
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Making light work: designing plasmonic structures for the selective photothermal methanation of carbon dioxide†
Yi Fen
Zhu
a,
Bingqiao
Xie
a,
Jodie A.
Yuwono
ac,
Priyank
Kumar
 a,
Abhinav S.
Sharma
b,
Michael P.
Nielsen
b,
Avi
Bendavid
d,
Rose
Amal
a,
Jason
Scott
*a and
Emma C.
Lovell
*a
a,
Abhinav S.
Sharma
b,
Michael P.
Nielsen
b,
Avi
Bendavid
d,
Rose
Amal
a,
Jason
Scott
*a and
Emma C.
Lovell
*a
aParticles and Catalysis Research Group, School of Chemical Engineering, The University of New South Wales, NSW 2052, Australia. E-mail: jason.scott@unsw.edu.au; e.lovell@unsw.edu.au
bSchool of Photovoltaic and Renewable Energy Engineering, The University of New South Wales, NSW 2052, Australia
cSchool of Chemical Engineering and Advanced Materials, The University of Adelaide, Adelaide, SA 5005, Australia
dCSIRO Manufacturing, Lindfield, NSW 2070, Australia
First published on 7th February 2024
Abstract
Effectively engaging light to induce catalytic activity requires the careful selection of a catalyst support with appropriate and beneficial properties. On this basis, black, plasmonic TiN was employed as a Ni catalyst support for the CO2 methanation reaction under illuminated-only conditions. The positive effects of light illumination were found to be defined by the Ni deposit size and the Ni–TiN interaction. At a high Ni loading (40 wt%, 70 wt%), simulated sunlight induces plasmonic heating through the TiN support which is sufficient to initially in situ reduce the Ni deposits and initiate CO2 methanation. Photothermal effects from TiN and the metallic Ni, combined with reaction exothermicity, then continue to further reduce the Ni and amplify the methanation reaction. At a lower Ni loading (10 wt%), the Ni deposits are smaller and more dispersed. In this case, the topmost Ni deposit surfaces are more strongly influenced by the TiN support due to their closer proximity to the metal–support interface. DFT calculations revealed that this condition can facilitate the migration of light induced plasmonic hot charge carriers from the TiN towards the exposed Ni surface, altering the surface charge of the Ni. The adsorption strength of *CO is subsequently enhanced to enable further reaction rather than desorption as product, thereby boosting CH4 selectivity. The findings discern between the different phenomena (plasmonic heating and hot electron migration) invoked by plasmonic excitation and offer new insight on the contribution these phenomena make to governing catalyst activity and selectivity.
Broader contextThe Sabatier reaction for CH4 synthesis is widely studied in the scope of CO2 valorisation. The use of sunlight as a direct energy input is motivated by the energy intensive nature of CO2 conversion. Intrinsic solar heating (photothermal) can directly offset large scale heating requirements. Moreover, electronic effects induced by illuminating a suitable catalyst support has the potential to invoke positive effects, mediating the way the reaction unfolds to boost CH4 selectivity over CO, a common by-product of CO2 methanation. Herein, for the first time, the application of titanium nitride-supported nickel (Ni–TiN) as a visible light responsive catalyst for selective CH4 synthesis from CO2 is explored. Tuning the Ni![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TiN ratio can facilitate either solar heating sufficient for in situ catalyst activation or participation of hot charge carriers leveraged from plasmonic TiN to enhance CH4 synthesis under visible light illumination. :TiN ratio can facilitate either solar heating sufficient for in situ catalyst activation or participation of hot charge carriers leveraged from plasmonic TiN to enhance CH4 synthesis under visible light illumination.
|
Introduction
Carbon dioxide (CO2), once captured from point sources or directly from the atmosphere, may be sequestered in deep ocean and underground reserves, an approach which necessitates transportation costs and storage sites.1 CO2 utilisation represents a complementary strategy, where the captured CO2 can be used as a carbon feedstock to synthesise value-added chemicals.2,3 The reaction of CO2 with H2 to form CH4 (namely CO2 methanation), which can be used as both a synthetic fuel and key chemical feedstock, has been extensively explored. The CO2 methanation reaction is exothermic by nature. However, activation of the stable CO2 molecule requires a high amount of energy, often in the form of heat. In the effort to approach net zero emissions, fossil derived energy must be minimised within the conversion process. Incorporating renewable energy, such as abundant sunlight, within the process is essential. The solar spectrum consists of ultraviolet (UV) (∼5%; <400 nm), visible light (∼45%; 400 nm–700 nm), and near infra-red (NIR) (∼50%; >700 nm) contributions.4 Each spectral light range, when harnessed using appropriate light responsive catalytic materials, can help promote CO2 reduction. The efficient and direct use of sunlight alleviates the reliance on external energy inputs. Catalyst selection fundamentally dictates the mechanism and wavelengths of light absorption, the material characteristic activated and the means by which the captured energy can influence the overall reaction.Conventional photocatalysis is facilitated by illuminating semiconducting materials using visible and/or UV light. Electrons and holes are generated which can migrate toward surface active sites to reduce and oxidise reactants, respectively.5 However, conventional photocatalysis is challenged by fast electron–hole recombination rates that often leads to low conversion.5 Longer wavelength contributions, such as NIR, possess lower photon energy whereby NIR photons absorbed by dark materials can be dissipated as heat to thermally assist catalytic reactions. For capturing visible light, materials with a plasmonic response in the visible region can generate hot charge carriers via a localised surface plasmon resonance (LSPR) effect. The plasmonic hot charge carriers can relax and thermalise (creating intense localised hot spots) to drive reactions and/or directly assist surface reactions in a nonthermal manner.6 For realistic and reliable applications of light driven/assisted CO2 methanation at large scale, the broad and efficient usage of the solar spectrum is crucial.
Active transitional metals, such as Ni, Ru and Co, with strong hydrogen dissociation abilities are frequently explored as catalyst on ceramic supports (oxides, carbides, nitride) for CO2 methanation.7–10 Whilst Ni, Ru and Co have demonstrated good light-for-heat tendencies for light-driven/light assisted applications, Ni is the most explored element due to low-cost and earth abundance.3,10–13 The application of Ni facilitates an economic and efficient approach for a deep hydrogenation of CO2 required for CH4 synthesis.
To uncover the possible benefits of visible light other than providing heat, several strategies have been effective for harnessing visible light-induced hot charge carriers. Promoting the basicity of a metal oxide support, for instance by La2O3 inclusion, has been shown to facilitate acidic CO2 molecule adsorption close to Ni catalyst deposits. Enhanced intermediate adsorption near the Ni–support interface then promoted photoactivation of the formate intermediate by leveraging light responsivity of the Ni deposits, thereby boosting CH4 synthesis.8,14 Incorporating noble metals (Au, Ag) with a plasmonic response in the vis-NIR range alongside Ni (i.e., plasmonic bi-metallic active sites) has also been explored for light-assisted CO2 methanation. Visible light-activated plasmonic hot charge carriers derived from the Au/Ag moieties were able to evoke light enhanced CO2 conversion.15,16 However, the tendency of Au and Ag to promote the reverse water gas shift reaction (RWGS) (CO2 + H2 ⇌ CO + H2O) drove the selectivity towards CO production to the detriment of CH4 selectivity. Consequently, engaging a metal catalyst which can enhance the nonthermal plasmonic effects must also invoke selectivity towards the methanation reaction.15
Light absorption and utilisation facilitated by the support material is also a means by which light can boost catalytic activity. Wu et al. implemented niobium carbide (Nb2C) and titanium carbides (Ti3C2) (black coloured MXenes) as photothermal supports for a Ni catalyst. Both support materials exhibited enhanced optical absorption resulting in heat generation to thermally drive CO2 methanation.17 A recent report on titanium nitride (TiN) supported Ru for CH4 synthesis demonstrated that TiN plays a significant role in assisting the overall light-to-heat conversion achieved by the catalyst. An exponential correlation between CH4 production and light intensity highlighted that the Ru–TiN catalyst enabled a thermally dominant pathway under simulated sunlight.10 TiN is a dark coloured ceramic material with a plasmonic response within the visible region that is comparable to Au.18,19 Vis-NIR light induced hot carrier production from TiN and subsequent injection into an interfaced metal and/or semiconductor have been investigated in the past in photonics,20,21 plasmon-enhanced electrochemical reaction, and photochemical reactions in aqueous conditions.22–24 Herein, we engage TiN as a plasmonic support material for a Ni catalyst to harness visible light to drive the CO2 methanation reaction. Coupling Ni–TiN enables the direct incorporation of sunlight as a sustainable energy vector in the conversion of waste CO2 to energy rich CH4. Ni–TiN catalysts with various Ni loadings are studied to elucidate the individual contributions of Ni and TiN towards catalyst activity and selectivity under visible light. The significance of Ni deposit size and ensuing Ni–TiN interaction on the phenomena (plasmonic heating or hot electron migration) invoked by the plasmonic TiN response is discerned and demonstrated to impact either reactant conversion or product selectivity during methanation. The findings offer new insights on the influence of plasmonic excitation derived from a plasmonic ceramic support (TiN) toward photothermal and visible light-assisted thermal catalytic systems. Based on the outcomes from this study, this class of material represents a promising candidate for catalyst design toward the utilisation of vis-NIR light as a primary energy input to offset energy requirements for CO2 conversion.
