
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicrowave-assisted carbon-confined iron nanoparticles for steering CO2 hydrogenation to heavy hydrocarbons†
Lisheng
Guo
 *ab,
Peipei
Ai
b,
Xinhua
Gao
c,
Hao
Wu
a,
Xianbiao
Wang
a,
Yasuharu
Kugue
d,
Jiaming
Liang
d,
Weizhe
Gao
d,
Xiaoyu
Guo
d,
Jian
Sun
*e,
Song
Sun
*a and
Noritatsu
Tsubaki
*d
*ab,
Peipei
Ai
b,
Xinhua
Gao
c,
Hao
Wu
a,
Xianbiao
Wang
a,
Yasuharu
Kugue
d,
Jiaming
Liang
d,
Weizhe
Gao
d,
Xiaoyu
Guo
d,
Jian
Sun
*e,
Song
Sun
*a and
Noritatsu
Tsubaki
*d
aSchool of Chemistry and Chemical Engineering, Anhui University, Hefei, Anhui 230601, China. E-mail: lsguo@ahu.edu.cn; suns@ustc.edu.cn
bState Key Laboratory of Clean and Efficient Coal Utilization, Taiyuan University of Technology, Taiyuan 030024, Shanxi, China
cState Key Laboratory of High-efficiency Utilization of Coal and Green Chemical Engineering, College of Chemistry & Chemical Engineering, Ningxia University, Yinchuan 750021, China
dDepartment of Applied Chemistry, School of Engineering, University of Toyama, Gofuku 3190, Toyama 930-8555, Japan. E-mail: tsubaki@eng.u-toyama.ac.jp
eDalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China. E-mail: sunj@dicp.ac.cn
First published on 12th April 2023
Abstract
The key to efficient thermocatalytic conversion of CO2 lies in the rational design of catalysts. In this work, Fe-based catalysts supported by different carbon materials (AC, GA, CNF, and MPC) were rapidly synthesized with the assistance of microwave heating. Compared with other supports, small-sized active precursors are coated by graphitic carbon layers for GA-supported Fe catalysts, which will slow down the sintering of particles. Small-sized Fe nanoparticles supported by GA present a benign carburization behavior and water removal ability, which play an important role in stabilizing active carbides and improving product selectivity. These features make GA a more promising support material. In addition, for the catalyst obtained by high-temperature calcination, the size of the active precursor is larger than that obtained by microwave heating, and it is easily coated by amorphous carbon shells, whereas for the small active phase precursor obtained by microwave heating treatment, it is encapsulated by graphitic carbons first, and then the amorphous carbon shells gradually appear around the active carbides with the increase in particle size. The carbon-confined environment provides a stable space for steering CO2 hydrogenation to heavy hydrocarbons. As a result, the rational catalyst exhibits favorable performance. Under relevant industrial conditions (320 °C, 2.0 MPa, 5 g h mol−1), the optimized K–Fe/GA-W-10 catalyst achieves a C5+ selectivity of 53.2% at CO2 conversion of 23.1% and presents a benign stability.
Broader contextThe transformation of CO2 into energy-rich fuels and valuable chemicals is a huge challenging and hot research topic. The critical bottleneck of this process lies in the difficult activation of inert CO2 molecules and poor chain growth ability. Efficient catalyst preparation is one of the key strategies to solve this problem and for potential industrial applications. At present, conventional catalyst synthesis requires high-temperature calcination to obtain the precursor, which usually consumes a good chunk of time. In addition, high-temperature calcination will also destroy the special structure of the catalyst. To overcome this huge challenge, we designed microwave-assisted carbon-confined iron nanoparticle catalysts for steering CO2 hydrogenation to heavy hydrocarbons. The high-temperature treatment lasts only a few seconds, and this method promotes the formation of ultra-high dispersion active metal sites. Moreover, the carbon-confined structure is conducive to CO2 conversion, and maintains the active phase stably. Consequently, the tailor-made catalyst presents benign performance. The present research provides new insights for the rapid fabrication of high-performance catalysts for practical industrial applications in catalytic conversions of C1 small molecules to high value-added chemicals. More importantly, the unique encapsulation and uniform dispersion of active sites from the microwave heating technology have good reference significance for other supported catalysts. |
Introduction
