
Engineering the oxygen-evolution activity by changing the A-site rare-earth elements in RSr3Fe1.5Co1.5O10−δ (R = La, Nd, Pr) Ruddlesden–Popper perovskites†
Wenyun
Zhu
a,
Jiani
Chen
a,
Dongliang
Liu
a,
Guangming
Yang
 a,
Wei
Zhou
a,
Ran
Ran
a,
Jie
Yu
*bc and
Zongping
Shao
*ad
a,
Wei
Zhou
a,
Ran
Ran
a,
Jie
Yu
*bc and
Zongping
Shao
*ad
aA State Key Laboratory of Materials-Oriented Chemical Engineering, College of Chemical Engineering, Nanjing Tech University, Nanjing, 210009, China
bSchool of Energy and Power, Jiangsu University of Science and Technology, Zhenjiang 212100, P. R. China
cDepartment of Building and Real Estate, Research Institute for Sustainable Urbanization (RISUD), Research Institute for Smart Energy (RISE), The Hong Kong Polytechnic University, Hung Hom, Kowloon, Hong Kong, 999077, P. R. China
dWA School of Mines: Minerals, Energy and Chemical Engineering, Curtin University, Perth 6102, Australia
First published on 3rd July 2023
Abstract
The design of high-performance and low-cost catalysts for the oxygen evolution reaction (OER) is paramount for storing and converting clean and renewable energy. Ruddlesden–Popper (RP)-structured perovskite oxides show promising potential for efficiently catalyzing the OER. In this study, a series of RP-type perovskites RSr3Fe1.5Co1.5O10−δ (R = La, Nd, Pr) are synthesized and investigated to correlate their structure and physical structure properties with OER activities. Among the synthesized materials, PrSr3Fe1.5Co1.5O10−δ shows the best OER performance, evidenced by the smallest overpotential (294 mV) as well as the lowest Tafel slope (63 mV dec−1). Such enhanced OER behavior is ascribed to larger electrochemically active areas, faster charge transfer rates, higher B-site valence state ions, more oxygen vacancies, and more favorable lattice oxygen oxidation (LOM) behavior. When applied in Zn–air batteries and water electrolyzers, PSFC also outperforms the benchmark catalyst RuO2, suggesting that PSFC has the potential to be an outstanding OER electrocatalyst for practical applications. This study highlights the significance of adjusting A-site elements for improving OER activities.
1. Introduction
Addressing the overuse of fossil fuels and associated environmental issues, various electrochemical energy storage and conversion systems, such as water splitting,1 fuel cells,2,3 and rechargeable metal–air batteries,4 are urgently required. The OER, referring to an electrochemical water oxidation pathway, plays a significant role in the above-mentioned energy and chemical transformation processes, while its sluggish reaction kinetics largely restricts the overall efficiency of these electrochemical systems.5,6 Hence, efficient OER electrocatalysts are highly needed to boost the reaction process. To date, the flagship OER electrocatalysts are still limited to precious metal-based oxides, such as RuO2 and IrO2,7–10 which have been hindered from commercial applications because of their high cost and scarcity.11 Toward this end, it is highly desirable to develop low-cost and high-performance catalytic materials to replace Ru and Ir.12,13Among numerous candidates, a plethora of functional perovskite oxides, including simple perovskites (with the general formula ABO3), double perovskites (A2B2O6), and Ruddlesden–Popper (RP) perovskites (AO(ABO3)n), have received extensive attention from researchers due to their rich structures, facile preparation, low cost, and superior OER performance, some of which even exceed the benchmark IrO2 or RuO2 under alkaline conditions.14–17 Particularly, owing to their compositional flexibility that allows heteroatom doping at A, B, and O sites, the phase structure and electronic state of perovskites can be well modulated, accordingly flourishing much room to rationally design better OER electrocatalysts. For instance, Mandal et al. prepared a class of La0.5Sr0.5Co0.8Fe0.2−xCuxO3−δ (x = 0–0.2) perovskites with different Cu amounts through the solution combustion method.18 Benefitting from the suitable B-site oxidation state and the increased oxygen vacancy concentration, the La0.5Sr0.5Co0.8Cu0.2O3−δ sample presented the most excellent OER performance in 1 M KOH.
In addition to the composition, diverse crystal structures may also determine the electrocatalytic performance. In particular, RP-type perovskite oxides have emerged as a new class of layered-structure materials with increasing research efforts due to their novel physical–chemistry properties. The RP-type perovskite has a general formula of An+1BnO3n+1 (n = 1, 2, 3, etc.), and is made up of alternative rock-salt layers (AO) and perovskite-like layers (ABO3) along the c-axis.19 When approximating the number of perovskite layers (n) to infinity, the RP-type perovskite turns into a simple perovskite, which illustrates the similarity between the RP-type perovskite and the simple perovskite, for instance, rich chemical compositions, changeable oxygen stoichiometries, and superior OER activities. Furthermore, the layered structure provides at least two chemical environments, perovskite slab and rock-salt slab, which can also bring about disparate cationic coordination environments and special anionic defects. Thus, the tunability of RP-type perovskites demonstrates more opportunities for the development of OER catalysts. Recently, Choi and his co-workers carried out a systematic work on the OER catalytic performance of RP structured Lan+1NinO3n+1 (n = 1, 2, 3, and 4).20 The abundant oxygen defects and charge carriers endowed La5Ni4O13−δ with a favorable electronic structure, resulting in an unusual lattice-oxygen oxidation mechanism. Miyahara et al. also found that an RP-type oxychloride Sr2Co0.8Fe0.2O3Cl outperformed the well-known active OER electrocatalyst Ba0.5Sr0.5Co0.8Fe0.2O3−δ, and minimized the potential gap of the OER and the oxygen reduction reaction (ORR) when compared to some previously reported conventional perovskite-type catalytic materials.21
MSr3Fe1.5Co1.5O10−δ (M = La, Ce, Pr, Nd, Pm, Sm, etc.), as a typical RP-type perovskite, has been reported with intriguing catalytic behavior in various research fields, such as fuel cells, water splitting, and CO2 reduction. RP-LaSr3(Co0.5Fe0.5)3O10−δ with an optimal doping ratio delivered increased oxygen vacancies, the favorable synergy between Fe and Co, and the facile oxygen redox capability, thus exhibiting satisfying ORR and OER activities.22 Besides, LaSr3Fe1.5Co1.5O10−δ with an optimized morphology shows boosted activity towards both the OER and the ORR due to more exposed active sites and enlarged electrochemical surface areas (ECSAs).23 Castillo et al. systematically studied RP-phase LnSr3Fe1.5Co1.5O10−δ with Ln = Pr, Nd, and La as the oxygen reduction electrode in solid oxide fuel cells and demonstrated that PrSr3Fe1.5Co1.5O10−δ offered the lowest polarization resistance.24 As is known, different A-site metal oxides possess variable electronic structures, thus eventually affecting the catalytic performance of the perovskites. A systematic study of A-site elements in MSr3Fe1.5Co1.5O10−δ perovskites will provide a highly useful guide for the design of RP-structure perovskites as efficient OER catalysts.
