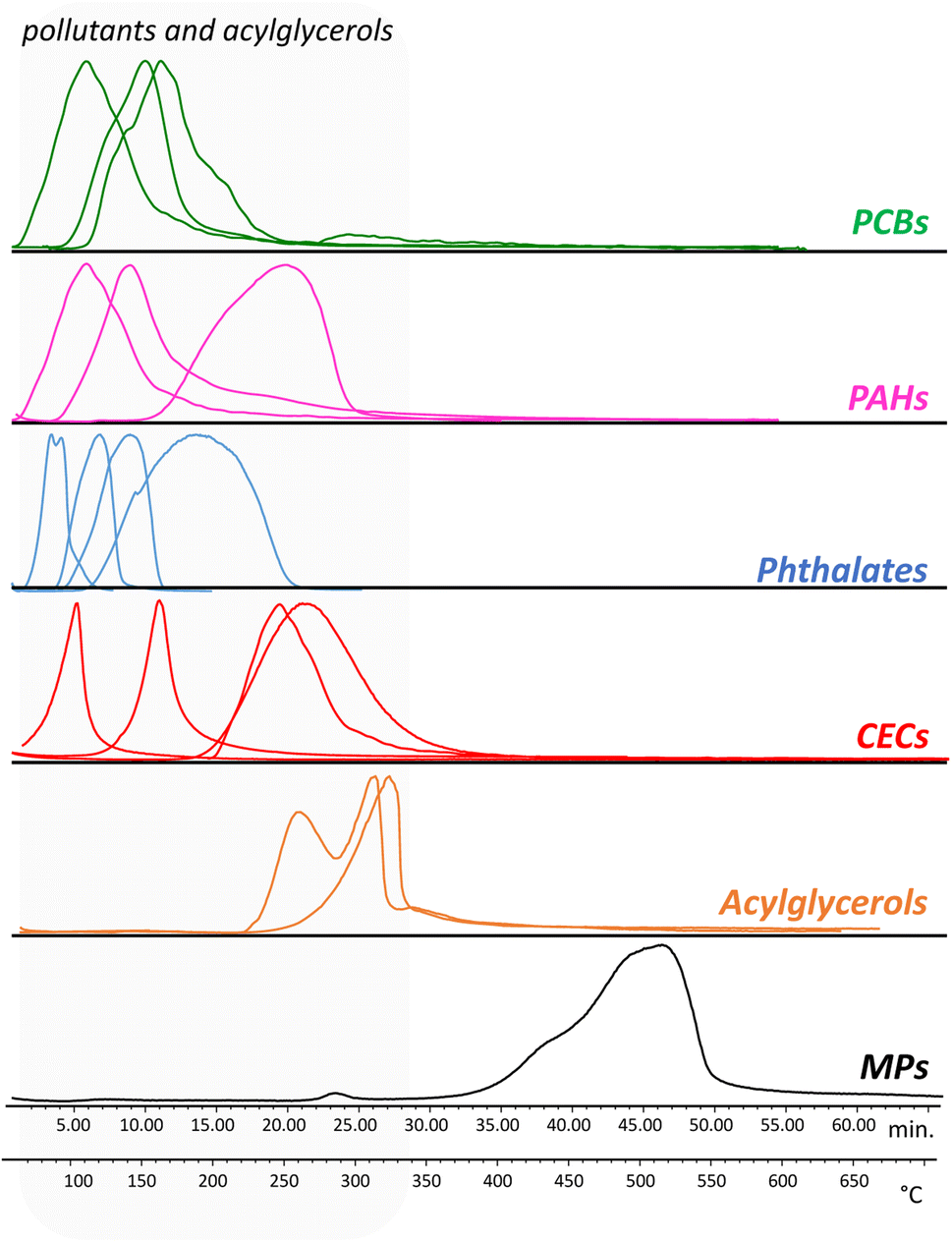 | ||
| Fig. 1 EGA-MS thermal profiles obtained for a selection of contaminants and for the microplastic standard. PCBs: PCB28, PCB180; PCB209; PAHs: pyrene, benzo(a)pyrene, indeno[1,2,3-C,D]pyrene; phthalates: dibutyl phthalate, benzyl butyl phthalate, bis(2-ethylhexyl) phthalate, bis(7-methyloctyl) phthalate; CECs: 2-ethylhexyl 4-methoxycinnamat, 3-tert-butyl-4-hydroxyanisole, diclofenac, methiocarb; acylglycerols: dipalmitin, tristearin. All the standards are listed in ascending order of degradation temperatures. | ||
The thermal desorption temperatures of the pollutants were in the range 50–350 °C: the PCBs and PAHs were characterized by desorption temperatures in the range 50–320 °C while the phthalate plasticizers were in the range 50–280 °C. The CECs were instead characterized by different desorption temperatures, with methiocarb in the range 110–200 °C, diclofenac 210–320 °C, 3-tert-butyl-4-hydroxyanisole 70–150 °C, and 2-ethylhexyl 4-methoxycinnamate 210–340 °C. As for the MPs standard, the degradation temperatures of all the polymers were in the range 350–600 °C. Based on these results, the temperature selected for the thermal desorption of the pollutants was 350 °C, while the pyrolysis was performed at 600 °C. Finally, two standards of acylglycerols (tripalmitin and dipalmitin) were analyzed in order to evaluate their thermal degradation temperature. The EGA-MS analysis highlighted that at a temperature of 350 °C, monopalmitin, dipalmitin, and tripalmitin generate their respective fatty acids acyl substituents and glycerol.