Almost a century on, massive CO2 emissions result in several environmental issues related to global warming, ocean acidification, etc., whereas thermocatalytic CO2 hydrogenation provides a promising route for CO2 elimination and reutilization.1–6 Selective hydrogenation of CO2-derived highly valuable products including carbon monoxide (CO), alcohols, alkenes, aromatics, oxygenates, and liquid hydrocarbons is necessary.7–11 Among them, liquid or long-chain hydrocarbons, as substitutable fuels, are one of the most desirable products.10,12–15 Recently, significant advances in the selective synthesis of liquid hydrocarbons from CO2 hydrogenation have been reported by constructing efficient functional catalysts.10,12,13 Yet, it remains a huge challenge to directly convert CO2 into liquid hydrocarbons, since CO2 molecules are thermodynamically stable and carbon chain propagation needs to overcome higher energy barriers. In terms of CO2 hydrogenation, raw molecules can be catalyzed to valuable products over modified metal oxides or multi-site composite functional catalysts.16–18Iron as a typical catalyst can in situ form two types of active sites under CO2/H2 conditions, including Fe3O4 for reverse water–gas shift (RWGS) and carbides (FexCy) for chain growth, and they have been widely applied for CO2 hydrogenation via modified Fischer–Tropsch synthesis (FTS). Generally, a single iron catalyst without any regulation presents a poor catalytic performance such as low catalytic activity and high light hydrocarbon selectivity, far from needs and expectations. To address this issue, a series of tailor-made iron-based catalysts were synthesized by regulating COx adsorption, electronic environments of active sites, interaction between active metal and support materials, etc. A common means is to introduce electronic promoters in the iron-based system, usually referring to alkali metals (Li, Na, and K), alkaline-earth metals (Mg, Ca, and Sr), and transition metals (Cu, Mn, Zn, Zr, Mo, V, and Nb). Given the excellent promotional effects, K and Na promoters are widely selected for modifying iron-based catalysts to achieve benign catalytic performance. The existence of Na or K above can enhance the surface alkalinity, promoting the adsorption of COx molecules.19 With the incorporation of promoters such as Na, delocalized electrons are transferred from the electronic promoter to the interface of iron carbides, making the surface of iron carbides electron rich.20 CO activation on carbides occurs via Fe–C σ-bonding and the simultaneous back-donation of iron species valence electrons into the 2π* orbital of CO.21 The enhanced electron density of carbides derived from electron transfer is able to intensify the Fe–C bond and weakens the C–O bond.22 Besides, more carbides are formed with the introduction of alkali metals.23,24 Unlike the Na promoter, it is accepted that K is more suitable for the formation of long-chain hydrocarbons.19,24–27
Apart from the promoter modification, controlling the interaction between the active metal and the support is adopted as another important means.28,29 Conventional oxide supports such as Al2O3, SiO2, and TiO2 have been widely used in Fe-based catalysts for CO2 conversion. However, a strong interaction between Fe and the oxide support may form a hardly reducible iron species (for example, iron silicates) and inhibit the reduction and carburization of an active Fe phase during the CO2 hydrogenation. Furthermore, various types of carbonaceous supports such as carbon nanofibers (CNFs), mesoporous carbons (MPCs), activated carbons (ACs), and graphenes (GAs) have been regarded as promising catalytic supports due to their tunable surface properties, thermal stability, and weak metal–support interactions.22,30–33 Although some advances have been reported, carbonaceous materials as promising supports have not yet received enough attention in the process of CO2 hydrogenation to long-chain hydrocarbons, let along the differences of promotional effects among different carbonaceous supports. Previously, we have designed supported iron nanoparticles for FTS by utilizing “microwave dielectric heating” effects, in which iron species presents benign dispersion.34 Microwave energy, when incident on a lossy dispersive material, generates heat within the material via the interactions of the electromagnetic field with the material's molecular and electronic structures. According to the Poynting theorem, the total power entering a volume V through the surface S goes partially into increasing the field energy stored inside V and is partially lost into heat.35 It can be judged that the ability of a material to heat up in the presence of an electromagnetic wave depends on the nature of its interaction with electric and magnetic components of the field. Generally, carbonaceous materials (graphite, graphenes, etc.) are dielectric absorbers having a sp2 bonded carbon network, which contains copious amounts of delocalized π electrons, which in the presence of an external electric field, start moving in the direction of the field and generate an electric current. As the electrons cannot couple to the changes of phase of the electric field, energy in the form of heat is dissipated. This process is referred to as interfacial polarization (also known as the Maxwell–Wagner–Sillars effect).36–38
