DOI:
10.1039/D3SU00149K
(Tutorial Review)
RSC Sustain., 2023,
1, 1655-1671
High-pressure hydrogen generation from dehydrogenation of formic acid
Received
15th May 2023
, Accepted 9th August 2023
First published on 10th August 2023
Abstract
Recently, it has been demonstrated that high-pressure hydrogen gas was produced by dehydrogenation of formic acid (FA). This technology may reduce the overall energy and cost required for the multiple stages of compression of high-pressure hydrogen supply at present. Some successful results towards this goal have been achieved by the scientific community so far. In this review, we present the recent advances in the development of both homogeneous and heterogeneous catalysts for high-pressure gas generation (H2 and CO2) from formic acid.
 Soumyadip Patra | Soumyadip Patra completed his BSc in Chemistry from the University of Calcutta and subsequently did his MSc in Chemistry from Indian Institute of Technology (ISM) Dhanbad, India. Then, he joined the Catalysis group, Indian Institute of Technology, Indore as a PhD student under the supervision of Prof. Sanjay Kumar Singh. His doctoral thesis primarily focuses on the Development of Active Molecular Catalysts for Hydrogen Production from C-1 Based LOHCs. After completing his PhD, he moved to Japan and is presently working as a postdoctoral scientist under Prof. Yuichiro Himeda at AIST Tsukuba. His research interest lies in Reversible Hydrogen Storage in LOHCs. |
 Babulal Maji | Babulal Maji completed his Master of Science in Chemistry from IIT Kharagpur in 2016. He then received his PhD from IISER Bhopal, India with Prof. Joyanta Choudhury in 2022. During his PhD tenure, he developed a water soluble Ir-NHC catalyst for reversible hydrogen storage under ambient conditions in N-heteroarenes and HCOOH platforms. After that he joined the Global Zero Emission Research Centre (GZR), Advanced Industrial Science and Technology (AIST), Japan, with Prof. Himeda as Post-Doctoral Researcher in 2023. Currently, Maji is working on the development of active and robust catalysts for the generation of methanol from CO2 and dihydrogen from formic acid. |
 Hajime Kawanami | Hajime Kawanami is Chief Senior Researcher at the National Institute of Advanced Industrial Science and Technology (AIST), and Professor of University of Tsukuba, Collaborative Graduate School Program. He graduated from Tohoku University and received a PhD from Tohoku University in 1997. Then, he was Research Associate at Kindai University, and in 2001, he joined AIST. He is studying organic chemistry under high-pressure and supercritical fluid conditions, and one of his present interests is CO2 utilization using high-pressure technology. He recently received The Ichimura Prize in Science against Global Warming for Distinguished Achievement, in 2019. |
 Yuichiro Himeda | Yuichiro Himeda is a Prime Senior Researcher at the National Institute of Advanced Industrial Science and Technology (AIST). He works on the development of state-of-the-art catalysis for H2 storage and CO2 reduction. His research interests include the development of catalysts based on new concepts, activation of small molecules, and CO2 utilization for energy storage. |
Sustainability spotlight
To achieve the goal of a carbon-neutral society, the gradual shift to sustainable and renewable energy sources is of prime importance. In this regard, hydrogen (H2) gas owing to its high gravimetric energy density is considered as one of the most promising energy carriers for the future. However, the difficulties associated with the storage and transportation of gaseous H2 due to its low volumetric energy density limit its usage. Therefore, liquid-phase hydrogen carriers have gained considerable attention as a promising way of storing H2. Formic acid (FA), having a H2 content of 4.4 wt%, can release H2 with the help of a suitable catalyst under mild reaction conditions. Furthermore, some research groups including us have successfully developed catalysts capable of producing high-pressure gas from FADH. On the other hand, the high-pressure hydrogen supply is facing difficulties due to the costly gas-compression and cumbrous handling, which seems to be hindering the widespread use of hydrogen fuelling stations. We hope that the scientific developments compiled in this review would pave the way for the development of high-performance catalytic systems for high-pressure H2 generation from FA and become an innovative technology in hydrogen fuelling stations in the near future. Our work emphasizes the importance of the following UN sustainable development goals: affordable and clean energy (SDG 7), industry, innovation, and infrastructure (SDG 9), and climate action (SDG 13).
|
1. Introduction
In recent years, the scientific community has devoted considerable attention to the development of alternate and sustainable energy technologies in response to the rising levels of different forms of pollution worldwide and alarming levels of global warming associated with it. Hydrogen (H2) is considered as a prime candidate to replace the fossil fuel-based economy because it possesses much higher gravimetric energy density as compared to most natural gases and it reacts with oxygen in fuel cells to generate electricity with the formation of water as the only byproduct.1–6 Therefore, it is a very effective and clean alternate energy carrier for the sustainable development of our society but there are certain challenges we must overcome for the implementation of a hydrogen-based economy. H2 has a very low volumetric energy density which makes it difficult to store and transport safely.1,7,8 Therefore, the development of safe and efficient H2 storage materials is of prime importance in the gradual shift from a fossil fuel-based economy to a hydrogen-based one.9,10| | | HCOOH(l) → H2(g) + CO2(g) ΔH° = +31.2 kJ mol−1, ΔG° = −32.9 kJ mol−1 | (1) |
| | | MHCO2(aq) + H2O(l) → H2(aq) + MHCO3(aq) ΔH° = +20.5 kJ mol−1, ΔG° = +0.72 kJ mol−1 | (2) |
Formic acid (FA; HCOOH) is the simplest carboxylic acid available in nature. It is a colourless liquid with a pungent odour, moderate corrosiveness, and relatively low toxicity. However, it is widely used as a food additive, preservative, dye, rubber, leather, textile, and bactericide. On the other hand, FA which contains 4.4 wt% of H2 is considered as one of the attractive H2 storage materials because of its liquid nature, making its storage and transportation relatively easier as compared to gaseous H2. The dehydrogenation of FA (FADH) to CO2 and H2 is endothermic but an exergonic reaction (eqn (1)), where FA stores chemical energy and subsequently, it can generate high-pressure H2 during the dehydrogenation reaction. In contrast, formate does not have sufficient energy to release high-pressure H2 by dehydrogenation (eqn (2)).11,12
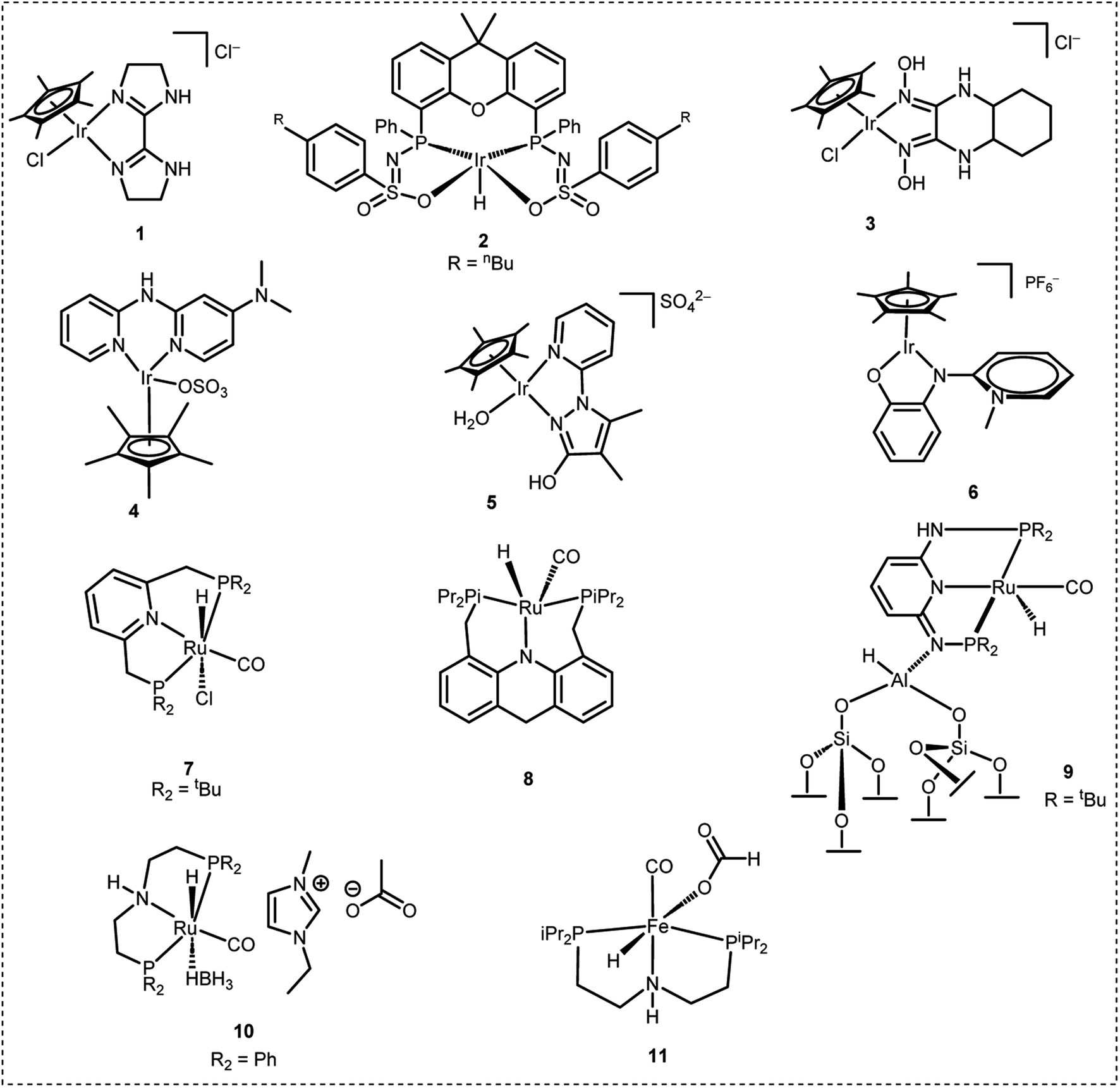
H2 can be released from FA on demand with the help of suitable catalysts under mild reaction conditions.13–21 This has drawn the attention of many researchers and several very active homogeneous as well as heterogeneous catalysts have been developed over the past 15 years which efficiently dehydrogenate FA to H2 and CO2 selectively.22–50 In this context, iridium based catalytic systems were found to be most effective. For instance, in 2015, Li et al. developed a Cp*-Ir(III) catalyst containing a bisimidazoline ligand (1 in Fig. 1) which displayed an exceptionally high TOF of 487![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 h−1 for FADH in water.26 In the same year, Reek et al. developed an Ir-bisMETAMORphos complex (2 in Fig. 1) for the base-free dehydrogenation of formic acid. Mechanistic studies revealed that this type of ligand can act as an internal base and aids in the stabilization of the rate-determining transition state through hydrogen bonding.44 In 2018, Li et al. explored some Cp*-Ir(III) catalysts with a glyoxime ligand framework (3 in Fig. 1) and achieved a TON of almost 5 million in FADH.27 In 2019, Fischmeister et al. reported the base-free dehydrogenation of formic acid with an iridium(III) Cp*(dipyridylamine) catalyst (4 in Fig. 1) and achieved a TOF of 13292 h−1 at 100 °C. The dipyridylamine ligands possibly operated via outer-sphere interactions.45 In the same year, we developed the Cp*-Ir(III) catalyst with a pyridyl pyrazole ligand framework (5 in Fig. 1) which was stable in water at reflux temperature (100 °C) and FA was completely consumed during the dehydrogenation reaction. The active catalyst displayed long-term stability towards FADH (35 days) at 70 °C to achieve a TON of 10 million.32 Recently, Albrecht et al. developed a Cp*-Ir(III) catalyst containing a phenoxy substituted pyridylidene-amine (PYE) ligand framework (6 in Fig. 1) to achieve a TOF of 280000 h−1 for FADH at 100 °C in DMSO. TONs of up to 3 million were achieved with this simple catalytic system.31 Among the other metal-based catalysts active for FADH, Ru-based complexes were also explored. For instance, Dupont et al. reported FADH with the Ru complex [{RuCl2(p-cymene)}2] dissolved in the ionic liquid (IL) 1-(2-(diethylamino)ethyl)-3-methylimidazolium chloride at 80 °C without any additional bases. It was found that the IL stabilizes the active catalytic species and the immobilized amine group acts as a crucial promoter for FADH.46 Pidko et al. reported a highly active Ru-PNP based pincer complex (7 in Fig. 1) for the reversible (de)hydrogenation reaction with a very high TON/TOF in the presence of a DMF/Et3N mixture.47 Grützmacher et al. reported efficient base-free dehydrogenation of formic acid by the ruthenium complex RuH2(PPh3)4 at 60 °C. TOFs of up to 36000 h−1 could be achieved with this catalytic system.48 Subsequently, Milstein et al. also developed a 9H-acridine Ru-based PNP pincer complex (8 in Fig. 1) with excellent stability and activity in the presence of neat formic acid.33 In 2022, Yaacoub et al. have reported a ruthenium PN3P pincer complex immobilized on a fibrous silica nanosphere with strong Lewis acidity (9 in Fig. 1) to efficiently catalyze FADH with a TON of 600000 and recyclability of up to 45 cycles.49 Recently, Nielsen et al. explored Ru-PNP complexes (10 in Fig. 1) in combination with an ionic liquid (1-ethyl-3-methylimidazolium acetate, EMIM OAc) for FADH, achieving an overall TON exceeding 18 million.50 In 2014, Hazari and co-workers reported a non-noble metal Fe based PNP-pincer complex (11 in Fig. 1) for FADH to achieve TONs of up to 983642 using LiBF4 as an additive.41,42 From the point of view of implementation, DENS is a Netherlands based start-up company which aims to explore the possibilities of formic acid as a potential hydrogen carrier for sustainable energy fuel. They have developed a hydrozine generator which can be utilized to supply energy. Although there is considerable progress in the development of catalysts for H2 generation from FA, the results are still limited from the practical point of view where we need to focus on the high-pressure gas release from FADH and separation of the H2 and CO2. This would be an important step towards the implementation of a hydrogen-based economy.
500 h−1 for FADH in water.26 In the same year, Reek et al. developed an Ir-bisMETAMORphos complex (2 in Fig. 1) for the base-free dehydrogenation of formic acid. Mechanistic studies revealed that this type of ligand can act as an internal base and aids in the stabilization of the rate-determining transition state through hydrogen bonding.44 In 2018, Li et al. explored some Cp*-Ir(III) catalysts with a glyoxime ligand framework (3 in Fig. 1) and achieved a TON of almost 5 million in FADH.27 In 2019, Fischmeister et al. reported the base-free dehydrogenation of formic acid with an iridium(III) Cp*(dipyridylamine) catalyst (4 in Fig. 1) and achieved a TOF of 13292 h−1 at 100 °C. The dipyridylamine ligands possibly operated via outer-sphere interactions.45 In the same year, we developed the Cp*-Ir(III) catalyst with a pyridyl pyrazole ligand framework (5 in Fig. 1) which was stable in water at reflux temperature (100 °C) and FA was completely consumed during the dehydrogenation reaction. The active catalyst displayed long-term stability towards FADH (35 days) at 70 °C to achieve a TON of 10 million.32 Recently, Albrecht et al. developed a Cp*-Ir(III) catalyst containing a phenoxy substituted pyridylidene-amine (PYE) ligand framework (6 in Fig. 1) to achieve a TOF of 280000 h−1 for FADH at 100 °C in DMSO. TONs of up to 3 million were achieved with this simple catalytic system.31 Among the other metal-based catalysts active for FADH, Ru-based complexes were also explored. For instance, Dupont et al. reported FADH with the Ru complex [{RuCl2(p-cymene)}2] dissolved in the ionic liquid (IL) 1-(2-(diethylamino)ethyl)-3-methylimidazolium chloride at 80 °C without any additional bases. It was found that the IL stabilizes the active catalytic species and the immobilized amine group acts as a crucial promoter for FADH.46 Pidko et al. reported a highly active Ru-PNP based pincer complex (7 in Fig. 1) for the reversible (de)hydrogenation reaction with a very high TON/TOF in the presence of a DMF/Et3N mixture.47 Grützmacher et al. reported efficient base-free dehydrogenation of formic acid by the ruthenium complex RuH2(PPh3)4 at 60 °C. TOFs of up to 36000 h−1 could be achieved with this catalytic system.48 Subsequently, Milstein et al. also developed a 9H-acridine Ru-based PNP pincer complex (8 in Fig. 1) with excellent stability and activity in the presence of neat formic acid.33 In 2022, Yaacoub et al. have reported a ruthenium PN3P pincer complex immobilized on a fibrous silica nanosphere with strong Lewis acidity (9 in Fig. 1) to efficiently catalyze FADH with a TON of 600000 and recyclability of up to 45 cycles.49 Recently, Nielsen et al. explored Ru-PNP complexes (10 in Fig. 1) in combination with an ionic liquid (1-ethyl-3-methylimidazolium acetate, EMIM OAc) for FADH, achieving an overall TON exceeding 18 million.50 In 2014, Hazari and co-workers reported a non-noble metal Fe based PNP-pincer complex (11 in Fig. 1) for FADH to achieve TONs of up to 983642 using LiBF4 as an additive.41,42 From the point of view of implementation, DENS is a Netherlands based start-up company which aims to explore the possibilities of formic acid as a potential hydrogen carrier for sustainable energy fuel. They have developed a hydrozine generator which can be utilized to supply energy. Although there is considerable progress in the development of catalysts for H2 generation from FA, the results are still limited from the practical point of view where we need to focus on the high-pressure gas release from FADH and separation of the H2 and CO2. This would be an important step towards the implementation of a hydrogen-based economy.