3.2 Calibration curves
Calibration curves for the pollutants were built using different diluted solutions containing each a different class of pollutants. The solutions for the phthalate plasticizers and the CECs were prepared in acetone with an average concentration of 50 ppm, while for the PAHs and PCBs two different commercial standard solutions were used, prepared in isooctane with a concentration of 10 ppm. Different volumes of standard solutions were added directly in the pyrolysis cup and dried under nitrogen flow prior the analysis. The calibration curves were built in the range 0.05–1.0 μg. 2 μL of a solution containing 50 ppm of anthracene-d10 and dibutyl phthalate-3,4,5,6-d4, and 4 μL of HMDS were added in the cup before the analysis and used as internal standards and derivatizing agent respectively. The areas of all the standards were integrated on the SIM chromatogram (ESI†). All the calibration curves showed a good linearities with R2 values in the range 0.9547–0.9991. No traces of thermal degradation products were detected in the chromatograms, confirming that the thermal desorption conditions were sufficiently mild to avoid the decomposition of the pollutants.The calibration curves for the microplastics were built using the microplastic calibration standard set. Different amounts of standard were weighted in the pyrolysis cup and added with 2 μL of a solution containing 50 ppm of anthracene-d10 as internal standard. The polymers mass range were: 11–39 μg for PE, 2.8–9.6 μg for PP, 0.6–2.0 for PS, 1.2–4.3 μg for ABS, 1.4–5.0 μg for SBR, 0.6–1.9 μg for PMMA, 0.3–1.0 μg for PC, 4.1–14.5 μg for PVC, 1.1–3.9 μg for PET, 0.3–1.2 μg for N6, and 1.2–4.3 μg for N66. Quantitation of each polymer was achieved by integrating the peak areas of specific pyrolytic markers (ESI†). All calibration curves showed good linearity with R2 values in the range 0.9821–0.9996. The calibration curves obtained for both the pollutants and the polymers are summarized in Table 1.
| Slope | Intercept | R 2 | |
|---|---|---|---|
| Pollutants | |||
| 2,4,4′-Trichlorobiphenyl (PCB28) | 1.19 × 107 | 1.44 × 105 | 0.9965 |
| 2,2′,5,5′-Tetrachlorobiphenyl (PCB52) | 8.61 × 106 | 7.99 × 104 | 0.9971 |
| 2,2′,4,5,5′-Pentachlorobiphenyl (PCB101) | 1.19 × 107 | 2.23 × 105 | 0.9769 |
| 2,2′,3,4,4′,5′-Hexachlorobiphenyl (PCB138) | 8.47 × 106 | −1.52 × 104 | 0.9986 |
| 2,2′,4,4′,5,5′-Hexachlorobiphenyl (PCB153) | 7.58 × 106 | −2.99 × 104 | 0.9990 |
| 2,2′,3,4,4′,5,5′-Heptachlorobiphenyl (PCB180) | 8.28 × 106 | 2.27 × 105 | 0.9638 |
| Decachlorobiphenyl (PCB209) | 6.37 × 106 | 9.45 × 104 | 0.9820 |
| Phenanthrene | 2.60 × 107 | 5.92 × 105 | 0.9670 |
| Anthracene | 3.08 × 107 | −1.27 × 105 | 0.9916 |
| Fluoranthene | 3.48 × 107 | 1.24 × 106 | 0.9978 |
| Pyrene | 3.77 × 107 | 1.32 × 106 | 0.9886 |
| Benz(a)anthracene | 3.57 × 107 | −1.42 × 105 | 0.9907 |
| Chrysene | 3.60 × 107 | 2.07 × 105 | 0.9890 |
| Benzo(b)fluoranthene | 3.42 × 107 | −2.53 × 105 | 0.9940 |
| Benzo(k)fluoranthene | 3.70 × 107 | −1.26 × 104 | 0.9744 |