In this work, a series of carbon material (including GA, CNF, AC, and MPC)-supported iron nanoparticles were prepared for CO2 conversion, where the precursors were treated by rapid microwave heating (MH). The improved dispersion, carburization behavior, and water removal ability are responsible to the high selectivity of long-chain hydrocarbons. Thereinto, a small-sized Fe nanoparticle encapsulated in a carbon shell is a key feature structure, which effectively stabilizes the carbide active phase and provides a favorable carbon environment. Correspondingly, a GA-supported Fe nanoparticle catalyst reaches C5+ selectivity as high as 53.2% at CO2 conversion of 23.1% (Scheme 1).
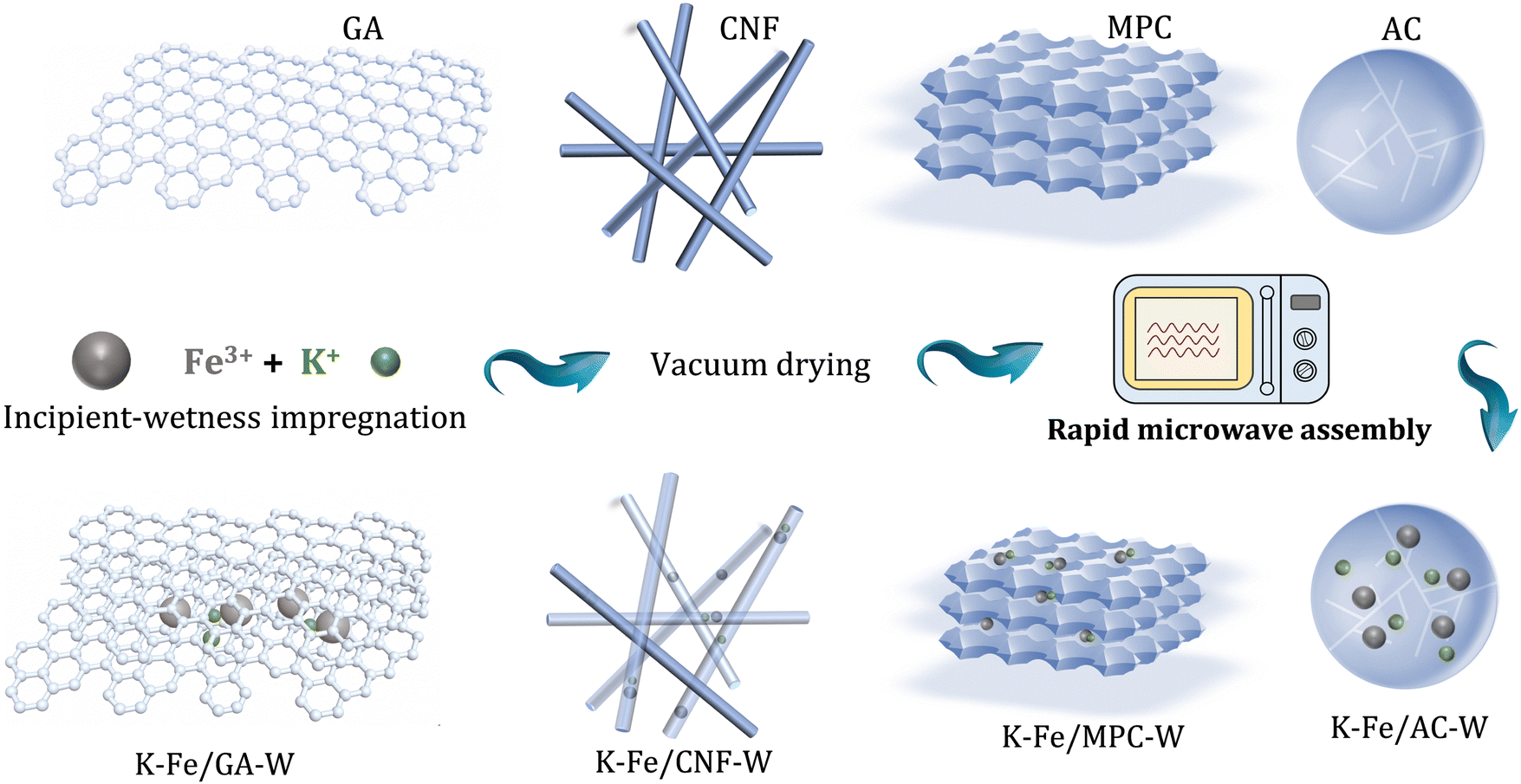 | ||
| Scheme 1 Schematic diagram of the synthesis of different catalysts via facile microwave heating treatment. | ||
Experimental
Catalyst preparation
Commercial GA, CNF, MPC, and AC were selected as carbonaceous supports. Prior to utilization, the carbon supports were filtered, washed with deionized water and then dried. Subsequently, 15 wt% Fe and 5 wt% K were introduced into the support by an incipient wetness impregnation method. It was then vacuum dried at 80 °C for 10 h to remove water. The obtained catalyst was treated with microwave for 10 seconds under the protection of N2 atmosphere (using a 700 W microwave oven). In each experimental synthesis, 0.5 g catalyst precursor was evenly placed in a Petri dish. Then it was moved into a microwave oven and placed in the center of the oven. It is worth noting that the whole microwave oven was placed in a glove box with nitrogen gas as the protective gas (Fig. S1, ESI†). Finally, the corresponding catalysts were marked as K–Fe/GA-W-10, K–Fe/CNF-W-10, K–Fe/AC-W-10, and K–Fe/MPC-W-10, respectively.In addition, in order to investigate the effect of microwave treatment time, the catalysts treated for different microwave time periods were prepared, marked as K–Fe/GA-W-t (t = 0, 5, and 15 s). Meanwhile, a K–Fe/GA catalyst was also fabricated via muffle calcination at different temperatures (T = 550, 700, or 850 °C for 5 h) under the protection of N2 atmosphere and labelled as K–Fe/GA-C-T.
By contrast, a SiO2-supported iron catalyst was also fabricated. Similar to carbon material-supported Fe catalysts, same contents of Fe and K were introduced into SiO2 by the same method. Considering that microwave heating could not heat SiO2, the obtained precursor was calcined in a muffle (550 °C for 5 h) under the protection of N2 atmosphere, and the obtained catalyst was labeled as K–Fe/SiO2-C-550.
Catalyst characterization
Ex situ powder X-ray diffraction (XRD) data were acquired using a Rigaku RINT 2400 X-ray Diffractometer with Cu Kα radiation (40 kV, 40 mA). A step size of 0.02° s−1 over a 2θ range of 20–80° was adopted.In situ XRD data were conducted using an Empyrean-100 PANalytical Empyrean-100 373 diffractometer with Cu Kα irradiation. The catalyst was first exposed to a H2 atmosphere for reduction, and the temperature was gradually increased from room temperature to 400 °C. After reduction at 400 °C for 90 min, the temperature was decreased to 320 °C. Subsequently, CO2 and H2 (H2/CO2 = 3) were introduced to the catalyst with a reaction pressure of 1.0 bar. The XRD patterns were collected over time.
N2 adsorption–desorption curves were measured at −196 °C using a NOVA 2200e apparatus to determine the specific surface area based on the Brunauer–Emmett–Teller (BET) method. Before measurement, the catalyst was outgassed under vacuum at 200 °C for 6 h.
A BELCAT-II-T-SP analyser was used to perform the H2-temperature programmed reduction (H2-TPR) and CO2-temperature programmed desorption (CO2-TPD). For H2-TPR, 50 mg catalyst was pretreated at 150 °C with Ar for 1 h. Subsequently, 5 vol% H2/Ar gas mixture was fed into the reactor when the temperature dropped to 50 °C. Finally, the H2-TPR trace was recorded from 50 to 800 °C at a heating rate of 10 °C min−1. As for CO2-TPD, 50 mg catalyst was reduced at 400 °C for 2 h in flowing 5 vol% H2/Ar. After pretreatment, the catalyst was flushed in an Ar flow for 30 min. Then, the temperature was decreased to 50 °C in the Ar flow and saturated with CO2 for 30 min. After flushing in the Ar flow for 30 min, CO2-TPD signals were recorded from 50 to 800 °C at a heating rate of 10 °C min−1.
X-ray photoelectron spectroscopy (XPS) analysis was performed using a Thermo Fisher Scientific ESCALAB 250Xi multifunctional X-ray photoelectron instrument.
Transmission electron microscopic (TEM) images of catalysts were acquired using a JEOL JEM-3200Fs at an accelerating voltage of 100 kV.
Raman spectroscopy measurements were taken using a HORIBA HR Evolution Raman spectrometer with a 2 mW 532 nm excitation laser. The exposure time for each line was 10 s, and the number of exposures was 2.
The static water contact angle was measured using a DSA100M (Theta Flex) in an environmental chamber saturated with water vapor under ambient conditions.