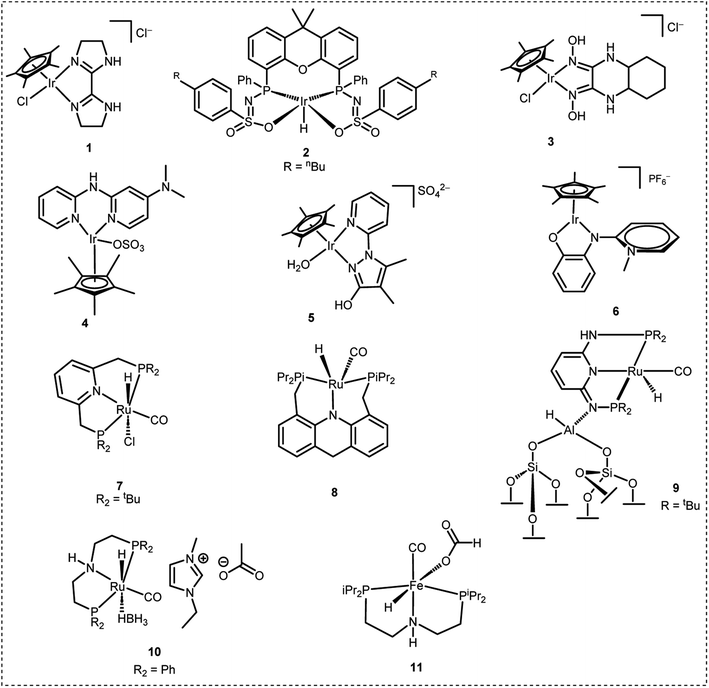 |
| | Fig. 1 Selective catalytic systems for hydrogen production from FA. | |
At present, the major infrastructure in the fuelling stations is dedicated to the mechanical compression equipment for multi-stage compression to achieve the final pressure required to refuel the hydrogen fuel cell electric vehicles at 70 MPa.51–53 The high-pressure H2 supply from liquid-phase hydrogen carriers without multiple stages of compression can significantly lower the investment, maintenance, and operating cost for the mechanical compressor.51 The advantage of FA over other liquid-phase hydrogen carriers is that FADH can provide H2 at high pressures of several hundred bars at near-ambient temperature. However, the separation of one equivalent of CO2 accompanied by the high-pressure H2 is a challenge associated with the implementation of this technology.54,55 Handling of high-pressure hydrogen and implementation of this technology always comes with the risk of explosion. For instance, in 2019, a hydrogen refuelling station in Sandvika, Norway exploded and burst into flames.56 Similarly, in the same year a hydrogen tank explosion occurred in Gangneung, South Korea which led to the death of two people.57 So, utmost precautions should be taken while handling high pressure gaseous hydrogen. As dihydrogen is liberated, a proper setup for safe ventilation of hydrogen gas into the atmosphere is required. Reactions should be carried out under a closed fume hood. The high-pressure reaction should be carried out in a suitable high pressure resistant stainless-steel reactor.
There are several homogeneous as well as heterogeneous catalysts that have been reported to date which can release high-pressure H2 from FA at relatively low reaction temperature.22,58–68 Furthermore, we have contributed towards subsequent separation of CO2 from a high-pressure gas mixture (H2 and CO2) by gas–liquid phase separation.54 In this review, we have tried to summarize the progress achieved in the high-pressure gas generation from FA so far which is in contrast to some recent review articles that focus on the mechanism of heterogeneously catalysed decomposition along with electrooxidation of formic acid and reactor set-ups for hydrogen production from formic acid decomposition.69–71 Special attention is given to our achievement at AIST.54,62–67
2. Homogeneous catalysts for high-pressure hydrogen generation from FA
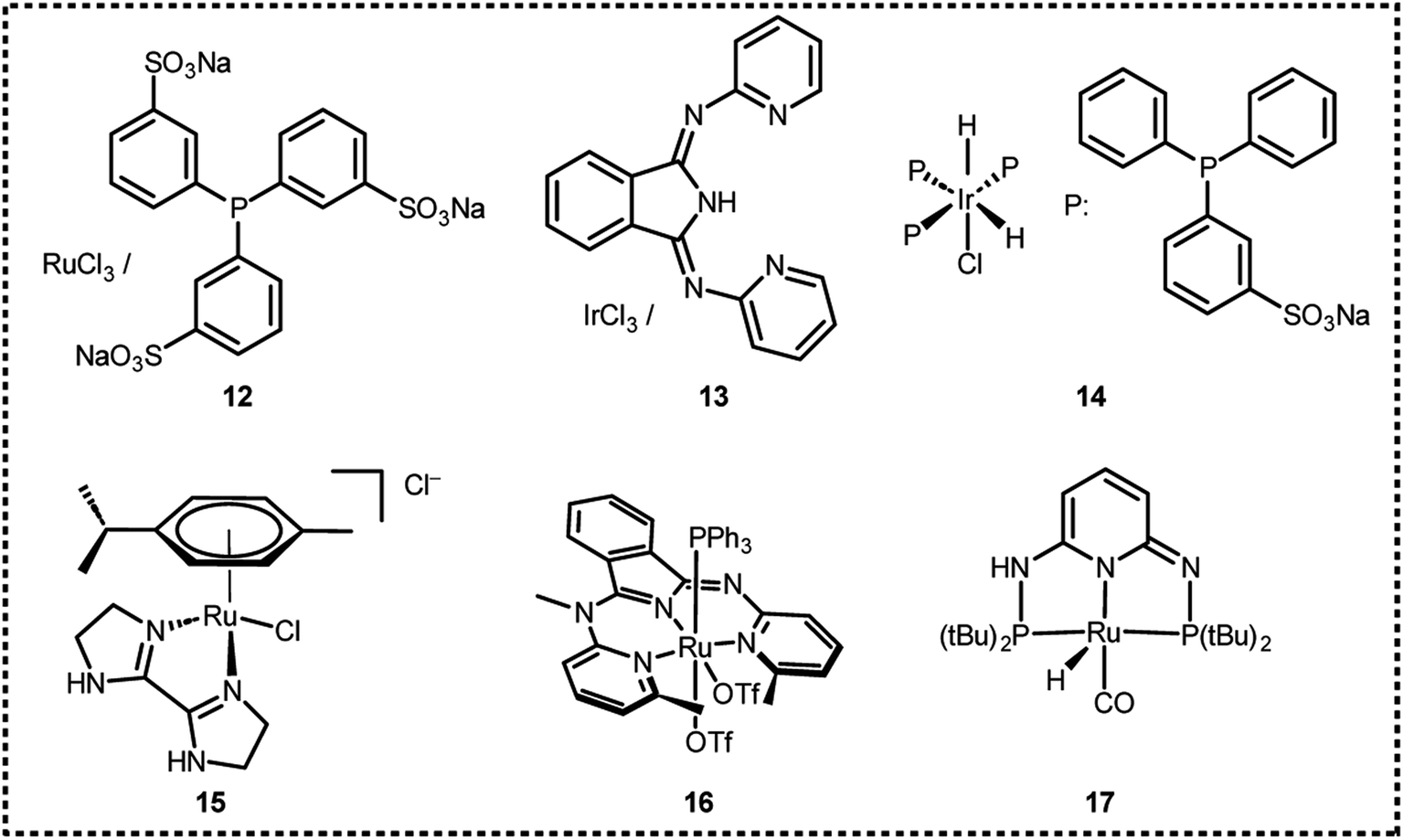
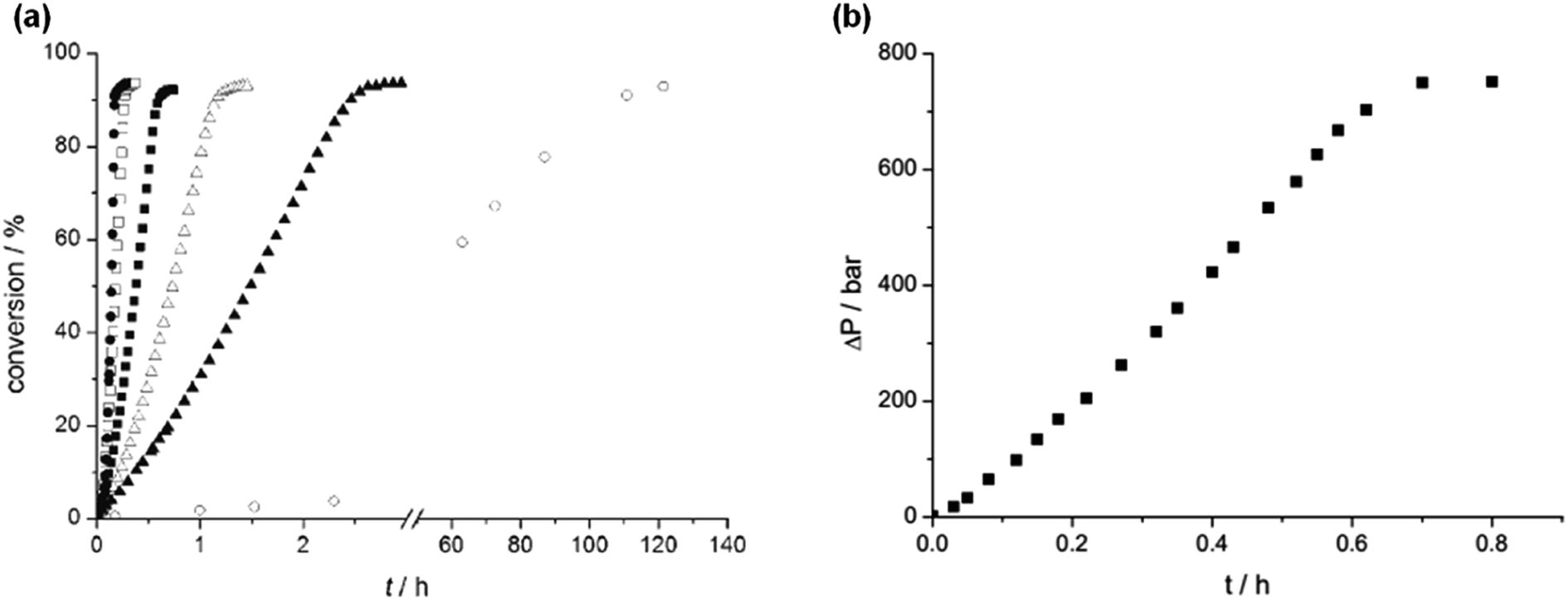
In 2008, Laurenczy et al. reported an efficient and robust catalytic system 12 (Fig. 2 and Table 1) for FADH using a water soluble meta-trisulfonated triphenylphosphine (m-TPPTS) ligand with [Ru(H2O)6]2+ or RuCl3 as the metal precursor.22 The generated H2/CO2 pressure from FADH reached up to 75 MPa without any inhibition of the catalytic activity (Fig. 3). Dehydrogenation was observed at a faster rate at high temperatures. The highly water-soluble homogeneous catalyst was found to be stable up to 170 °C and remained active even after one year in solution. No trace of CO (detection limit of 3 ppm) was detected by FTIR spectroscopy in the gaseous mixture. A continuous H2 generation system was developed by adding FA under pressure into a reactor. The gases generated were released at a constant pressure rate from the reactor. The purity of H2 obtained with this system makes it compatible with all types of fuel cells and the advantage of producing a constant pressure of H2 is that it can be used directly in electric engines without the technical difficulties associated with pressure drops in high-pressure cylinders.22
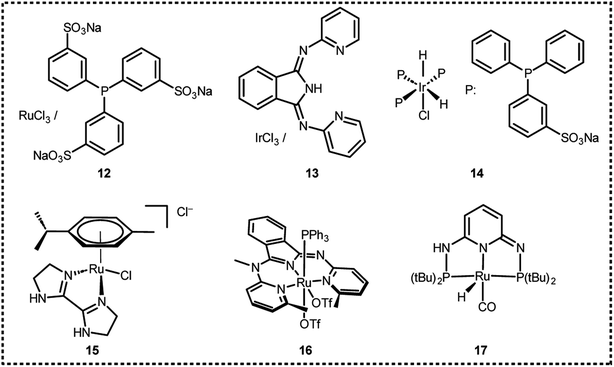 |
| | Fig. 2 Catalysts for high-pressure gas generation from FADH. | |
Table 1 Results of high-pressure gas generation from FADH with homogeneous catalysts
| Entry |
Catalyst |
Temp. (°C) |
Time (h) |
Gas pressure (MPa) |
Ref |
| 1 |
12
|
90 |
0.7 |
75 |
22
|
| 2 |
13
|
112 |
10 |
16 |
58
|
| 3 |
14
|
100 |
0.2 |
14 |
59
|
| 4 |
15
|
80 |
4 |
24 |
60
|
| 5 |
16
|
120 |
3 |
19.2 |
61
|
| 6 |
17
|
80 |
24 |
20 |
62
|
| 7 |
18
|
80 |
12 |
123 |
54
|
| 8 |
20
|
80 |
3 |
110 |
65
|
| 9 |
21
|
80 |
5 |
153 |
55
|
| 10 |
24
|
80 |
4 |
157 |
67
|
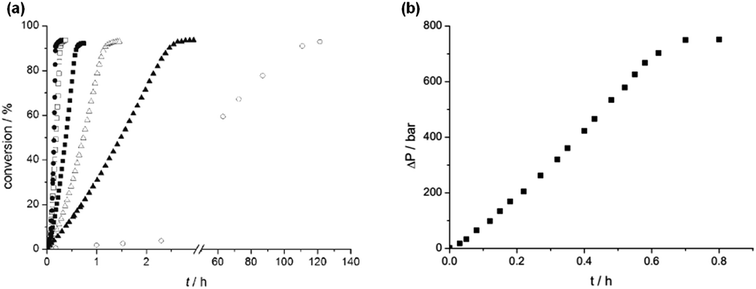 |
| | Fig. 3 (a) Effect of temperature on FADH in a closed batch system with catalyst 12 (25 °C (○), 70 °C (▲), 80 °C (△), 90 °C (■), 100 °C (□), 120 °C (●)); (b) kinetic trace of FADH in a closed system with a pressure increase to 75 MPa. Reproduced with permission.22 Copyright 2008, Wiley-VCH. | |
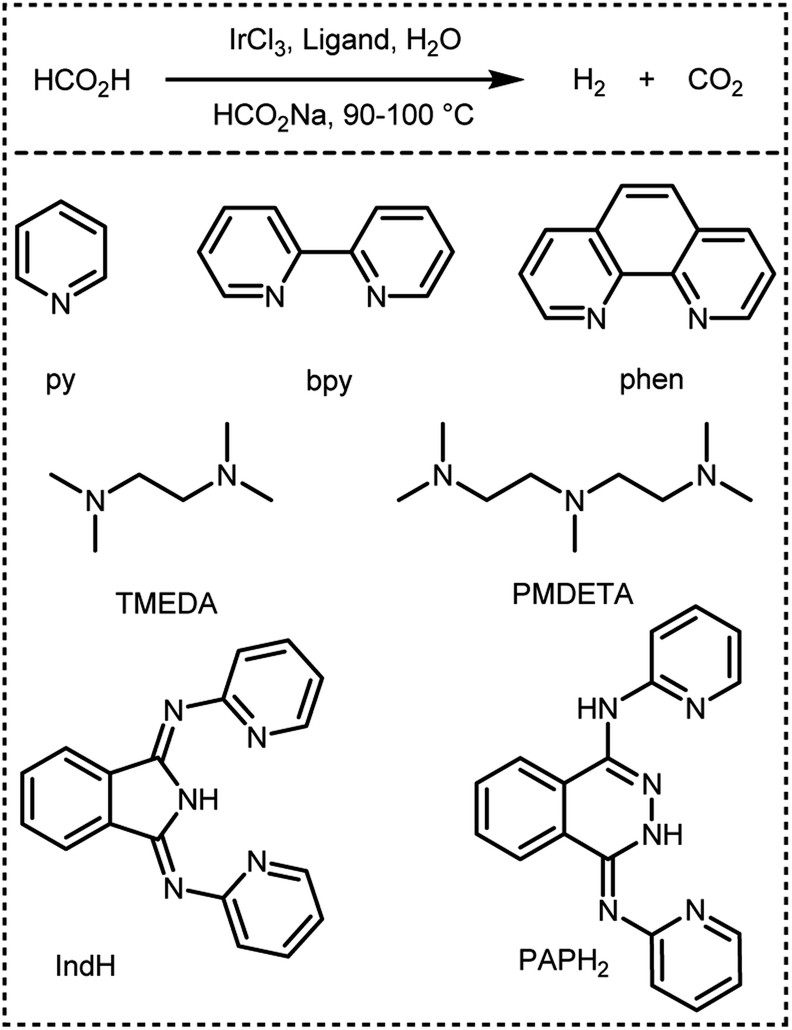
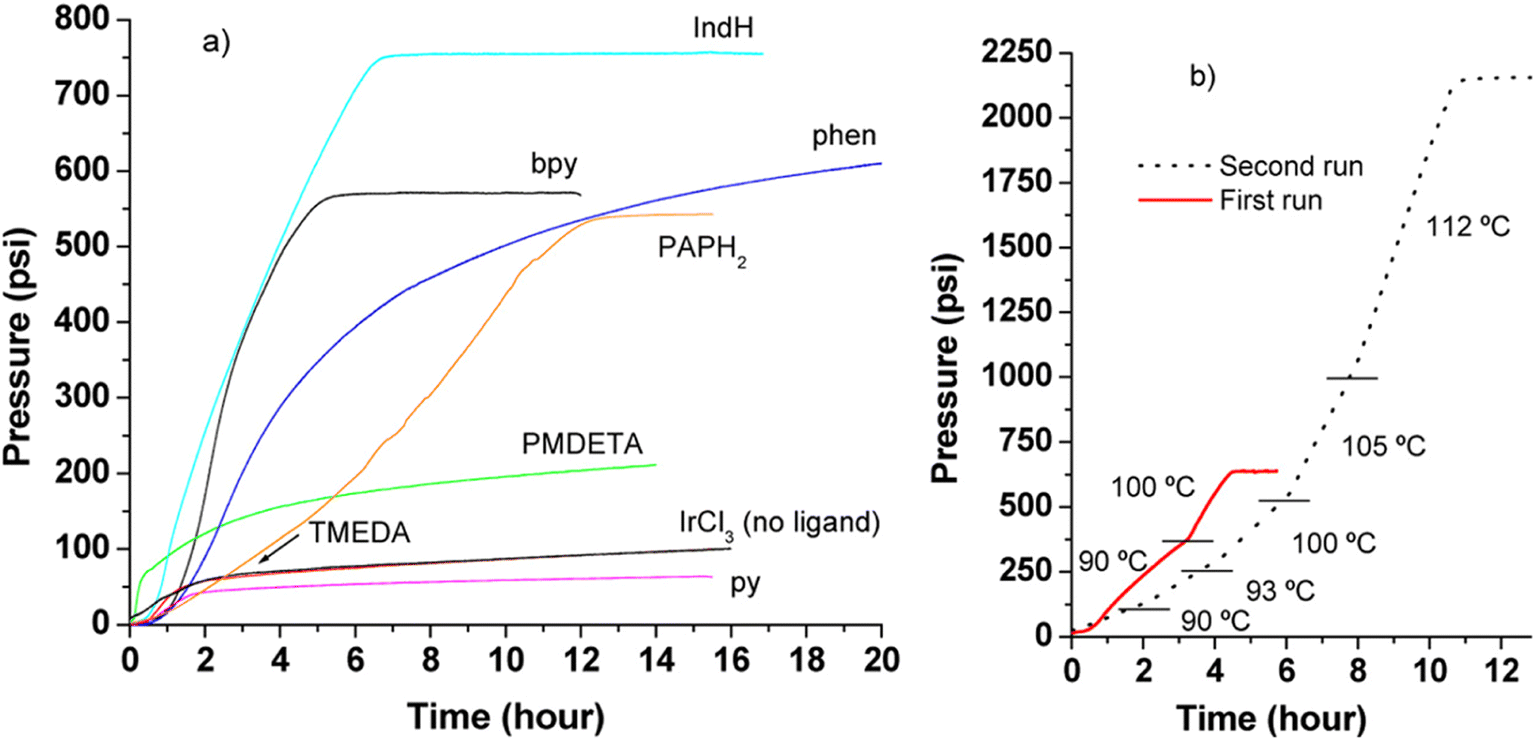
In 2016, Czaun et al. studied a robust catalytic system 13 (Fig. 2 and Table 1) consisting of IrCl3 and 1,3-bis(2′-pyridylimino)isoindoline (IndH) for FADH using [FA] = 3.6 M and [SF] = 0.4 M (SF: sodium formate) in water at 90–100 °C.58 In the preliminary experiments, the authors tested IrCl3 with various mono and bidentate N-donor ligands for FADH. 1,10-Phenanthroline (phen) was less active as compared to 2,2′-bipyridine (bpy) with IrCl3 (Scheme 1). The flexible aliphatic bidentate ligand tetramethylethylenediamine (TMEDA) in combination with IrCl3·H2O displayed very low activity and even lower activity was observed with the monodentate pyridine (py) ligand. The most promising results were obtained with a 1:1 mixture of IrCl3 and IndH in terms of both activity and selectivity (Fig. 4). When the metal to ligand ratio was changed to 1:2, a further increase in activity was observed in the first run but the reaction rate was found to be significantly lower in the second run. Based on these preliminary experiments, the IrCl3:IndH = 1:1 system was selected for further testing the catalytic performance in a continuous reactor setup. High gas-pressure up to 2200 psi (16 MPa) could be generated from FADH with the active catalyst.