Herein, we successfully synthesize a series of RP-phase RSr3Fe1.5Co1.5O10−δ (R = La, Nd, Pr) materials via a sol–gel route, namely LaSr3Fe1.5Co1.5O10−δ, NdSr3Fe1.5Co1.5O10−δ, and PrSr3Fe1.5Co1.5O10−δ (denoted as LSFC, NSFC, and PSFC), and conduct a comprehensive investigation on the impact of R metals, primarily on the OER catalytic properties under alkaline conditions. In the 0.1 M KOH electrolyte, PSFC delivers the best OER catalytic activity with the smallest overpotential of 294 mV at 10 mA cm−2 and a Tafel slope (63 mV dec−1), which also outperforms those of the benchmark RuO2 and most of the reported metal oxides. Moreover, a higher mass activity (MA) of 80.548 A gcatalyst−1 and a specific activity (SA) of 7.3742 A mcatalyst−2 for PSFC were also achieved among the as-prepared materials. Combining various experimental results, the outstanding performance of PSFC originated from a more rapid charge transfer rate, an enlarged ECSA, a higher Co/Fe valence state, and a richer oxygen vacancy content, which further triggered the favorable lattice oxygen oxidation mechanism processes. When tested in energy-related devices including rechargeable zinc–air batteries and water electrolyzers, PSFC outperforms the RuO2 benchmark, which suggests its great potential for practical applications.
2. Results and discussion
2.1 Phase and microstructure
Here, MSr3Fe1.5Co1.5O10−δ (M = La, Nd, Pr) are prepared using a typical one-pot sol–gel route, which ensured good element dispersion through the perovskite microstructure. Fig. 1a exhibits the room-temperature X-ray diffraction (XRD) patterns of LSFC, NSFC, and PSFC, which reveals the formation of single-phase structures for all as-obtained samples. Further Rietveld refinement demonstrates the tetragonal structure with a space group of I4/mmm. Their lattice parameters are a = b = 3.8086(2) Å, c = 27.86395(1) Å, and α = β = γ = 90°, as listed in Table S1 (ESI†). Besides, the average Co–O length between LSFC, NSFC, and PSFC shows a negligible difference, which cannot be mainly responsible for the following OER activity difference. A schematic structure for this kind of RP-type material (n = 3) is exhibited in Fig. 1b, consisting of perovskite ABO3 layers and rock-salt AO layers. The scanning electron microscopy (SEM) images of these compounds in Fig. S1 (ESI†) show the typical bulk morphology with particles on a micrometer scale. The bulk morphology is derived from high-temperature calcination, which is also reflected by the N2 adsorption/desorption isotherms (Fig. 1c). The surface areas of LSFC, NSFC, and PSFC are 10.015, 10.875, and 10.923 m2 g−1, respectively. The energy dispersive spectroscopy (EDS) result presented in Fig. S2 and Table S2 (ESI†) proves that the elemental composition of the samples basically conforms to the expected atomic ratio. Transmission electron microscopy (TEM) is further performed to show the phase structural features. The clear lattice fringes of 0.192 and 0.276 nm, shown in Fig. 1d–f, correspond to the d204 and d178 facets of LSFC, NSFC, and PSFC. Energy-dispersive X-ray spectroscopy (EDX) elemental mapping in Fig. 1g and Fig. S3 and S4 (ESI†) revealed that all elements are uniformly distributed.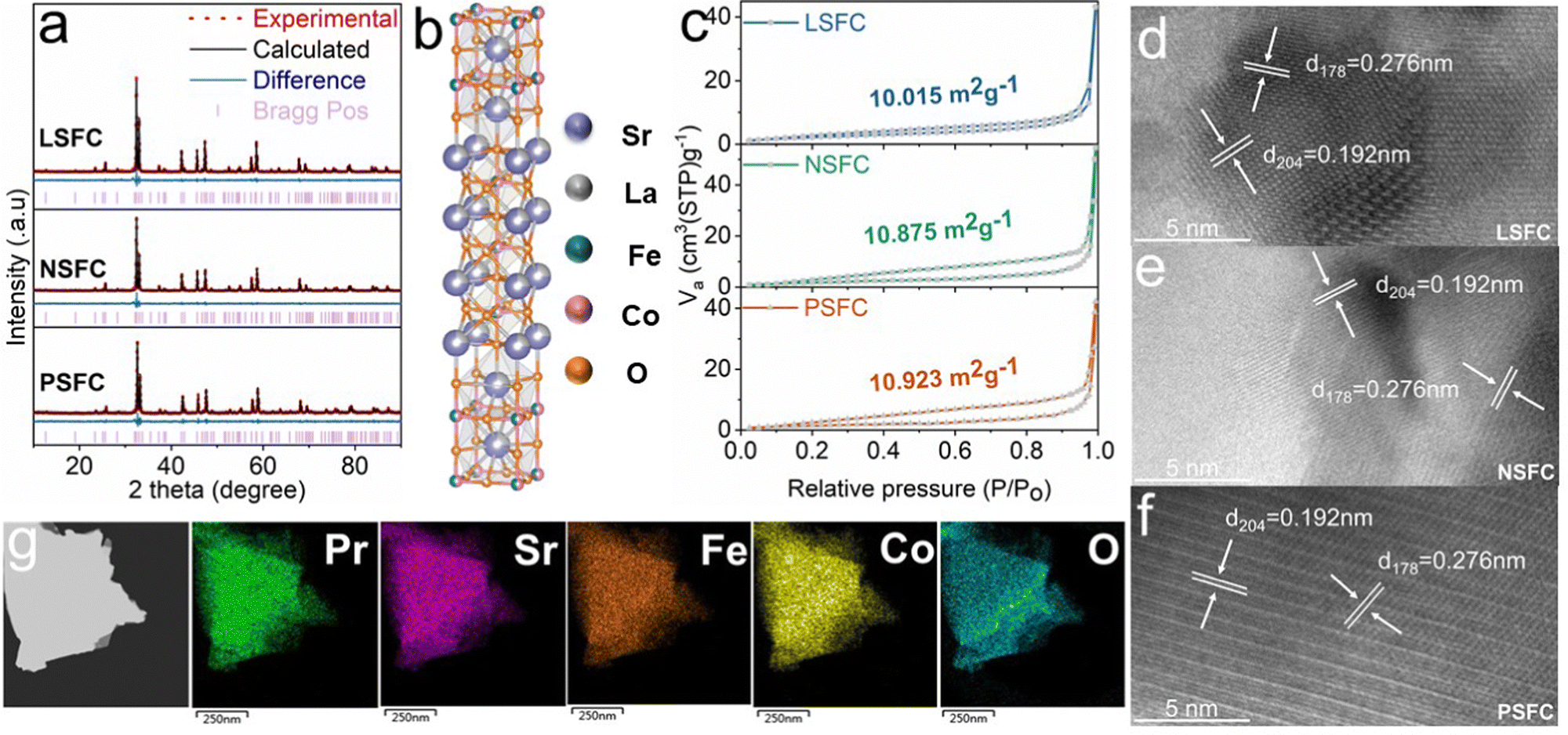 | ||
| Fig. 1 (a) Refined X-ray diffraction profiles of LSFC, NSFC, and PSFC. (b) Schematic presentation of LSFC. (c) Specific surface areas of LSFC, NSFC, and PSFC calculated based on the BET method from the N2 adsorption–desorption isotherms. (d–f) HRTEM images of LSFC, NSFC, and PSFC along the [178] and [204] zone axes. (g) Scanning TEM images and corresponding elemental mappings of PSFC. | ||
2.2. Electrocatalytic OER performance