Catalyst evaluation
The CO2 hydrogenation reaction was carried out in a fixed-bed reactor with the condition of 320 °C, 2.0 MPa and 4800 mL gcat−1 h−1. The catalyst was diluted with SiO2 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mass ratio of the catalyst:silica). Before the reaction, the catalyst was reduced by H2 at 400 °C for 10 h. After reduction, the given conditions were adopted to test the performance of CO2 hydrogenation. Gas chromatography (GC) was used to determine the conversion and product selectivities. The light hydrocarbons (C1–C7) were analyzed using an online GC with a flame ionization detector (FID). The outlet gas (Ar, CO, CH4 and CO2) was analyzed using another GC system with a thermal conductivity detector (TCD). The heavy hydrocarbons (C5+) were collected using an ice-trap and analyzed using an off-line GC with n-dodecane (0.1 g) as an internal standard. The calculation method for CO2 conversion, CO selectivity and hydrocarbon selectivity has been previously reported in the literature.22,39
:1 mass ratio of the catalyst:silica). Before the reaction, the catalyst was reduced by H2 at 400 °C for 10 h. After reduction, the given conditions were adopted to test the performance of CO2 hydrogenation. Gas chromatography (GC) was used to determine the conversion and product selectivities. The light hydrocarbons (C1–C7) were analyzed using an online GC with a flame ionization detector (FID). The outlet gas (Ar, CO, CH4 and CO2) was analyzed using another GC system with a thermal conductivity detector (TCD). The heavy hydrocarbons (C5+) were collected using an ice-trap and analyzed using an off-line GC with n-dodecane (0.1 g) as an internal standard. The calculation method for CO2 conversion, CO selectivity and hydrocarbon selectivity has been previously reported in the literature.22,39
Results and discussions
Oriented synthesis of heavy hydrocarbons from CO2 hydrogenation
CO2 hydrogenation performances were investigated over different material-supported Fe nanoparticles (K–Fe/AC-W-10, K–Fe/CNF-W-10, K–Fe/GA-W-10, K–Fe/MPC-W-10, and K–Fe/SiO2-C-550) with a condition of 320 °C, 2.0 MPa, 4800 mL gcat−1 h−1. The catalytic activities and product distributions are summarized in Table 1. The K–Fe/AC-W-10 catalyst exhibits 16.4% selectivity to CH4, 44.7% selectivity to C5+ as well as a moderate CO selectivity (40.2%) at CO2 conversion of 26.3%. Thereinto, unsaturated alkenes, especially long-chain hydrocarbons, are dominant in hydrocarbon products due to the promotional effect of the alkaline K promoter (Fig. S2, ESI†).24 Meanwhile, C5+ hydrocarbon distribution in liquid products was also analyzed (Fig. S3, ESI†). However, when the CNF replaces the AC, although C5+ hydrocarbon selectivity increased, the selectivity to by-product CO increased slightly (45.7%). In terms of Fe supported by MPC supports, it exhibits poor selectivity of C5+ hydrocarbon and high CO selectivity. In contrast, a Fe-based catalyst supported by the GA can maintain high C5+ selectivity. In order to further verify the better performance from the carbonaceous material-supported Fe catalysts, comparison is made with a reference K–Fe/SiO2-C-550 catalyst. In addition to the calcination method, the K–Fe/SiO2 catalyst is also subjected to a long MH time, and it is found that extending the MH time is also conducive to the formation of higher hydrocarbons (Table S1, ESI†). It suggests that MH is also effective for the metal species carried on the support materials. Notably, Fe supported by carbonaceous materials is more favorable for the efficient conversion of CO2 to valuable hydrocarbons than inert SiO2, and the CO2 hydrogenation performance depends on the types of carbon supports evidently. Besides, in terms of K–Fe/GA-W-10, the effects of the K promoter content on CO2 were also investigated and compared, as given in Table S2 (ESI†). As shown, with the increase in K promoter content, the selectivity of liquid hydrocarbon is further improved. When the content of K promoter is 7.5%, C5+ selectivity is as high as 58.6%. In addition, the superfluous K promoter will further reduce the C5+ selectivity. For K promoters, good interfacial contact helps to promote iron active species to catalyze CO2 to form valuable hydrocarbons. However, excessive K addition will lead to the active sites being covered, leading to poor performance (Table S2, ESI†). The C5+ product selectivity is compared with that of typical catalysts reported recently. The corresponding catalysts are simply K promoter–modified Fe catalysts supported by carbon-based materials. Obviously, the designed K–Fe/GA-W-10 catalyst shows a high C5+ selectivity compared with traditional catalysts (Table 1).| Catalyst | HTa | GHSVb | H2/CO2 | P/MPa | T/°C | CO2 Conv. | CO Sel. | CO-free HCs Sel. (mol%) | Yield (C5+)/% | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CH4 | C2–C4 | C5+ | |||||||||
| All the conversion and selectivity data are collected at a stable 6–8 h on stream.a Represents the heating time (HT) over 100 °C during catalyst preparation.b Unit in mL gcat−1 h−1. | |||||||||||
| K–Fe/SiO2-C-550 | 5 h | 4800 | 2.5 | 2.0 | 320 | 23.1 | 46.1 | 14.8 | 39.6 | 45.6 | 5.7 |
| K–Fe/AC-W-10 | 10 s | 4800 | 2.5 | 2.0 | 320 | 26.3 | 40.2 | 16.4 | 38.9 | 44.7 | 7.1 |
| K–Fe/CNF-W-10 | 10 s | 4800 | 2.5 | 2.0 | 320 | 25.6 | 45.7 | 15.2 | 33.0 | 51.8 | 7.2 |
| K–Fe/GA-W-10 | 10 s | 4800 | 2.5 | 2.0 | 320 | 23.1 | 42.9 | 13.7 | 33.1 | 53.2 | 7.0 |
| K–Fe/MPC-W-10 | 10 s | 4800 | 2.5 | 2.0 | 320 | 24.5 | 46.2 | 21.8 | 37.8 | 40.4 | 5.3 |
| K–Fe/SiO2-C-550 | >10 h | 4800 | 2.5 | 2.0 | 320 | 18.9 | 38.7 | 40.8 | 31.2 | 28.0 | 3.2 |
| FeK/MWNTs27 | >21 h | 9000 | 3.0 | 2.0 | 340 | 43.6 | 23.4 | 27.1 | 38.5 | 34.4 | 11.5 |
| FeK/MPC30 | >21 h | 8000 | 3.0 | 2.5 | 300 | 37.2 | 8.4 | 20.0 | 36.1 | 43.9 | 15.0 |
| KFe/NCNT31 | >30 h | 50000 |
3.0 | 2.5 | 360 | 31.8 | 74.6 | 76.1 | 20.1 | 3.8 | 0.3 |
| Fe/C–K2CO340 | >34 h | 2400 | 3.0 | 2.0 | 320 | 37.1 | 11.5 | 12.9 | 41.6 | 45.5 | 14.9 |
| FeK/C-1EDA41 | >24 h | 2400 | 3.0 | 1.0 | 300 | 20.1 | 31.7 | 17.2 | 43.3 | 39.5 | 5.4 |
In general, the obtained catalysts for the reaction need heating pre-treatment, such as calcination, to obtain the desired structure or phase composition. Previous reports have revealed that long-time high-temperature treatment results in several undesired transformations including active-phase sintering and structure failure, which will cause poor catalytic performance, in turn. Previously, it has been reported that the MH method as a facile operation is able to sharply shorten the heating time and promote the dispersion of active species.34 As a result, the high-temperature pretreatment time for a given catalyst was listed and compared in Table 1. Table 1 only lists the heating processing times above 100 °C. Intuitively, different from the conventional heating procedure, MH treatment can effectively shorten the heating time from several hours to seconds (10 s), which means a shorter preparation cycle and a lower preparation cost. To some extent, the K–Fe/GA-W-10 catalyst obtained by this MH method presents more potential application values.