| Benzo(a)pyrene | 3.61 × 107 | −1.13 × 106 | 0.9923 |
| Dibenz[a,h]anthracene | 8.51 × 107 | −2.82 × 106 | 0.9953 |
| Benzo[g,h,i]perylene | 3.45 × 107 | −7.76 × 105 | 0.9890 |
| Indeno[1,2,3-C,D]pyrene | 5.83 × 107 | 4.42 × 105 | 0.9972 |
| Methiocarb | 7.36 × 106 | −6.82 × 104 | 0.9875 |
| Diclofenac | 5.84 × 106 | −1.59 × 105 | 0.9547 |
| 3-tert-Butyl-4-hydroxyanisole | 2.37 × 105 | −8.81 × 103 | 0.9914 |
| 2-Ethylhexyl 4-methoxycinnamate | 2.67 × 107 | −6.26 × 105 | 0.9656 |
| Dibutyl phthalate | 2.80 × 107 | −1.01 × 106 | 0.9937 |
| Bis(8-methylnonyl) phthalate | 1.84 × 107 | −1.54 × 106 | 0.9584 |
| Bis(7-methyloctyl) phthalate | 2.16 × 107 | −1.78 × 106 | 0.9694 |
| Benzyl butyl phthalate | 1.49 × 107 | −1.12 × 106 | 0.9832 |
| Bis(2-ethylhexyl) phthalate | 1.94 × 107 | −1.45 × 106 | 0.9825 |
|
|||
| Polymers | |||
| Polyethylene (PE) | 2.55 × 104 | −7.83 × 104 | 0.9953 |
| Polypropylene (PP) | 3.83 × 104 | −4.28 × 103 | 0.9995 |
| Polystyrene (PS) | 2.15 × 105 | 1.21 × 104 | 0.9996 |
| Acrylonitrile butadiene styrene (ABS) | 1.03 × 105 | −7.19 × 104 | 0.9821 |
| Styrene-butadiene rubber (SBR) | 9.79 × 104 | −1.01 × 103 | 0.9993 |
| Polymethylmethacrylate (PMMA) | 9.30 × 105 | −5.89 × 104 | 0.9991 |
| Polycarbonate (PC) | 1.04 × 106 | −2.63 × 105 | 0.9913 |
| Polyvinyl chloride (PVC) | 1.58 × 105 | −2.14 × 103 | 0.9966 |
| Polyethylene terephthalate (PET) | 4.36 × 105 | 2.54 × 104 | 0.9992 |
| Nylon 6 (N6) | 3.66 × 105 | −2.03 × 104 | 0.9965 |
| Nylon 66 (N66) | 1.63 × 105 | −5.16 × 104 | 0.9956 |
3.3 Microwave-assisted extraction approach
The optimization of the extraction procedure and the evaluation of the extraction recoveries of the different classes of pollutants were performed by spiking a commercial lyophilized flour produced with the bivalve mollusk of Perna Canaliculus. Blank analyses were carried out to rule out the possible presence of relevant concentrations of pollutants that could affect the recoveries evaluation. Different extraction times and temperatures were tested to maximize the extraction yields for the different classes of pollutants. The optimization of the method allowed us to define the best experimental conditions for the extractions at 80 °C for 60 min. Table 2 reports the method features optimized for the recovery and quantification of the pollutants, while the total ion (TIC) and SIM chromatograms obtained at the best extraction conditions are reported in Fig. 2.| Compounds | Recovery (%) | CV% | LOD (ng) | LOQ (ng) |
|---|---|---|---|---|
| 2,4,4′-Trichlorobiphenyl (PCB28) | 97 | 5 | 0.003 | 0.008 |
| 2,2′,5,5′-Tetrachlorobiphenyl (PCB52) | 97 | 8 | 0.003 | 0.008 |
| 2,2′,4,5,5′-Pentachlorobiphenyl (PCB101) | 122 | 13 | 0.033 | 0.100 |
| 2,2′,3,4,4′,5′-Hexachlorobiphenyl (PCB138) | 109 | 7 | 0.008 | 0.025 |
| 2,2′,4,4′,5,5′-Hexachlorobiphenyl (PCB153) | 99 | 11 | 0.005 | 0.014 |
| 2,2′,3,4,4′,5,5′-Heptachlorobiphenyl (PCB180) | 104 | 15 | 0.002 | 0.007 |
| Decachlorobiphenyl (PCB209) | 87 | 5 | 0.001 | 0.002 |
| Phenanthrene | 118 | 12 | 0.001 | 0.004 |
| Anthracene | 101 | 9 | 0.004 | 0.013 |