 |
| | Scheme 1 Dehydrogenation of FA in the presence of IrCl3 and different N-donor ligands reported by Czaun et al.58 | |
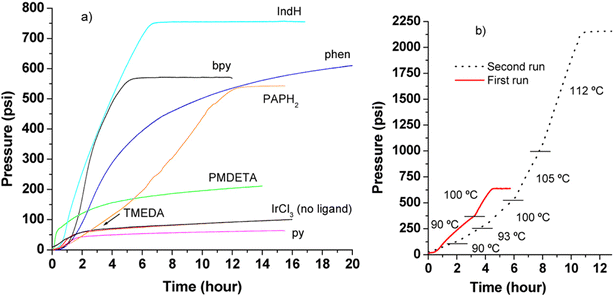 |
| | Fig. 4 Pressure versus time diagram for FADH. (a) First runs, catalyst precursors: IrCl3 and various N-donor ligands, T = 100 °C. (b) IrCl3 in the presence of IndH, first and second runs. [IrCl3]0 = [Ligand]0 = 3.0 mM. Dashes divide the temperature segments, in the first run: 90 and 100 °C; in the second run: 90, 93, 100, 105, and 112 °C, respectively. Reproduced with permission.58 Copyright 2016, American Chemical Society. | |
The continuous FADH reactor was coupled with a polymer exchange membrane (PEM) fuel cell to demonstrate continuous electricity generation by FADH. The catalyst was active after 20 days of continuous use and retained its activity even after one year of its original preparation. GC-TCD analysis of the produced gas mixture revealed an equimolar (1:1) ratio of H2/CO2 and the absence of CO. Through this work, the authors successfully demonstrated that the chemical energy stored in FA can be converted to electricity continuously with the help of a hydrogen-air PEM fuel cell.
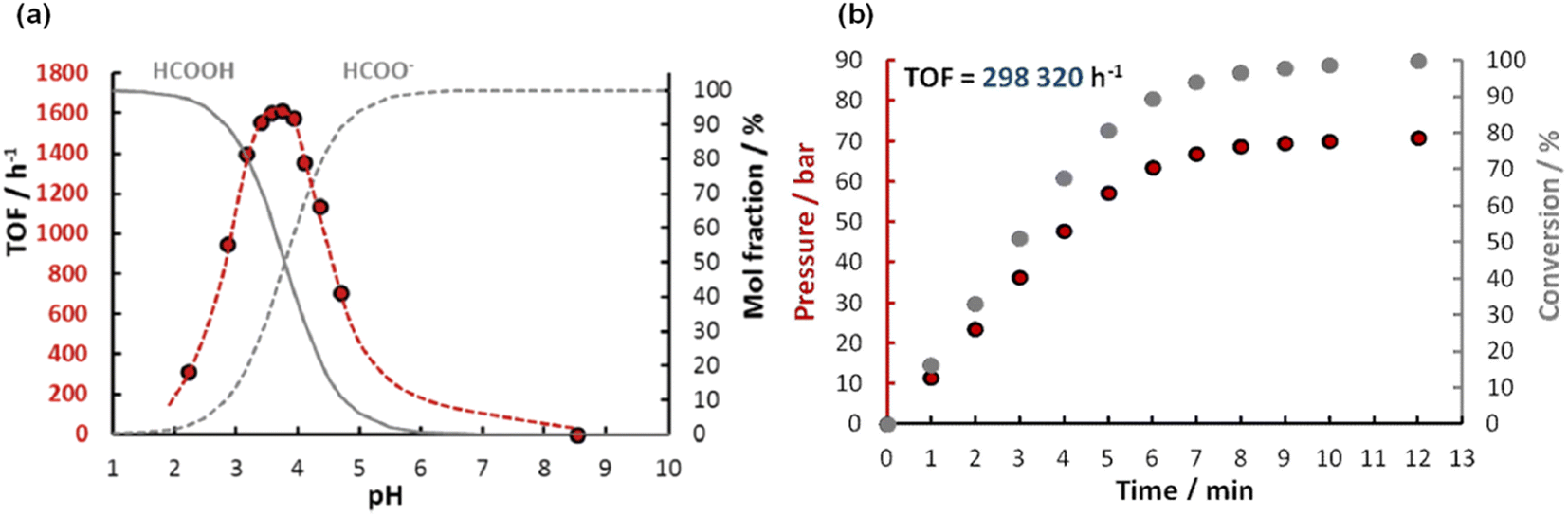
In the same year, Joo et al. developed a new water-soluble catalyst cis-mer-[IrH2Cl(mtppms)3] (mtppms = monosulfonated triphenylphosphine) 14 (Fig. 2 and Table 1) for FADH in water and examined it at a wide range of temperatures.59 The active complex 14 catalyzed the FADH to selectively yield a 1:1 gaseous mixture of H2 and CO2 without any significant CO contamination (<10 ppm). The rate of FADH was pH dependent and the maximum TOF was achieved at a pH of 3.75. FADH was subsequently performed in a 100 mL Parr reactor and the resulting increase in pressure was monitored. A maximum TOF of 298000 h−1 in FADH could be achieved with this catalytic system and high-pressure gas generation up to 14 MPa was observed after full conversion of FA (Fig. 5).
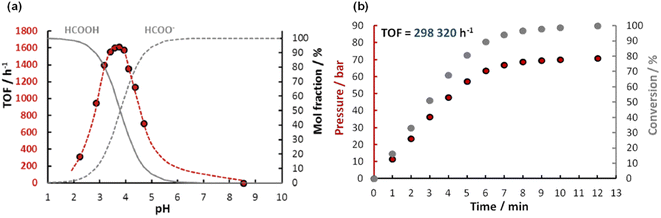 |
| | Fig. 5 (a) Effect of pH on FADH catalysed by 14 (red dots; nIr = 9.9 × 10−6 mol; nHCOOH+HCOONa = 1.00 × 10−3 mol; V = 5.0 mL; T = 60 °C), and the calculated molar distribution of HCOOH (full line) and HCOO− (broken line), respectively. (b) Time course of FADH in a closed reactor; nIr = 5 × 10−6 mol; nHCOONa = 5 × 10−2 mol; nHCOOH = 1.44 × 10−1 mol; V = 20.0 mL; T = 100 °C. Reproduced with permission.59 Copyright 2016, Royal Society of Chemistry. | |
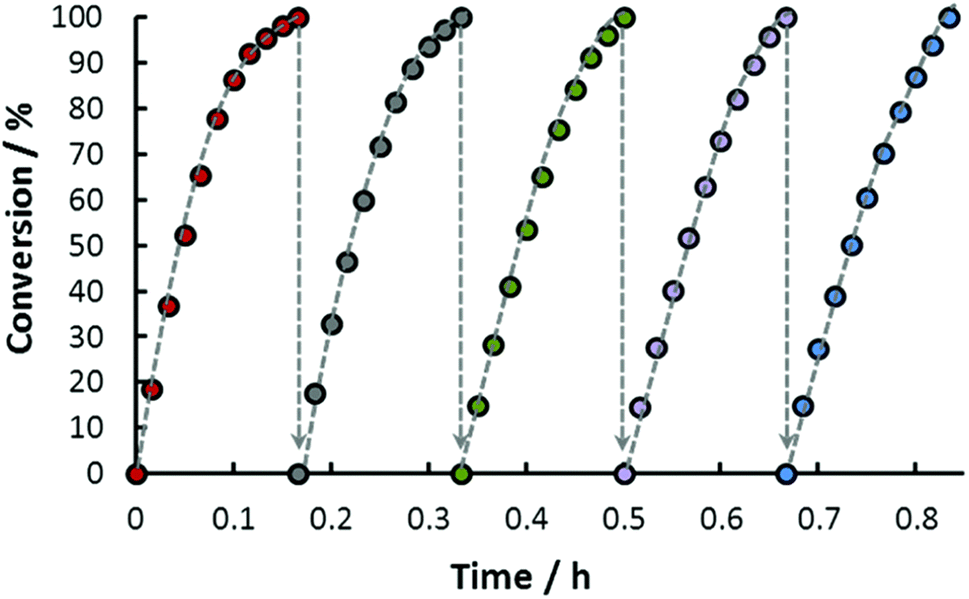
Recyclability experiments were performed with the catalytic system where the catalyst was active for 5 consecutive runs without any significant loss of activity at 100 °C. About 8 MPa gas pressure was produced in each cycle (Fig. 6). The catalyst was also examined for long-term stability where a total TON of 674000 was achieved in 40 h at 115 °C.59
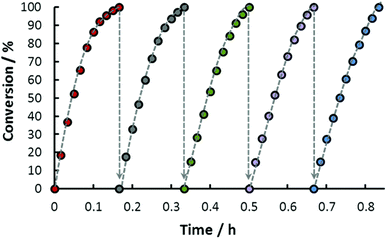 |
| | Fig. 6 Repeated use of the same solution of 14 for FADH (nIr = 9.8 × 10−6 mol; nHCOONa = 5 × 10−2 mol; nHCOOH = 1.33 × 10−1 mol per cycle; V = 20.0 mL; T = 100 °C). Reproduced with permission.59 Copyright 2016, Royal Society of Chemistry. | |
In 2016, Huang et al. also reported an arene-Ru(II) complex bearing an N,N′-diimine ligand 15 (Fig. 2 and Table 1) for high-pressure gas generation (H2 and CO2) of 24 MPa from FADH at 80 °C.60 The use of ruthenium-based catalysts presents a much more economical option as compared to iridium-based catalysts.
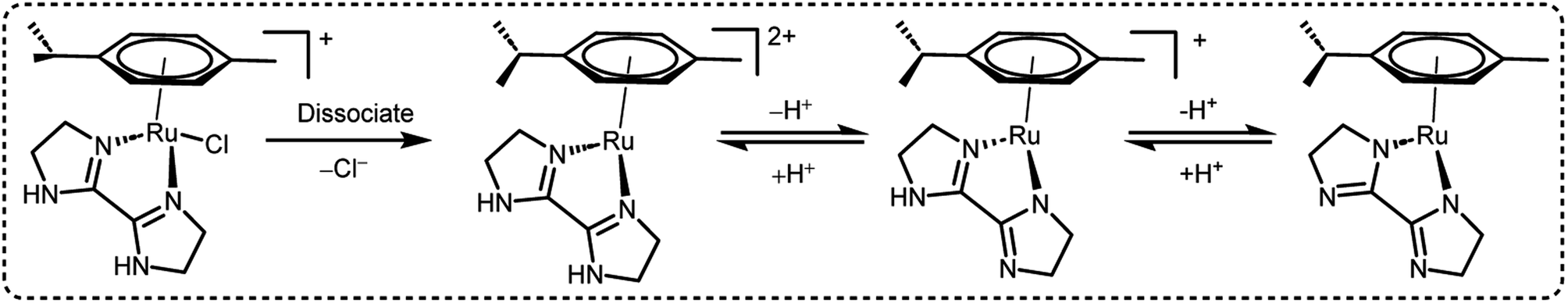
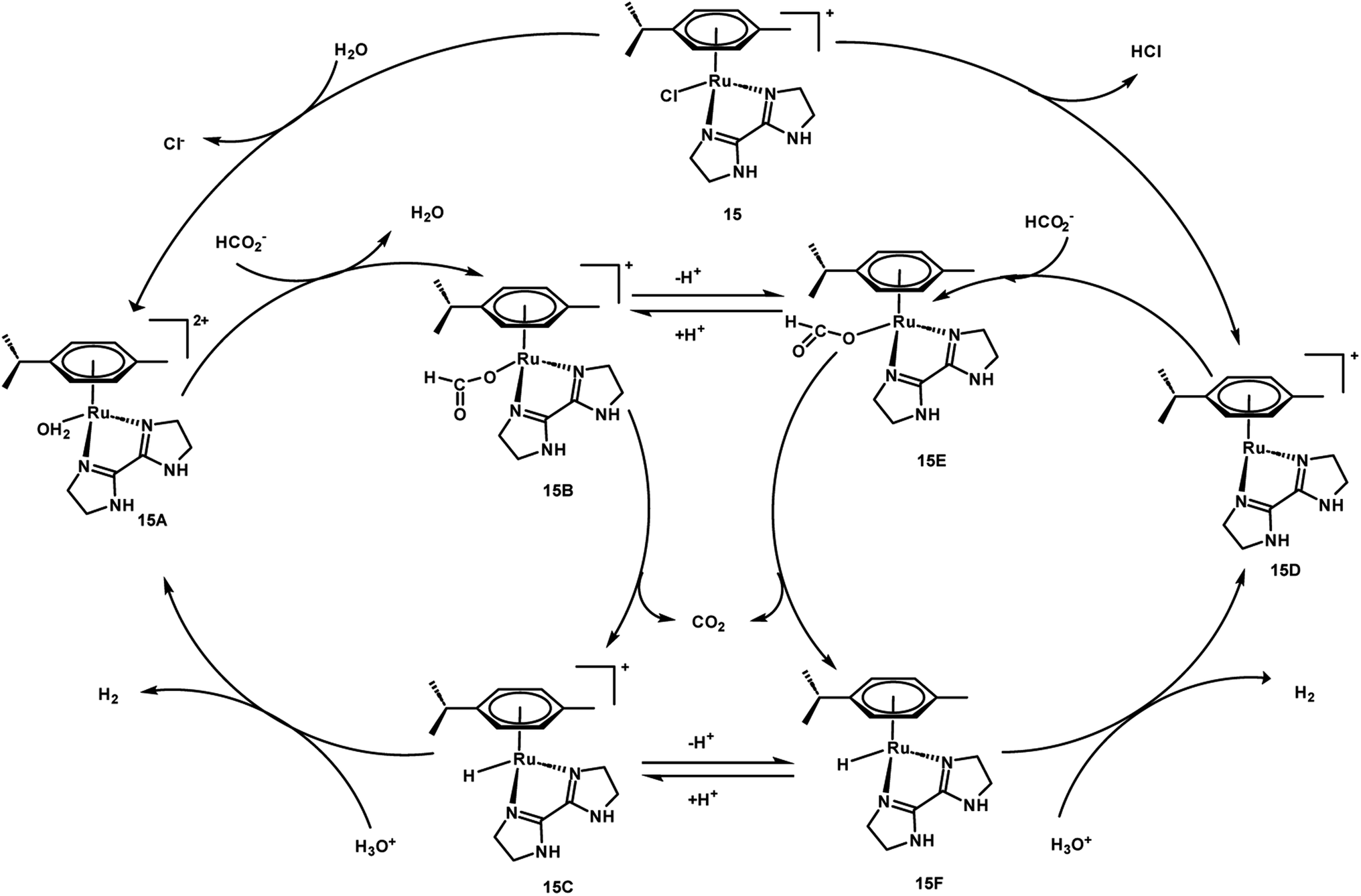
A preliminary investigation of FADH using different FA/SF ratios in an aqueous solution at 90 °C led to the achievement of a maximum TOF value of 12000 h−1 at an FA:SF ratio of 1:5. The authors conducted various NMR and MS studies to detect the plausible active organometallic intermediates in the catalytic cycle (Scheme 2). Based on the observations, a plausible reaction mechanism was proposed which comprised of two different pathways depending on the presence and absence of SF. In the absence of SF, the Ru-aqua complex 15A is generated and subsequently the water ligand is replaced by formate to give complex 15B. The Ru-hydride species 15C is generated by the decarboxylation of formate (15B). The Ru-aqua complex could be regenerated with the generation of H2 in the presence of hydronium (H3O+) ions in the reaction medium. In the presence of SF, it was proposed that complex 15 could be deprotonated with the loss of chloride anions to give intermediate species 15D. Subsequent coordination of formate to 10 generates complex 15E, which could be in equilibrium with 15B and may be decarboxylated to form the Ru-hydride intermediate 15F (Scheme 3).60
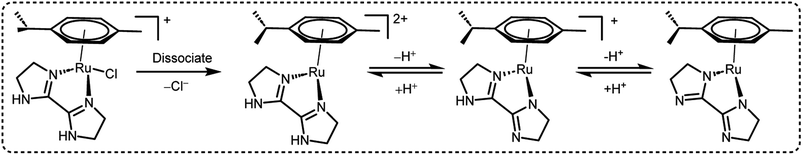 |
| | Scheme 2 Active intermediate species of 15 in FADH as reported by Huang et al.60 Reproduced with permission. Copyright 2016, American Chemical Society. | |
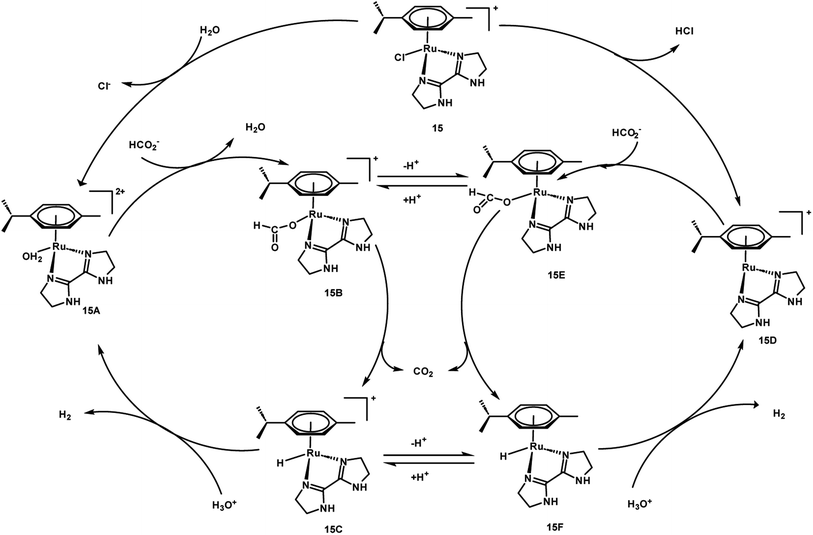 |
| | Scheme 3 Proposed mechanism for FADH with 15. Reproduced with permission.60 Copyright 2016, American Chemical Society. | |
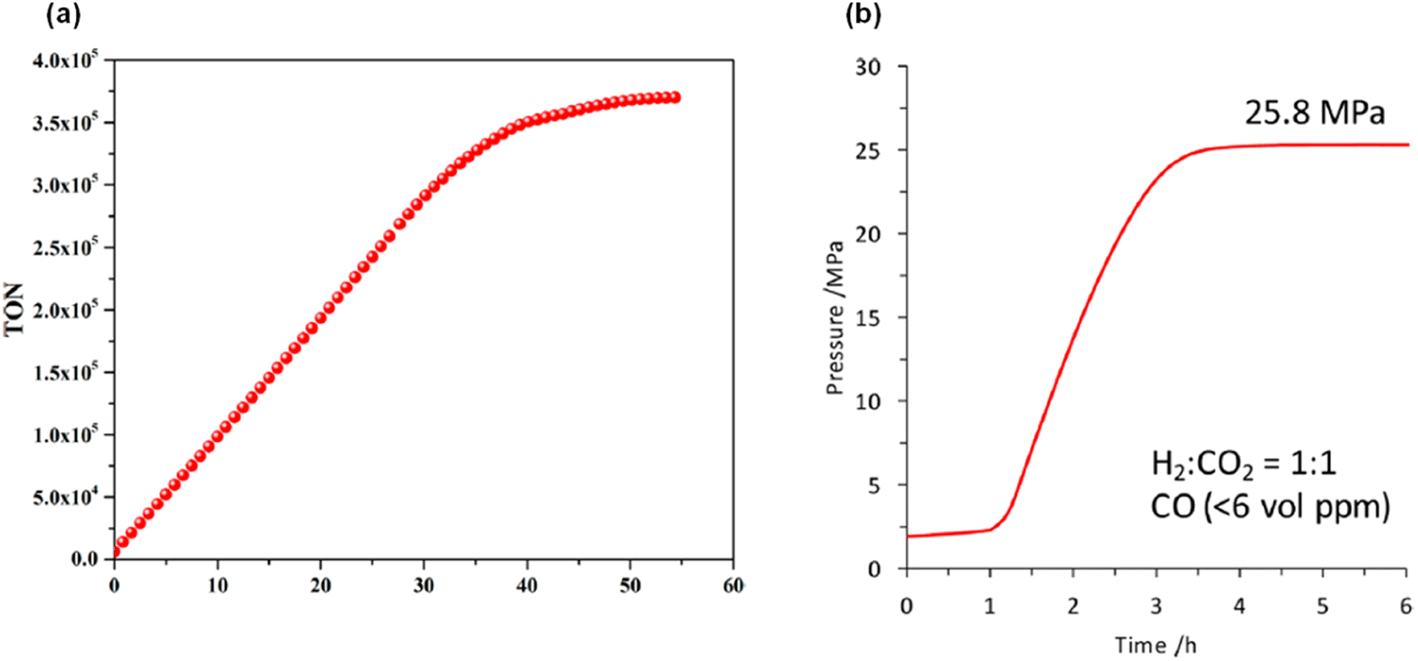
A maximum TON of 350000 was achieved in 35 hours by adding FA at a constant rate (Fig. 7a). The authors used SF free 6.5 M FA solution in the presence of 15 to generate the high-pressure (24 MPa) gaseous mixture (H2 + CO2) within 4 hours (Fig. 7b). CO was not detected in the analysis of the gas mixture by GC-TCD, making it suitable for use in fuel cells.