Meanwhile, the influences of MH time (0–15 s) and calcination temperature (550–850 °C) on CO2 hydrogenation performance over GA-supported Fe catalysts were also investigated (Fig. 1). As for the K–Fe/GA-W-0 catalyst without any heating treatment, it presents a poor C5+ hydrocarbon selectivity. With the prolongation of MH time, the catalytic activity increases slightly while C5+ hydrocarbon selectivity increases obviously. When the microwave time reaches 10 seconds, CO2 hydrogenation shows good performance. On the contrary, calcined catalysts at high temperatures show inferior catalytic hydrogenation performance. Moreover, calcination at high temperatures (850 °C) is not conducive to the formation of C5+ hydrocarbon. Catalytic stability is an essential index for the designed catalyst. As depicted in Fig. 1(c), the K–Fe/GA-W-10 catalyst presents benign stability over 200 h on stream. The C5+ selectivity stably maintains at 53% throughout the test. These results further indicate that the catalyst prepared by MH has a better application prospect than that of the catalyst prepared by conventional calcination in terms of preparation cost and cycle.
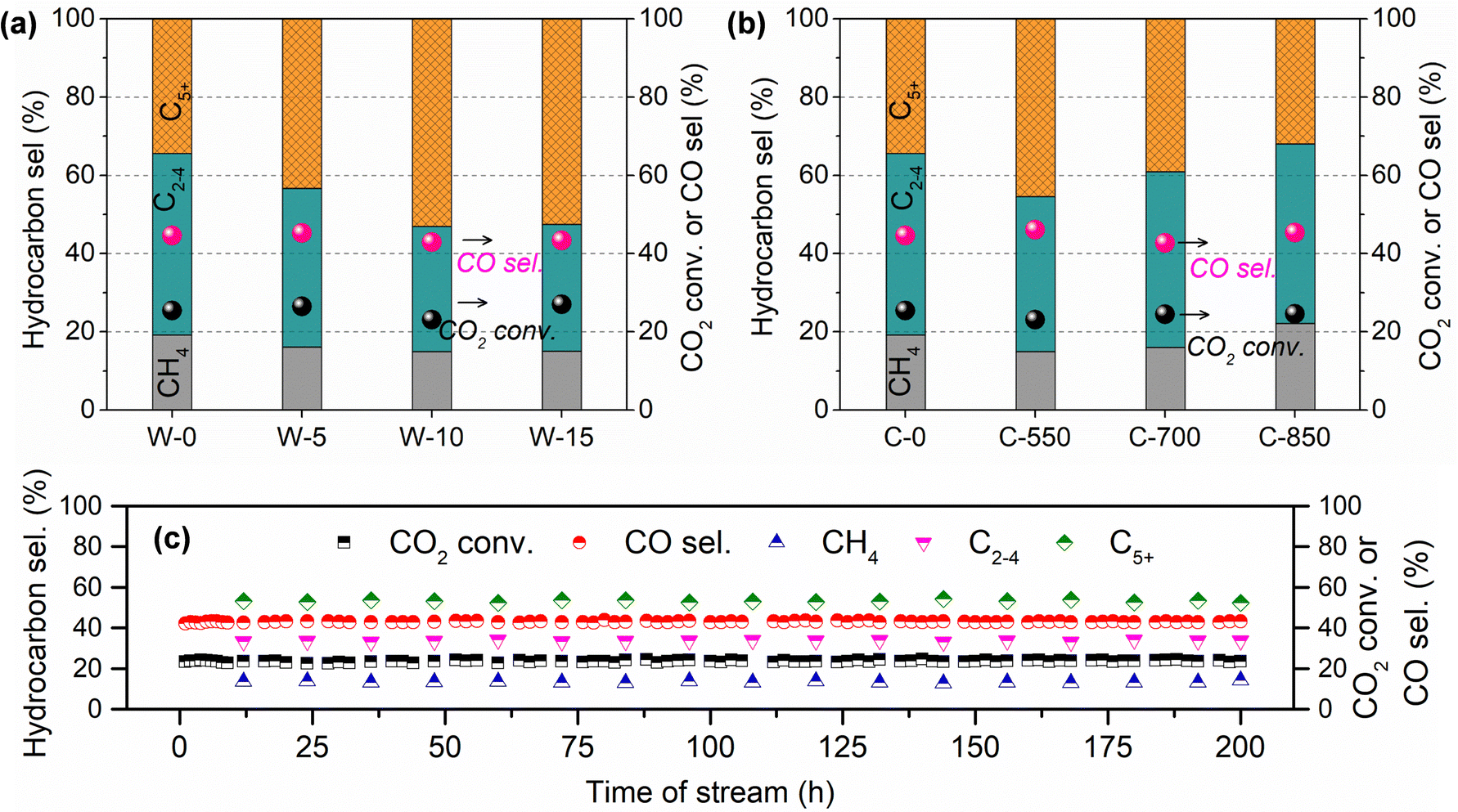 | ||
| Fig. 1 Effects of microwave time (0–15 s) and calcination temperature (550–850 °C) on CO2 hydrogenation performance: (a) microwave time, (b) calcination temperature, and (c) catalytic stability. W-0 and C-0 represent the K–Fe/GA sample without any high-temperature treatment, that is, the sample after vacuum drying. Pink and black spheres stand for CO2 conversion and CO selectivity. Reaction conditions: H2/CO2/Ar = 67.6/27.1/5.3, P = 2.0 MPa, GHSV, 4800 mL gcat−1 h−1. | ||
Structural characterization and phase compositions
Fig. S4 and Table S3 (ESI†) depict and summarize the specific surface areas of different Fe-based catalysts based on N2 physisorption measurement. In Table S3 (ESI†), the specific surface areas for the different Fe-based catalysts follow the sequence of K–Fe/AC (375 m2 g−1) > K–Fe/GA-W-10 (91 m2 g−1) > K–Fe/MPC-W-10 (69 m2 g−1) > K–Fe/SiO2-C-550 (20 m2 g−1) > K–Fe/CNF-W-10 catalyst (19 m2 g−1). Fig. S5a (ESI†) shows the ex situ XRD patterns of the as-prepared catalysts. For carbon material-supported Fe catalysts, the characteristic diffraction peaks of Fe3O4 (JCPDS, 89-0688), an active phase for RWGS reaction, are detected. By contrast, Fe2O3 (JCPDS, 89-0598) with a high oxidation state rather than Fe3O4 is found in the K–Fe/SiO2-C-550 catalyst. These results indicate that the carbon-supported Fe catalyst is easier to form active phase by MH treatment. Fig. S5b (ESI†) shows the ex situ XRD patterns of the spent catalysts. For K–Fe/SiO2 catalysts, the main phase after 6 h reaction is metallic Fe (JCPDS, 85-1410). However, the diffraction patterns of Fe2C (JCPDS, 37-0999) and Fe5C2 (JCPDS, 89-2544) are detected in the used K–Fe/AC-W-10 sample. Single Fe5C2 (JCPDS, 89-2544) occurs in the K–Fe/CNF-W-10 spent sample. In terms of K–Fe/GA-W-10, the peaks of Fe2C (JCPDS, 37-0999) and Fe5C2 (JCPDS, 89-2544) can be also obviously distinguished. When the support is MPC, the main phase