| Fluoranthene | 92 | 15 | 0.001 | 0.004 |
| Pyrene | 94 | 5 | 0.001 | 0.004 |
| Benz(a)anthracene | 91 | 6 | 0.001 | 0.004 |
| Chrysene | 88 | 5 | 0.006 | 0.018 |
| Benzo(b)fluoranthene | 88 | 7 | 0.001 | 0.002 |
| Benzo(k)fluoranthene | 107 | 12 | 0.001 | 0.003 |
| Benzo(a)pyrene | 87 | 11 | 0.001 | 0.003 |
| Dibenz[a,h]anthracene | 101 | 10 | 0.000 | 0.001 |
| Benzo[g,h,i]perylene | 107 | 12 | 0.001 | 0.003 |
| Indeno[1,2,3-C,D]pyrene | 87 | 15 | 0.002 | 0.007 |
| Methiocarb | 110 | 10 | 0.044 | 0.134 |
| Diclofenac | 108 | 7 | 0.011 | 0.032 |
| 3-tert-Butyl-4-hydroxyanisole | 102 | 18 | 0.441 | 1.336 |
| 2-Ethylhexyl 4-methoxycinnamate | 115 | 8 | 0.023 | 0.069 |
| Dibutyl phthalate | 90 | 7 | 0.098 | 0.298 |
| Bis(8-methylnonyl) phthalate | 119 | 5 | 0.014 | 0.044 |
| Bis(7-methyloctyl) phthalate | 114 | 6 | 0.049 | 0.150 |
| Benzyl butyl phthalate | 90 | 11 | 0.083 | 0.250 |
| Bis(2-ethylhexyl) phthalate | 100 | 14 | 0.076 | 0.230 |
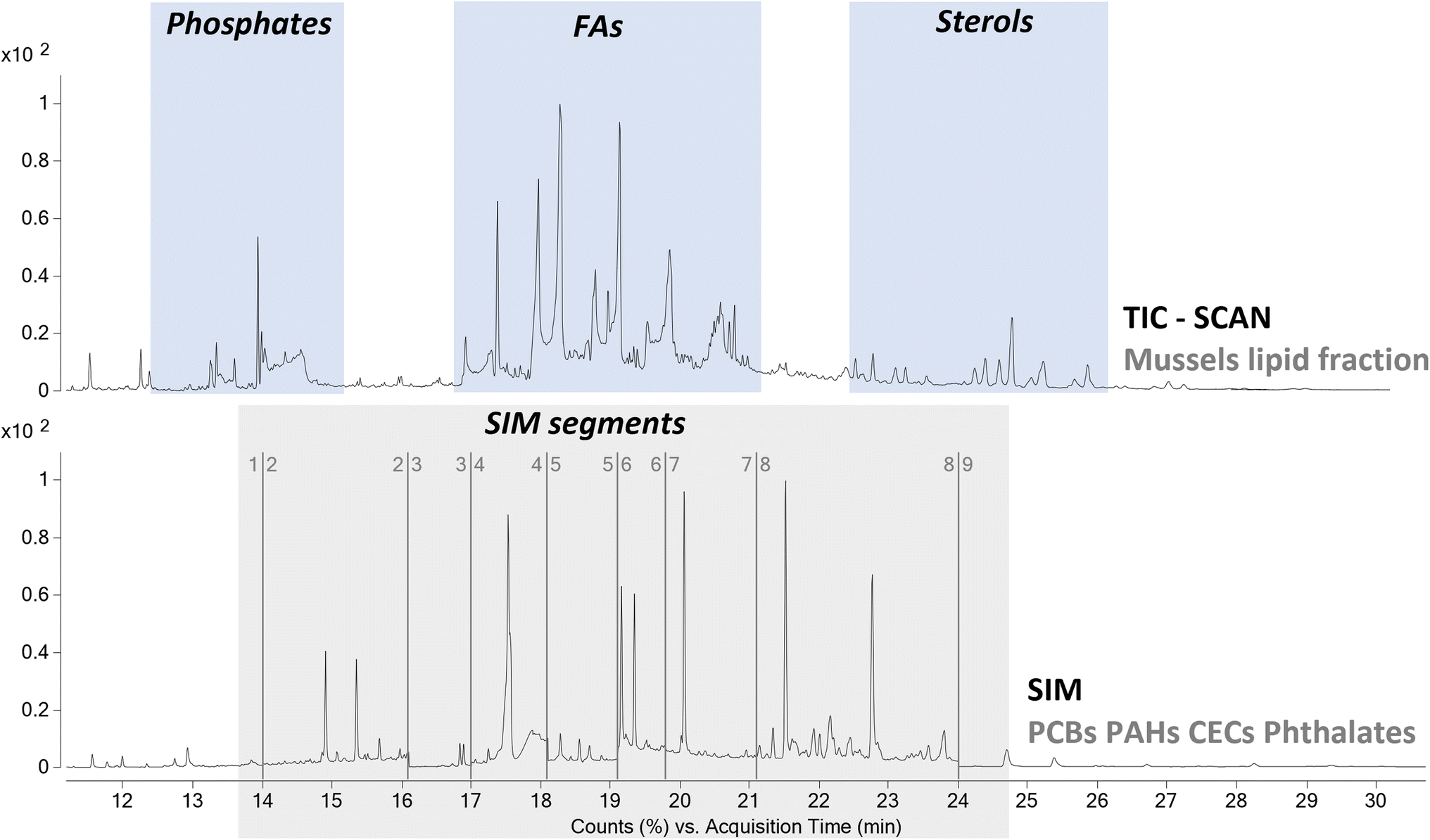 | ||
| Fig. 2 Py-GC-MS chromatogram obtained for the mussels spiked with the standard solution of pollutants at the best extraction conditions; total ion chromatogram (TIC), above; selected ion monitoring (SIM) chromatogram, below. | ||
From a qualitative point of view the TIC was characterized by the presence of a high abundance of desorption products associated to the lipidic content of the mussels. Thus, the extraction protocol used to remove the pollutants proved to be effective also in the extraction of the acylglycerol portion of the mussels. As highlighted in Section 3.1, the selected temperature of 350 °C for the thermal desorption of the contaminants was also suitable for the pyrolysis of these species to the corresponding fatty acids and glycerol. The degradation of acylglycerols to fatty acids led to a simplification of the chromatographic profile, reducing the possible interferences of matrix components on the detection of analytes of interest. The use of HMDS as derivatizing agent to obtain the corresponding trimethylsilyl esters, allowed us to perform a comprehensive characterization of all the lipid species. In detail, the analysis highlighted the presence of cholesteryl benzoate, cholesta-3,5-diene, and cholesterol as most abundant sterols. The analysis also highlighted the presence of several fatty acids and polyunsaturated fatty acids, whit tetradecanoic acid (myristic), hexadecenoic acid (palmitic), octadecenoic acid (oleic), octadecanoic acid (stearic), and eicosapentaenoic acid as most abundant fatty acids. Finally, the analysis also showed the presence of phosphates. These results agreed with the literature, proving that the analytical procedure can provide also useful information not only on the pollutants but also on the lipid content of the mussels. Alternatively, a fraction of the extract can be analyzed with complementary analytical approaches, such as reverse phase liquid chromatography,37 in order to obtain a more complete overview of the chemical composition of the lipid fraction of the mussels.