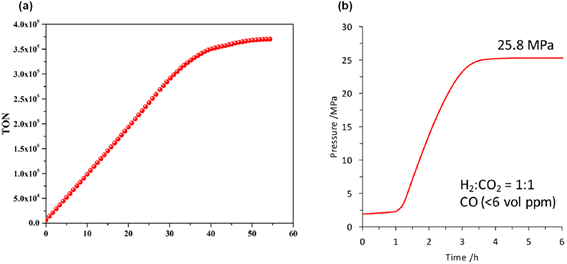 |
| | Fig. 7 (a) Optimized performance for FADH. Reaction conditions: 15 (0.50 μmol), 90 °C, FA (2.0 M, 10.0 mL), [FA]:[SF] = 1:5; FA was added to the FA/SF solution at 0.2 mL h−1. (b) Time-dependent gas evolution through FADH in the presence of 15. The reaction was carried out at 80 °C in an autoclave (internal volume is 7.0 mL) with 2 MPa of He gas, FA aqueous solution (6.5 mol L−1, 4.0 mL), and catalyst (8.0 μmol, 2.0 mmol L−1). Reproduced with permission.60 Copyright 2017, American Chemical Society. | |
In 2018, Szymczak et al. investigated several ruthenium(II) complexes supported by bis(pyridyl)isoindoline (BPI) and terpyridine (TPY) pincer scaffolds for CO2 hydrogenation as well as high-pressure gas generation from FADH.61 The authors investigated the impact of ligand charge, steric bulkiness and the bite angle on the catalytic activity which are essential parameters to establish ligand design guidelines for (de)hydrogenation reactions. The effect of substituents at the 2-position of the pyridine arm in Ru(II)-BPI complexes was investigated for the (de)hydrogenation reactions. Catalytic activity for the (de)hydrogenation reactions was enhanced upon replacement of –H with –Me or –O− groups.
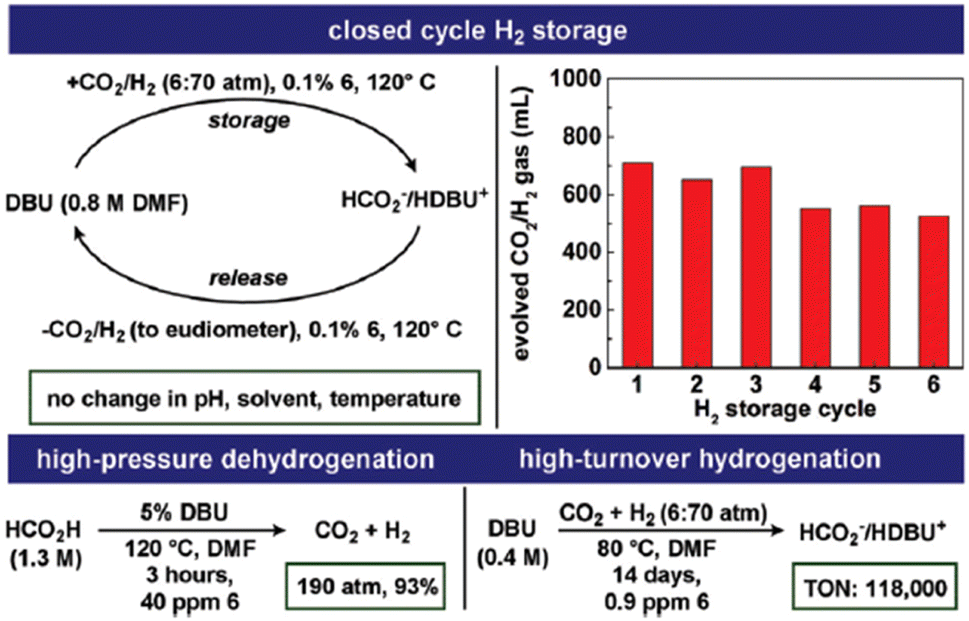
The catalyst 16 (Fig. 2 and Table 1) was found to be most active in the reversible CO2 hydrogenation reaction. Catalyst 16 was utilized to generate high-pressure H2/CO2 gas from FADH in a sealed vessel. A 1.3 M DMF solution of FA containing DBU as the base was heated in the presence of a catalytic amount of 16 to generate 19.2 MPa pressure (H2 + CO2) in the reactor at 120 °C. The high catalytic activity of 16 was applied to a closed chemical H2 storage system where initially CO2 was hydrogenated to FA in a reactor and subsequently the reactor was cooled, depressurized and reheated at 120 °C to release the stored H2/CO2 gas at ambient pressure. The cycle could be repeated six times (Fig. 8). At the end of the sixth cycle, the analysis of the evolved gas mixture by GC-TCD revealed no detectable quantities of CO (detection limit: 0.01%).
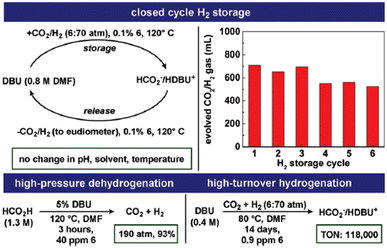 |
| | Fig. 8 Operation of a closed-cycle H2 storage device using 16 as a catalyst, high-pressure FADH, and high-turnover CO2 hydrogenation. Reproduced with permission.61 Copyright 2018, Royal Society of Chemistry. | |
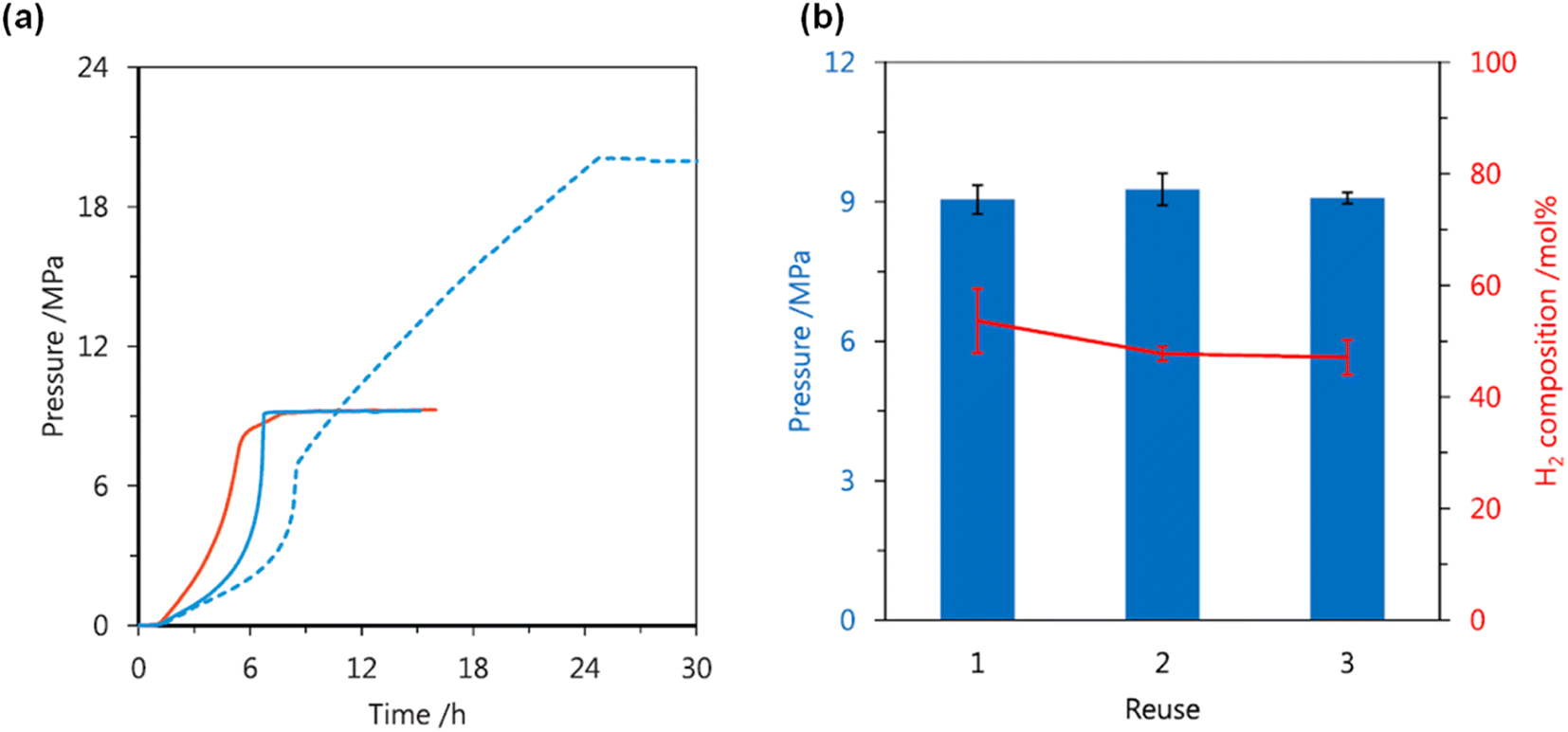
In 2019, Huang and Kawanami explored a Ru complex bearing a dearomatized pyridine-based pincer PN3P ligand 17 in high-pressure H2 generation by selective FADH.62 Initially, the authors explored catalyst 17 for FADH in DMSO. They observed that catalyst deactivation occurred under the high-pressure conditions in DMSO. To overcome this drawback, the authors employed 1,4-dioxane as the solvent, which is polar, aprotic and hydrophilic. A maximum gas pressure of 20 MPa could be achieved from FADH at 80 °C with this catalytic system.