is single Fe2C (JCPDS, 37-0999). Previously, it has been reported that Fe2C could be transformed into Fe5C2 during the FTS reaction at elevated temperatures.42 Obviously, the formed carbide phases after reaction depend on the type of support materials and operation conditions. In general, CO2 hydrogenation forms CO over Fe3O4 and then it is catalyzed to heavy hydrocarbons over carbide phases via the FTS route.Fig. 2 and Fig. S6 (ESI†) show the in situ XRD patterns of different catalysts under the same operating conditions. For the K–Fe/AC-W-10 catalyst, new iron phases ascribed to FeO (JCPDS, 74-1886) and metallic Fe (JCPDS, 85-1410) appear (Fig. 2(a), 400-0). It provides a direct evidence that Fe3O4 was reduced to FeO and then to Fe. As the reduction time expands, the diffraction peak intensity of Fe gradually increases. However, the intensity of metallic Fe decreases when the reactant gas is introduced into the system and a new phase corresponding to Fe3O4 appears. Fe2C and Fe5C2 appear with the proceeding of the reaction. Apart from the obvious peaks of Fe3O4, the peaks of Fe could be clearly distinguished. For K–Fe/CNF-W-10, the diffraction intensity of Fe changes slightly with the extension of reduction process, and a new carbide (Fe5C2) is formed when the reactant gas is introduced. Meanwhile, Fe has the strongest diffraction intensity than that of other phases. It is worth noting that when the reaction proceeds to a certain extent, the change in the diffraction intensity of metallic Fe is almost negligible (Fig. 2(a) and (b)). As far as K–Fe/GA-W-10 is concerned, the content of Fe increases continuously with the proceeding of reduction. However, the diffraction intensity of Fe is sharply decreased after the introduction of reactant gas, and Fe2C is formed under the atmosphere. This indicates that carbides are easier to form in this catalyst. With the reaction proceeding, the dominant phases are Fe3O4 and Fe2C, and the diffraction intensity of Fe becomes very low. For K–Fe/MPC-W-10, the diffraction intensity of Fe increases gradually during the reduction process. When the reactant gas is introduced, Fe is oxidized, resulting in the formation of Fe3O4, and carbides gradually appear as the reaction proceeds. Different from the above-mentioned catalysts, the diffraction peak of Fe3O4 increases gradually with the proceeding of the reaction (Fig. 2(d)). By contrast, the iron species in the K–Fe/SiO2-C-550 catalyst are difficult to be reduced and used for carbonization. The main phases are also Fe3O4 and Fe2O3 (Fig. S6, ESI†).
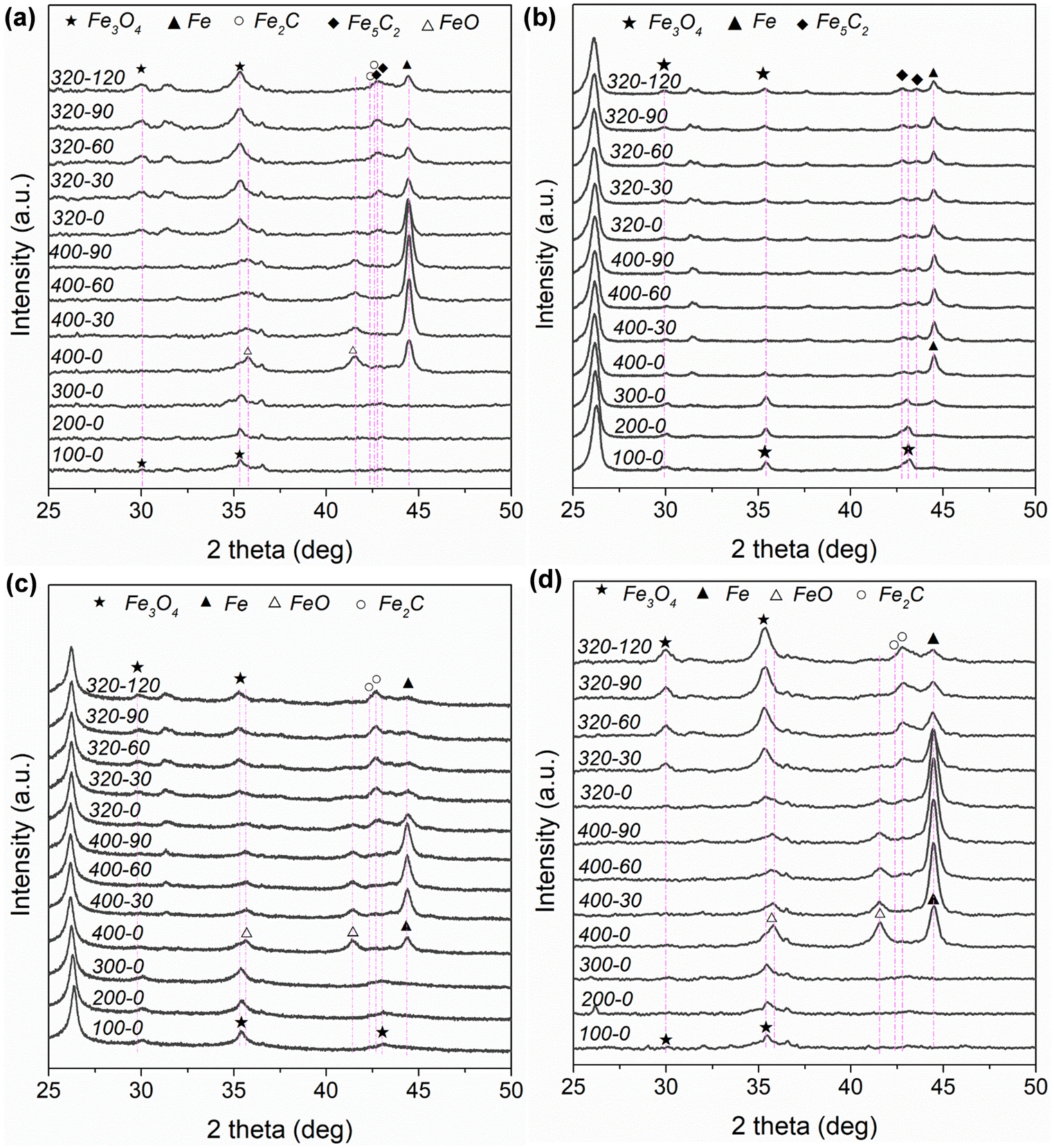 | ||
| Fig. 2 In situ XRD patterns of different catalysts under given conditions: (a) K–Fe/AC-W-10, (b) K–Fe/CNF-W-10, (c) K–Fe/GA-W-10, and (d) K–Fe/MPC-W-10. Notes: the reduction temperature was raised from room temperature to 400 °C and reduced for 90 min in a H2 atmosphere, then the reaction gas was switched to reaction and phase changes were in situ detected. In terms of 320–120, 320 represents 320 °C and 120 stands for 120 min at 320 °C. | ||
Influences of supports and heating treatment operations