Results and discussion
Catalyst characterisation
A suite of xNi–TiN samples (x = 0, 0.1, 0.4, 0.7 reflecting 0, 10, 40 and 70 wt% Ni loadings, respectively) were prepared via conventional wet impregnation. The as-prepared samples were characterised for structure and composition at the surface and in the bulk (Fig. 1). The actual Ni loading (wt%) as determined by ICP-OES aligns well with the nominal values (Table S1, ESI†).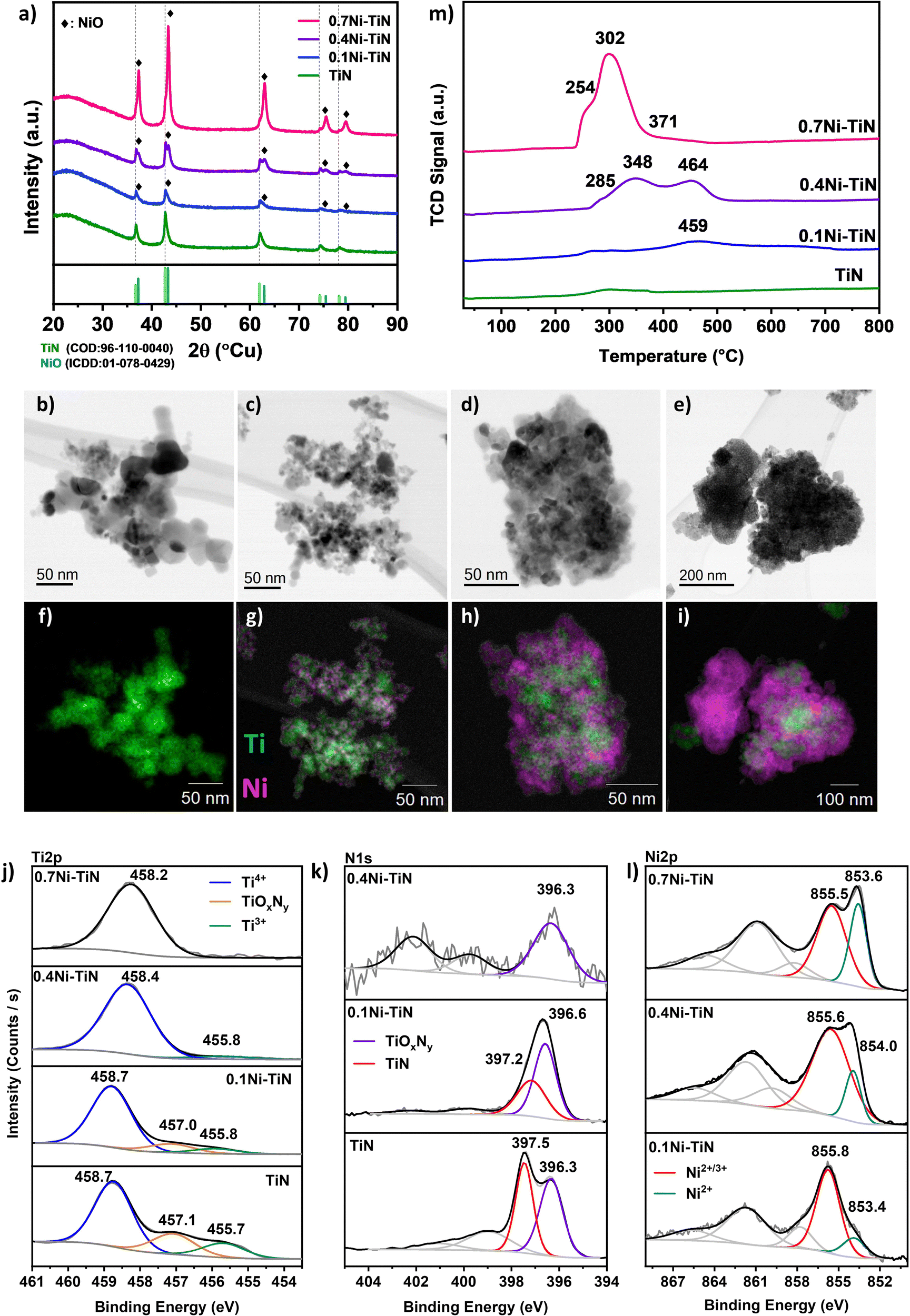 | ||
| Fig. 1 Characteristics of neat TiN, 0.1Ni–TiN, 0.4Ni–TiN, and 0.7Ni–TiN: (a) XRD spectra; (b)–(i) STEM micrographs and corresponding EDS mapping showing Ni (magenta) and Ti (green) content; XPS spectra at (j) Ti2p, (k) N1s, and (l) Ni2p spectral range (where applicable); (m) H2-TPR profiles. | ||
X-ray diffraction (XRD) was conducted to understand the crystalline phases present in the synthesised Ni–TiN samples. Fig. 1(a) displays the profiles for neat TiN and the impregnated samples. All samples show the characteristic TiN (COD: 96-110-0040) peaks corresponding to TiN(111); (200); (220); (311); (222) planes at 36.7°; 42.6°; 61.8°; 74.1°; 78.1°, respectively, confirming the presence of crystalline TiN and its preservation after Ni impregnation for all samples. As shown in Table 1, the crystallite size of TiN increases upon Ni loading which may be attributed to sintering during calcination. For all Ni-loaded samples, NiO (peaks at 37.3°; 43.3°; 62.9°; 75.4°; 79.4° corresponding to the (111); (200); (220); (311); (222) planes, respectively) can be identified, confirming Ni deposition as NiO (ICDD: 01-078-0429). The NiO crystallite sizes (Table 1) indicate that increasing Ni loading results in an increase in crystallite size. This is particularly apparent for the 0.7Ni–TiN sample which possesses a significantly larger NiO crystallite size (17.6 nm). Note that estimating the NiO crystallite size for the 0.1Ni–TiN sample was difficult due to overlap of the NiO peak with the TiN peaks.
Scanning transmission electron microscopy (STEM) was conducted to understand the Ni and TiN interaction as well as Ni deposit sizes and distributions. Fig. 1(b)–(e) show the brightfield STEM micrographs and (Fig. 1(f)–(i)) STEM-EDS elemental mapping for each sample. From the micrographs, it is evident that the Ni deposit distribution over TiN changes significantly as a function of Ni loading. The neat TiN samples exhibit a homogenous Ti distribution which is consistent with the XRD results suggesting the presence of only TiN crystals. The 0.1Ni–TiN samples exhibited good Ni dispersion over the TiN support with evidence of distinct cluster-like Ni deposits being formed over the TiN. On increasing the Ni loading to 40 wt% (0.4Ni–TiN) the TiN is loaded with aggregated Ni deposits. As shown in Fig. 1(i), at the maximum Ni loading (0.7Ni–TiN), large clusters of NiO independent of TiN are evident. This is consistent with the XRD results which suggest large NiO deposits (17.3 nm) are present. As well as distinct, bulk NiO particles, there is also evidence of TiN encrusted/anchored by large Ni deposits. HR-TEM micrographs of the reduced xNi–TiN catalysts (Fig. S6–S8, ESI†) further reveal the mode of Ni deposit presence after catalyst activation (NiO reduction to metallic Ni). A correlation between Ni loading and Ni deposit size after reduction and during methanation is apparent. Ni deposits over TiN remain cluster-like for 0.1Ni–TiN, while 0.4Ni–TiN and 0.7Ni–TiN contain large metallic Ni deposits to give Ni-rich surfaces. Ni particles independent of the TiN support are also present for 0.7Ni–TiN.
X-ray photoelectron spectroscopy (XPS) was used to probe the surface chemistry of each sample, as shown in Fig. 1(j)–(l). Three key peaks are evident in the Ti2p region across the samples. Within the Ti2p region of TiN (Fig. 1(j)), the peak located at 458.7 eV is assigned to the Ti4+ species from TiO2, whereas the peak located at 455.7 eV represents Ti3+ species from TiN.10,25–27 The peak located between these two terminal phases (at 457.1 eV) can be ascribed to a transitional oxynitride (TiOxNy) species native to TiN samples.25 In the case of neat TiN, all three Ti species are present. Combined with the XRD findings (which indicate only a TiN presence) it is apparent the bulk material is crystalline TiN which has a degree of surface oxidation. When Ni is present on the TiN, the relative presence of the Ti4+ peak increases with increasing Ni loading. The increasing presence of Ti4+ reflects an increasing surface oxide presence which may be attributable to: (i) exposure of the sample to air during handling; (ii) additional oxidation by residual water (from the impregnation process) during the high temperature treatment stage. Additionally, as the Ni loading increases the relative contribution of the Ti-species in the XPS surface atomic composition (Table 1) decreases. The diminishing presence of Ti-species is a consequence of the increasing Ni coverage of the TiN support with increasing Ni loading.
The corresponding N1s spectra (Fig. 1(k)) of the neat TiN can be resolved into two peaks at ∼397.5 eV and ∼396.3 eV, identified as Ni3− from TiN and nonstoichiometric N from TiOxNy, respectively.25–29 Similar to the Ti2p results, as the Ni loading increases, the relative intensity of the N1s spectra is diminished, as a result of the increasing presence of Ni. For 0.1Ni–TiN, the N1s oxynitride peak is more prominent. No N1s peaks were detected within the N1s spectral region for 0.7Ni–TiN, in line with the Ti2p spectrum. In all cases, the observed titanium oxide and oxynitrides are likely limited to the sample surface, as the XRD results indicated the TiN content was preserved without any Ti oxides being present.
The Ni2p spectra (Fig. 1(l)) of the impregnated samples verify the presence of oxidised Ni species. The peak located within 853–854 eV is attributable to Ni2+, consistent with the bulk NiO identified by XRD. The higher energy peak located at 855–856 eV may be associated with transitional Ni2+/3+ species, likely present as oxide/oxyhydroxides.30–33 As the Ni loading increases, the peak attributed to Ni2+/3+ shifts toward a lower binding energy, whilst its relative intensity to the Ni2+ peak decreases. With the increase in Ni deposit size, the relative sampling depth into the Ni deposits would differ accordingly, being able to reach closer to the core of smaller deposits.25 Collectively, it is evident that the Ni species at the surface of samples with lower Ni loading (particularly in the case of 0.1Ni–TiN) have a higher oxidation state. Gage et al. reported a strong metal support interaction for Ni–TiN samples.25 Such a scheme may facilitate a high dispersion of small Ni deposits with better Ni-on-TiN interfacing, which is consistent with the 10% Ni loading within this work. On this basis, Ni species that are more susceptible to oxidation would remain closer to the catalyst surface, resulting in deposits at higher oxidation states as is evident in XPS. This contrasts with 0.4Ni–TiN and 0.7Ni–TiN where the Ni deposits behave more like bulk aggregates as support influence on the surface-exposed Ni species is attenuated with increasing deposit size.
H2-TPR was used to determine sample reducibility (Fig. 1(m)). Negligible H2 consumption was observed for the neat TiN suggesting the TiN support surface has low reducibility. Introducing Ni increased H2 consumption. The TPR profile for 0.7Ni–TiN exhibits a primary reduction peak at ∼300 °C, accompanied by two shoulders at 255 °C and 370 C. The 0.4Ni–TiN shows two reduction peaks at higher temperatures, 350 °C and 465 °C, preceded by a shoulder at 285 °C. The 0.1Ni–TiN has a broad peak centred around 460 °C. The reduction profiles of the Ni-loaded samples demonstrate a general shift toward lower reduction temperatures with increasing Ni loading, suggesting a difference in the dominant Ni oxide species present for each loading. The dominance of low temperature reduction peaks (200–300 °C) for 0.7Ni–TiN is likely due to a large amount of support-independent NiO aggregates (seen in STEM (Fig. 1(e) and (i))) combined with aggregated Ni deposits distant from the Ni–TiN interface. At a 40 wt% Ni loading there is an increase in TiN supported NiO deposits, lowering Ni reducibility. The presence of well dispersed Ni species at higher oxidation states on 0.1Ni–TiN (from XPS) suggests an even stronger Ni interaction with the TiN support surface. The dominance of strongly interacting species lessens Ni reducibility giving the broad reduction peaks centred at 460 °C.