The high-pressure gas generation could be repeated three times with selectivity of an equimolar ratio of H2 and CO2 and without any significant loss in the catalytic activity (Fig. 9).62
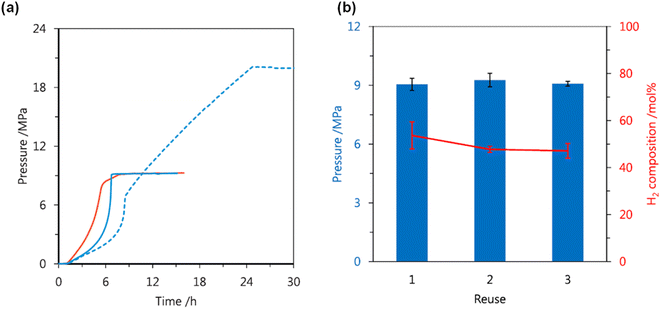 |
| | Fig. 9 (a) Effect of solvent on the gas pressure generated by FADH at 80 °C using 17: red solid line, 3.7 mol L−1 FA in DMSO; blue solid line, 3.7 mol L−1 FA in 1,4-dioxane; blue dotted line, 6.4 mol L−1 FA in 1,4-dioxane. (b) Reuse of 17 for the gas pressure and composition generated by FADH at 80 °C in 1,4-dioxane. Additional FA was added to the reaction solution after depressurization to atmospheric pressure. Reproduced with permission.62 Copyright 2019, Elsevier. | |
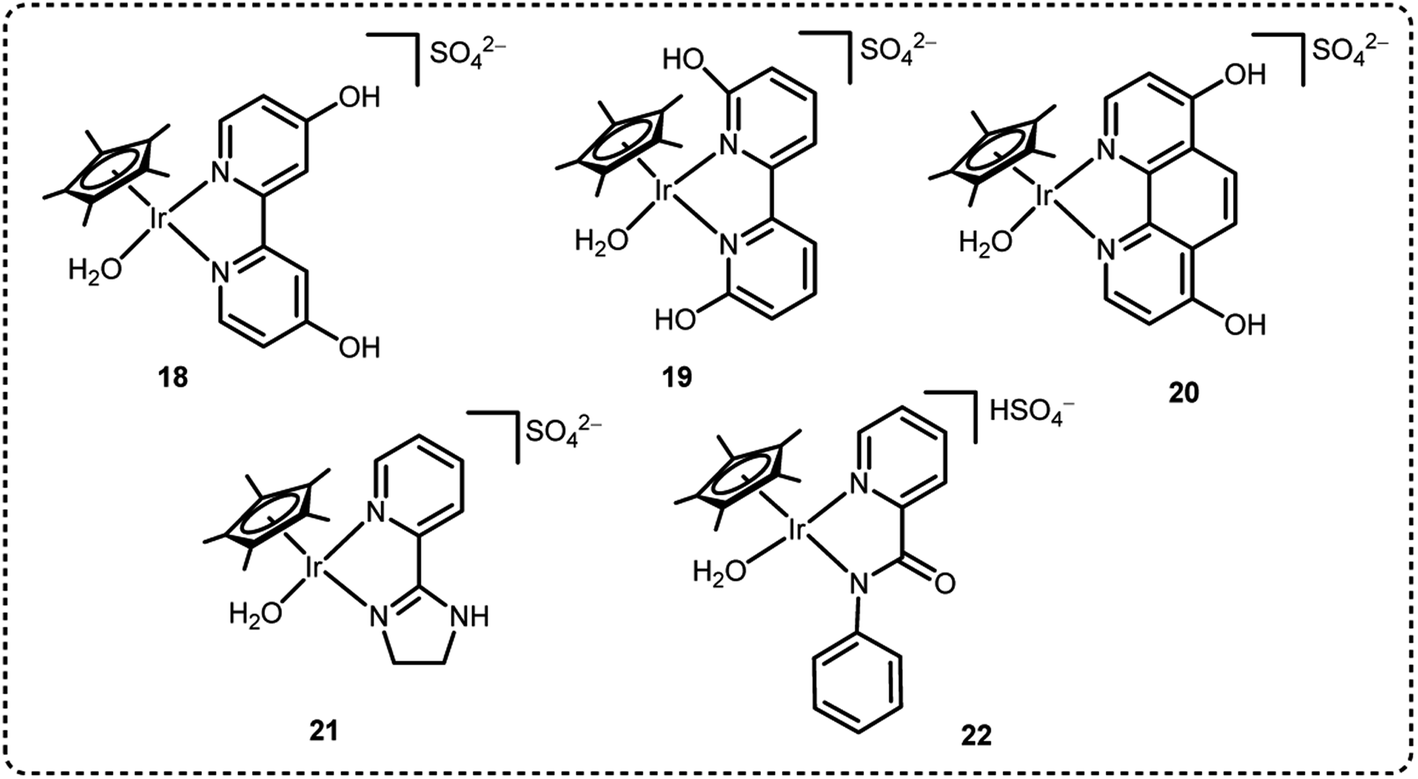
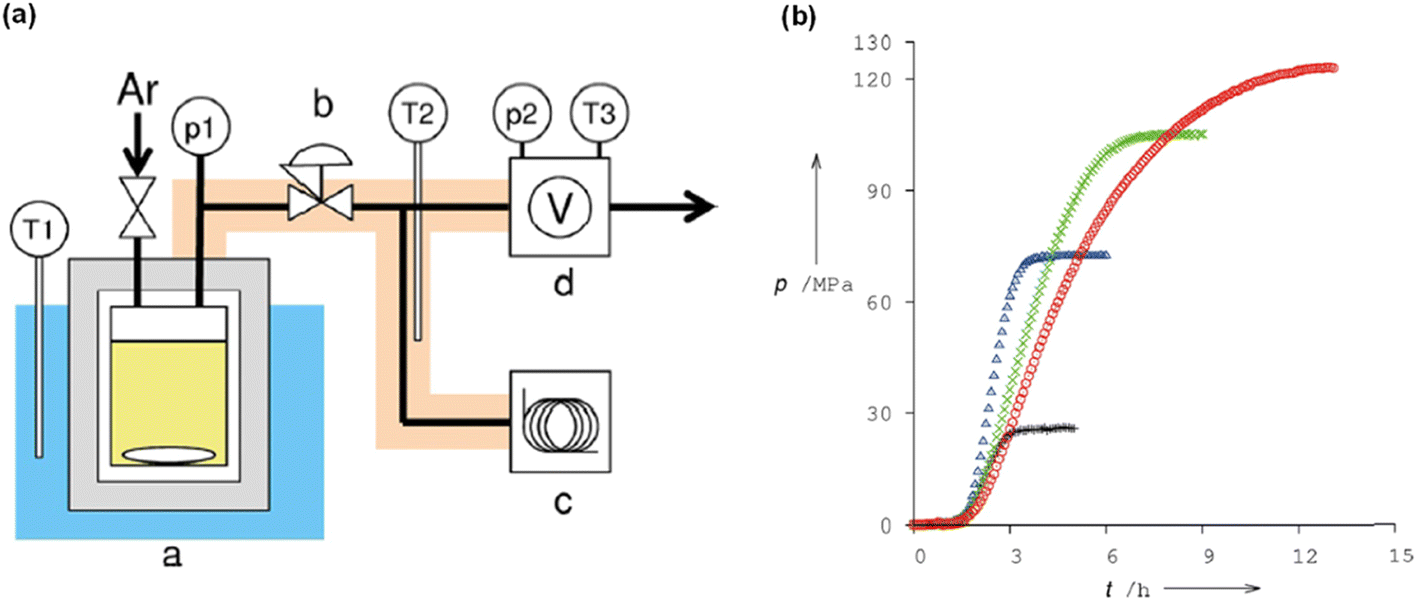
We have been engaged in the development of efficient catalysts for CO2 hydrogenation and FADH reactions over two decades.19,24,25,32 We have developed several benchmark half-sandwich iridium complexes for high-pressure gas generation from FADH over the course of time. The catalyst 18 was investigated for high-pressure gas generation by FADH in a closed reactor (Fig. 10). A pressure of 106 MPa was attained by the generated gases from the dehydrogenation of a 15 M FA solution at 80 °C. Subsequently, the pressure remained constant for a few hours indicating that chemical equilibrium was attained. The generated high-pressure gas comprised of an equimolar mixture of H2 and CO2 with almost no CO contamination (<6 vol ppm). Over 90% of the initial FA was converted into the gaseous products. When a 20 M FA solution was used as the initial solution, high pressure of generated gases up to 123 MPa could be achieved at 80 °C (Fig. 11).54
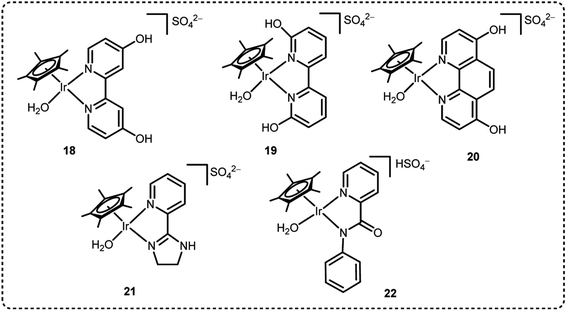 |
| | Fig. 10 Half-sandwich Ir-complexes explored for high-pressure gas generation by FADH. | |
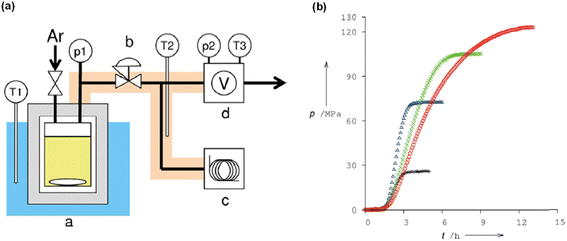 |
| | Fig. 11 (a) Schematic diagram of the apparatus for monitoring the pressure evolution during FADH: (a) high-pressure reactor made of 316 stainless steel, (b) stop valve or back-pressure regulator, (c) gas chromatograph, and (d) flow meter. Pressure and temperature are recorded at p1–p2 and T1–T3, respectively. (b) Time course of the generated pressure by FADH using 18 at various initial concentrations of FA: 4 M (black plus), 10 M (blue triangle), 15 M (green cross), and 20 M (red circle). Reproduced with permission.54 Copyright 2016, Wiley-VCH. | |
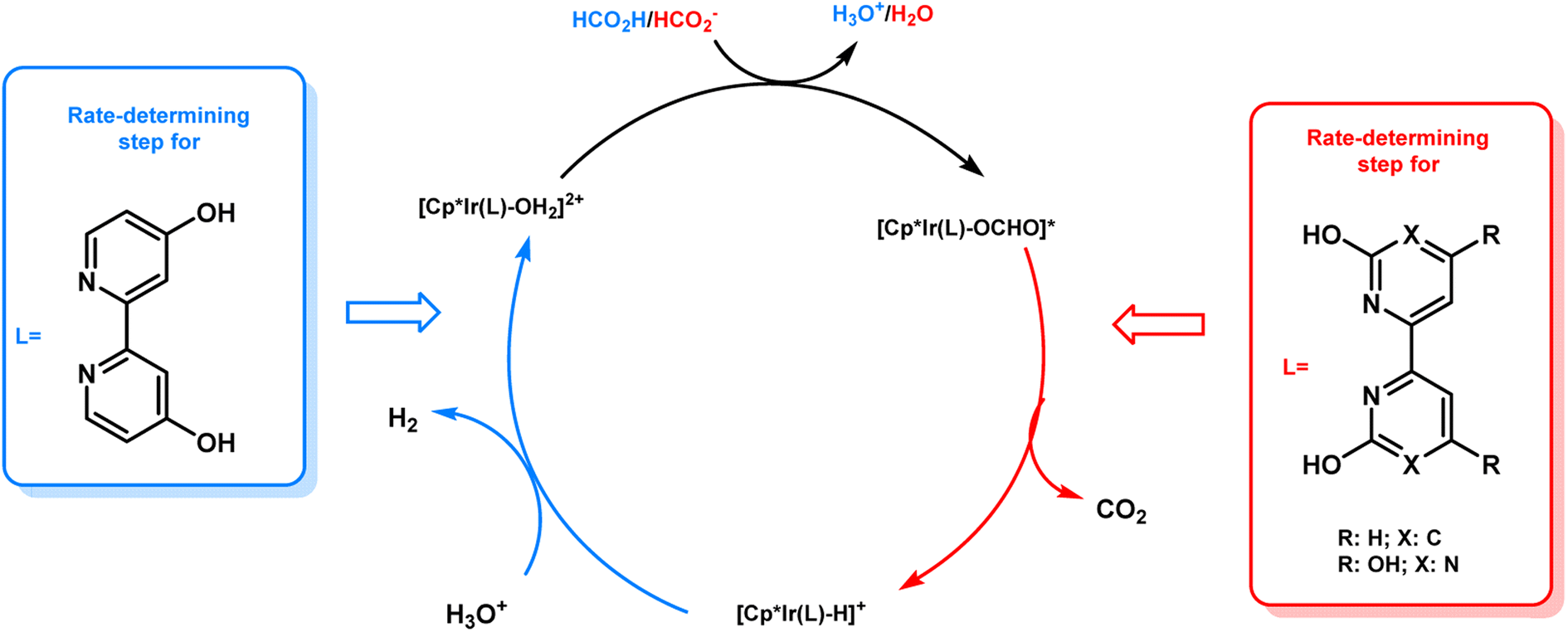
From the results of the kinetic isotope effect, the β-hydride elimination step was proposed to be the rate determining step in the catalytic cycle of FADH in the presence of 18 and gas pressure had no significant effect on the reaction mechanism. When the structural analogue 19 was investigated for FADH to examine the effect of the position of –OH groups on the bipyridine ring, it was found that catalyst 19 could also successfully generate high-pressure H2 from FADH and its catalytic activity was higher as compared to 18 due to a proton relay mechanism with water and the –OH groups in close vicinity of the metal center.64 From the kinetic isotope effect studies, it could be inferred that the rate-determining steps (RDS) in the reaction mechanism were different depending on the positions of the hydroxyl (–OH) groups in the bipyridine ring. FADH generally proceeds via three elementary reactions: (i) formation of a metal-formato complex by the replacement of the initial ligand with a formate ion, (ii) formation of a metal-hydride complex by decarboxylation of the formate and (iii) hydrogen generation by reaction of the metal-hydride complex and a hydronium ion (H3O+). The RDS of 18 was (iii) whereas the RDS of 19 was presumed to be (ii) based on the decrease of the energy barrier of (iii) due to the pendant base effect of the OH group in close vicinity of the metal center. This suggestion was supported by theoretical (DFT) calculations (Scheme 4). Although, the addition of sodium formate (SF) increased the rate of dehydrogenation reaction, but the attained final pressure from the FA/SF 1:1 solution was much lower than the base free FA solution. The rate of high-pressure gas generation gradually decreased with the repetitive use of catalyst 19, but the high-pressure gas was generated at a constant rate during the reuse of 18. During the catalytic dehydrogenation reaction with 19 at high pressure, the high-pressure H2 generated might be responsible for the deactivation of the catalyst by causing the decomposition of 19 into a bipyridine ligand and an Ir-trihydride complex through possible isomerization of the ligand. It was confirmed that the position of the –OH groups on the bipyridine ligand had a vital role in the catalyst stability at high pressures due to the different rates of deactivation of catalysts 18 and 19 (Fig. 12).
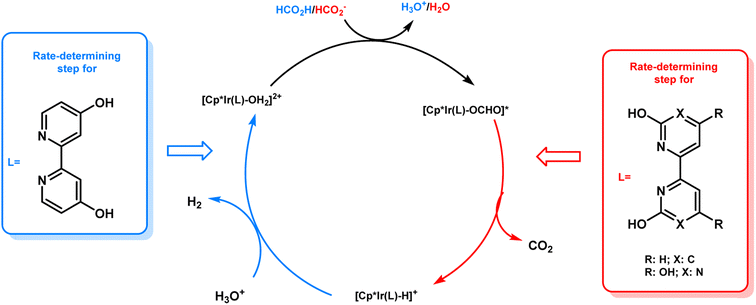 |
| | Scheme 4 Proposed mechanism and difference in the rate-determining steps for catalysts 18 and 19. Reproduced with permission.19 Copyright 2019, Wiley-VCH. | |
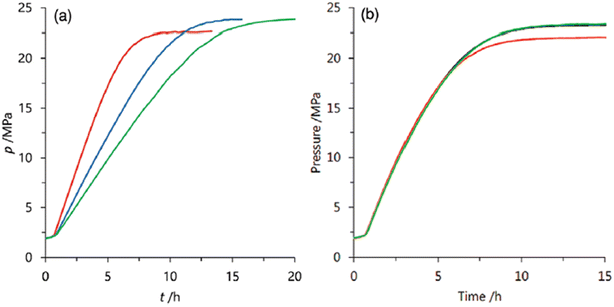 |
| | Fig. 12 Repeated use of (a) 19 and (b) 18 for high-pressure gas generation from FADH (red: first, blue: second, green: third reaction cycle). Reproduced with permission.64 Copyright 2017, Wiley-VCH. | |
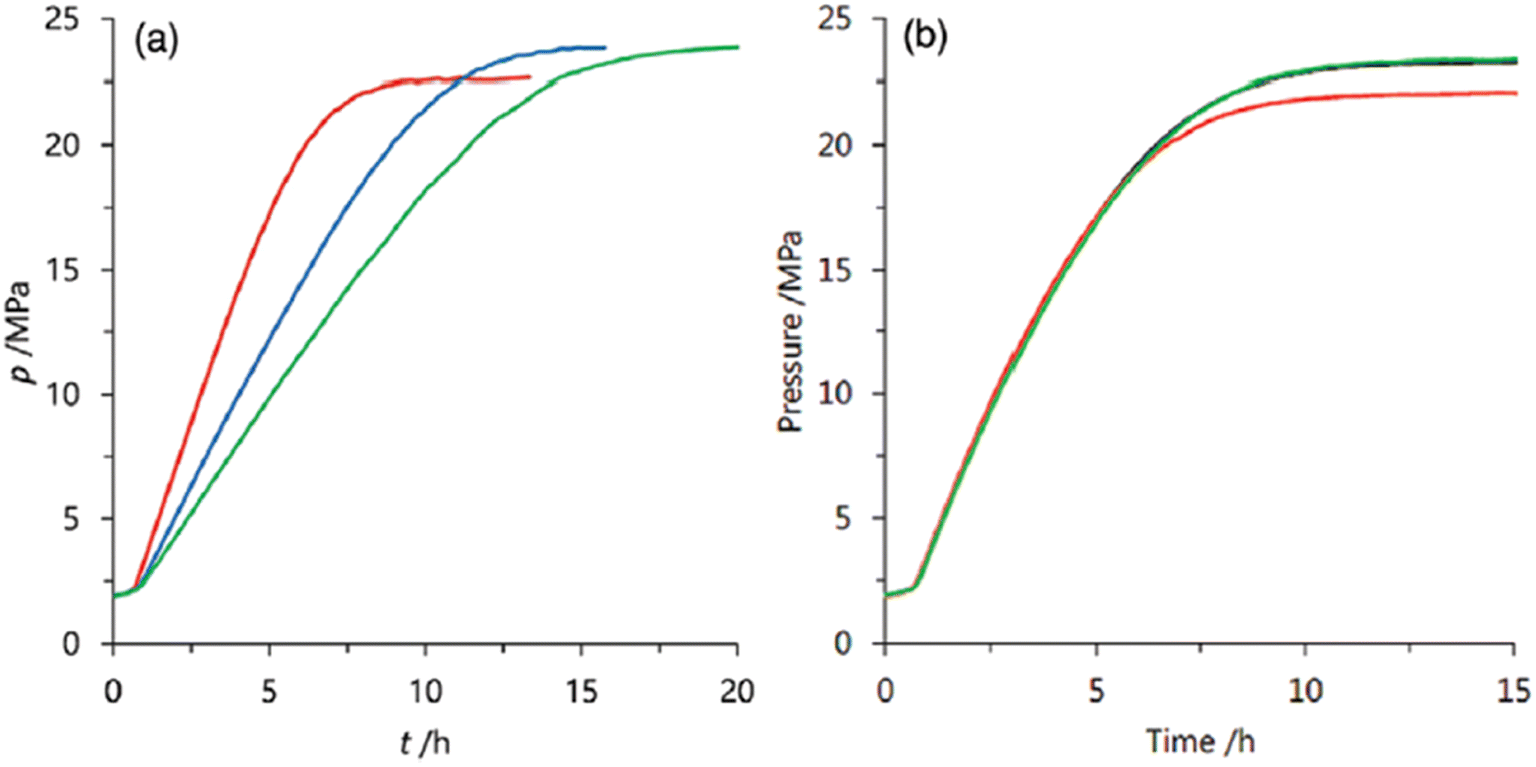
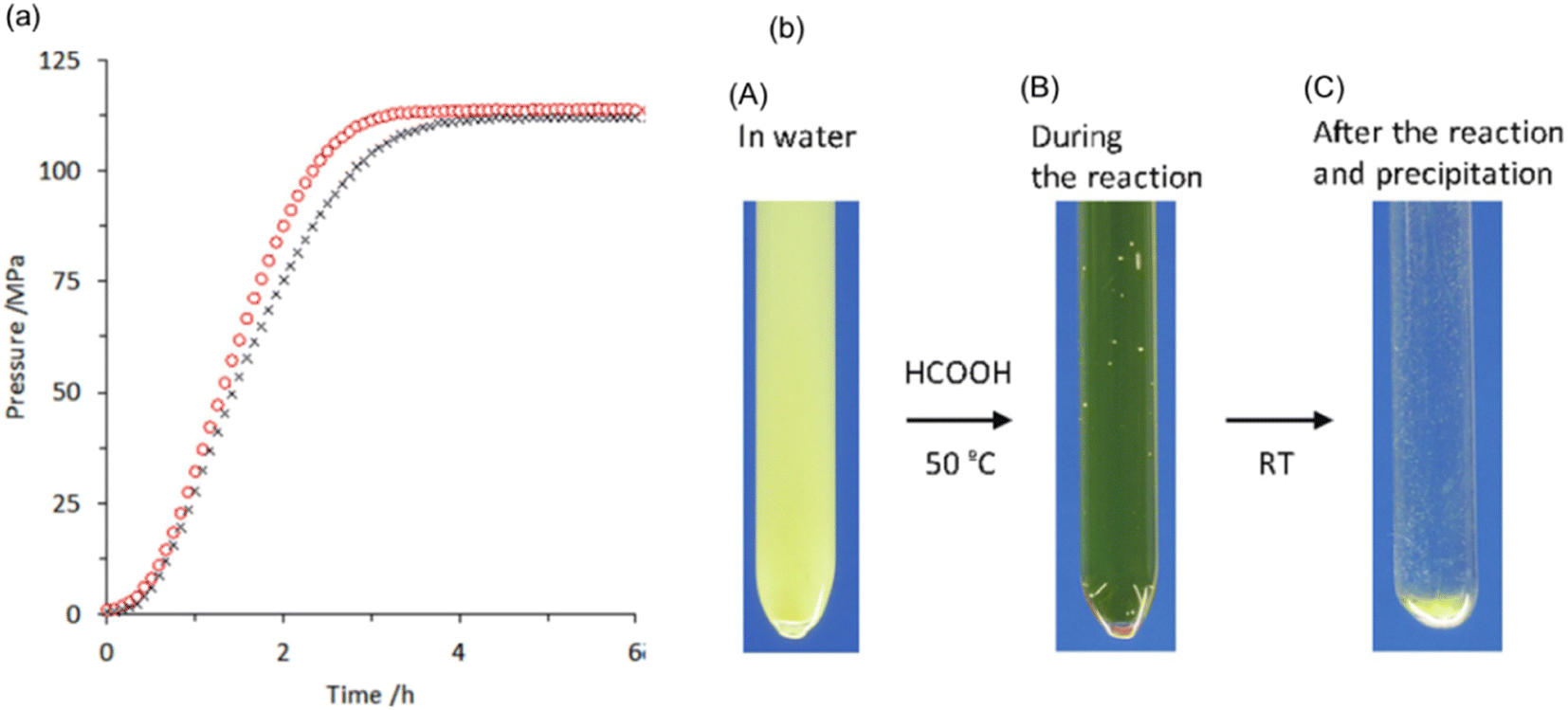
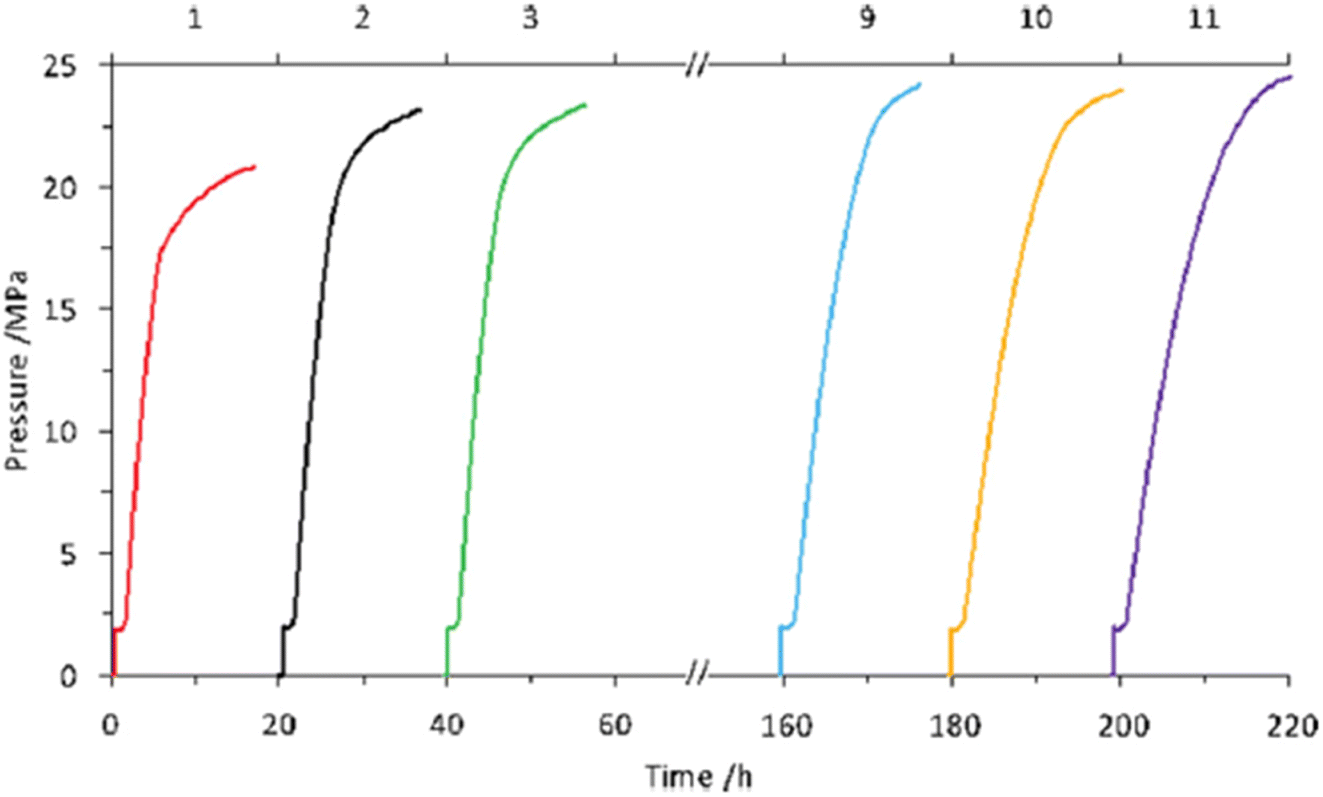
We also explored the half sandwich Ir complex 20 with a dihydroxy phenanthroline ligand for generation of high-pressure H2 by FADH. 20 shows slightly higher activity and much higher stability for FADH under high-pressure reaction conditions as compared to 18. High-pressure gases of 110 MPa could be generated from FADH with 20.65 After the reaction, the catalyst 20 could be easily recovered by simple filtration as it is precipitated out from the solution owing to its pH-dependent solubility (Fig. 13). This is an exceptional case for a homogeneous catalyst. 20 was active even after the 11th cycle of high-pressure gas generation showing excellent recyclability (Fig. 14).