The TEM images of the as-prepared catalysts are compared to study the differences in microstructures of active species (Fig. S7, ESI†). Iron species can be uniformly distributed in the catalyst regardless of the types of Fe-based catalyst supported by carbon materials. Meanwhile, the lattice spacings of 0.253 and 0.296 nm could be attributed to the (311) and (220) planes of Fe3O4 (Fig. S7, ESI†). However, for K–Fe/CNF-W-10 and K–Fe/GA-W-10 samples, iron species are obviously covered by a graphitic carbon layer (Fig. S7b and c, ESI†). In particular, K–Fe/GA-W-10 presents a small-size distribution with ca. 5 nm. The HAADF-TEM images and the corresponding elemental mapping images indicate that the elements are evenly distributed without any high-temperature treatment (K–Fe/GA-W-0, Fig. 3(a)). In contrast, the element distributions in the K–Fe/GA-W-10 sample after MH treatment are more uniform (K–Fe/GA-W-10, Fig. 3(b), and Fig. S8, ESI†).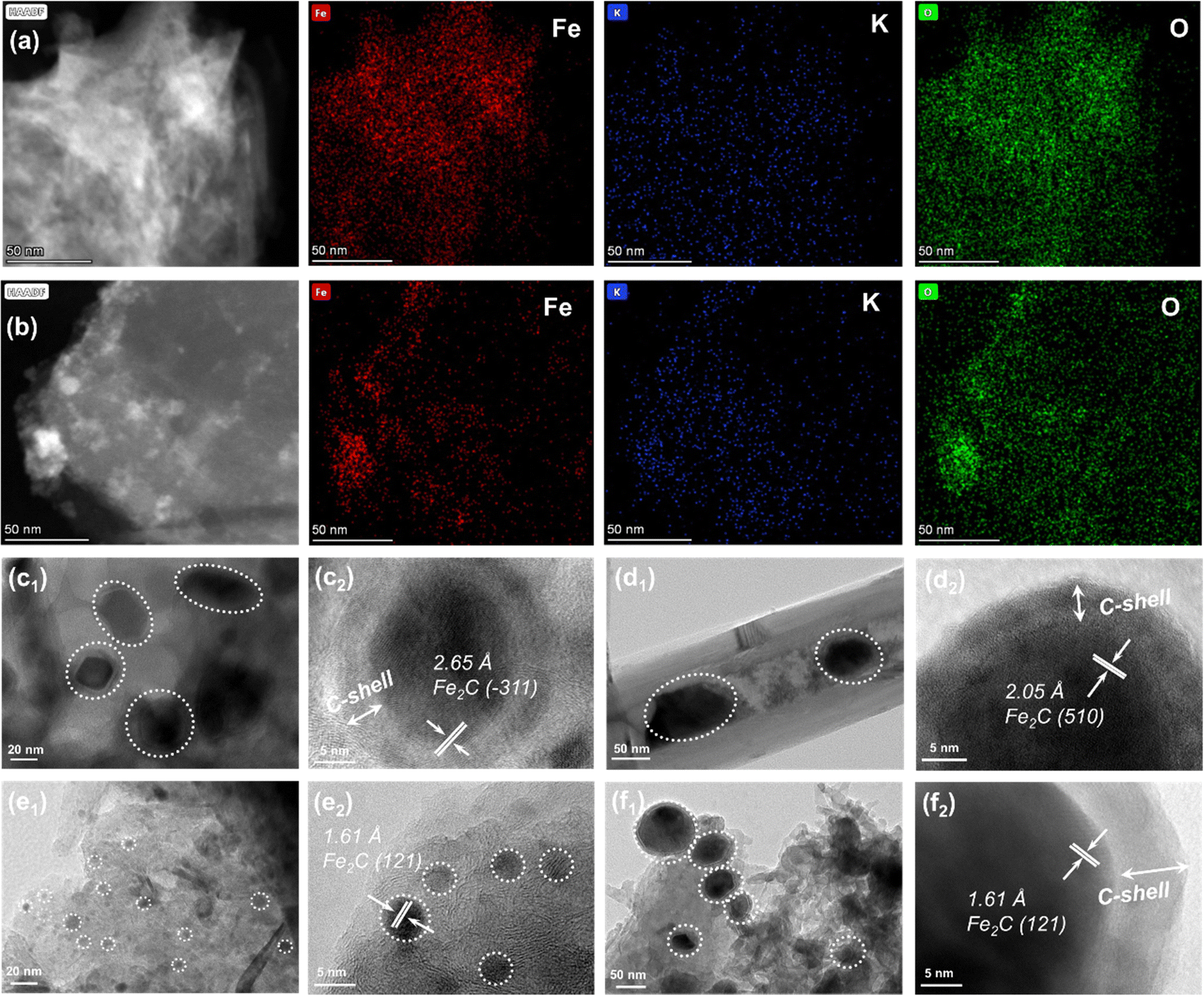 | ||
| Fig. 3 HAADF-TEM images and the corresponding elemental mapping images of (a) K–Fe/GA-W-0 and (b) K–Fe/GA-W-10 catalysts. TEM and HR-TEM images of spent catalysts: (c) K–Fe/AC-W-10, (d) K–Fe/CNF-W-10, (e) K–Fe/GA-W-10, and (f) K–Fe/MPC-W-10 catalysts. | ||
Fig. 3(c)–(f) and Fig. S9, S10 (ESI†) show the TEM images and the corresponding crystallite sizes of the used samples. After the reaction, K–Fe/AC-W-10 exhibits a core–shell structure, in which the shell is a carbon layer and the core is carbides. The lattice planes of Fe2C (−311) are detected as the main active phases. The particle size of iron species is about 20 nm (Fig. 3(a) and Fig. S10a, ESI†). A similar structure also appears in the K–Fe/CNF-W-10 sample with a larger size (50 nm), and the main phase is Fe5C2 (510). However, for the used K–Fe/GA-W-10 sample, there is no core–shell structure. Notably, iron species present small particle sizes, usually less than 5 nm, which is different from that of other catalysts. For the remaining catalysts, the particle sizes of iron active species increase significantly after the reaction (Fig. S7 and S10, ESI†). In addition, the main phase is Fe2C (121) for K–Fe/GA-W-10. For the K–Fe/MPC-W-10 catalyst, the core–shell structure also appears after the reaction, and the particle sizes change from 40 to 100 nm (Fig. 3(d)). Similar to the K–Fe/GA-W-10 sample, Fe2C (121) remains the dominant phase, which is well consistent with the XRD analysis (Fig. S5, ESI† and Fig. 3(d)). Besides, based on the XRD data shown in Fig. 2, the particle sizes of different catalysts in various time periods are compared and listed in Table S4 (ESI†). Obviously, with the process of reduction, the particle size of iron species increased gradually, and then the catalytic particle meditation decreased when switching to reaction gas. With the extension of the reaction time, the active phase particles gradually increased and stabilized. Similar to the TEM results, the GA-supported catalyst had the smallest particle size of iron species. These findings indicate that the utilization of GA support is conducive to the carburization of Fe3O4, producing small particles of active phases and providing more contact interfaces between active sites and feed gases. Moreover, GA-supported Fe nanoparticles are more conducive to maintaining the small particle size. By contrast, for the used K–Fe/SiO2-C-550, Fe3O4 (220) rather than carbides as the active phase is detected and the sizes counter at 100 nm, which is also confirmed by XRD results (Fig. S6 and S9, ESI†).