To assess the OER electrocatalytic performance of LSFC, NSFC and, PSFC in 0.1 M KOH solution, we employed a classical thin-film rotating disk electrode technique. For comparison, the analogous measurements were also carried out on the benchmark RuO2 catalyst. Fig. 2a exhibits the representative polarization curves of these as-synthesized samples. Obviously, PSFC delivers the most outstanding OER activity among the four samples in the aspect of the smallest overpotential and the largest current response. Of note, the overpotential to yield a 10 mA cm−2 current density (η10) is considered as a key parameter of electrocatalysts due to their pertinence with conversion efficiency. Remarkably, PSFC displays a fairly low η10 of 294 mV, when compared to the η10 values of LSFC (341 mV) and NSFC (375 mV), which is also superior to that of RuO2. In addition, the kinetics of these three samples can be reflected by the Tafel plots in Fig. 2b. PSFC displays a lower Tafel slope (63 mV dec−1) relative to LSFC (91 mV dec−1) and NSFC (112 mV dec−1), indicating faster reaction kinetics. The overpotential η10 and the Tafel slope of various representative electrocatalysts in the 0.1 M KOH solution are presented in Fig. 2e and Table S3 (ESI†), which highly demonstrates that PSFC stands out as the best-performing OER electrocatalyst among other reported catalysts so far. To further evaluate the activity, we normalized their OER activity to the mass and specific surface area, which are named mass activity (MA) and specific activity (SA), respectively. Considering that the SA can reflect the intrinsic activity of the catalysts and the MA is of great significance for practical applications, the comparison of the SA and MA among the three samples is highly meaningful. As shown in Fig. 2c, PSFC achieves a MA of 80.548 A gcatalyst−1 at 1.55 V, which is ∼4 and ∼2.7 times that of NSFC (19.8161 A gcatalyst−1) and LSFC (29.7778 A gcatalyst−1), respectively. Besides, the SA of PSFC (7.3742 A mcatalyst−2) is also significantly higher than those of NSFC (1.8222 A mcatalyst−2) and LSFC (2.9733 A mcatalyst−2). Both of them confirmed the better intrinsic activity of PSFC. | ||
| Fig. 2 (a) OER polarization curves of LSFC, NSFC, PSFC, and commercial IrO2 catalysts in O2-saturated 0.1 M KOH. (b) Tafel plots of LSFC, NSFC, and PSFC. (c) Mass activities based on the oxide weight and BET surface area-normalized intrinsic activities of the catalysts at 1.55 V. (d) Chronopotentiometric curves of the catalysts at 10 mA cm−2. (e) OER activity comparisons of η10 with other reported state-of-the-art catalysts in 0.1 M KOH. | ||
Apart from activity, operating stability, as another evaluation standard of the practical application, is also of great importance. Therefore, chronopotentiometry (CP) testing was conducted to disclose the long-term durability of LSFC, NSFC, and PSFC. Remarkably, the PSFC electrode could be stably operated at 10 mA cm−2 for about 66 hours as shown in Fig. 2d, with slight potential increments of about 91 mV for LSFC, 89 mV for NSFC, and 72 mV for PSFC, better than RuO2, the potential of which was increased by about 140 mV after only 48 h. Besides, the samples after the CP testing were further characterized to unveil the micro-structural robustness. As demonstrated in Fig. S5 (ESI†), the XRD data suggested that the original phase structure was sustained for all post-OER LSFC, NSFC, and PSFC materials. The EDS of the catalysts after the OER suggests that the atomic composition does not obviously change after the OER (Fig. S2, ESI†). Besides, Fig. S6 (ESI†) shows the comparison of XPS curves before and after the CP testing. The XPS peaks of Fe and Co shift to higher energy after the OER, indicating the increased valence state of Fe and Co, which is common for OER catalysts.25 Then, we conducted inductively coupled plasma-mass spectroscopy (ICP-MS) to disclose the element dissolution. As shown in Table S4 (ESI†), after OER activation, the leached ions are mainly Fe ions, and Co ions are not detected in their electrolytes. For the leaching of Fe ions, NSFC is most obvious and PSFC has hardly any leaching (Fig. S7, ESI†).