From a quantitative point of view, even if the analyses were carried out using a single quadrupole analyzer, the use of a combination of SIM and full scan acquisitions allowed us to evaluate the presence of all the classes of compounds spiked in the samples. The method features were evaluated according to the ICH guidelines. The recoveries of all the compounds were in the range 87–122%, while the coefficients of variation were all lower than 18%. Only the recoveries of naphthalene, acenaphthylene, acenaphthene, and dimethyl phthalate were characterized by values lower than 60%, with very high coefficient of variation and thus a poor reproducibility. This aspect could be related to the volatility of the analytes, that could be partially lost during the drying step of the procedure, after the extraction.
The limits of detection were evaluated on the procedural blanks and were in the range 0.001–0.441 ng, while the limits of quantification were in the range 0.001–1.360 ng. These values were obtained by adding in the pyrolysis cup a volume of 140 μL of solvent, corresponding to the same max volume used for the mussel extracts. Thus, the LODs and LOQs were expressed as the minimum amount of pollutant detectable in the pyrolysis cup. While for most of the contaminants the values were comparable, the CECs and phthalate plasticizers showed higher values. In detail the highest limits were obtained for methiocarb, 3-tert-butyl-4-hydroxyanisole, dibutyl phthalate, benzyl butyl phthalate, and bis(2-ethylhexyl) phthalate. Although these values were up to hundred times higher than those detected for PCBs, and PAHs, they were still lower than 1.5 ng, making the method still capable of detecting them at trace levels.
3.4 Microwave-assisted digestion approach
The effects of the microwave-assisted digestion for the analysis of microplastics were tested directly on the microplastic calibration standard used for the pyrolysis calibration, using hydrochloric and nitric acids, and sodium hydroxide as reagents. In the same way as the analysis of non-polymeric pollutants in Section 3.3, blank analyses were performed on non-spiked mussel flour to ensure that no peak ascribable to polymer markers could be detected in the matrix. The use of the standard microplastic mixture allowed us to evaluate the recoveries of MPs after the different digestion approaches using plastic particles with sizes more in agreement with environmental microplastics. Working with polymers with these reduced dimensions enabled us to obtain more accurate analytical data that better reflect the behavior of MPs in environmental samples. To evaluate the recoveries for the different procedures (Table 3), we used the set of calibration curves built with the same microplastics standard.| Polymer | LOD (μg) | LOQ (μg) | HCl digestion | HNO3 digestion | Spiked Mussels | ||
|---|---|---|---|---|---|---|---|
| Recovery (%) | CV% | Recovery (%) | CV% | Recovery (%) | |||
| PE | 0.05 | 0.16 | 99 | 1 | 110 | 12 | 87 |
| PP | 0.08 | 0.24 | 96 | 1 | 100 | 18 | 104 |
| PS | 0.02 | 0.06 | 96 | 3 | 73 | 16 | 88 |
| ABS | 0.04 | 0.11 | 106 | 1 | 71 | 17 | 105 |
| SBR | 0.08 | 0.25 | 104 | 2 | 32 | 32 | 98 |
| PMMA | 0.001 | 0.003 | 103 | 2 | 107 | 23 | 109 |
| PC | 0.01 | 0.03 | 93 | 6 | 41 | 35 | 114 |
| PVC | 0.02 | 0.07 | 113 | 16 | 129 | 46 | 138 |
| PET | 0.13 | 0.40 | — | — | — | — | — |
| N6 | 0.02 | 0.07 | 34 | 9 | 36 | 20 | — |
| N66 | 0.05 | 0.16 | 16 | 11 | 19 | 37 | — |
In the preliminary test performed using sodium hydroxide for the alkaline digestion critical challenges were encountered during the filtration step, which were caused by the high content of saponifiable lipids in the matrix. As a result, this pretreatment cannot be used as a standalone sample purification method for microplastic analysis. For this reason, nitric and hydrochloric acids were tested to perform an acidic digestion.