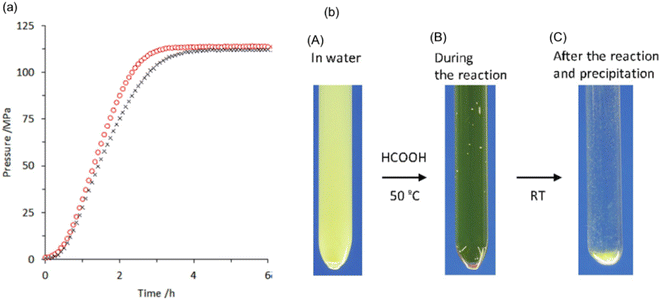 |
| | Fig. 13 (a) Time course of high-pressure gas evolution from FADH in the presence of catalysts 18 (black cross) and 20 (red circle). Reaction conditions: autoclave: 24 mL internal volume, FA aqueous solution: 16 mol L−1, 13 mL, catalyst: 2.0 mmol L−1, reaction temperature: 80 °C. (b) Images of the reactant (catalyst 20: 8 μmol, water: 3 mL, FA (100%): 1 mL) during the reaction at different stages: (A) before the reaction at RT, pH 6.8; (B) during reaction after addition of FA at 50 °C under high-pressure conditions (22 MPa), pH 0.9; and (C) after the reaction and precipitation of the catalyst after cooling down to RT, pH 1.9. Reproduced with permission.65 Copyright 2016, Wiley-VCH. | |
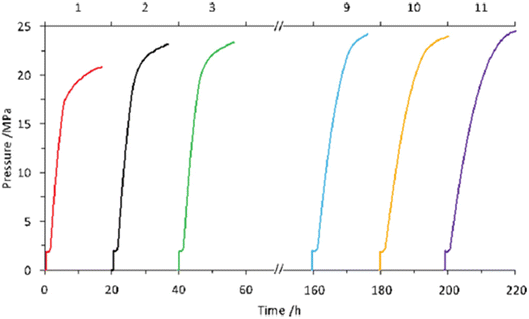 |
| | Fig. 14 Recycling experiments of high-pressure gas evolution from FA in the presence of catalyst 20. The catalyst was recycled after the former reaction without any purification except filtration. Reaction conditions: 50 °C, 2 MPa He, FA aqueous solution (7 mol L−1, 4 mL), catalyst (2 mmol L−1, 8 μmol). FA was added after depressurization (1 mL). The upper horizontal axis represents the time of high-pressure gas release. Reproduced with permission.65 Copyright 2016, Wiley-VCH. | |
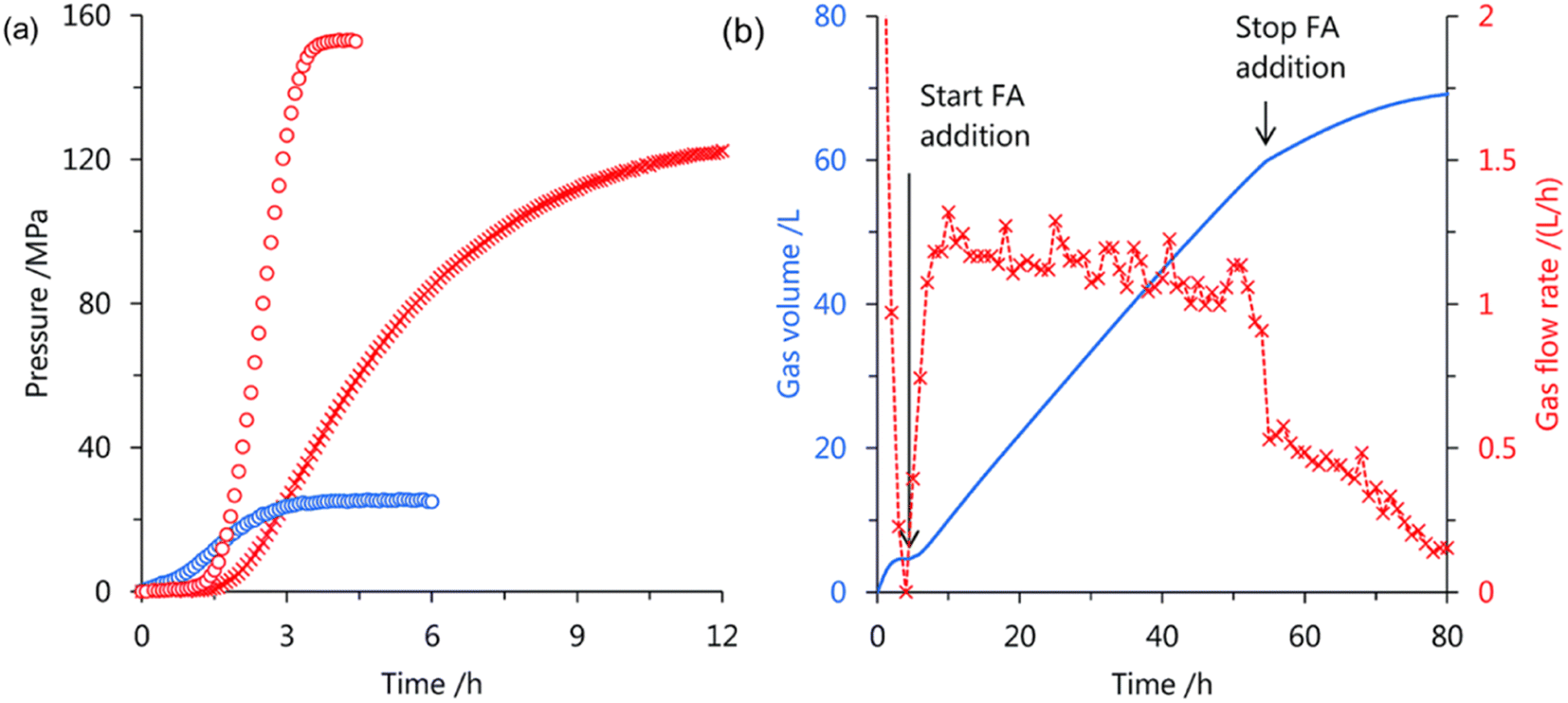
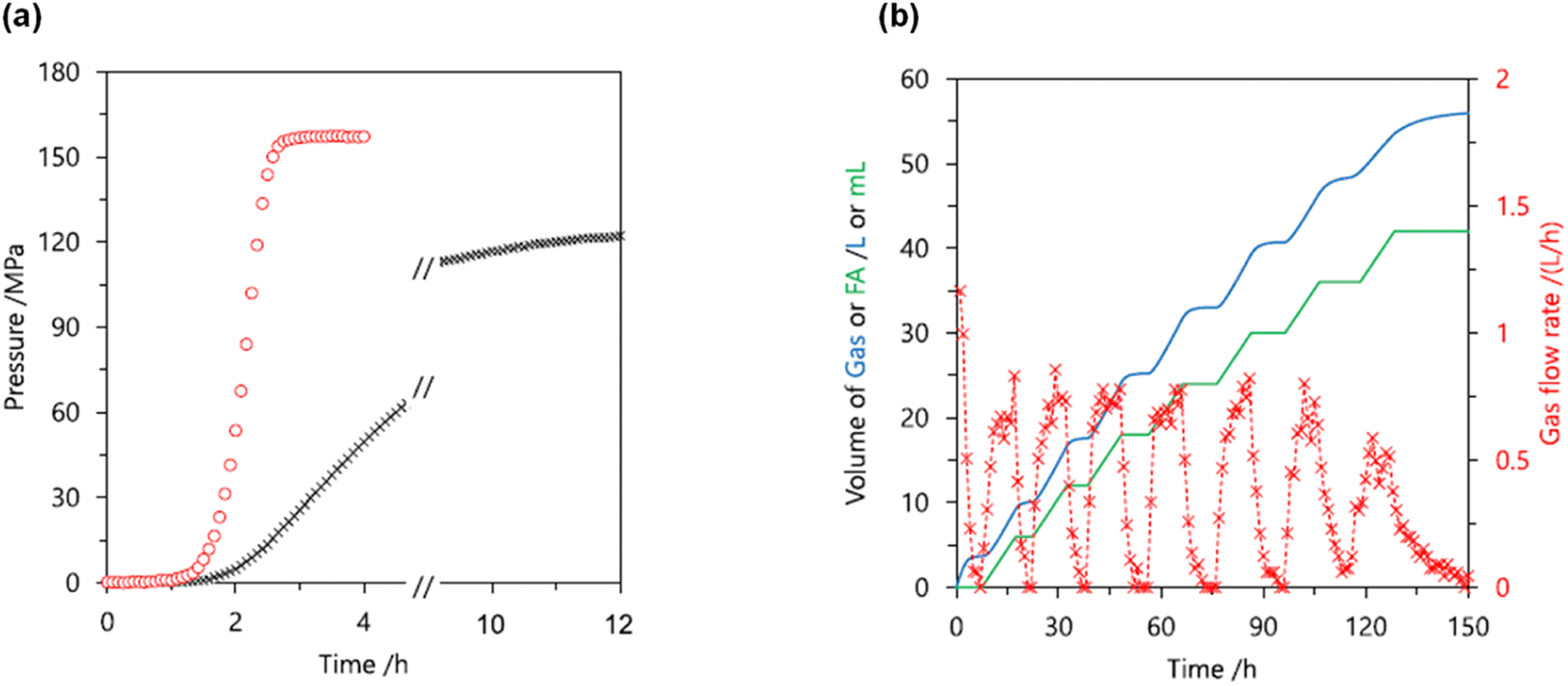
Later, we investigated a half-sandwich Ir complex bearing a pyridyl-imidazoline ligand 21 for high-pressure gas generation from FA. High-pressure gas up to 153 MPa could be generated from 20 M aqueous FA solution in 5 hours at 80 °C using 21 (Fig. 15).55 The high-pressure gas was composed of an equimolar ratio of H2 and CO2 with negligible concentration of CO (<6 vol ppm). The catalytic activity of 21 can be retained at very high pressures most probably because the chelating conformation remains intact when the imidazoline moiety rotates around the pyridyl-imidazoline single bond. The presence of bases accelerated the rate of FADH but very poor conversion of FA was achieved. Continuous production of high-pressure H2 by FADH was performed using catalyst 21 by addition of neat FA into the reaction solution at a constant rate by a high-pressure liquid pump every time the gas generation stopped. The added FA was continuously and selectively dehydrogenated to H2 and CO2 under the high-pressure conditions. The rate of generation of high-pressure gas could also be controlled by adjusting the rate of addition of FA.55
 |
| | Fig. 15 (a) Time course of high-pressure gas generation by FADH using catalysts 18 (cross) and 21 (circle); 80 °C and 20 mol L−1 FA aqueous solution (red), 40 °C and 5 mol L−1 FA aqueous solution (blue). Reaction conditions: FA aqueous solution (13 mL), catalyst (2 mmol L−1). (b) Continuous FADH at high pressure: gas volume (blue; solid line), gas rate (red; cross). Reaction conditions: 60 °C, 40 MPa, initial FA aqueous solution (8 mol L−1, 40 mL), FA addition (1.2 mL h−1, 50 h), catalyst 21 (16 μmol). Time starts after reaching the pressure. Reproduced with permission.55 Copyright 2018, Royal Society of Chemistry. | |
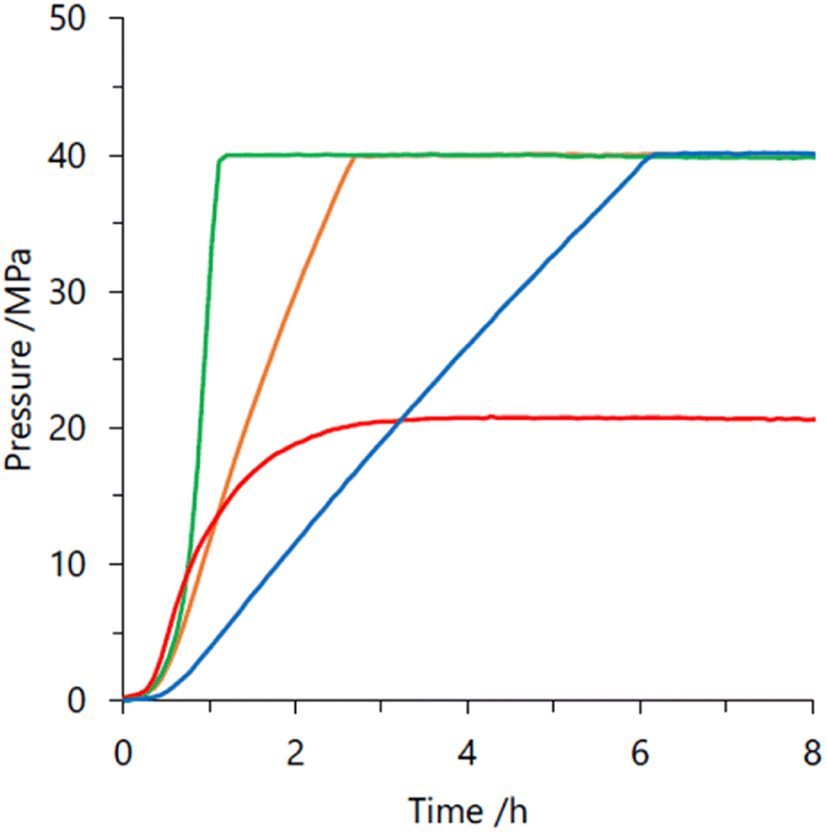
In 2019, we explored some Cp*-Ir complexes with different kinds of N,N′-bidentate donor ligands for high pressure gas production from FADH.66 The activity and stability of the complexes 1, 5, 21 and 22 for high-pressure gas generation from FADH were systematically investigated. A 16 M (40 mL) aqueous solution of FA containing 16 μmol of complexes 1, 5, 21 and 22 was used for the high-pressure reactions at 60 °C. The generated gas pressure reached 40 MPa after 1 h, 3 h and 6 h with catalysts 1, 21 and 5, respectively. Catalyst 22 could only generate up to 20 MPa gas pressure after 10 h under analogous reaction conditions (Fig. 16). However, 40 MPa gas pressure could be achieved with 22 in 2 h while using a lower concentration of 8 M FA. It was inferred that although catalyst 22 was highly active, its deactivation occurred quickly under high pressure and high FA concentration conditions. Analysis of the reaction aliquot by ESI-MS indicated the formation of an Ir-trihydride complex ([(Cp*Ir)2H3]+) which could be responsible for the deactivation of 22. The residual concentration of FA (i.e., FA conversion) was calculated after the dehydrogenation reactions. It was found that the remaining concentration of FA was much higher for complex 1 as compared to 5 and 21. Thus, it was inferred that catalysts 5 and 21 have higher catalytic activity and stability for FADH than 1 under high-pressure reaction conditions indicating the vital role of the ligand in the catalytic stability. From this study, it was concluded that catalyst 1 having a biimidazoline ligand shows the highest activity followed by 22, 21 and 5 but catalytic stability is in the order of 21 > 5 > 1 > 22.66
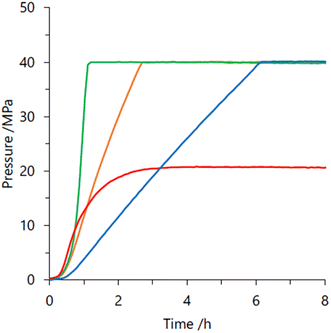 |
| | Fig. 16 Time course of the generated gas-pressure by FADH using different Ir complexes: 1 (green), 5 (blue), 21 (orange) and 22 (red). Reaction conditions: 60 °C, aqueous FA (16 M, 40 mL), complex (0.4 mM). Time is defined as the time after heating the reaction solution. Reproduced with permission.66 Copyright 2019, Wiley-VCH. | |
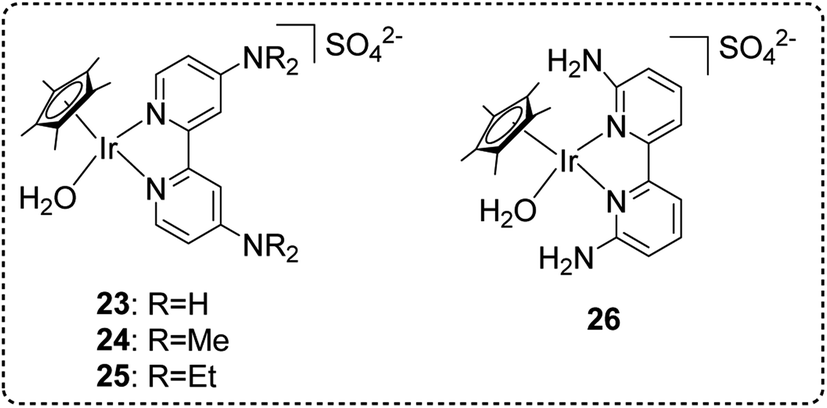
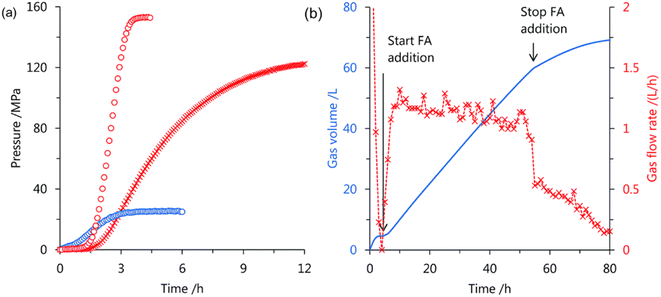
Very recently, we have developed a Cp*-Ir complex with amino groups at the 4,4′-positions on the bipyridine ligand 23–26 (Fig. 17) for high-pressure gas generation from FADH.67 The effect of different substituents at different positions on the bipyridine ring was investigated and it was found that the Ir complexes with 2,2′-bipyridine ligands bearing amino or alkylamino groups at the 4,4′-positions displayed higher activity and stability for catalytic FADH as compared to our previously reported complexes containing 4,4′-hydroxy substituted bipyridine ligand 18. Additionally, complex 23 with amino groups at the 4,4′-position of the bipyridine ring afforded higher catalytic stability under high-pressure conditions as compared to complex 26 with amino groups at the 6,6′-position of the ring. The optimization of catalyst performance in FADH was conducted using different catalysts under atmospheric pressure. Catalyst 23 was found to be more effective in FADH as compared to catalyst 18. Complexes 24 and 25 with electron donating dimethylamino and diethylamino groups slightly enhanced the catalytic activity. The reaction mechanism was studied by NMR analysis and it was inferred that the stability of the hydride intermediate is affected by the position of the amino substituents on the bipyridine ring and is the key factor to determine the catalytic activity. In the high-pressure gas generation from FADH experiments, catalyst 24 produced a very high gas pressure of 157 MPa in 4 hours from an initial 21 M FA solution at 80 °C (Fig. 18a). The same molar ratio of H2 and CO2 was detected by GC-TCD from the analysis of the high-pressure gas mixture without any CO (<6 vol ppm) contamination. The catalytic activity of catalyst 23 was investigated for continuous H2 generation by FADH both at atmospheric pressure and 20 MPa pressure (Fig. 18b). The results indicated that complex 23 was able to successfully generate high-pressure gas with a constant pressure of 20 MPa over six cycles of FA addition and retained its stability for 60 hours under high-pressure conditions. These results indicated that 23 has better stability as compared to our previously established Cp*-Ir (pyridyl-imidazoline) catalyst 21 under high pressure as the latter complex lost its stability after 30 hours at 20 MPa pressure. To the best of our knowledge, the 157 MPa gas pressure generated by this catalytic system at 80 °C is the highest gas pressure generated from catalytic FADH to date.67
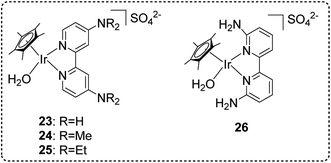 |
| | Fig. 17 Cp*-Ir complexes with amino-substituted bipyridine ligands explored for high-pressure gas generation by FADH.67 | |
 |