2.3. The activity origin and OER mechanism
It is widely accepted that the electronic configurations of B and O sites in perovskites play a paramount part in catalyzing the OER.26 Here, X-ray photoelectron spectroscopy (XPS) was first employed to detect the chemical states of Fe, Co, and O elements on the surface of samples. XPS spectra and the corresponding deconvolution results of LSFC, NSFC, and PSFC are shown in Fig. 3a–c. They display the peak at binding energies of ∼710 eV, ∼780 eV, and ∼532 eV, which can be assigned to Fe 2p, Co 2p, and O 1s, respectively. The Fe 2p3/2 XPS spectra of LSFC, NSFC, and PSFC all can be deconvoluted into two peaks of Fe3+ and Fe4+.27 As shown in Fig. 3a and Table S5 (ESI†), the proportion of Fe4+ arranges in the order of PSFC > LSFC >NSFC. Similar to Fe peaks, the Co 2p XPS spectra can also be deconvoluted into two peaks,28 indicating the coexistence of Co3+ and Co4+ in the catalysts. The corresponding deconvolution curves reveal that PSFC possesses the highest content of Co4+ and Fe4+ species (Fig. 3a, b and, Table S5, ESI†). Previous research has proved that high-valence metal ions were beneficial for the adsorption of oxygen-containing intermediates and significantly enhanced the activity of the OER, which consist of the conclusion we draw here.29,30Fig. 3c exhibits the O 1s XPS spectra of LSFC, NSFC, and PSFC. They all can be deconvoluted into lattice oxygen species (∼529 eV for OL), oxygen vacancies (∼531.3 eV for OV), and hydroxyl groups or the surface-adsorbed oxygen (∼532.6 eV for Oabs).31,32 As shown in Fig. 3d and Table S5 (ESI†), the content of oxygen vacancy is calculated by the relative area of its fitted sub-peaks. Obviously, PSFC has the highest relative content of OV species among the three catalysts, which previously have been demonstrated as highly active sites.33 Because oxygen vacancies modify the interaction processes (absorption and desorption) of the electrocatalysts with the reactants as well as the bulk material properties (such as the electronic structure and electronic conductivity) thus affecting the OER performance.34 For example, Zhuang et al. found that oxygen vacancies were favorable for the formation of OH− and O2− intermediates on the surface Co3+ close to an oxygen vacancy from DFT calculations.35 Asnavandi et al. also demonstrated that cations associated with oxygen vacancies were more active by combining the experimental findings and DFT calculations.36 Furthermore, Bao et al. found that the oxygen vacancies could promote the adsorption of H2O molecules onto the surface of the catalysts by DFT, which could increase the reactivity of active sites and the OER performance.37 Through these previous works, a reasonable conjecture can be obtained that oxygen vacancies in our samples can also improve the OER performance by affecting the cation activity of Co3+/4+ and Fe3+/4+ close to it and facilitating the adsorption of the intermediate, thus promoting the OER performance. | ||
| Fig. 3 (a–c) Fe 2p, Co 2p and O 1s XPS spectra of LSFC, NSFC, and PSFC. (d) The proportion of oxygen vacancies in LSFC, NSFC, and PSFC. (e) Linear fitting of the capacitive currents versus CV scan rates of LSFC, NSFC, and PSFC. (f) EIS spectra of LSFC, NSFC, and PSFC. | ||
For further illustration of the improvement of the OER performance, we investigated the ECSA and electrochemical impendence spectra (EIS) of these samples, which are important factors in determining the overall catalytic activity. The ECSA was reckoned using a double layer capacitance (Cdl) based on cyclic voltammetry (CV) testing under different scanning rates from 0.02 V s−1 to 0.1 V s−1 as shown in Fig. S8 (ESI†). In Fig. 3e, the normalized Cdl values comply with the following sequence: PSFC > LSFC > NSFC and the Cdl value of PSFC is over twice greater than that of NSFC, which conforms to the trends of OER properties and demonstrates that PSFC could provide more active sites to efficiently enhance the OER activity.38 The EIS measurement is used to represent the charge transfer resistance. Fig. 3f presents the Nyquist plots and discloses the corresponding Rct values of these catalysts under OER conditions, which are determined by the size of the semicircle in the high-frequency range. Obviously, PSFC shows a far smaller Rct value when compared with LSFC and NSFC, revealing more rapid charge transfer and more facile OER kinetics, which enables faster transportation of reactants from the electrolytes to the catalytic active sites of the electrocatalyst.29
As previously pointed out, PSFC delivered the best OER activity and stability. To deepen our understanding of the activity origin of PSFC, we investigated and gleaned the reaction mechanism. To date, two quintessential mechanisms are widely accepted for OER catalysis. One is the conventional adsorbate evolution mechanism (AEM) based on the active metal sites, and the other one is the lattice oxygen oxidation mechanism, named LOM.39–41 In the AEM, the OER process is largely hindered by the scaling relationships of the adsorption energy difference between the *OOH and *OH intermediates, resulting in a minimum theoretical overpotential of approximately 0.37 V for the OER (Fig. 4a up). However, the LOM breaks the limitation of the AEM by coupling the O–O radicals directly, thus enabling the design of OER electrocatalysts with a higher efficiency (Fig. 4a down). Previous reports have proved that the high-valence metal ions and short molecule-level distances in oxides are the crucial factors to switch the OER mechanism from the AEM to the LOM. Considering the similar Co–O bond length for three samples, it can be inferred that PSFC with higher Co valence obeys the LOM more efficiently.42–44
 | ||
| Fig. 4 (a) Schematics of the AEM and LOM pathways. (b) OER polarization curves of PSFC under KOH solutions at different pH values. (c) OER activities of LSFC, NSFC, and PSFC at 1.6 V versus the RHE as a function of pH values. (d) OER polarization curves of PSFC in 1 M KOH and 1 M TMAOH hydroxide solutions. (e) The summary of the current density decay and the Tafel slope decay of various catalysts in 1 M KOH and TMAOH electrolytes, respectively. | ||
Catalysts following the LOM mechanism generally show pH-dependent activity, which can be demonstrated by testing their activity in KOH electrolytes at different pH values.29,43,44 As shown in Fig. 4b and Fig. S9a, S9b (ESI†), LSFC, NSFC, and PSFC show obvious pH-dependent activity, with a positive relationship between the activity and pH value, suggesting that the lattice oxygen participated in the OER process. It is noteworthy that the increased current density of PSFC is prominently larger than those of LSFC and NSFC under a potential of 1.6 V vs. RHE, indicating the most apparent lattice-oxygen participation behavior of PSFC. Beyond this, the slope of logjversus pH values can be used to evaluate the proton reaction order in the LOM process.29,43 In Fig. 4c, the slope of PSFC is larger than those of LSFC and NSFC, which is consistent with the conclusion that PSFC displays extremely apparent lattice oxygen participation behavior.