UV-vis spectroscopy was conducted to probe the optical properties of the different samples. Fig. 2(a) shows that TiN possessed broad-band absorbance within the visible range (405–740 nm) that extended into NIR region above 750 nm. This is characteristic for TiN samples with unbound electrons responsive to visible light illumination and is responsible for the LSPR effect within TiN.18 Absorbance by the TiN in the UV region is mainly attributed to the photoexcitation of valence electrons from N to Ti.18 Following Ni deposition, vis-NIR absorption from the TiN plasmonic effect is muted. This can be attributed to the presence of surface NiO which does not strongly absorb visible-light.34,35 The samples with a lower Ni loading provided better visible light absorption due to a lower NiO influence. Correspondingly, a colour change from black (neat TiN) to dark grey (0.7Ni–TiN) was apparent with increasing Ni loading (Fig. S10, ESI†). UV absorption below 350 nm became more prominent with Ni loading, attributable to the NiO having significant UV absorption at around 300 nm.34,35
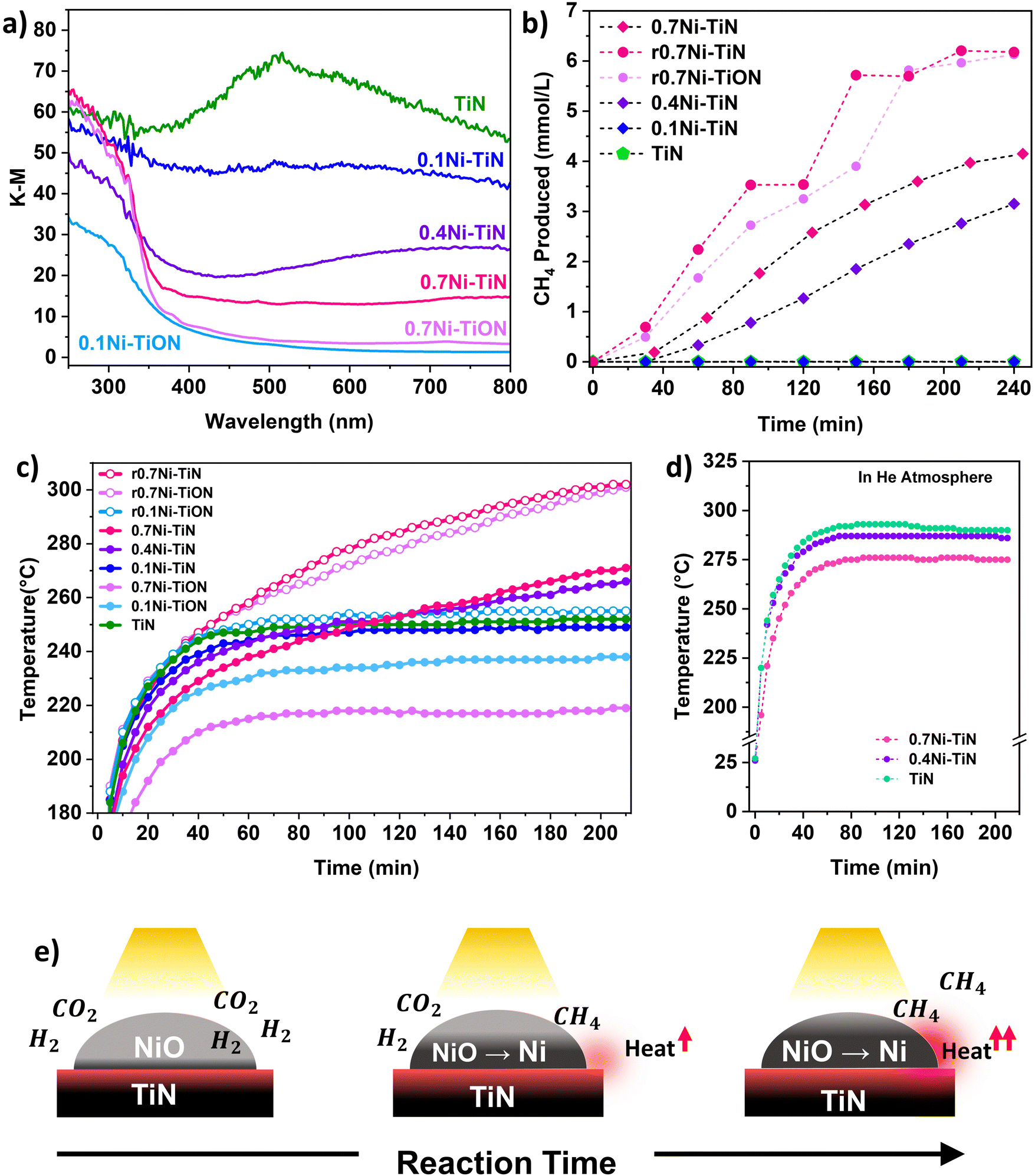 | ||
| Fig. 2 (a) UV-vis absorption spectra for neat TiN, 0.1Ni–TiN, 0.4Ni–TiN, 0.7Ni–TiN and oxidised controls (0.1Ni–TiON, 0.7Ni–TiON); (b) photothermal CH4 production by the various samples; (c) corresponding temperature profiles under photothermal conditions (Xe lamp at 20A setting); (d) xNi–TiN temperature profiles under inert He atmosphere (∼80 kPa); (e) proposed photothermally-driven in situ Ni reduction mechanism. | ||
Photothermal CO2 methanation
The performance of the Ni–TiN catalysts for photothermal CO2 methanation (eqn (1)) was assessed within a batch reactor under Xe lamp illumination (300–1100 nm) without any other external heat input or use of light filters.| CO2 + 4H2 → CH4 + 2H2O ΔH298K = −164 kJ mol−1 | (1) |
The initial heat generated within the reductive reaction environment instigates the in situ reduction of NiO to metallic Ni, which provides the active sites needed for the methanation reaction to commence. The exothermicity of the reaction further increases catalyst bed temperature, which supplemented the in situ reduction to generate additional metallic Ni sites. XRD spectra of the spent 0.4Ni–TiN and 0.7Ni–TiN catalysts under photothermal conditions (Fig. S11, ESI†) substantiates the presence of metallic Ni during methanation on exposure to photothermal conditions. The in situ reduction resulted in a colour change in the spent 0.7Ni–TiN and 0.4Ni–TiN from grey/dark grey to black (Fig. S10, ESI†) with metallic Ni recovery which would concomitantly increase the overall vis-NIR light absorption. This in situ reduction process is illustrated in Fig. 2(e). A temperature ‘snowballing’ effect has been previously reported for photothermal methanation over a Ni/CexTiyO2 catalyst which demonstrated a strong dependence on Ni reducibility.9 NiO reduction to metallic Ni must occur to enable the H2 dissociation required for CH4 production, as NiO is not active towards CO2 methanation.11,36 The temperature within the batch reactor (as governed by photothermal effects induced by the catalyst) and Ni reducibility work cooperatively to initiate Ni reduction and facilitate subsequent reaction. Herein, TiN thermalization initiates the NiO reduction process. The importance of NiO reducibility is highlighted in this work where the Ni reduction temperature differed significantly across samples. Overall, the 0.4Ni–TiN and 0.7Ni–TiN possessed higher reducibility and more active sites relative to 0.1Ni–TiN, thus exhibiting a greater likelihood for CO2 activation induced by the photothermal effect on TiN.
Control experiments were performed to understand the thermal contributions from light-induced heating (by TiN and Ni) and the exothermic methanation reaction. The TiN, 0.4Ni–TiN, and 0.7Ni–TiN samples were placed in a He atmosphere and exposed to the same operating conditions as the photothermal reactions. The resulting temperature profile Fig. 2(d) shows an increase in maximum bed temperature with increasing TiN content (decreasing Ni content). The disparity mimics the trend in temperature profiles during the first hour in Fig. 2(c), which was mainly governed by the optical property of the samples (as opposed to heat generated directly from the exothermic reaction). The findings illustrate the strong light-to-heat conversion capability of the neat TiN, demonstrating the potential of TiN as a plasmonic catalyst support.
To further understand the thermal contributions of TiN, oxidised control samples (0.7Ni–TiON, 0.1Ni–TiON) were synthesised. The oxidised samples were brown in colour (Fig. S10, ESI†) and exhibit TiO2-related (anatase and rutile) peaks in their XRD spectra (Fig. S13, ESI†) as well as poorer visible light absorption relative to their TiN-supported counterparts (Fig. 2(a)). TiN oxidation (to TiON) significantly decreases the vis-NIR light absorption by the support (Fig. S12, ESI†). Compared to the TiN-supported catalysts, the oxidized samples provide lower catalyst bed temperatures under the same illumination conditions (0.1Ni–TiON: ∼237 °C (stabilised); 0.7Ni–TiON: ∼192 °C (t = 20 min), and 217 °C (stabilised) compared to 0.1Ni–TiN: ∼248 °C (stabilised); and 0.7Ni–TiN; ∼212 °C (t = 20 min, prior to significant reaction)). The lower temperatures are insufficient to trigger significant NiO reduction (Fig. 1(m) and Fig. S16, ESI†) and the subsequent methanation reaction (Fig. S17, ESI†). The results provide direct evidence on the critical role of TiN-induced photothermal heating for eliciting in situ Ni activation for the ensuing methanation reaction over the 0.4Ni–TiN and 0.7Ni–TiN catalysts.
To understand the role of metallic Ni, the 0.7Ni–TiN and 0.7Ni–TiON catalysts were pre-reduced ex situ (H2:N2 = 1:1; 450 °C; 1 h) prior to the methanation reaction. The pre-reduced catalysts (denoted as r0.7Ni–TiN and r0.7Ni–TiON) were black in colour, unlike the as prepared 0.7Ni–TiN (dark grey) and 0.7Ni–TiON (light brown). The increased blackness indicates a greater extent of Ni reduction, which was confirmed by the presence of Ni0 states (852.4 eV) from XPS (Fig. S15), ESI† and a significant decrease in the H2-TPR peak intensity (Fig. S16, ESI†). Under photothermal reaction conditions, the presence of metallic Ni in the pre-reduced samples produced temperature profiles that were significantly higher than the unreduced ones. Consequently, r0.7Ni–TiN and r0.7Ni–TiON demonstrated a steeper increase in reaction temperature (Fig. 2(c)), as well as significantly higher CH4 production relative to the as prepared 0.7Ni–TiN (Fig. 2(b)). The results show that, once activated, light-induced heating on metallic Ni significantly increases the temperature of the catalyst bed.35 The difference in 0.7Ni–TiN performance with and without pre-reduction suggests that for systems reliant on TiN thermalisation for initial catalyst activation (0.4Ni–TiN and 0.7Ni–TiN), a more gradual Ni-reduction and methanation procedure occurs.
For the photothermally driven system, especially evidenced in the case of 0.4Ni–TiN and 0.7 Ni–TiN catalysts, the plasmonic TiN support supplies the initial heat energy that is essential for initiating Ni catalyst activation and CO2 methanation reaction. The exothermic CO2 methanation reaction is then able to provide more heat to promote Ni catalyst activation and CO2 methanation. By pre-reducing the catalysts, heat generation can be improved, boosting the reactivity of the system. Additionally, the Ni:TiN weight ratio must be considered for tuning Ni reducibility which is governed by the metal–support interaction and will influence the capacity to invoke the photothermal effect for CO2 methanation.
Decoupling the effects of light and heat
To further understand the role of light on CO2 methanation over Ni–TiN, continuous flow reactions using a Harrick reactor system with external heating at a range of temperatures (150–450 °C), under dark (i.e., thermal catalysis; non-illuminated) and visible light illuminated conditions (visible light-assisted thermal catalysis; white LED light, 420 nm < λ < 710 nm, ∼0.90 W cm−2) were conducted. This procedure highlights the influence of visible light illumination on the methanation performance against non-illuminated conditions.8,15,16,37 All samples were reduced in situ (450 °C, 1 h, H2:N2 = 1:1) prior to reaction. All pre-reduced samples supported by TiN appeared black in colour. After pre-reduction, a 1:4:1 flow of CO2:4H2:N2 at 24 mL min−1 (N2 as an internal standard) was introduced with the results shown in Fig. 3. The neat TiN offers negligible activity (Fig. 3(a)) with very limited CH4 production. For all Ni-containing samples, CO2 conversion increases with increasing temperature. The only detected products were CH4 and CO with the CO produced from the competing RWGS.