| | Fig. 18 (a) Time course of the generated pressure by FADH at 80 °C in a highly concentrated FA solution using the Ir complexes (2 mM): ×, 18 in 20 M FA; ○, 24 in 21 M FA. (b) Continuous FADH using 23 (16 μmol) at 50 °C and 20 MPa. Blue line, generated gas volume; green line, added FA volume; ×, flow rate of generated gas. Time elapsed after reaching the pressure of 20 MPa. FA was added at high pressure at a rate of 0.6 mL h−1 over 10 h, and the FA addition was repeated seven times. Reproduced with permission.67 Copyright 2020, American Chemical Society. | |
3. Heterogeneous catalysts for high-pressure hydrogen generation from FA
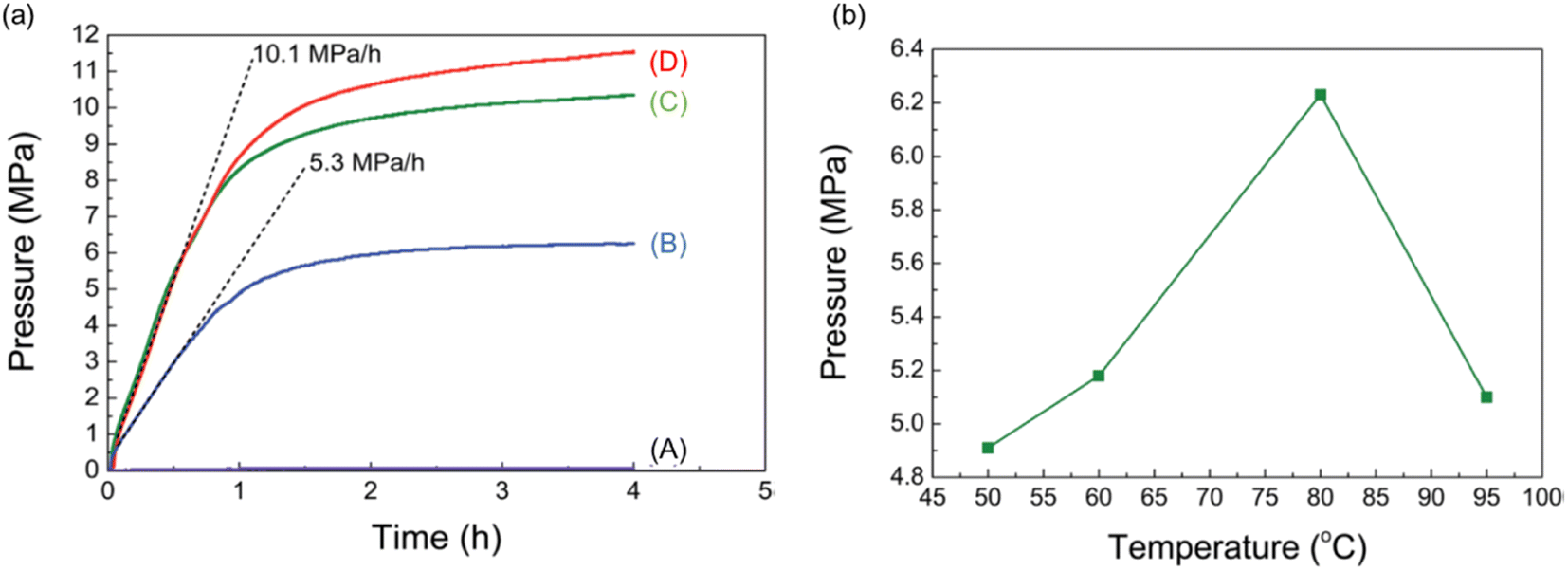
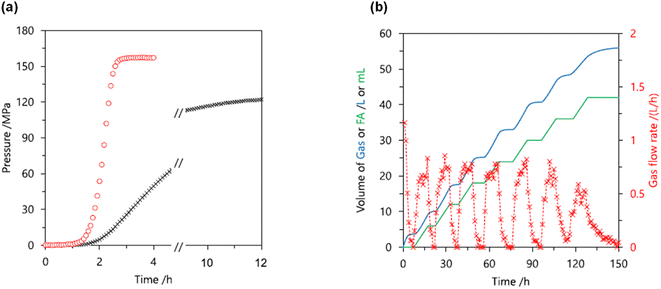
Heterogeneous catalysts have become a popular choice among researchers due to their easy separation and recovery after the catalytic reaction. This is essential for upgrading the catalyst from the research laboratory to industrial scale. Most studies of FADH with heterogeneous catalysts focus on H2 generation at atmospheric pressure. Therefore, one of the targets here is to develop heterogeneous catalysts for high-pressure gas generation by FADH. In 2015, Xu et al. reported a diamine alkalized reduced graphene oxide (rGO) supported palladium nanoparticle catalyst for efficient FADH (TOF = 3810 h−1) at 50 °C and atmospheric pressure.39 Recently, Kawanami and Xu investigated the application of this same catalytic system in high-pressure FADH in a suitable reaction setup.68 In the initial optimization experiments of this work, different loadings of the Pd/PDA-rGO catalysts were investigated for the dehydrogenation of an aqueous solution of FA:SF = 1:1 (6.7 M concentration) at 80 °C (Fig. 19a). The initial experiments revealed that higher gas pressure was generated by FADH with higher catalyst loadings. Increasing the reaction temperature above 80 °C had a detrimental effect on the high-pressure gas generation (Fig. 19b).
 |
| | Fig. 19 (a) Gas pressure versus time (solid lines) and pressure increasing rate (dashed lines) obtained from FADH with different amounts of catalyst (solution: 6.7 mol L−1 FA + 6.7 mol L−1 SF; liquid volume: 6 mL; temperature: 80 °C; catalyst amount: (A) 0 mg, (B) 10 mg, (C) 20 mg, (D) 50 mg). (b) Gas pressures obtained at different temperatures from FADH (solution: 6.7 mol L−1 FA + 6.7 mol L−1 SF; liquid volume: 6 mL; catalyst: 10 mg; time: 15 h). Reproduced with permission.68 Copyright 2017, Royal Society of Chemistry. | |
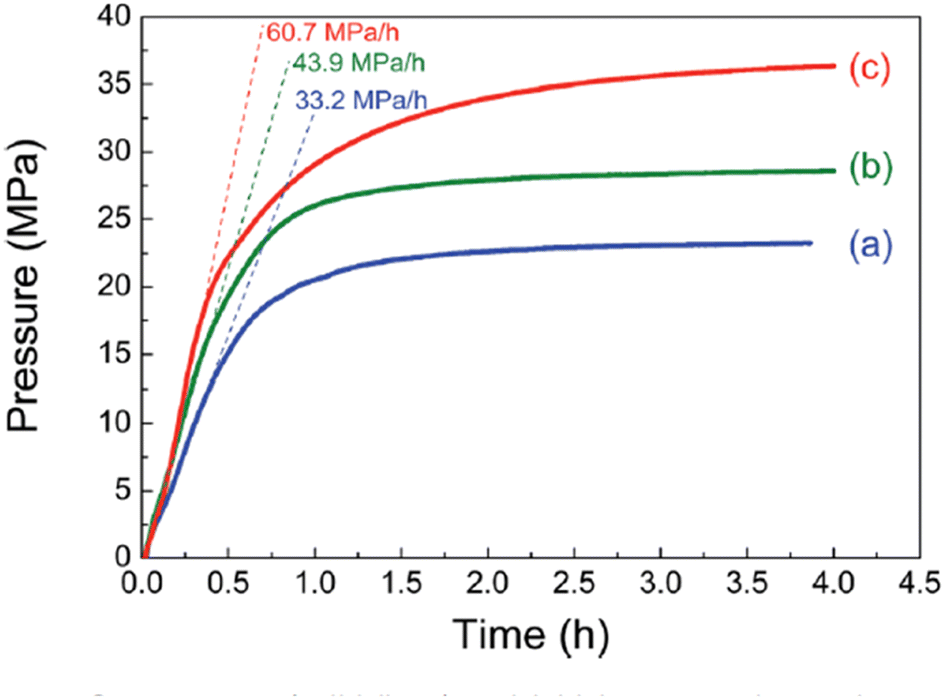
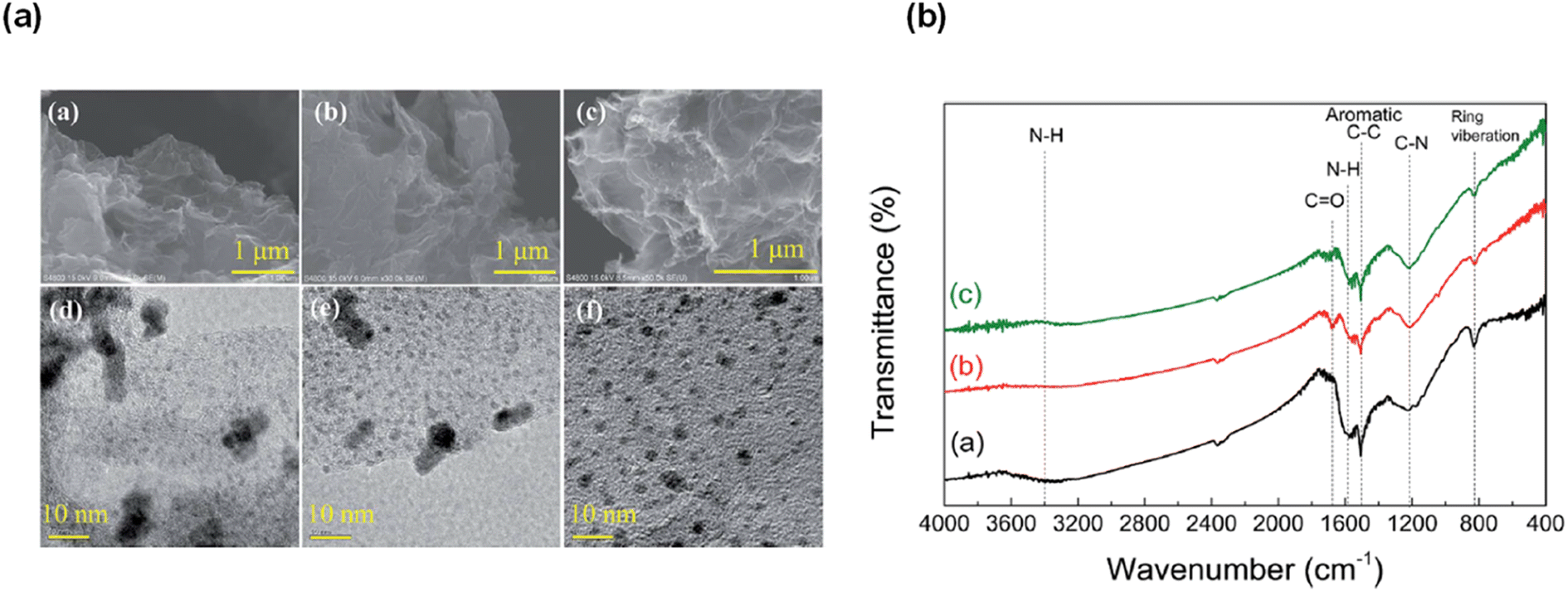
The highest gas pressure of 36.3 MPa was achieved from FADH with 100 mg catalyst loading at 80 °C (Fig. 20). An initial gas pressure rate of 60.7 MPa h−1 could be achieved. The industrial applicability of this heterogeneous catalytic system was examined by performing the catalyst recyclability experiments. It was found that the gas pressures achieved with the recovered catalyst were relatively lower than the fresh ones under similar reaction conditions which suggested a deactivation after the reaction. From XRD, TEM and SEM analyses, the morphology of the catalysts was relatively unchanged before and after the reaction (Fig. 21a). FT-IR analyses indicated that the asymmetric C–N stretching (1220 cm−1) and aromatic C–C stretching (1510 cm−1) vibration peaks remained relatively unchanged after the reaction, while the peaks at 1575 and 3400 cm−1 decreased corresponding to N–H bending and stretching respectively. The new absorption peak at 1680 cm−1 found in the FT-IR spectra after the reaction might correspond to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration in amide (Fig. 21b). This evidence suggest that the amine group is partially transformed into amide after the reaction and is probably the reason behind the deactivation of the catalyst. No peak around 1675 cm−1 was found in the FT-IR spectrum of the catalyst after FADH reaction under atmospheric pressure conditions. This suggested that high-pressure gas could be responsible for the transformation of amine to amide in the Pd/PDA-rGO catalyst. Attempts to reactivate the catalyst by hydrolysis of the amide group into amine and calcination of the catalyst at high temperature were not very successful. It was inferred that the unstable supporting material PDA-rGO under high pressure causes the deactivation of the catalyst.68
O stretching vibration in amide (Fig. 21b). This evidence suggest that the amine group is partially transformed into amide after the reaction and is probably the reason behind the deactivation of the catalyst. No peak around 1675 cm−1 was found in the FT-IR spectrum of the catalyst after FADH reaction under atmospheric pressure conditions. This suggested that high-pressure gas could be responsible for the transformation of amine to amide in the Pd/PDA-rGO catalyst. Attempts to reactivate the catalyst by hydrolysis of the amide group into amine and calcination of the catalyst at high temperature were not very successful. It was inferred that the unstable supporting material PDA-rGO under high pressure causes the deactivation of the catalyst.68
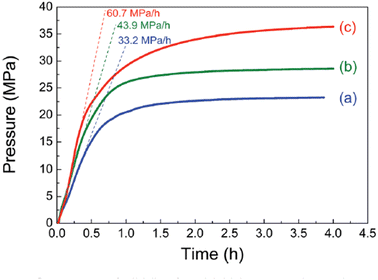 |
| | Fig. 20 Gas pressure (solid lines) and initial pressure increasing rate (dashed lines) versus time obtained from FADH with the mixture of SF at 80 °C. (a) Catalyst amount: 50 mg, [FA]: 2 M; (b) catalyst amount: 100 mg, [FA]: 4 M; (c) catalyst amount: 100 mg, [FA]: 6.7 M. Reproduced with permission.68 Copyright 2018, Royal Society of Chemistry. | |
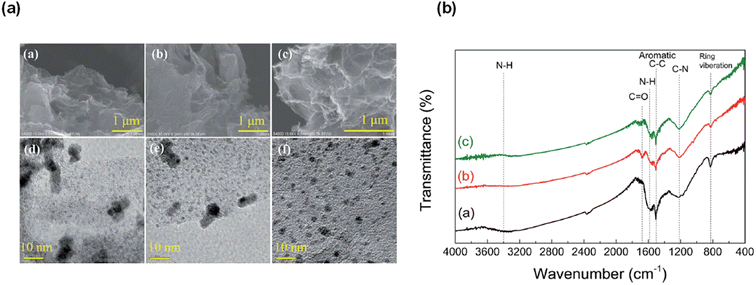 |
| | Fig. 21 (a) SEM and TEM images of the Pd/PDA-rGO catalyst before ((a) SEM; (d) TEM) and after reaction ((b) SEM; (e) TEM), and after being re-activated and reused 3 times ((c) SEM; (f) TEM) (reaction conditions: 2 mol L−1 formic acid + 2 mol L−1 sodium formate; 20 mg catalyst; 80 °C; 4 h). (b) FT-IR spectra of the Pd/PDA-rGO: (a) before the reaction, (b) after pressurized reaction, and (c) after unpressurized reaction (catalyst: 20 mg; temperature: 80 °C; time: (b) 4 h, (c) 35 min; solution: (b) 2 mol L−1 formic acid + 2 mol L−1 sodium formate, (c) 2.2 mol L−1 formic acid + 2.2 mol L−1 sodium formate; liquid volume: (b) 6 mL, (c) 3 mL). Reproduced with permission.68 Copyright 2018, Royal Society of Chemistry. | |
4. Purification of the high-pressure gas mixture
Along with the development of several active catalysts for high-pressure gas generation by FADH, we focused on the purification of the gas mixture to ensure its applicability in electric fuel cell vehicles.54,55 The separation of H2 and CO2 was achieved by changing the physical fluid state from a supercritical phase to a gas–liquid phase maintaining the high-pressure conditions. The generated high-pressure gas mixture (H2 and CO2) at 80 °C exists in the supercritical phase as it has a lower critical point at 17 °C at 30 MPa pressure. By cooling down the gas mixture to a lower temperature below 17 °C at 30 MPa, the fluid phase changes from a supercritical phase to a gas–liquid phase without depressurization. Therefore, the generated high-pressure gas mixture (H2 and CO2) at 80 °C and 30 MPa was cooled down to −15 °C to obtain the gas–liquid phase and high-pressure H2 (69 mol%) could be recovered. The purity of H2 increased to 85 mol% when the temperature was further lowered to −51 °C (Fig. 22).54 In case of catalyst 17, the high-pressure gas released from FADH at 60 °C was cooled to low temperatures (−78 °C), while maintaining the pressure.55 While cooling the separator from −60 °C to −70 °C, a pressure drop was observed from 13 to 12 MPa which indicated the solidification of CO2. More than 95 mol% H2 gas was continuously obtained by gas composition analysis during pressurization from 11 MPa to atmospheric pressure while keeping the separator cool. Almost pure CO2 was obtained by just closing and heating the separator to room temperature after the depressurization process. Thus, the sequential production and separation of high-pressure H2 and CO2 was achieved which is very essential for H2 delivery to the hydrogen fuelling stations.