Except for the pH-dependent activity, the LOM can be indirectly proved by peroxo-like (O22−) and superoxide-like (O2−) negative species. The free energy between the O22− and O2− species is 0.22 eV, namely that O22− is more stable than O2− in thermodynamics, and it is possible to probe the O22− species directly during the OER process. So, we introduced the O22− sensitive tetramethylammonium cation (TMA+) into the electrolyte as a chemical detector to track the O22− species during the LOM process.45–47 We compared the OER activity of LSFC, NSFC, and PSFC electrodes in 1 M KOH and 1 M tetramethylammonium hydroxide (TMAOH) electrolytes, respectively. As shown in Fig. 4d and Fig. S9c, d (ESI†), the OER activity of all three samples decreased because the indispensable intermediate O22− for the LOM is captured by TMA+. As presented in Fig. 4e, PSFC displays the most obvious decrease in the current density in 1 M TMAOH when compared with that 1 M KOH, ulteriorly proving that PSFC shows the most obvious LOM behavior.48
2.4. Practical applications in water splitting and Zn–air batteries
The OER process is the key anodic half-reaction of the water electrolyzer and occurs during the charging process of a rechargeable Zn–air battery, which determines the overall efficiencies of the above energy devices. In this work, the best-performed PSFC was selected as the anode in the water splitting device and the air electrode in Zn–air batteries for investigating its performance in real applications. We first assembled a two-electrode water electrolyzer pairing PSFC and Pt/C loaded on Ni foam as the anodic and cathodic electrodes, respectively (Fig. 5a). For comparison, a water electrolyzer with RuO2 as the anode was further constructed. Fig. 5b shows that the PSFC system only needs a voltage of about 1.628 V to yield a water-splitting current response of 20 mA cm−2, and exhibits better performance than RuO2. Moreover, the alkaline electrolyzer by coupling PSFC and Pt/C exhibits high stability, which shows undetectable voltage fluctuations after 90 h operation (Fig. 5c). Furthermore, we fabricated a Zn–air battery (ZAB) that employs PSFC + Pt/C as an air cathode (Fig. 5d). In addition, commercial RuO2 was selected as the control sample. For the ZAB based on PSFC + Pt/C, a high open circuit potential (OCP) of 1.5 V is observed, slightly lower than the OCP of the battery with RuO2 + Pt/C (1.59 V). The discharge polarization curves and the corresponding power density are presented in Fig. S10 (ESI†). The battery equipped with PSFC + Pt/C arrived at its peak power density of about 40 mW cm−2 at 60 mA cm−2, comparable to that of RuO2 + Pt/C (arriving at its peak of about 46 mW cm−2 at 70 mA cm−2). In Fig. 5e, the ZAB with PSFC + Pt/C offered a small charge/discharge voltage gap, which was slightly narrower than that of the ZAB with RuO2 + Pt/C, indicating its excellent rechargeability. Besides, as shown in Fig. 5f, the ZAB with PSFC + Pt/C showed remarkable stability in a prolonged cycling test. After 420 charge/discharge cycles (around 140 hours), the voltage gap of the PSFC + Pt/C cell enlarged by 0.187 V, which was significantly smaller than the result of the ZAB with the RuO2 + Pt/C air electrode (0.228 V). Considering the above benign performance, PSFC can be a promising OER catalyst for practical alkaline water electrolysis and Zn–air batteries. | ||
| Fig. 5 (a) A schematic configuration of the water electrolyzer. (b) Polarization curves of bare Ni foam//Ni foam, Pt/C//PSFC, and Pt/C(−)//RuO2(+) for overall water splitting in 0.1 M KOH at a scan rate of 5 mV s−1. (c) Chronopotentiometry curve of water electrolysis using PSFC and RuO2 as anodes respectively at a constant current density of 20 mA cm−2 in 0.1 M KOH. (d) A schematic configuration of Zn–air batteries. (e) Charge–discharge polarization curves of PSFC and Pt/C + RuO2 based Zn–air batteries. (f) Long-term galvanostatic charge–discharge cycling profiles of ZABs with the Pt/C + RuO2 electrocatalyst and PSFC electrocatalyst at 2 mA cm−2. | ||
3. Conclusions
In summary, a series of layered RP perovskites, LSFC, NSFC, and PSFC, were investigated to reveal the correlation between their structure and physical properties and the catalytic activity for the OER by tuning A sites. Their activities are in the order of PSFC > LSFC > NSFC. Besides, we proved that the larger surface areas, faster charge transfer rates, higher valence state ions, and abundant oxygen vacancies gave rise to the highest OER activity of PSFC among these three samples. In addition, compared with LSFC and NSFC, further mechanistic experiments showed that PSFC underwent the more favorable LOM process. Also, when acting as the anodic electrode in an overall water-splitting system, PSFC drove a current density of 20 mA cm−2 at a small operation voltage of merely 1.628 V and displayed robust stability after 90 h continuous electrolysis operation, and as the air cathode in ZAB, it also showed benign stability for its narrow voltage gap after 420 continuous charge/discharge cycles. These merits revealed the potential of PSFC as a promising OER catalyst for use in various energy storage/conversion applications. This research underlines the significance of tuning A-site elements as an effective regulation tool for improving OER activities and points out directions for subsequent research in RP materials as OER electrocatalysts, marking a significant advance in the design and fabrication of earth-abundant and low-cost catalytic materials for full water splitting and Zn–air batteries.Author contributions
Supervision, funding acquisition, and methodology: Guangming Yang, Wei Zhou, Jie Yu, and Zongping Shao; software: Dongliang Liu; investigation, visualization and roles/writing – original draft: Wenyun Zhu; writing – review and editing: Jie Yu and Jiani Chen.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China under no. 21878158.Notes and references
- S. Wang, Q. Jiang, S. Ju, C.-S. Hsu, H. M. Chen, D. Zhang and F. Song, Identifying the geometric catalytic active sites of crystalline cobalt oxyhydroxides for oxygen evolution reaction, Nat. Commun., 2022, 13, 6650 CrossRef CAS PubMed
.