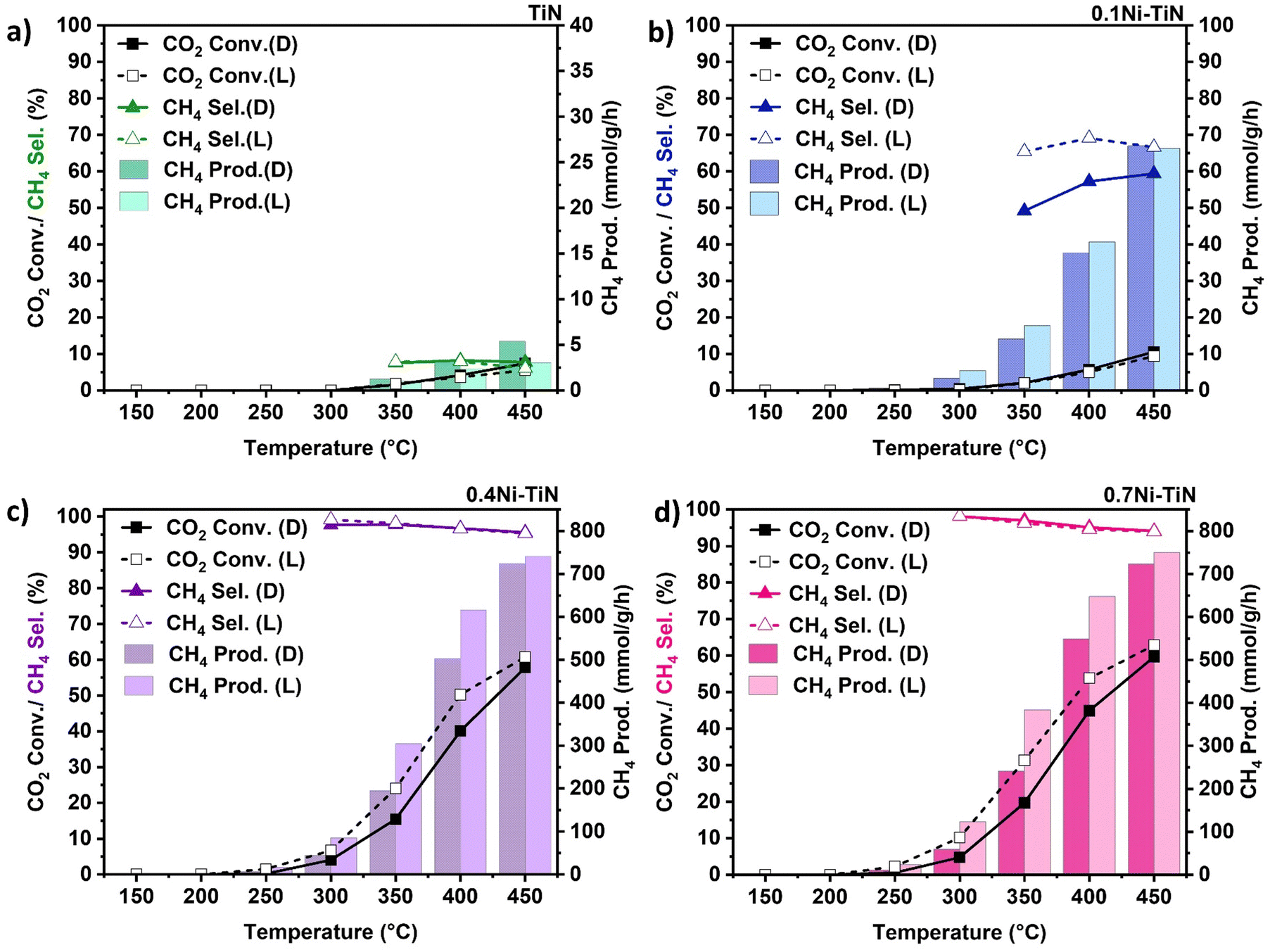 | ||
| Fig. 3 CO2 methanation reaction activity (conversion (Conv.), CH4 production (Prod.)) and CH4 selectivity (Sel.) with temperature over (a) neat TiN; (b) 0.1Ni–TiN, (c) 0.4Ni–TiN, and (d) 0.7Ni–TiN under non-illuminated (D) and illuminated (L) conditions conducted within a Harrick reactor (24 mL min−1, CO2:N2:H2 = 1:1:4, 150 °C–450 °C). | ||
In the case of high Ni loadings (0.4Ni–TiN, 0.7Ni–TiN) visible light illumination boosts CO2 conversion (Fig. 3(c) and (d)) across the entire temperature range, without impacting selectivity. The conversion enhancement is diminished at higher temperatures (e.g., 450 °C) and CH4 selectivity remains consistent (>93%) across both dark and illuminated trials. The pre-reduced 0.4Ni–TiN and 0.7Ni–TiN samples both contain abundant metallic Ni, inheriting superior light-for-heat capabilities.11 As such, the apparent conversion enhancement is likely correlated to additional thermal effects localised at the illuminated catalyst bed surface, leading to a local temperature gradient. The increased surface temperature will heighten the methanation rate, giving the enhanced conversion. The bulk temperature of the catalyst bed remains regulated by the reactor feedback controller, meaning the light-to-heat conversion was likely localised to the catalyst surface/metal deposits in the form of hot-spotting.
The influence of light on the high Ni loading samples was further explored by assessing the performance of the oxidised TiN (TiON supported) samples. Pre-reduced 0.7Ni–TiON also facilitated a light enhancement as illustrated by an elevated CO2 conversion (Fig. S14, ESI†). The observed ‘light enhancement’ in conversion was likely driven by the metallic Ni, as visible light absorption by TiN weakens after oxidation (Fig. 2(a)). On this basis, it is apparent that in Ni dominant systems (0.4Ni–TiN and 0.7Ni–TiN), light is largely harnessed by the metallic Ni in the form of heat to drive/supplement CO2 methanation. The difference in performance for the pre-reduced 0.4Ni–TiN and 0.7Ni–TiN was not significant considering a 30 wt% difference in Ni loading. This is attributed to the excessive Ni presence promoting the formation of bulk Ni aggregates, while only surface exposed Ni species are accessible as active sites for reactant interaction. The Ni-weight normalised CH4 production rate further highlights the difference in active site accessibility (1711 mmol gNi−1 h−1 and 997 mmol gNi−1 h−1, for 0.4Ni–TiN and 0.7Ni–TiN, respectively at 400 °C under illumination; 1397 mmol gNi−1 h−1 and 845 mmol gNi h−1, for 0.4Ni–TiN and 0.7Ni–TiN, respectively at 400 °C without illumination). Whilst the impact of the high metal loading is diminished in the pre-reduced, flow system, there remains an impact in the photothermal system. This can be attributed to the difference in reducibility with the in situ activation under strict photothermal conditions.
Interestingly, illumination of the 0.1Ni–TiN produced a different impact during the continuous flow reactions. In this case (Fig. 3(b)), CO2 conversion is constant for both the dark and illuminated conditions, however a significant change in CH4 selectivity is evident. For instance, illumination at 400 °C results in an increase in CH4 selectivity from 57.3% to 69.1%, whilst CO2 conversion remained constant. The contrast in light response relative to the higher Ni loaded samples (i.e., no change in conversion and a notable shift in selectivity) demonstrates contribution by the nonthermal effects of light. The 0.1Ni–TiN sample possesses surface Ni species which are both accessible to the reactants and influenced by the plasmonic TiN support (vide supra). The interaction suggests a potential pathway for the migration of hot charge carriers, generated within TiN under visible light illumination, to participate in the surface reaction, activating a pathway that favors CH4 synthesis. It is important to note that the reaction over 0.1Ni–TiON does not lead to significant methanation activity, with both conversion and CH4 selectivity laying below 10% across the 150 °C–450 °C temperature range. This indicates the importance of the influence of TiN, being pronounced in terms of promoting CH4 synthesis at low Ni loadings.
Transient absorption spectroscopy (TAS) was employed for neat TiN and pre-reduced 0.1Ni–TiN (r0.1Ni–TiN), dispersed in glycerol, to identify the dominant effect of illumination within the samples (Fig. S18, ESI†). The observed relaxation occurred in the order of tens to hundreds of picoseconds (ps), suggesting strong electron–phonon coupling leading to rapid lattice heating.38,39 This demonstrates excellent light-to-heat conversion by neat TiN. The result aligns with the observed increase in catalyst bed temperature with decreased Ni loading under inert conditions amongst the prepared (NiO loaded) samples. The contribution of TiN towards the initial catalyst bed thermalisation under photothermal conditions is evident. The result further cements TiN as a critical thermal element with greater heat contribution relative to NiO which supplements Ni reduction.
The inclusion of Ni leads to a red shift in the spectral response which likely accounts for enhanced optical absorption from metallic Ni (once NiO reduction takes place) at vis-NIR wavelengths (Fig. S18a and b, ESI†). Kinetics decay fit at 450 nm demonstrate that heat dissipation in 0.1Ni–TiN is much slower relative to neat TiN (Fig. S18c and d, ESI†). This may suggest a decrease in the overall thermal conductivity leading to reduced heat loss in Ni loaded samples. The decrease in thermal conductivity along with a strong light-to-heat effect derived from metallic Ni contributes to an enhanced thermal effect. An increase in the thermal effect with metallic Ni content under visible light illumination aligns well with the observed CO2 conversion enhancement over 0.4Ni–TiN and 0.7Ni–TiN which resembled the effect of direct temperature increase under strictly thermal (non-illumined) conditions. However, the sole contribution of light-induced heating cannot reconcile the significant light enhancement in CH4 selectivity that occurred specifically over 0.1Ni–TiN. It is postulated that nonthermal contributions, such as small population of hot electrons that potentially influences and alters the methanation pathway cannot be completely ruled out.
The performance comparison under non-illuminated and illuminated conditions highlights the thermal and/or nonthermal effects harnessed by Ni–TiN composites under visible light illumination that are advantageous for CO2 methanation. Both 0.4Ni–TiN and 0.7Ni–TiN represent samples with high Ni content (Ni-dominant systems) where visible light was likely harnessed in the form of heat to enhance CO2 conversion. Conversely, for 0.1Ni–TiN with a comparatively low Ni content, visible light illumination led to enhanced CH4 selectivity. The influence of visible light illumination on CO2 methanation performance appears to differ for samples depending on high or low Ni loadings. To understand the origin of the distinct influence of visible light, 0.1Ni–TiN and 0.7Ni–TiN have been selected for detailed mechanistic studies.
Mechanistic investigation
To better understand how the observed enhancements are driven by light, intrinsic mechanistic differences over the two surfaces with low (10 wt%) and high (70 wt%) loadings must be distinguished. in situ diffuse reflectance infrared Fourier transform spectroscopy (in situ DRIFTS) was used to probe the reaction intermediates and understand possible reaction pathways present over the 0.1Ni–TiN, and 0.7Ni–TiN catalysts under non-illuminated conditions. Further, density functional theory (DFT) calculations were used to understand the differing roles of light.During the in situ DRIFTS studies, as TiN and reduced xNi–TiN are black in colour, the samples were diluted with KBr (0.75 wt% sample content) to improve the signal to noise ratio in the resultant spectra. Additionally, in situ DRIFTS spectra of the neat TiN were obtained (diluted at 1:5 weight ratio TiN:synthetic diamond) to elucidate the intrinsic role of the TiN support towards reactant adsorption and activation. Prior to reaction, the diluted samples were reduced in situ with H2:He (1:1) at 450 °C for 1 h. in situ DRIFTS spectra intrinsic to the KBr, and synthetic diamond diluents were also obtained as a reference (Fig. S19, ESI†).
In the case of neat TiN, during initial CO2 adsorption (no accompanying H2 or diluent gas introduced) a peak resembling linear *CO 2076 cm−1 is apparent (Fig. 4(c)), though being weakly adsorbed.40 No clear intermediate peaks over TiN are observed under reaction conditions. Unlike basic supports, the TiN surface does not significantly contribute to CO2 adsorption in forming carbonate/bicarbonate/carboxylate/formate-type intermediates. The direct contribution of the TiN support to CO2 activation is likely small, which is consistent with the negligible methanation performance over neat TiN.