The MPs recoveries obtained with nitric acid varied widely for each polymer. As expected, recoveries were low for polyamides (N6, N66) and rubber (SBR) due to their poor chemical resistance in acidic and strong oxidative environments. However, low recoveries were also obtained for PS, ABS, and PC. Furthermore, the coefficient of variation for the recoveries of all polymers was higher than 12%, with values up to 46% obtained for PVC. On the other hand, the recovery of PET was too high (up to four times higher respect to the initial standard amount), suggesting possible interference deriving from the procedure: the quantification was performed using benzoic acid as a pyrolysis marker, this marker could derive also from the oxidation of other polymers present in the mix, such as PS, that was in part lost during the digestion (recovery 73%).
The MPs recoveries obtained using hydrochloric acid for the digestion, excluding the polyamides that are partially hydrolyzed during the pretreatment, were in the range 96–106%. Similar to what was obtained with nitric acid, the recoveries for PET were up to two times higher than the initial amount of polymer present in the standard, suggesting also in this case a significant contribution of benzoic acid from the oxidation/degradation of other polymers than PET. The CV% obtained for the polymers were lower than 16%. Compared to the digestion performed using nitric acid, the HCl digestion provided better analytical performances. Additionally, the solutions and residues were more easily manageable during the filtration process. Considering these advantages, we decided to perform the method validation following the ICH guidelines on the HCl procedure.
We evaluated the limits of detection and quantification based on the procedural blanks, and the results are reported in Table 3.
Finally, the MPs recoveries were also determined by spiking the commercial Perna Canaliculus flour. Excluding the polyamides and the PET, all the recoveries for the polymers were in the range 87–114%, with coefficients of variation similar to those obtained for the analysis on the microplastic calibration standard. Only PVC was characterized by a recovery of about 140% (while it was 113% in absence of matrix). This result can be indicative of a matrix effect on the analysis since the quantification was performed using naphthalene as a marker. Unfortunately, this polymer is characterized by a pyrolysis profile mainly constituted by aromatic compounds, limiting the selection of the pyrolysis marker to perform quantitative analysis. Naphthalene can derive from the pyrolysis of different natural sources (e.g. residues of proteinaceous material from the digestion38), and thus can influence the analytical result.
4. Conclusions
The increasing awareness on the environmental impact of microplastics and their interactions with other contaminants leads to the necessity of multi-target analytical protocols. We believe that the method presented in this work constitutes a reliable tool for the detection and quantitation of both low-molecular weight contaminants and microplastics.The use of microwave-assisted extraction allowed us to avoid the conventional, time-intensive pretreatment procedures for microplastics analysis. The analysis of low-molecular weight contaminants and microplastics in two separate runs also provided simpler chromatographic profiles, increasing the method performances and reducing the risk of matrix interference. The promising results obtained for a chemically complex sample such as seafood suggests that this method could also be applied to other matrices, such as water- and sediment-based ones.
Additional improvements of the method are required to obtain higher extraction recoveries, especially for polymers such as PET and nylon. A screening of possible alternative pyrolytic markers could also improve the accuracy of the method towards PET, avoiding the risk of overestimation when oxidized aromatic species are present in the sample. Future studies could also aim at combining the two microwave extraction steps into one, further reducing the time and volume of chemicals required for sample preparation. As this method constitutes an innovation in the field of microplastic analysis, validation by interlaboratory test could also be considered in the future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research was supported by University of Pisa Project PRA_2022_58 “Microwave assisted approaches for heritage science, environment, and energy” and by Milestone Srl (Sorisole, BG, Italy).References
- L. F. Amato-Lourenço, L. dos Santos Galvão, L. A. de Weger, P. S. Hiemstra, M. G. Vijver and T. Mauad, An emerging class of air pollutants: potential effects of microplastics to respiratory human health?, Sci. Total Environ., 2020, 749, 141676 CrossRef PubMed.
- M. Narayanan, Origination, fate, accumulation, and impact, of microplastics in a marine ecosystem and bio/technological approach for remediation: a review, Process Saf. Environ. Prot., 2023, 177, 472–485 CrossRef CAS.
- J. G. B. Derraik, The pollution of the marine environment by plastic debris: a review, Mar. Pollut. Bull., 2002, 44, 842–852 CrossRef CAS PubMed.
- A. Bianco, F. Sordello, M. Ehn, D. Vione and M. Passananti, Degradation of nanoplastics in the environment: reactivity and impact on atmospheric and surface waters, Sci. Total Environ., 2020, 742, 140413–140421 CrossRef CAS PubMed.
- G. Biale, J. La Nasa, M. Mattonai, A. Corti, V. Vinciguerra, V. Castelvetro and F. Modugno, A Systematic Study on the Degradation Products Generated from Artificially Aged Microplastics, Polymers, 2021, 13, 1997 CrossRef CAS PubMed.
- G. Biale, J. La Nasa, M. Mattonai, A. Corti, V. Castelvetro and F. Modugno, Seeping plastics: potentially harmful molecular fragments leaching out from microplastics during accelerated ageing in seawater, Water Res., 2022, 219, 118521 CrossRef CAS PubMed.