- S. Y. Gomez and D. Hotza, Current developments in reversible solid oxide fuel cells, Renew. Sust. Energ. Rev., 2016, 61, 155–174 CrossRef CAS
- Q. Huang, S. Jiang, Y. Wang, J. Jiang, Y. Chen, J. Xu, H. Qiu, C. Su and D. Chen, Highly active and durable triple conducting composite air electrode for low-temperature protonic ceramic fuel cells, Nano Res., 2023, 1–9 Search PubMed
- S.-W. Ke, W. Li, Y. Gu, J. Su, Y. Liu, S. Yuan, J.-L. Zuo, J. Ma and P. He, Covalent organic frameworks with Ni-Bis(dithiolene) and Co-porphyrin units as bifunctional catalysts for Li-O2 batteries, Sci. Adv., 2023, 9, 2398 CrossRef PubMed
- Y. Wen, C. Liu, R. Huang, H. Zhang, X. Li, F. P. Garcia de Arquer, Z. Liu, Y. Li and B. Zhang, Introducing Bronsted acid sites to accelerate the bridging-oxygen-assisted deprotonation in acidic water oxidation, Nat. Commun., 2022, 13, 4871 CrossRef CAS PubMed
- H. Sun, X. Xu, H. Kim, W. Jung, W. Zhou and Z. Shao, Electrochemical Water Splitting: Bridging the Gaps between Fundamental Research and Industrial Applications, Energy Environ. Mater., 2022, 12441 Search PubMed
- H. Jin, X. Liu, P. An, C. Tang, H. Yu, Q. Zhang, H.-J. Peng, L. Gu, Y. Zheng, T. Song, K. Davey, U. Paik, J. Dong and S.-Z. Qiao, Dynamic rhenium dopant boosts ruthenium oxide for durable oxygen evolution, Nat. Commun., 2023, 14, 354 CrossRef CAS PubMed
- K. Du, E. Gao, C. Zhang, Y. Ma, P. Wang, R. Yu, W. Li, K. Zheng, X. Cheng, D. Tang, B. Deng, H. Yin and D. Wang, An iron-base oxygen-evolution electrode for high-temperature electrolyzers, Nat. Commun., 2023, 14, 253 CrossRef CAS PubMed
- J. Wang, H. Yang, F. Li, L. Li, J. Wu, S. Liu, T. Cheng, Y. Xu, Q. Shao and X. Huang, Single-site Pt-doped RuO2 hollow nanospheres with interstitial C for high-performance acidic overall water splitting, Sci. Adv., 2022, 8, 9271 CrossRef PubMed
- J. Yu, Y. Dai, Q. He, D. Zhao, Z. Shao and M. Ni, A mini-review of noble-metal-free electrocatalysts for overall water splitting in non-alkaline electrolytes, Mater. Rep.: Energy, 2021, 1, 100024 CAS
- J. Yu, Z. Li, T. Liu, S. Zhao, D. Guan, D. Chen, Z. Shao and M. Ni, Morphology control and electronic tailoring of CoxAy (A = P, S, Se) electrocatalysts for water splitting, Chem. Eng. J., 2023, 460, 141674 CrossRef CAS
- C. Wang, C. Li, J. Liu and C. Guo, Engineering transition metal-based nanomaterials for high-performance electrocatalysis, Mater. Rep.: Energy, 2021, 1, 100006 CAS
- H. Sun, H. Kim, S. Song and W. Jung, Copper foam-derived electrodes as efficient electrocatalysts for conventional and hybrid water electrolysis, Mater. Rep.: Energy, 2022, 2, 100092 CAS
- Y. Zhu, W. Zhou, Y. Zhong, Y. Bu, X. Chen, Q. Zhong, M. Liu and Z. Shao, A Perovskite Nanorod as Bifunctional Electrocatalyst for Overall Water Splitting, Adv. Energy Mater., 2017, 7, 1602122 CrossRef
- J. Yu, Q. He, G. Yang, W. Zhou, Z. Shao and M. Ni, Recent Advances and Prospective in Ruthenium-Based Materials for Electrochemical Water Splitting, ACS Catal., 2019, 9, 9973–10011 CrossRef CAS
- Y. Zhu, D. Liu, H. Jing, F. Zhang, X. Zhang, S. Hu, L. Zhang, J. Wang, L. Zhang, W. Zhang, B. Pang, P. Zhang, F. Fan, J. Xiao, W. Liu, X. Zhu and W. Yang, Oxygen activation on Ba-containing perovskite materials, Sci. Adv., 2022, 8, 4072 CrossRef PubMed
- M. Retuerto, L. Pascual, J. Torrero, M. A. Salam, A. Tolosana-Moranchel, D. Gianolio, P. Ferrer, P. Kayser, V. Wilke, S. Stiber, V. Celorrio, M. Mokthar, D. G. Sanchez, A. S. Gago, K. A. Friedrich, M. A. Pena, J. A. Alonso and S. Rojas, Highly active and stable OER electrocatalysts derived from Sr2MIrO6 for proton exchange membrane water electrolyzers, Nat. Commun., 2022, 13, 7935 CrossRef CAS PubMed
- R. Mandal, Y. Mahton, C. Sowjanya, K. Sanket, S. K. Behera and S. K. Pratihar, Electrocatalytic behaviour of Cu-substituted La0.5Sr0.5Co0.8Fe0.2-xCuxO3−δ (x = 0-0.2) perovskite oxides, J. Solid State Chem., 2023, 317, 123668 CrossRef CAS