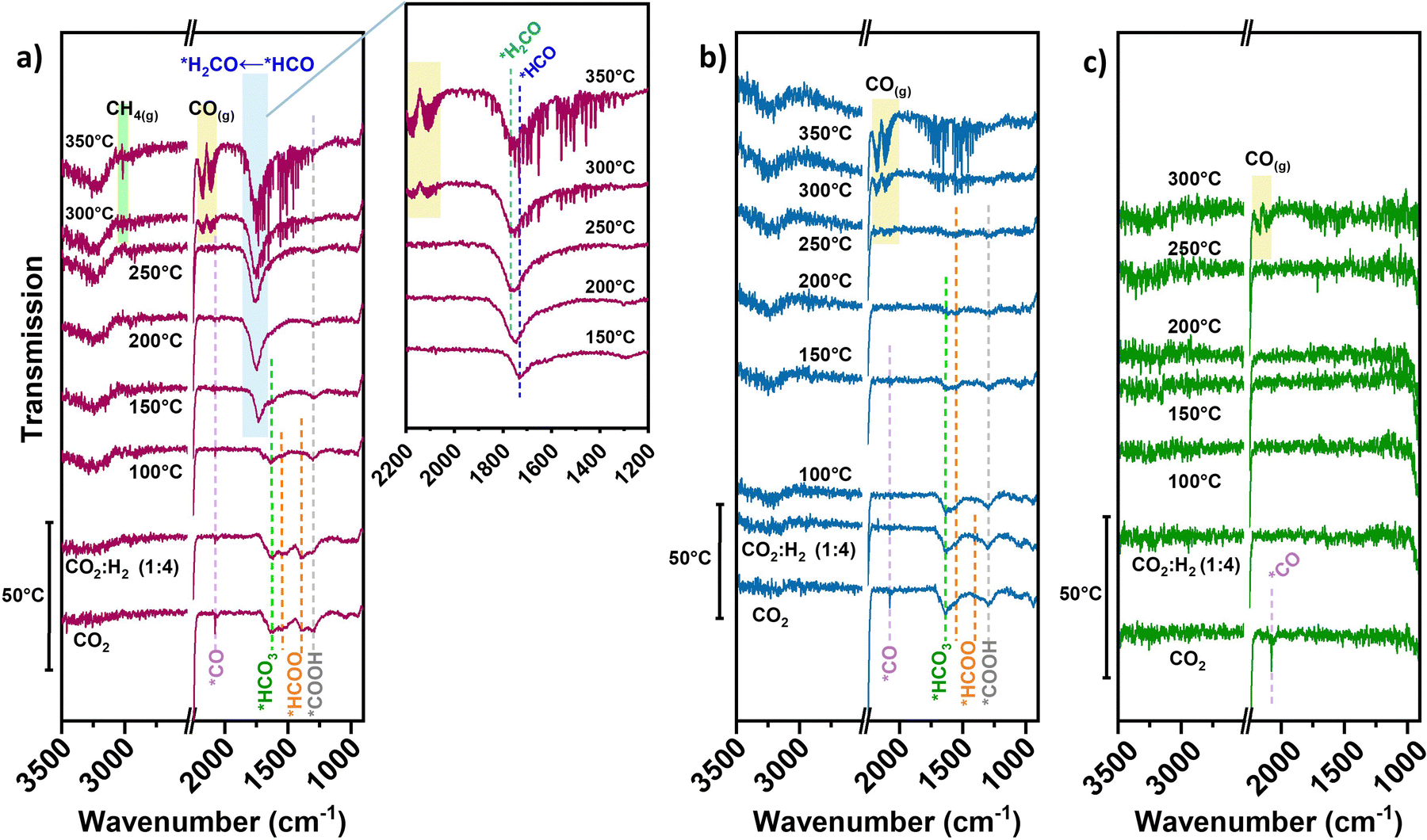 | ||
| Fig. 4 in situ DRIFTS spectra for (a) 0.7Ni–TiN, (b) 0.1Ni–TiN, and (c) neat TiN. | ||
For the pre-reduced 0.7Ni–TiN, CO2 introduction at 50 °C evokes a range of different peaks within the 1800–1200 cm−1 range (Fig. 4(a)). The peak at 1296 cm−1 may be attributed to carboxylate (*COOH) species.41,42 The gradual decrease in *COOH intensity upon H2 introduction and subsequent temperature increase demonstrates its consumption to produce other species over 0.7Ni–TiN. The peak located at 1632 cm−1 is attributed to bicarbonate (*HCO3) and the peaks at 1544 cm−1 and 1387 cm−1 to formate (*HCOO) species.14,43 Ni-coordinated *COOH species can also produce peaks at ∼1620 cm−1 and ∼1390 cm−1 which overlap with the current *HCO3/*CO3-related peaks.44,45
Increasing the temperature from 50 °C to 100 °C invokes depletion of the *HCOO, while the intensity of *HCO3 peak at 1632 cm−1 is also dampened. At 150 °C, a dominant peak at 1729 cm−1 resembling formyl (*HCO) adsorbates appears.46 The observed exchange between *HCOO and *HCO suggests a consumption of *HCOO through decomposition to *HCO species under elevated temperatures (eqn (2)). Further increasing the temperature to 200 °C and above results in a broadening of the previously-formyl peak towards 1770 cm−1 resembling formaldehyde (*H2CO).47 On this basis, a gradual and sequential hydrogenation of *HCO to other hydrogenated carbonyl species (*HnCO) (eqn (3)) may form a key part of the hydrogenation mechanism over 0.7Ni–TiN.
| *HCOO + H2 → *HCO + H2O | (2) |
| *HCO + *H → *H2CO | (3) |
| *HnCO + 2*H → CHn + H2O | (4) |
| CHn + (4 − n)*H → CH4 | (5) |
The activity results in Fig. 3(a) indicated that neat TiN lacks hydrogenation capability which resulted in high CO selectivity. Furthermore, illumination did not offer significant changes in performance. A Ni presence was crucial for both hydrogenation and light enhancement. To date, systems comprising Ni catalysts on oxide supports, such as TiO2, Al2O3 or SiO2, have not demonstrated a significant light response, with the exception of cases where basicity (e.g., CeO2 support, La2O3 promoted oxides) is prevalent.14,15 For these materials, enhanced CH4 synthesis was attributed to photoexcited charges from the surface metal, activating abundant *HCOO species adsorbed close to the catalyst deposits, which was facilitated by support basicity.8,14,37 In the case of TiN, with no evidence of basic properties, the influences from surface oxide-assisted adsorption, and/or photoexcited charges from Ni are not likely major factors for delivering the light-enhanced CH4 selectivity over 0.1Ni–TiN. Herein, Ni as a critical active site for hydrogenation may gain plasmonic hot charge carriers derived from TiN to influence the reaction.
DFT calculations were conducted to identify likely reaction pathways over the Ni surface involving the intermediates observed from in situ DRIFTS. The outcomes were used to establish the electronic interaction/charge distribution between the TiN support and Ni deposits at ground state and photo-exited states. Considering the Ni-dominant nature of 0.7Ni–TiN and the presence of TiN-independent Ni aggregates, relevant surface Ni sites were modelled as the Ni(111) surface. In contrast, 0.1Ni–TiN was modelled by a Ni cluster/TiN arrangement. The insets in Fig. 5(a) and (b) illustrate the two constructed models.
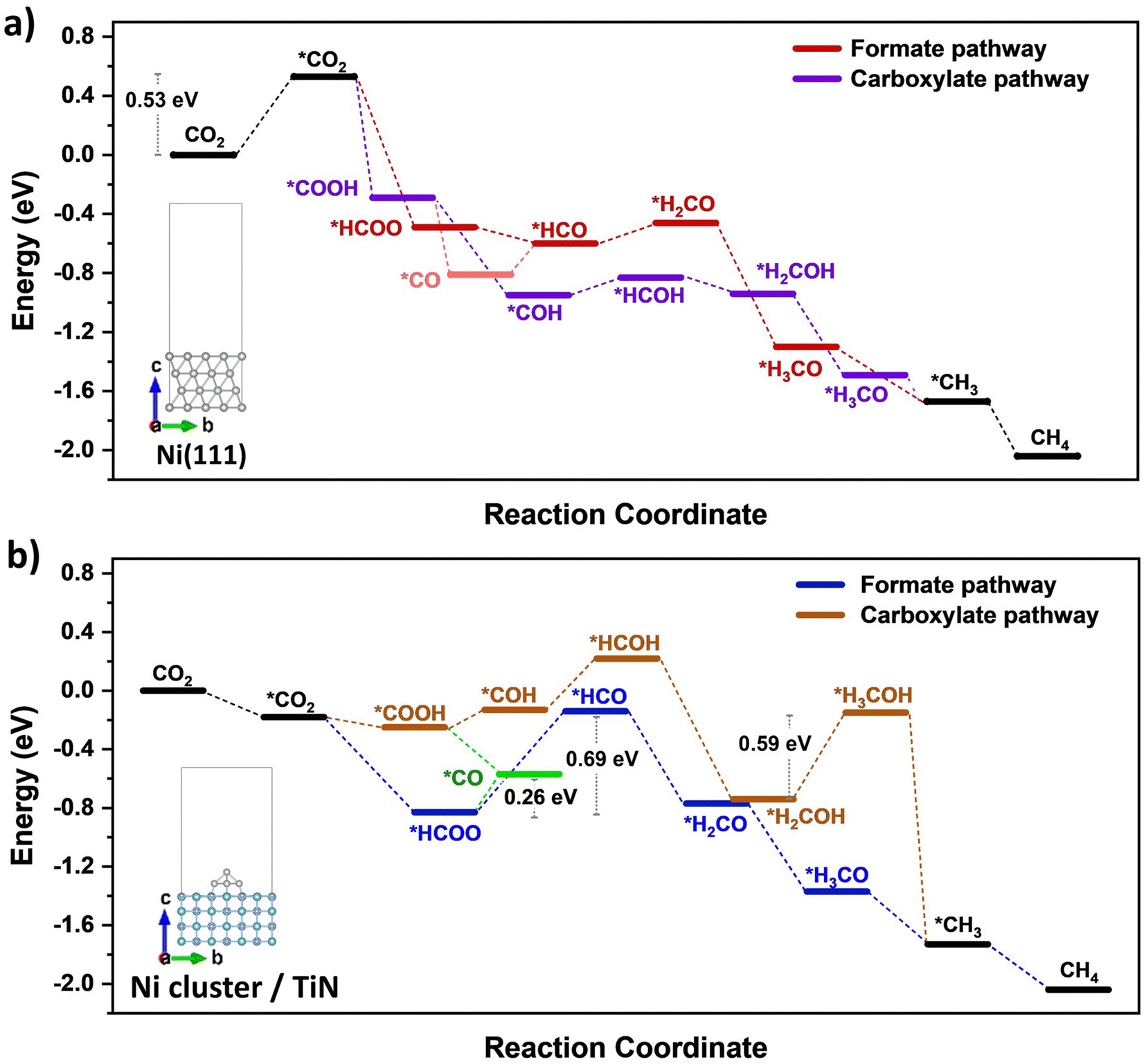 | ||
| Fig. 5 Calculated free energy profile for methanation under the formate and carboxylate-assisted routes over (a) Ni(111) (0.7Ni–TiN model); and (b) Ni cluster/TiN (0.1Ni–TiN model); modelled cell shown as inset. | ||
The calculated free energy diagram for Ni(111) (Fig. 5(a)) supports the occurrence of formate and carboxylate-assisted pathways. The latter may decompose to form *CO, or *COH. Fig. 5(a) shows that CO2 adsorption assumed the highest energy barrier (0.53 eV), after which, successive hydrogenation of the adsorbed CO2 would naturally enable progression of both pathways toward methanation. For Ni cluster/TiN (Fig. 5(b)), *HCOO hydrogenation to *HCO possessed the highest energy barrier under the formate pathway (0.69 eV). Comparatively, the energy barrier for *HCOO decomposition to *CO is 0.26 eV, presenting a more plausible route. Under a carboxylate pathway, following *COOH formation the subsequent hydrogenation into *HCOH are endothermic processes, where an exothermic *COOH decomposition to *CO is likely favored.