- M. Capolupo, A. Rafiq, I. Coralli, T. Alessandro, P. Valbonesi, D. Fabbri and E. Fabbri, Bioplastic leachates characterization and impacts on early larval stages and adult mussel cellular, biochemical and physiological responses, Environ. Pollut., 2023, 319, 120951 CrossRef CAS PubMed.
- G. Liu, Z. Zhu, Y. Yang, Y. Sun, F. Yu and J. Ma, Sorption behavior and mechanism of hydrophilic organic chemicals to virgin and aged microplastics in freshwater and seawater, Environ. Pollut., 2019, 246, 26–33 CrossRef CAS PubMed.
- V. Soursou, J. Campo and Y. Picó, A critical review of the novel analytical methods for the determination of microplastics in sand and sediment samples, TrAC, Trends Anal. Chem., 2023, 166, 117190–117204 CrossRef CAS.
- L. Simon-Sánchez, M. Grelaud, C. Lorenz, J. Garcia-Orellana, A. Vianello, F. Liu, J. Vollertsen and P. Ziveri, Can a Sediment Core Reveal the Plastic Age? Microplastic Preservation in a Coastal Sedimentary Record, Environ. Sci. Technol., 2022, 56, 16780–16788 CrossRef PubMed.
- W. Dellisanti, M. M.-L. Leung, K. W.-K. Lam, Y. Wang, M. Hu, H. S. Lo and J. K. H. Fang, A short review on the recent method development for extraction and identification of microplastics in mussels and fish, two major groups of seafood, Mar. Pollut. Bull., 2023, 186, 114221 CrossRef CAS PubMed.
- S. Fraissinet, A. Pennetta, S. Rossi, G. E. De Benedetto and C. Malitesta, Optimization of a new multi-reagent procedure for quantitative mussel digestion in microplastic analysis, Mar. Pollut. Bull., 2021, 173, 112931 CrossRef CAS PubMed.
- A. Dehaut, L. Hermabessiere and G. Duflos, Current frontiers and recommendations for the study of microplastics in seafood, TrAC, Trends Anal. Chem., 2019, 116, 346–359 CrossRef CAS.
- F. Ribeiro, E. D. Okoffo, J. W. O'Brien, S. Fraissinet-Tachet, S. O'Brien, M. Gallen, S. Samanipour, S. Kaserzon, J. F. Mueller, T. Galloway and K. V. Thomas, Quantitative Analysis of Selected Plastics in High-Commercial-Value Australian Seafood by Pyrolysis Gas Chromatography Mass Spectrometry, Environ. Sci. Technol., 2020, 54, 9408–9417 CrossRef CAS PubMed.
- L. Nalbone, F. Cincotta, F. Giarratana, G. Ziino and A. Panebianco, Microplastics in fresh and processed mussels sampled from fish shops and large retail chains in Italy, Food Control, 2021, 125, 108003 CrossRef CAS.
- S. Webb, H. Ruffell, I. Marsden, O. Pantos and S. Gaw, Microplastics in the New Zealand green lipped mussel Perna canaliculus, Mar. Pollut. Bull., 2019, 149, 110641 CrossRef CAS.
- A. I. Catarino, R. Thompson, W. Sanderson and T. B. Henry, Development and optimization of a standard method for extraction of microplastics in mussels by enzyme digestion of soft tissues, Environ. Toxicol. Chem., 2017, 36, 947–951 CrossRef CAS PubMed.
- A. Gomiero, P. Strafella, K. B. Øysæd and G. Fabi, First occurrence and composition assessment of microplastics in native mussels collected from coastal and offshore areas of the northern and central Adriatic Sea, Environ. Sci. Pollut. Res., 2019, 26, 24407–24416 CrossRef CAS PubMed.
- Y. Liu, R. Li, J. Yu, F. Ni, Y. Sheng, A. Scircle, J. V. Cizdziel and Y. Zhou, Separation and identification of microplastics in marine organisms by TGA-FTIR-GC/MS: a case study of mussels from coastal China, Environ. Pollut., 2021, 272, 115946 CrossRef CAS PubMed.
- J. La Nasa, G. Biale, D. Fabbri and F. Modugno, A review on challenges and developments of analytical pyrolysis and other thermo-analytical techniques for the quali-quantitative determination of microplastics, J. Anal. Appl. Pyrolysis, 2020, 149, 104841 CrossRef CAS.
- I. Goßmann, R. Süßmuth and B. M. Scholz-Böttcher, Plastic in the air?! – Spider webs as spatial and temporal mirror for microplastics including tire wear particles in urban air, Sci. Total Environ., 2022, 832, 155008 CrossRef PubMed.
- Y.-J. Huang, B.-S. Lin, C.-L. Lee and P. Brimblecombe, Enrichment behavior of contemporary PAHs and legacy PCBs at the sea-surface microlayer in harbor water, Chemosphere, 2020, 245, 125647 CrossRef CAS PubMed.