 |
| | Fig. 22 Phase behaviour of the generated gas from FA in the separator at various conditions: (a) 0.1 MPa and 35 °C, (b) 30 MPa and 35 °C, and (c) 30 MPa and −10 °C. Generation conditions (80 °C, 30 MPa), aqueous solution of FA (12 M, 40 mL), catalyst 14 (0.2 × 10−3 M, 8 μmol). The black vertical tube in the view cell is the inlet tube of the generated gas. Reproduced with permission.54 Copyright 2016, Wiley-VCH. | |
5. Conclusion
At present, the most common industrial methods of formic acid production include hydrolysis of methyl formate and oxidation of biomass. The direct hydrogenation of CO2 into FA is more attractive because it serves the dual purpose of CO2 utilization and hydrogen storage in liquid form. The development of efficient catalysts for the high-pressure gas generation from formic acid is always challenging due to the highly acidic reaction conditions which often lead to the decomposition of the active catalytic species. Therefore, very efficient and stable catalysts under acidic pH are needed to serve this purpose and hopefully achieve the 70 MPa pressure (without CO2) required to refuel hydrogen fuel cell electric vehicles. In addition, CO contamination is always an issue in FADH at higher temperatures which can poison the fuel cells. To minimize this disadvantage, selective catalysts should be developed that dehydrogenate FA to H2 and CO2 and recently the use of membrane microreactors instead of batch reactors has been shown to facilitate the continuous removal of CO during heterogeneously catalysed decomposition of FA.72 In this review, we have summarized the recent developments in catalytic high-pressure gas generation from FADH with a special focus on our research. Both homogeneous and heterogeneous catalysts were developed which could produce high-pressure gas above 150 MPa and 35 MPa, respectively, by FADH. Some of our developed catalysts displayed long-term stability in water under high-pressure reaction conditions as compared to most other catalysts which decompose after a short period of time. In addition, high-pressure H2 was easily separated from CO2 by changing the fluid phase at lower temperatures without depressurization. Thus, a technology for the continuous production of high-pressure H2 under ambient reaction conditions was developed without any mechanical compressor. The developed catalytic processes should be scaled up and corresponding life cycle assessment (LCA) studies must be performed before implementation of this technology. The potential high-pressure H2 release from a liquid-phase H2 carrier like FA is an excellent technological option for long-term H2 storage, long-distance hydrogen transport, and delivery. The chemical compressor based on FADH without a mechanical compressor has the potential to substantially reduce the energy demand and infrastructure cost of hydrogen fuelling stations. We believe that the scientific developments compiled in this review would pave the way for the development of more robust catalytic systems for high-pressure gas generation from high concentrations of FA in the near future.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the New Energy and Industrial Technology Development Organization (NEDO) and JSPS KAKENHI (grant number 23H00315).
References
- N. Armaroli and V. Balzani, ChemSuschem, 2011, 4, 21–36 CrossRef CAS PubMed.
- G. M. Whitesides and G. W. Crabtree, Science, 2007, 315, 796–798 CrossRef CAS PubMed.
- P. Moriarty and D. Honnery, Int. J. Hydrogen Energy, 2009, 34, 31–39 CrossRef CAS.
- P. Moriarty and D. Honnery, Int. J. Hydrogen Energy, 2010, 35, 12374–12380 CrossRef CAS.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- J. A. Turner, Science, 2004, 305, 972–974 CrossRef CAS PubMed.
- U. Eberle, M. Felderhoff and F. Schuth, Angew. Chem., Int. Ed., 2009, 48, 6608–6630 CrossRef CAS PubMed.
- A. F. Dalebrook, W. J. Gan, M. Grasemann, S. Moret and G. Laurenczy, Chem. Commun., 2013, 49, 8735–8751 RSC.
- H. L. Jiang, S. K. Singh, J. M. Yan, X. B. Zhang and Q. Xu, Chemsuschem, 2010, 3, 541–549 CrossRef CAS PubMed.
- K. Sordakis, C. H. Tang, L. K. Vogt, H. Junge, P. J. Dyson, M. Beller and G. Laurenczy, Chem. Rev., 2018, 118, 372–433 CrossRef CAS PubMed.
- A. Boddien, F. Gärtner, C. Federsel, P. Sponholz, D. Mellmann, R. Jackstell, H. Junge and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 6411–6414 CrossRef CAS PubMed.
- F. Xu, W. Huang, Y. Wang, D. Astruc and X. Liu, Inorg. Chem. Front., 2022, 9, 3514–3521 RSC.
- M. Grasemann and G. Laurenczy, Energy Environ. Sci., 2012, 5, 8171–8181 RSC.
- R. van Putten, T. Wissink, T. Swinkels and E. A. Pidko, Int. J. Hydrogen Energy, 2019, 44, 28533–28541 CrossRef CAS.
- D. Mellmann, P. Sponholz, H. Junge and M. Beller, Chem. Soc. Rev., 2016, 45, 3954–3988 RSC.
- J. Eppinger and K. W. Huang, ACS Energy Lett., 2017, 2, 188–195 CrossRef CAS.
- H. Zhong, M. Iguchi, M. Chatterjee, Y. Himeda, Q. Xu and H. Kawanami, Adv. Sustainable Syst., 2018, 2, 1700161 CrossRef.
- I. Dutta, S. Chatterjee, H. Cheng, R. K. Parsapur, Z. Liu, Z. Li, E. Ye, H. Kawanami, J. S. C. Low, Z. Lai, X. J. Loh and K.-W. Huang, Adv. Energy Mater., 2022, 12, 2103799 CrossRef CAS.
- N. Onishi, M. Iguchi, X. C. Yang, R. Kanega, H. Kawanami, Q. Xu and Y. Himeda, Adv. Energy Mater., 2019, 9, 1801275 CrossRef.
- N. Onishi, R. Kanega, H. Kawanami and Y. Himeda, Molecules, 2022, 27, 455 CrossRef CAS PubMed.
- M. X. Liu, Y. K. Xu, Y. Meng, L. J. Wang, H. Wang, Y. C. Huang, N. Onishi, L. Wang, Z. J. Fan and Y. Himeda, Adv. Energy Mater., 2022, 12, 2200817 CrossRef CAS.
- C. Fellay, P. J. Dyson and G. Laurenczy, Angew. Chem., Int. Ed., 2008, 47, 3966–3968 CrossRef CAS PubMed.
- B. Loges, A. Boddien, H. Junge and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 3962 CrossRef CAS PubMed.
- Y. Himeda, Green Chem., 2009, 11, 2018–2022 RSC.
- J. F. Hull, Y. Himeda, W. H. Wang, B. Hashiguchi, R. Periana, D. J. Szalda, J. T. Muckerman and E. Fujita, Nat. Chem., 2012, 4, 383–388 CrossRef CAS PubMed.
- Z. J. Wang, S. M. Lu, J. Li, J. J. Wang and C. Li, Chem.–Eur. J., 2015, 21, 12592–12595 CrossRef CAS PubMed.
- S. M. Lu, Z. Wang, J. Wang, J. Li and C. Li, Green Chem., 2018, 20, 1835 RSC.
- Q. G. Liu, X. F. Yang, Y. Q. Huang, S. T. Xu, X. Su, X. L. Pan, J. M. Xu, A. Q. Wang, C. H. Liang, X. K. Wang and T. Zhang, Energy Environ. Sci., 2015, 8, 3204–3207 RSC.
- N. Wang, Q. M. Sun, R. S. Bai, X. Li, G. Q. Guo and J. H. Yu, J. Am. Chem. Soc., 2016, 138, 7484–7487 CrossRef CAS PubMed.
- X. C. Yang, P. Pachfule, Y. Chen, N. Tsumori and Q. Xu, Chem. Commun., 2016, 52, 4171–4174 RSC.
- N. Lentz and M. Albrecht, ACS Catal., 2022, 12, 12627–12631 CrossRef CAS.
- N. Onishi, R. Kanega, E. Fujita and Y. Himeda, Adv. Synth. Catal., 2019, 361, 289 CrossRef CAS.
- S. Kar, M. Rauch, G. Leitus, Y. Ben-David and D. Milstein, Nat. Catal., 2021, 4, 193–201 CrossRef CAS PubMed.
- D. Wei, R. Sang, P. Sponholz, H. Junge and M. Beller, Nat. Energy, 2022, 7, 438 CrossRef CAS.
- J. J. A. Celaje, Z. Lu, E. A. Kedzie, N. J. Terrile, J. N. Lo and T. J. Williams, Nat. Commun., 2016, 7, 11308 CrossRef CAS PubMed.
- K. Mori, Y. Futamura, S. Masuda, H. Kobayashi and H. Yamashita, Nat. Commun., 2019, 10, 4094 CrossRef PubMed.
- Q. L. Zhu, N. Tsumori and Q. Xu, J. Am. Chem. Soc., 2015, 137, 11743–11748 CrossRef CAS PubMed.
- Q. L. Zhu, F. Z. Song, Q. J. Wang, N. Tsumori, Y. Himeda, T. Autrey and Q. Xu, J. Mater. Chem. A, 2018, 6, 5544–5549 RSC.
- F.-Z. Song, Q.-L. Zhu, N. Tsumori and Q. Xu, ACS Catal., 2015, 5, 5141–5144 CrossRef CAS.
- F. Bertini, I. Mellone, A. Ienco, M. Peruzzini and L. Gonsalvi, ACS Catal., 2015, 5, 1254–1265 CrossRef CAS.
- E. A. Bielinski, P. O. Lagaditis, Y. Zhang, B. Q. Mercado, C. Würtele, W. H. Bernskoetter, N. Hazari and S. Schneider, J. Am. Chem. Soc., 2014, 136, 10234–10237 CrossRef CAS PubMed.
- J. B. Curley, N. E. Smith, W. H. Bernskoetter, N. Hazari and B. Q. Mercado, Organometallics, 2018, 37, 3846–3853 CrossRef CAS.
- A. Léval, A. Agapova, C. Steinlechner, E. Alberico, H. Junge and M. Beller, Green Chem., 2020, 22, 913 RSC.
- S. Oldenhof, M. Lutz, B. de Bruin, J. I. van der Vlugt and J. N. Reek, Chem. Sci., 2015, 6, 1027 RSC.
- S. Wang, H. Huang, T. Roisnel, C. Bruneau and C. Fischmeister, ChemSusChem, 2019, 12, 179–184 CrossRef CAS PubMed.
- J. D. Scholten, M. H. G. Prechtl and J. Dupont, ChemCatChem, 2010, 2, 1265–1270 CrossRef CAS.
- G. A. Filonenko, R. van Putten, E. N. Schulpen, E. J. M. Hensen and E. A. Pidko, ChemCatChem, 2014, 6, 1526–1530 CrossRef CAS.
- C. Prichatz, M. Trincado, L. Tan, F. Casas, A. Kammer, H. Junge, M. Beller and H. Grützmacher, ChemSusChem, 2018, 11, 3092–3095 CrossRef CAS PubMed.
- L. Yaacoub, I. Dutta, B. Werghi, B. W. J. Chen, J. Zhang, E. A. Hamad, E. P. Ling Ang, E. Pump, A. B. Sedjerari, K.-W. Huang and J.-M. Basset, ACS Catal., 2022, 12, 14408–14417 CrossRef CAS.
- L. Piccirilli, B. Rabell, R. Padilla, A. Riisager, S. Das and M. Nielsen, J. Am. Chem. Soc., 2023, 145, 5655–5663 CrossRef CAS PubMed.
- K. Muller, K. Brooks and T. Autrey, Energy Fuels, 2018, 32, 10008–10015 CrossRef CAS.
- K. Müller, K. Brooks and T. Autrey, Energy Fuels, 2017, 31, 12603–12611 CrossRef.
- K. Reddi, A. Elgowainy and E. Sutherland, Int. J. Hydrogen Energy, 2014, 39, 19169–19181 CrossRef CAS.
- M. Iguchi, Y. Himeda, Y. Manaka, K. Matsuoka and H. Kawanami, Chemcatchem, 2016, 8, 886–890 CrossRef CAS.
- M. Iguchi, M. Chatterjee, N. Onishi, Y. Himeda and H. Kawanami, Sustainable Energy Fuels, 2018, 2, 1719–1725 RSC.
-
Explosion at hydrogen station – NRK Norway, https://www.nrk.no/norge/eksplosjon-ved-hydrogenstasjon-1.14582914, accessed 07-07-2023.
-
Center for Hydrogen Safety, https://www.aiche.org/chs/conferences/international-center-hydrogen-safety-conference/2019/proceeding/paper/review-hydrogen-tank-explosion-gangneung-south-korea, accessed 07-07-2023.
- M. Czaun, J. Kothandaraman, A. Goeppert, B. Yang, S. Greenberg, R. B. May, G. A. Olah and G. K. S. Prakash, ACS Catal., 2016, 6, 7475–7484 CrossRef CAS.
- G. Papp, G. Olveti, H. Horvath, A. Katho and F. Joo, Dalton Trans., 2016, 45, 14516–14519 RSC.
- C. Guan, D. D. Zhang, Y. P. Pan, M. Iguchi, M. J. Ajitha, J. S. Hu, H. F. Li, C. G. Yao, M. H. Huang, S. X. Ming, J. R. Zheng, Y. Himeda, H. Kawanami and K. W. Huang, Inorg. Chem., 2017, 56, 438–445 CrossRef CAS PubMed.
- J. B. Geri, J. L. Ciatti and N. K. Szymczak, Chem. Commun., 2018, 54, 7790–7793 RSC.
- M. Iguchi, C. Guan, K. W. Huang and H. Kawanami, Int. J. Hydrogen Energy, 2019, 44, 28507–28513 CrossRef CAS.
- M. Iguchi, H. Zhong, Y. Himeda and H. Kawanami, Chem. –Eur. J., 2017, 23, 17017–17021 CrossRef CAS PubMed.
- M. Iguchi, H. Zhong, Y. Himeda and H. Kawanami, Chem. –Eur. J., 2017, 23, 17788–17793 CrossRef CAS PubMed.
- M. Iguchi, Y. Himeda, Y. Manaka and H. Kawanami, Chemsuschem, 2016, 9, 2749–2753 CrossRef CAS PubMed.
- M. Iguchi, N. Onishi, Y. Himeda and H. Kawanami, ChemPhysChem, 2019, 20, 1296–1300 CrossRef CAS PubMed.
- H. Kawanami, M. Iguchi and Y. Himeda, Inorg. Chem., 2020, 59, 4191 CrossRef CAS PubMed.
- H. Zhong, M. Iguchi, F. Z. Song, M. Chatterjee, T. Ishizaka, I. Nagao, Q. Xu and H. Kawanami, Sustainable Energy Fuels, 2017, 1, 1049–1055 RSC.
- X. Liu, T. Jacob and W. Gao, J. Energy Chem., 2022, 70, 292–309 CrossRef CAS.
- S. Hafeez, E. Harkou, A. Spanou, S. M. Al-Salem, A. Villa, N. Dimitratos, G. Manos and A. Constantinou, Mater. Today Chem., 2022, 26, 1011120 Search PubMed.
- B. Wang, S. Yang, Z. Yu, T. Zhang and S. Liu, Mater. Today Commun., 2022, 31, 103617 CrossRef CAS.
- S. Hafeez, S. M. Al-Salem, A. Bansode, A. Villa, N. Dimitratos, G. Manos and A. Constantinou, Ind. Eng. Chem. Res., 2022, 61, 1655–1665 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Babulal
Maji
,
Babulal
Maji