Calculated adsorption energies (Table S2, ESI†) show that under neutral charge conditions, *CO can adsorb more strongly onto Ni(111) surfaces (−1.38 eV) relative to Ni cluster/TiN (−1.13 eV). The higher *CO adsorption strength over 0.7Ni–TiN benefits further conversion (e.g., via *CHO) under ample *H toward CH4 synthesis. In contrast, desorption as gaseous CO occurs more readily over 0.1Ni–TiN. It supports the notion that the *HCOO/*COOH, and *CO as generated over 0.7Ni–TiN and 0.1Ni–TiN would follow different pathways, such that CH4, and CO are favored over respective Ni sites.
Bader charge analysis revealed that for the Ni(111) surface, the addition of an electron had little effect on the charge of the topmost Ni layer (−0.019 → −0.050) (Table S3, ESI†). Under illumination, the Ni surfaces of 0.7Ni–TiN can be expected to remain relatively neutral in charge. This means that the observed conversion enhancement under illumination is not likely a result of the light-induced electronic alterations on Ni surface. Here, the strong light-for-heat capabilities observed under photothermal condition presents a more plausible cause. Bader charge analysis of the ground state model simulating 0.1Ni–TiN (Ni cluster/TiN model) demonstrates the charge distribution between Ni and TiN within the system, from which the direction of charge flow can be inferred. TiN possesses a positive charge (+0.253) while the surface of Ni clusters possess a negative charge (−0.147), suggesting a tendency for electrons to migrate from TiN and accumulate at the surface of Ni clusters. Under photoexcited conditions, modelled by the introduction of an electron (total charge of −1 to the system), the Ni surface becomes more negatively charged (−0.217). Interestingly, under photoexcited conditions, the adsorption energy of *CO over the Ni cluster/TiN was strengthened (−1.13 eV → −1.30 eV), whilst that over Ni(111) remained relatively unchanged (−1.38 eV → −1.35 eV) (Table S4, ESI†). The outcomes suggest that for visible light illuminated 0.1Ni–TiN, light excited plasmonic hot electrons from TiN may migrate towards Ni clusters and modulate Ni surface charge density. This enhances *CO stability to shift the subsequent pathway away from direct CO desorption, and further towards CH4 synthesis. The results suggest the occurrence of a light-induced nonthermal effect within a predominantly thermally driven system (suggested by TAS).
By assessing the activity performance along with in situ DRIFTS and DFT outcomes, the reaction scheme illustrated in Fig. 6 captures the proposed mechanism over 0.7Ni–TiN and 0.1Ni–TiN samples and the influence of light on these two systems. Namely, 0.7Ni–TiN, capable of supplying ample dissociated hydrogen, likely supports methanation via *COOH and *HCOO intermediates. The latter would veer towards CO consecutive hydrogenation into *HCO/*H2CO species, which then slowly and selectively hydrogenate to CH4. Bader charge analysis revealed that the observed conversion enhancement over 0.7Ni–TiN was likely an effect of additional heat induced by the high Ni content, rather than light induced electronic alterations. This increased heat generation accelerates conversion of the more stable species such as *H2CO enroute to CH4 formation. In the case of 0.1Ni–TiN, CO2 conversion via both *HCOO and *COOH intermediates are possible, though the decomposition of both intermediates to *CO would be favored. The adsorption strength of the *CO is weak over the Ni sites of 0.1Ni–TiN, which aids CO desorption as a major product, consistent with the high CO selectivity for the sample under dark conditions. Under illumination, the plasmonic charge carriers generated on TiN may migrate towards, and manifest at, Ni surfaces. The negatively charged Ni surface enhances *CO adsorption strength, where surface stabilised *CO is more viable for further hydrogenation, leading to enhanced CH4 synthesis. Herein, the use of a plasmonic support demonstrates a direction of the harvested solar energy in the form of heat and/or hot electrons to prompt surface mechanisms which alter the state of Ni deposits in favor of CH4 synthesis.
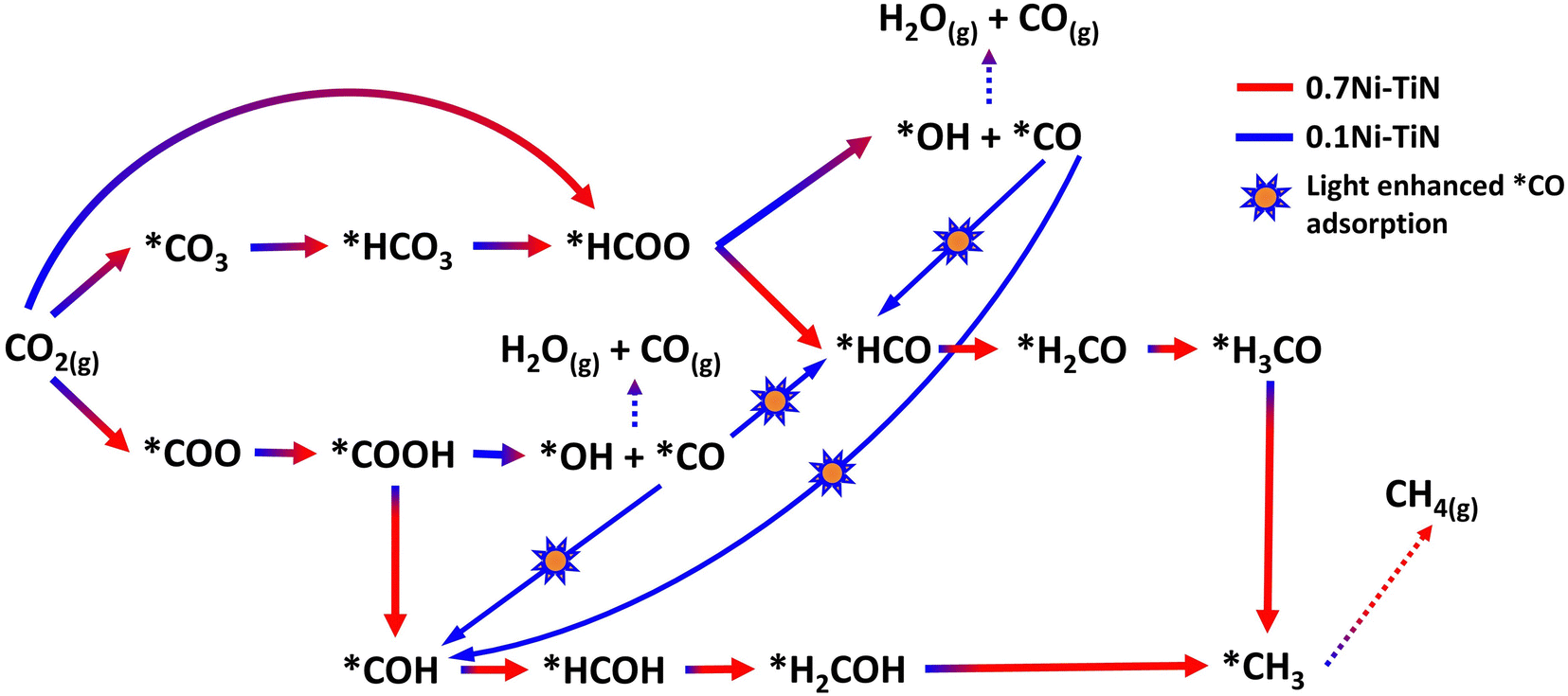 | ||
| Fig. 6 Proposed reaction mechanism over 0.7Ni–TiN and 0.1Ni–TiN. | ||
Conclusions
The findings from the study on the Ni–TiN catalyst reveal a new pathway to harness the beneficial attributes of light (heat and/or hot electrons) for CO2 methanation with the use of a plasmonic support. While TiN as a support does not actively participate in the reaction, it is able to facilitate visible light absorption. The dominant positive effect of the incident light is reliant on the interaction between surface Ni and the TiN support, which is influenced by the Ni/TiN ratio. At a high Ni loading (40–70 wt%), highly reducible Ni aggregates, whose electronical property are less influenced by the TiN support, were formed. The initial photothermal effect generated by TiN was sufficient to initiate in situ Ni activation under the reaction atmosphere. The formed metallic Ni species, possessing strong vis-IR absorbance capability, were then able to instigate the exothermic methanation reaction. Both the metallic Ni and the exothermic reaction can subsequently generate more heat, further boosting both Ni reduction and the reaction. For the high Ni-loaded samples, light was primarily harnessed as heat. In contrast, at low Ni loading (10 wt%), the Ni deposits were smaller and dispersed. As a result, the topmost Ni surfaces are more strongly influenced by the TiN support due to their closer proximity to the metal–support interface. These Ni deposits possessed higher oxidation states, and in situ Ni activation and methanation did not occur under strictly photothermal conditions despite the greater TiN content. The comparatively stronger interaction between Ni and TiN may instead enable the transfer of a small population of hot electrons from the plasmonic TiN to Ni under visible light-assisted thermal conditions (external heating and white LED illumination). This concomitantly alters the charge density of exposed Ni surfaces, strengthening *CO adsorption to enable further reaction rather than desorption as CO, thereby boosting CH4 selectivity.Coupling TiN with Ni in this work represents a unique strategy where the TiN catalyst support is engaged to access incident light in multiple ways to boost thermal catalytic CO2 methanation performance. Incident light energy captured by the TiN plasmon offers the added advantage of enhancing *CO adsorption by modifying the Ni surface charge to encourage further hydrogenation (thereby boosting CH4 selectivity) alongside the typical conversion of light to heat to invoke thermal catalysis.
To date, visible light-enhanced CO2 methanation has relied on two strategies: (1) employing a basic support to improve the formation of formate intermediates close to metal deposits, where photoexcited charge carriers from the metal then acts to promote formate activation for CH4 synthesis;8,14 or (2) using a plasmonic noble metal to boost CO2 conversion.15,16 The origin and influence of the nonthermal effect presented here is distinct from other studies as the plasmonic effect derives from the support (i.e., TiN) rather than the metal catalyst. By using plasmonic TiN the inclusion of noble metals (e.g., Au, Ag) that would otherwise impact CH4 selectivity by favoring CO is circumvented. Overall, the work offers a new strategy for effectively harnessing light for photo-and-thermal catalytic systems whereby the plasmonic TiN support is used to boost CO2 methanation via enhanced light-induced heating and/or plasmon-induced electronic modification on Ni active sites.
Experimental
Synthesis
Neat TiN materials were prepared via transferred arc plasma technique.18 Ti metal (99.995%) was nitrided under a He and N2 atmosphere inside a vacuum chamber. Plasma arc was ignited by striking the precursor anode with a tungsten cathode. TiN nanoparticles were collected off the chamber walls as product.xNi–TiN samples containing x% Ni loading (x = 10, 40, 70) were synthesised via conventional wet impregnation to deposit Ni onto plasma arc synthesised neat TiN. Calculated amounts of nickel(II) nitrate hexahydrate (98%, Ajax Finechem) were used to create synthesis mixtures containing 40 mg mL−1 of neat TiN. Synthesis mixtures were stirred using a magnetic stirrer, with N2 bubbling through the suspension for 1.5 h. Stirred suspensions were dried in a vacuum oven at 80 °C for 5 h. The oven dried synthesis mixtures were loaded into a tubular reactor and purged overnight in N2, followed by calcination under flowing N2 at 450 °C (5 °C min−1) for 4 h to synthesise xNi–TiN samples.
xNi–TiON (x = 0.1, 0.7) sample synthesis followed a different calcination procedure. The calcination of oven dried synthesis mixture was performed in a Carbolite horizontal tube furnace without initial N2 purging. N2 flow and temperature ramping (4 h at 450 °C, ramping at 5 °C min−1) commenced simultaneously to synthesize the TiON-supported samples.