- M. M. Mwanza, E. N. Ndunda, G. O. Bosire, V. O. Nyamori and B. S. Martincigh, Advances in sample pretreatment and detection of PCBs in the environment, J. Hazard. Mater. Adv., 2021, 4, 100028 CrossRef CAS.
- J. La Nasa, G. Biale, F. Modugno, A. Ceccarini and S. Giannarelli, Magic extraction: solid-phase extraction and analytical pyrolysis to study polycyclic aromatic hydrocarbon and polychlorinated biphenyls in freshwater, Environ. Sci. Pollut. Res., 2022, 29, 64252–64258 CrossRef CAS PubMed.
- E. Manoli and C. Samara, Polycyclic aromatic hydrocarbons in natural waters: sources, occurrence and analysis, TrAC, Trends Anal. Chem., 1999, 18, 417–428 CrossRef CAS.
- C. Qu, S. Albanese, A. Lima, D. Hope, P. Pond, A. Fortelli, N. Romano, P. Cerino, A. Pizzolante and B. De Vivo, The occurrence of OCPs, PCBs, and PAHs in the soil, air, and bulk deposition of the Naples metropolitan area, southern Italy: Implications for sources and environmental processes, Environ. Int., 2019, 124, 89–97 CrossRef CAS PubMed.
- J. La Nasa, G. Biale, M. Mattonai and F. Modugno, Microwave-assisted solvent extraction and double-shot analytical pyrolysis for the quali-quantitation of plasticizers and microplastics in beach sand samples, J. Hazard. Mater., 2021, 401, 123287 CrossRef CAS PubMed.
- F. Akoueson, C. Chbib, S. Monchy, I. Paul-Pont, P. Doyen, A. Dehaut and G. Duflos, Identification and quantification of plastic additives using pyrolysis-GC/MS: a review, Sci. Total Environ., 2021, 773, 145073 CrossRef CAS PubMed.
- L. Hermabessiere, A. Dehaut, I. Paul-Pont, C. Lacroix, R. Jezequel, P. Soudant and G. Duflos, Occurrence and effects of plastic additives on marine environments and organisms: a review, Chemosphere, 2017, 182, 781–793 CrossRef CAS PubMed.
- M. O. Barbosa, N. F. F. Moreira, A. R. Ribeiro, M. F. R. Pereira and A. M. T. Silva, Occurrence and removal of organic micropollutants: an overview of the watch list of EU Decision 2015/495, Water Res., 2016, 94, 257–279 CrossRef CAS PubMed.
- D. Fabbri, A. G. Rombolà, I. Vassura, C. Torri, S. Franzellitti, M. Capolupo and E. Fabbri, Off-line analytical pyrolysis GC–MS to study the accumulation of polystyrene microparticles in exposed mussels, J. Anal. Appl. Pyrolysis, 2020, 149, 104836 CrossRef CAS.
- V. V. Ribeiro, C. R. Nobre, B. B. Moreno, D. Semensatto, C. Sanz-Lazaro, L. B. Moreira and Í. B. Castro, Oysters and mussels as equivalent sentinels of microplastics and natural particles in coastal environments, Sci. Total Environ., 2023, 874, 162468 CrossRef CAS PubMed.
- Y. Cho, W. J. Shim, M. Jang, G. M. Han and S. H. Hong, Nationwide monitoring of microplastics in bivalves from the coastal environment of Korea, Environ. Pollut., 2021, 270, 116175 CrossRef CAS PubMed.
- M. Cole, Y. Artioli, R. Coppock, G. Galli, R. Saad, R. Torres, T. Vance, A. Yunnie and P. K. Lindeque, Mussel power: scoping a nature-based solution to microplastic debris, J. Hazard. Mater., 2023, 453, 131392 CrossRef CAS PubMed.
- L. Hermabessiere and C. M. Rochman, Microwave-Assisted Extraction for Quantification of Microplastics Using Pyrolysis–Gas Chromatography/Mass Spectrometry, Environ. Toxicol. Chem., 2021, 40, 2733–2741 CrossRef CAS PubMed.
- M. Matsueda, M. Mattonai, I. Iwai, A. Watanabe, N. Teramae, W. Robberson, H. Ohtani, Y.-M. Kim and C. Watanabe, Preparation and test of a reference mixture of eleven polymers with deactivated inorganic diluent for microplastics analysis by pyrolysis-GC–MS, J. Anal. Appl. Pyrolysis, 2021, 154, 104993 CrossRef CAS.
- J. La Nasa, I. Degano, L. Brandolini, F. Modugno and I. Bonaduce, A novel HPLC-ESI-Q-ToF approach for the determination of fatty acids and acylglycerols in food samples, Anal. Chim. Acta, 2018, 1013, 98–109 CrossRef CAS PubMed.
- S. Orsini, E. Bramanti and I. Bonaduce, Analytical pyrolysis to gain insights into the protein structure. The case of ovalbumin, J. Anal. Appl. Pyrolysis, 2018, 133, 59–67 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3va00216k |
| ‡ Co-first author. |
| This journal is © The Royal Society of Chemistry 2024 |