- T. Mizoguchi, H. Sonoki, E. Niwa, S. Taminato, D. Mori, Y. Takeda, O. Yamamoto and N. Imanishi, K2NiF4 type oxides, Ln2-xSrxNiO4+δ (Ln = La and Pr; x = 0-1.4) as an oxygen electrocatalyst for aqueous lithium-oxygen rechargeable batteries, Solid State Ionics, 2021, 369, 115708 CrossRef CAS
- S. R. Choi, J. I. Lee, H. Park, S. W. Lee, D. Y. Kim, W. Y. An, J. H. Kim, J. Kim, H. S. Cho and J. Y. Park, Multiple perovskite layered lanthanum nickelate Ruddlesden–Popper systems as highly active bifunctional oxygen catalysts, Chem. Eng. J., 2021, 409, 128226 CrossRef CAS
- Y. Miyahara, T. Fukutsuka, T. Abe and K. Miyazaki, Dual-Site Catalysis of Fe-Incorporated Oxychlorides as Oxygen Evolution Electrocatalysts, Chem. Mater., 2020, 32, 8195–8202 CrossRef CAS
- S. Liu, C. Sun, J. Chen, J. Xiao and J. L. Luo, A High-Performance Ruddlesden–Popper Perovskite for Bifunctional Oxygen Electrocatalysis, ACS Catal., 2020, 10, 13437–13444 CrossRef CAS
- Y. Kato, M. Rahman and T. Takeguchi, Optimization of La, Sr, Fe, Co-Doped Perovskite Oxides Particle for Bifunctional Oxygen Electrocatalysis in Alkaline Zinc-Air Battery, ECS Meeting Abstracts, 2021, 3, 295 CrossRef
- J. Vega-Castillo and F. Prado, Study of the n = 3 Ruddlesden–Popper phases LnSr3Fe1.5Co1.5O10-δ (Ln = La, Pr, Nd) as oxygen reduction electrodes by impedance spectroscopy, Solid State Ionics, 2018, 325, 228–237 CrossRef CAS
- Y. Dai, J. Yu, J. Wang, Z. Shao, D. Guan, Y. C. Huang and M. Ni, Bridging the Charge Accumulation and High Reaction Order for High-Rate Oxygen Evolution and Long Stable Zn-Air Batteries, Adv. Funct. Mater., 2022, 32, 2111989 CrossRef CAS
- H. Wang, J. Qi, N. Yang, W. Cui, J. Wang, Q. Li, Q. Zhang, X. Yu, L. Gu, J. Li, R. Yu, K. Huang, S. Song, S. Feng and D. Wang, Dual-Defects Adjusted
Crystal-Field Splitting of LaCo(1-x)Ni(x)O(3-delta) Hollow Multishelled Structures for Efficient Oxygen Evolution, Angew. Chem., Int. Ed., 2020, 59, 19691–19695 CrossRef CAS PubMed
- X. Wu, J. Yu, G. Yang, H. Liu, W. Zhou and Z. Shao, Perovskite oxide/carbon nanotube hybrid bifunctional ectrocatalysts for overall water splitting, Electrochim. Acta, 2018, 286, 47–54 CrossRef CAS
- Q. Lu, J. Yu, X. Zou, K. Liao, P. Tan, W. Zhou and Z. Shao, Self-Catalyzed Growth of Co, N-Codoped CNTs on Carbon-Encased CoSx Surface: A Noble-Metal-Free Bifunctional Oxygen Electrocatalyst for Flexible Solid Zn-Air Batteries, Adv. Funct. Mater., 2019, 29, 1904481 CrossRef
- H. Zhang, Y. Gao, H. Xu, D. Guan, Z. Hu, C. Jing, Y. Sha, Y. Gu, Y.-C. Huang, Y.-C. Chang, C.-W. Pao, X. Xu, J.-F. Lee, Y.-Y. Chin, H.-J. Lin, C.-T. Chen, Y. Chen, Y. Guo, M. Ni, W. Zhou and Z. Shao, Combined Corner-Sharing and Edge-Sharing Networks in Hybrid Nanocomposite with Unusual Lattice-Oxygen Activation for Efficient Water Oxidation, Adv. Funct. Mater., 2022, 32, 2207618 CrossRef CAS
- D. Q. Guan, K. F. Zhang, Z. W. Hu, X. H. Wu, J. L. Chen, C. W. Pao, Y. N. Guo, W. Zhou and Z. P. Shao, Exceptionally Robust Face-Sharing Motifs Enable Efficient and Durable Water Oxidation, Adv. Mater., 2021, 33, 2103392 CrossRef CAS PubMed
- Y. Dai, J. Yu, Z. Zhang, C. Chen, P. Tan, W. Zhou and M. Ni, Interfacial La Diffusion in the CeO2/LaFeO3 Hybrid for Enhanced Oxygen Evolution Activity, ACS Appl. Mater, 2021, 13, 2799–2806 CrossRef CAS PubMed
- H. Zhang, D. Guan, X. Gao, J. Yu, G. Chen, W. Zhou and Z. Shao, Morphology, crystal structure and electronic state one-step co-tuning strategy towards developing superior perovskite electrocatalysts for water oxidation, J. Mater. Chem. A, 2019, 7, 19228–19233 RSC
- Y. Zhu, W. Zhou, J. Yu, Y. Chen, M. Liu and Z. Shao, Enhancing Electrocatalytic Activity of Perovskite Oxides by Tuning Cation Deficiency for Oxygen Reduction and Evolution Reactions, Chem. Mater., 2016, 28, 1691–1697 CrossRef CAS