Characterisation
XRD was conducted on a PANalytical Xpert multipurpose system with a Cu-Kα X-ray radiation at a wavelength of 0.15406 nm, generated at a Cu anode under the supply with a current at 40 mA and 45 kV. The mean crystallite size (D) for Ni and TiN crystallites in both samples were estimated using the Scherrer equation below based on the full width at half maximum (β) and Bragg angle (θ) obtained from XRD. The shape factor (K) was taken as 0.9. X-ray wavelength (λ) for Cu used was 0.15406 nm.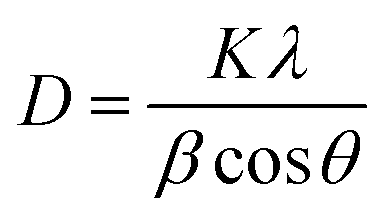 | (6) |
For STEM and HRTEM measurements, individual samples were dispersed within methanol via sonication. The sonicated mixture was then drop casted onto Cu-lacey carbon TEM grids and air dried. The sample laden Cu-lacey carbon TEM grids were pretreated overnight in vacuum to remove surface contaminants. STEM and HRTEM were conducted using a JEOL JEM-F200 at various magnifications.
To determine the ability for light absorption, UV-vis absorption spectra were obtained by subjecting samples to irradiation from 250 to 800 nm. This was achieved using a PerkinElmer Lambda 1050 UV/Vis spectrophotometer coupled with a Praying Mantis accessory (Harrick Scientific Products Inc.). Sample reflectance was measured, with the raw data being processed via Kubelka–Munk transformation to model absorption characteristics.
Assessment of catalyst reducibility via H2-TPR was carried out on a Micromeritics Autochem II 2920. Approximately 10 mg of sample was loaded onto a bed of quartz wool inside a quartz U-tube and heated to 150 °C (10 °C min−1) for 0.5 h in Ar (20 mL min−1) for pre-treatment. After cooling in Ar to room temperature, H2-TPR was performed under 10% H2/Ar (20 mL min−1) at a temperature ramp of 5 °C min−1 to 800 °C.
XPS was carried out in a Thermo ESCALAB250Xi X-ray photoelectron spectrophotometer (Thermo Scientific, UK). Mono-chromated Al K alpha (energy 1486.68 eV) was used as the X-ray source powered at 120 W (13.8 × 8.7 mA). Spot size at 500 micrometres was used, and the photoelectron take-off angle was set at 90° relative to the surface plane. Pass energies were defined at 100 eV for survey scans, and 20 eV for region scans. The binding energy was referenced against C 1s (284.8 eV) for adventitious hydrocarbon.
The transient absorption spectroscopy (TAS) measurements were performed using a HELIOS Transient Absorption Spectrometer, driven by a Spectra Physics Solstice amplified Ti: Sapphire laser operating at 800 nm with an output train of nominally ∼100 fs pulses at 1 kHz. The Solstice output was split, with some used to generate a supercontinuum probe (420 nm to 770 nm) and the remaining power directed to a Light Conversion TOPAS Optical Parametric Amplifier to produce the 400 nm pump beam. TAS measurements were performed in the transmission geometry with a depolarised pump beam of wavelength 400 nm (at intensity of ∼90 mW cm−2, corresponding to fluence of ∼9 × 10−5 J cm−2), kept at ∼10 times the probe size. The samples used for TAS measurements were prepared by sonicating (within an ice bath) 5 mg of sample powder in 4 mL of glycerol for ∼2.5 h to enable dispersion.
Catalytic testing
Illuminated and dark reactions under constant heat supply
Externally heated activity tests were completed under both dark and illuminated conditions to decouple the effect of light and heat. All reactions were carried out inside a Harrick reactor with an 8 mm quartz aperture using ∼10 mg of catalysts loaded onto a stainless-steel wire mesh creating a flat catalyst bed. The reactor cover was then secured, with the aperture being covered loosely by a piece of Al foil to prevent ambient light exposure. Prior to reaction, the sample was reduced under a 1:1 H2/N2 gas flow (24 mL min−1), heated to 450 °C (5 °C min−1) and held for 1 h before cooling to 150 °C at a rate of 5 °C min−1.
Reactions were carried out using a stoichiometric reactant mixture of CO2 (4 mL min−1) and H2 (16 mL min−1), accompanied by a N2 (4 mL min−1) standard all injected via mass flow controllers. The reactor system was heated at 5 °C min−1 to raise the temperature from 150 °C to 450 °C. The reactor temperature was held constant for 45 min at each 50 °C increment to monitor performance. The product gas passed through a cold trap after reactor exit for moisture removal. A fixed volume of product gas was sampled at 12-minute intervals throughout the experiment via a timed 6-port valve. Gas samples were analysed via an online GC unit equipped with an Agilent Technologies column (CarboPLOT P7), a temperature conductivity detector (TCD) for H2 and N2 detection and a flame ionisation detector (FID) for CO2, CH4 and CO detection.
Non-illuminated trials were carried out with the Harrick aperture covered using Al foil. Illuminated trials were carried out using white light emitted from an LED panel (31.4 V and 2.80 A, ∼0.9 W cm−2) mounted over the Harrick aperture. The light panel was attached to a heat sink equipped with a fan to assist heat dissipation. The reactor setup was then enclosed using black cardboard to prevent light leakage. A process flow diagram of the entire reactor set-up, and labelled photograph of the Harrick reactor detailing light placement is provided in Fig. S3 and S4 (ESI†), respectively.
Peak areas from the FID and TCD were normalised against the inert N2 fixed at 4 mL min−1 to account for volume changes in the gas mixture during reaction. Normalised peaks areas were converted into experimental component flow rates. CO2 conversion and CH4 selectivity were calculated based on respective flowrates using equations.
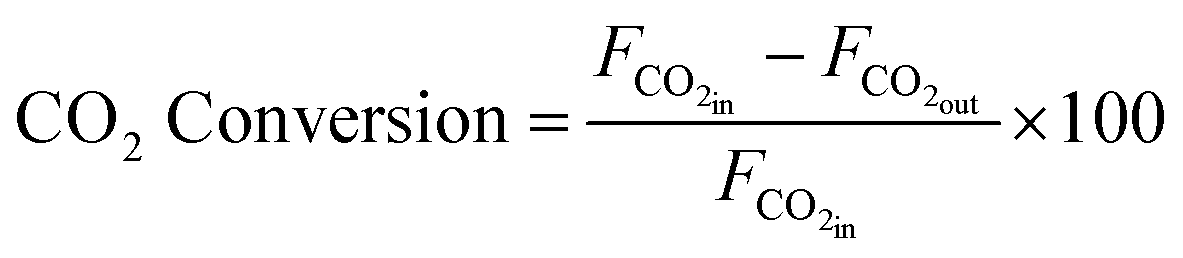 | (7) |
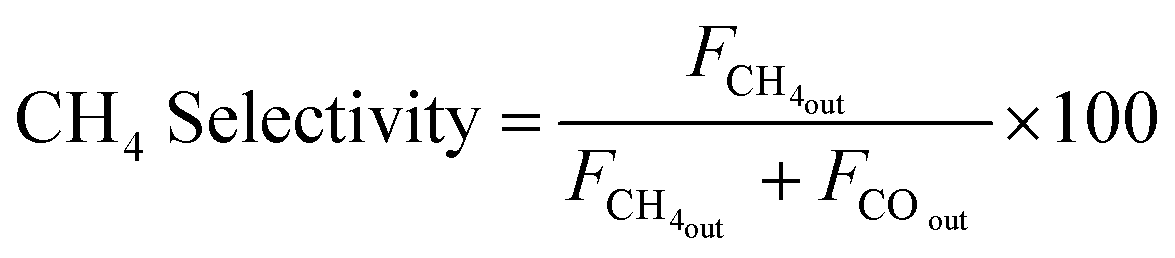 | (8) |
In situ DRIFTS
in situ DRIFTS was performed using a Brucker VERTEX 70v FTIR spectrometer with N2 cooled mid-infrared (MIR) source equipped with KBr optics, and a RockSolid interferometer previously described elsewhere.14 Samples were first diluted (100:0.75 ratio for KBr:sample or 100:20 ratio for diamond:sample, by weight) prior to analysis to improve signal strength due to high vis-NIR light absorption by the black samples after reduction. Approximately 50 mg of neat diluent were first loaded into the in situ Harrick cell fitted with ZnSe windows within the Praying Mantis accessory (Harrick Scientific Inc.) with the remaining volume being filled by the diluted sample. Prior to reaction, all diluted samples were reduced in situ under the same temperature and flow conditions as the Harrick reaction (450 °C, 1 h, H2:N2 = 1:1, 24 mL min−1). The reactor temperature was then decreased at 50 °C intervals to obtain background spectra at each temperature after stabilisation. Under typical reaction conditions (CO2:H2:He = 1:4:1, ∼12 mL min−1), transmission spectra at each temperature (typically 50–450 °C, at 50 °C intervals) were collected with corresponding temperature backgrounds applied. Each spectrum was collected from 4000 cm−1 to 600 cm−1 (64 scans, resolution of 2 cm−1).
Modelling and DFT calculations
A Ni(111) surface and a Ni pyramidical nanocluster were used to represent Ni-rich (0.7Ni–TiN) and Ni-poor (0.1Ni–TiN) surfaces, respectively, during the modelling. The Ni(111) surface was selected to model Ni sites on the 0.7Ni–TiN based on a low surface energy, which indicates the required energy formation of this Ni surface among other surfaces.51 Photoexcitation conditions were represented based on the assumption of electron excitation/transfer within the system upon illumination. This was mimicked by adding electrons to the system and analysing the impact on the electronic structure.52DFT calculations were performed using the Projector Augmented Wave (PAW)53,54 method as implemented in the Vienna Ab initio Simulation Package (VASP).55,56 The calculations were completed with a plane-wave cut-off energy of 500 eV and Gamma k-points mesh of 5 × 5 × 1. The slab method with a vacuum thickness of 20 Å was used to model Ni(111) and Ni-cluster/TiN(200) surfaces with four atomic layer thickness. Ni-cluster is modelled with five atomic pyramid structures on top of TiN(200) surface. The electronic self-consistent calculation was converged to 1 × 10−5 eV and ionic relaxation steps were performed using the conjugate-gradient method (IBRION = 2) and continued until the total force on each atom dropped below a tolerance of 2 × 10−2 eV Å−1. The generalised gradient approximation (GGA) was used for the exchange correlation functionals as parameterized by Perdew–Burke–Ernzerhof (PBE).57 Dispersion energy corrections were performed using the Grimme method. Bader charge analysis were performed to calculate the charge distribution within the systems. The zero-point energy and entropy corrections for molecules studied were considered on the calculation of free-energy diagram.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The work was supported by the Australian Research Council (ARC) Training Centre for Global Hydrogen Economy (IC200100023), ARC Discovery Project (DP230101861), and ARC DE230100789. TiN samples synthesised at CSIRO were supplied by Dr Avi Bendavid. HRTEM, STEM and EDS mapping were performed at University of Wollongong by Dr Lachlan Smillie. The authors further acknowledge the use of facilities and services within the Mark Wainwright Analytical Centre (MWAC).References
- A. A. Olajire, J. CO2 Util., 2013, 3–4, 74–92 CrossRef CAS.
- J. Zhao, Q. Yang, R. Shi, G. I. N. Waterhouse, X. Zhang, L.-Z. Wu, C.-H. Tung and T. Zhang, NPG Asia Mater., 2020, 12, 5 CrossRef CAS.
- J. Zhao, R. Shi, G. I. N. Waterhouse and T. Zhang, Nano Energy, 2022, 102, 107650 CrossRef CAS.
- Y. Wang, L. Wang, Z. Liu, E. Ye, J. H. Pan, G. Guan and Z. Li, Appl. Catal., A, 2022, 644, 118836 CrossRef CAS.
- W. Tu, Y. Zhou and Z. Zou, Adv. Mater., 2014, 26, 4607–4626 CrossRef CAS PubMed.
- B. Xie, E. Lovell, T. H. Tan, S. Jantarang, M. Yu, J. Scott and R. Amal, J. Energy Chem., 2021, 59, 108–125 CrossRef CAS.
- A. Quindimil, U. De-La-Torre, B. Pereda-Ayo, A. Davó-Quiñonero, E. Bailón-García, D. Lozano-Castelló, J. A. González-Marcos, A. Bueno-López and J. R. González-Velasco, Catal. Today, 2020, 356, 419–432 CrossRef CAS.
- S. Ullah, E. C. Lovell, T. H. Tan, B. Xie, P. V. Kumar, R. Amal and J. Scott, Appl. Catal., B, 2021, 294, 120248 CrossRef CAS.
- S. Jantarang, E. C. Lovell, T. H. Tan, J. Scott and R. Amal, Prog. Nat. Sci.: Mater. Int., 2018, 28, 168–177 CrossRef CAS.
- D. Mateo, J. C. Navarro, I. S. Khan, J. Ruiz-Martinez and J. Gascon, Molecules, 2022, 27, 2701 CrossRef CAS PubMed.
- D. Mateo, N. Morlanes, P. Maity, G. Shterk, O. F. Mohammed and J. Gascon, Adv. Funct. Mater., 2021, 31, 2008244 CrossRef CAS.
- Y. He, Y. Zhou, J. Feng and M. Xing, EFM, 2022, 1, 204–217 Search PubMed.
- C.-Y. Sun, Z.-W. Zhao, H. Liu and H.-Q. Wang, Rare Met., 2022, 41, 1403–1405 CrossRef CAS.
- T. H. Tan, B. Xie, Y. H. Ng, S. F. B. Abdullah, H. Y. M. Tang, N. Bedford, R. A. Taylor, K.-F. Aguey-Zinsou, R. Amal and J. Scott, Nat. Catal., 2020, 3, 1034–1043 CrossRef CAS.
- J. N. G. Stanley, I. García-García, T. Perfrement, E. C. Lovell, T. W. Schmidt, J. Scott and R. Amal, Chem. Eng. Sci., 2019, 194, 94–104 CrossRef CAS.
- I. García-García, E. C. Lovell, R. J. Wong, V. L. Barrio, J. Scott, J. F. Cambra and R. Amal, ACS Sustainable Chem. Eng., 2020, 8, 1879–1887 CrossRef.
- Z. Wu, C. Li, Z. Li, K. Feng, M. Cai, D. Zhang, S. Wang, M. Chu, C. Zhang, J. Shen, Z. Huang, Y. Xiao, G. A. Ozin, X. Zhang and L. He, ACS Nano, 2021, 15, 5696–5705 CrossRef CAS PubMed.
- S. Yick, A. Murdock, P. Martin, D. Kennedy, T. Maschmeyer and A. Bendavid, Nanoscale, 2018, 10, 7566–7574 RSC.
- A. Alvarez Barragan, N. V. Ilawe, L. Zhong, B. M. Wong and L. Mangolini, J. Phys. Chem. C, 2017, 121, 2316–2322 CrossRef CAS.
- S. L. Shinde, S. Ishii and T. Nagao, ACS Appl. Mater. Interfaces, 2019, 11, 21965–21972 CrossRef CAS PubMed.
- S. Ishii, S. L. Shinde, W. Jevasuwan, N. Fukata and T. Nagao, ACS Photonics, 2016, 3, 1552–1557 CrossRef CAS.
- K.-G.-G. C. De Silva, M. Finale and S. Chowdhury, Mater. Res. Bull., 2022, 152, 111834 CrossRef CAS.
- J. Zhang, A. Reda Woldu, X. Zhao, X. Peng, Y. Song, H. Xia, F. Lu, P. K. Chu and L. Hu, Appl. Surf. Sci., 2022, 598, 153745 CrossRef CAS.
- A. A. Barragan, S. Hanukovich, K. Bozhilov, S. S. R. K. C. Yamijala, B. M. Wong, P. Christopher and L. Mangolini, J. Phys. Chem. C, 2019, 123, 21796–21804 CrossRef CAS.
- S. H. Gage, C. Ngo, V. Molinari, M. Causà, R. M. Richards, F. S. Gentile, S. Pylypenko and D. Esposito, J. Phys. Chem. C, 2018, 122, 339–348 CrossRef CAS.
- N. C. Saha and H. G. Tompkins, J. Appl. Phys., 1992, 72, 3072–3079 CrossRef CAS.
- B. Siemensmeyer, K. Bade and J. W. Schultze, Ber. Bunsenges. Phys. Chem., 1991, 95, 1461–1469 CrossRef CAS.
- A. Trenczek-Zajac, M. Radecka, K. Zakrzewska, A. Brudnik, E. Kusior, S. Bourgeois, M. C. M. de Lucas and L. Imhoff, J. Power Sources, 2009, 194, 93–103 CrossRef CAS.
- A. Glaser, S. Surnev, F. Netzer, N. Fateh, G. Fontalvo and C. Mitterer, Surf. Sci., 2007, 601, 1153–1159 CrossRef CAS.
- A. P. Grosvenor, M. C. Biesinger, R. S. C. Smart and N. S. McIntyre, Surf. Sci., 2006, 600, 1771–1779 CrossRef CAS.
- E. L. Ratcliff, J. Meyer, K. X. Steirer, A. Garcia, J. J. Berry, D. S. Ginley, D. C. Olson, A. Kahn and N. R. Armstrong, Chem. Mater., 2011, 23, 4988–5000 CrossRef CAS.
- N. Weidler, J. Schuch, F. Knaus, P. Stenner, S. Hoch, A. Maljusch, R. Schäfer, B. Kaiser and W. Jaegermann, J. Phys. Chem. C, 2017, 121, 6455–6463 CrossRef CAS.
- K. Naim Ahmad, S. Samidin, F. Salleh, W. Nor Roslam Wan Isahak, A. Al-Amiery, M. Shahbudin Masdar, M. Rahimi Yusop, W. Khalid Al-Azzawi, M. Irwan Rosli and M. Ambar Yarmo, ChemistrySelect, 2022, 7, e202202394 CrossRef CAS.
- M. El-Kemary, N. Nagy and I. El-Mehasseb, Mater. Sci. Semicond. Process., 2013, 16, 1747–1752 CrossRef CAS.
- Z. Li, R. Shi, J. Zhao and T. Zhang, Nano Res., 2021, 14, 4828–4832 CrossRef CAS.
- A. Cárdenas-Arenas, H. S. Cortés, E. Bailón-García, A. Davó-Quiñonero, D. Lozano-Castelló and A. Bueno-López, Fuel Process. Technol., 2021, 212, 106637 CrossRef.
- S. Ullah, E. C. Lovell, R. J. Wong, T. H. Tan, J. Scott and R. Amal, ACS Sustainable Chem. Eng., 2020, 8, 5056–5066 CrossRef CAS.
- B. T. Diroll, S. Saha, V. M. Shalaev, A. Boltasseva and R. D. Schaller, Adv. Opt. Mater., 2020, 8, 2000652 CrossRef CAS.
- B. Doiron, Y. Li, R. Bower, A. Mihai, S. Dal Forno, S. Fearn, L. Hüttenhofer, E. Cortés, L. F. Cohen, N. M. Alford, J. Lischner, P. Petrov, S. A. Maier and R. F. Oulton, ACS Appl. Mater. Interfaces, 2023, 15, 30417–30426 CrossRef CAS PubMed.
- X. Yan, W. Sun, L. Fan, P. N. Duchesne, W. Wang, C. Kübel, D. Wang, S. G. H. Kumar, Y. F. Li, A. Tavasoli, T. E. Wood, D. L. H. Hung, L. Wan, L. Wang, R. Song, J. Guo, I. Gourevich, F. M. Ali, J. Lu, R. Li, B. D. Hatton and G. A. Ozin, Nat. Commun., 2019, 10, 2608 CrossRef PubMed.
- Y. Yu, Y. M. Chan, Z. Bian, F. Song, J. Wang, Q. Zhong and S. Kawi, Int. J. Hydrogen Energy, 2018, 43, 15191–15204 CrossRef CAS.
- J. Graciani, K. Mudiyanselage, F. Xu, A. E. Baber, J. Evans, S. D. Senanayake, D. J. Stacchiola, P. Liu, J. Hrbek, J. F. Sanz and J. A. Rodriguez, Science, 2014, 345, 546–550 CrossRef CAS PubMed.
- B. Xie, R. J. Wong, T. H. Tan, M. Higham, E. K. Gibson, D. Decarolis, J. Callison, K.-F. Aguey-Zinsou, M. Bowker, C. R. A. Catlow, J. Scott and R. Amal, Nat. Commun., 2020, 11, 1615 CrossRef CAS PubMed.
- N. Schreiter, J. Kirchner and S. Kureti, Catal. Commun., 2020, 140, 105988 CrossRef CAS.
- C. Vogt, E. Groeneveld, G. Kamsma, M. Nachtegaal, L. Lu, C. J. Kiely, P. H. Berben, F. Meirer and B. M. Weckhuysen, Nat. Catal., 2018, 1, 127–134 CrossRef CAS.
- R.-P. Ye, Q. Li, W. Gong, T. Wang, J. J. Razink, L. Lin, Y.-Y. Qin, Z. Zhou, H. Adidharma, J. Tang, A. G. Russell, M. Fan and Y.-G. Yao, Appl. Catal., B, 2020, 268, 118474 CrossRef CAS.
- A. Sápi, G. Halasi, J. Kiss, D. G. Dobó, K. L. Juhász, V. J. Kolcsár, Z. Ferencz, G. Vári, V. Matolin, A. Erdőhelyi, Á. Kukovecz and Z. Kónya, J. Phys. Chem. C, 2018, 122, 5553–5565 CrossRef.
- H. L. Huynh, J. Zhu, G. Zhang, Y. Shen, W. M. Tucho, Y. Ding and Z. Yu, J. Catal., 2020, 392, 266–277 CrossRef CAS.
- J. Ren, H. Guo, J. Yang, Z. Qin, J. Lin and Z. Li, Appl. Surf. Sci., 2015, 351, 504–516 CrossRef CAS.
- X. Jia, X. Zhang, N. Rui, X. Hu and C.-J. Liu, Appl. Catal., B, 2019, 244, 159–169 CrossRef CAS.
- W.-B. Zhang, C. Chen and S.-Y. Zhang, J. Phys. Chem. C, 2013, 117, 21274–21280 CrossRef CAS.
- G. E. P. O’Connell, T. H. Tan, J. A. Yuwono, Y. Wang, A. Kheradmand, Y. Jiang, P. V. Kumar, R. Amal, J. Scott and E. C. Lovell, Appl. Catal., B, 2024, 343, 123507 CrossRef.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ey00315a |
| This journal is © The Royal Society of Chemistry 2024 |