- K. Zhu, F. Shi, X. Zhu and W. Yang, The roles of oxygen vacancies in electrocatalytic oxygen evolution reaction, Nano Energy, 2020, 73, 104761 CrossRef CAS
- L. Zhuang, L. Ge, Y. Yang, M. Li, Y. Jia, X. Yao and Z. Zhu, Ultrathin Iron-Cobalt Oxide Nanosheets with Abundant Oxygen Vacancies for the Oxygen Evolution Reaction, Adv. Mater., 2017, 29, 1606793 CrossRef PubMed
- M. Asnavandi, Y. Yin, Y. Li, C. Sun and C. Zhao, Promoting Oxygen Evolution Reactions through Introduction of Oxygen Vacancies to Benchmark NiFe-OOH Catalysts, ACS Energy Lett., 2018, 3, 1515–1520 CrossRef CAS
- J. Bao, X. Zhang, B. Fan, J. Zhang, M. Zhou, W. Yang, X. Hu, H. Wang, B. Pan and Y. Xie, Ultrathin Spinel-Structured Nanosheets Rich in Oxygen Deficiencies for Enhanced Electrocatalytic Water Oxidation, Angew. Chem., Int. Ed., 2015, 54, 7399–7404 CrossRef CAS PubMed
- J. Yu, Y. Zhong, X. Wu, J. Sunarso, M. Ni, W. Zhou and Z. Shao, Bifunctionality from Synergy: CoP Nanoparticles Embedded in Amorphous CoOx Nanoplates with Heterostructures for Highly Efficient Water Electrolysis, Adv. Sci., 2018, 5, 1800514 CrossRef PubMed
- M. Lu, Y. Zheng, Y. Hu, B. Huang, D. Ji, M. Sun, J. Li, Y. Peng, R. Si, P. Xi and C. Yan, Artificially steering electrocatalytic oxygen evolution reaction mechanism by regulating oxygen defect contents in perovskites, Sci. Adv., 2022, 8, 3563 CrossRef PubMed
- S. Lee, Y. Lee, G. Lee and A. Soon, Activated chemical bonds in nanoporous and amorphous iridium oxides favor low overpotential for oxygen evolution reaction, Nat. Commun., 2022, 13, 3171 CrossRef CAS PubMed
- Z. He, J. Zhang, Z. Gong, H. Lei, D. Zhou, N. Zhang, W. Mai, S. Zhao and Y. Chen, Activating lattice oxygen in NiFe-based (oxy) hydroxide for water electrolysis, Nat. Commun., 2022, 13, 2191 CrossRef CAS PubMed
- J. Mefford, X. Rong, A. Abakumov, W. Hardin, S. Dai, A. Kolpak, K. Johnston and K. Stevenson, Water electrolysis on La1-xSrxCoO3-δ perovskite electrocatalysts, Nat. Commun., 2016, 7, 11053 CrossRef CAS PubMed
- H. Zhang, D. Guan, Z. Hu, Y. Huang, X. Wu, J. Dai, C. Dong, X. Xu, H. Lin, C. Chen, Z. Zhou and Z. Shao, Exceptional lattice-oxygen participation on artificially controllable electrochemistry-induced crystalline-amorphous phase to boost oxygen-evolving performance, Appl. Catal., B, 2021, 297, 120484 CrossRef CAS
- A. Grimaud, O. Diaz-Morales, B. Han, W. Hong, W. Hong, W. Giordano, K. Stoerzinger, M. Koper and S. Yang, Activating lattice oxygen redox reactions in metal oxides to catalyse oxygen evolution, Nat. Chem., 2017, 9, 457–465 CrossRef CAS
- Z. Huang, S. Xi, J. Song, S. Dou, X. Li, Y. Du, C. Diao, Z. Xu and X. Wang, Tuning of lattice oxygen reactivity and scaling relation to construct better oxygen evolution electrocatalyst, Nat. Commun., 2021, 12, 3992 CrossRef CAS PubMed
- C. Wang, P. Zhai, M. Xia, Y. Wu, B. Zhang, Z. Li, L. Ran, J. J. Gao, X. Zhang, Z. Fan, L. Sun and J. Hou, Engineering Lattice Oxygen Activation of Iridium Clusters Stabilized on Amorphous Bimetal Borides Array for Oxygen Evolution Reaction, Angew. Chem., Int. Edit., 2021, 60, 27126–27134 CrossRef CAS PubMed
- M. Zhao, X. Zheng, C. Cao, Q. Lu, J. Zhang, H. Wang, Z. Huang, Y. Cao, Y. Wang and Y. Deng, Lattice oxygen activation in disordered rocksalts for boosting oxygen evolution, Phys. Chem. Chem. Phys., 2023, 25, 4113–4120 RSC
- L. Tang, Y. Yang, H. Guo, Y. Wang, M. Wang, Z. Liu, G. Yang, X. Fu, Y. Luo, C. Jiang, Y. Zhao, Z. Shao and Y. Sun, High Configuration Entropy Activated Lattice Oxygen for O2 Formation on Perovskite Electrocatalyst, Adv. Funct. Mater., 2022, 32, 2112157 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3qm00472d |
| This journal is © the Partner Organisations 2023 |