The reduction and CO2 adsorption properties were investigated by H2-TPR and CO2-TPD experiments, respectively (Fig. S11 and S12, ESI†). Different from the inert SiO2 support, carbon material-supported Fe nanoparticles are easier to be reduced. Thereinto, the iron species in the K–Fe/AC-W-10 catalyst are more easily reduced, followed by K–Fe/GA-W-10 and K–Fe/MPC-W-10, and finally the K–Fe/CNF-W-10 sample (Fig. S11, ESI†), which is consistent with the results from in situ XRD patterns (Fig. 2). As from H2-TPR, improved reduction behavior can promote the formation of Fe phase and facilitate the in situ formation of active phase. In general, from CO2-TPD, the desorption temperature below 650 °C could be assigned to CO2 chemisorbed peaks and physical adsorption peaks, while the peaks above 650 °C are due to the degradation of carbonaceous materials. Correspondingly, according to the results of CO2-TPD, CO2 raw molecules could interact well with iron species over K–Fe/GA-W-10 and K–Fe/MPC-W-10 catalysts compared to the remaining samples (Fig. S12, ESI†).
The effects of MH time (0–15 s) on the catalyst structures were investigated (Fig. S13, ESI†). For the sample after vacuum drying without any MH treatment, it shows large bulk irregular size structures. With the extension of MH time, bulk structures become small-sized particles, given that the microwave heating action is strengthened. When the microwave time exceeds 10 s, the catalyst particles are obviously reduced and further change is not obvious (ca. 5 nm), and more graphitic carbon layers appear around the metal species. This indicates that the suitable MH time is beneficial for the formation of small-sized iron species. In addition, TEM was performed on the used catalysts treated with different microwave durations (Fig. S14, ESI†). It can be found that a suitable microwave heat treatment duration (10 s) can effectively tune the particle size of active species. The chain growth reaction is a structurally sensitive reaction, and the smaller particle size is helpful to improve the catalytic performance. Meanwhile, in order to further compare the difference between MH and traditional heating methods such as calcination, the microwave and calcined samples were compared (Fig. S15, ESI†). Apparently, high calcination temperatures (700 and 850 °C) render the formation of large particles (Fig. S15b and c, ESI†). Although an appropriate low calcination temperature (550 °C) brings uniform particles, MH treatment is more conducive to the formation of small-sized iron species with high exposed active surface. In addition, the samples with various MH time periods present similar XRD patterns, indicating that MH can effectively promote the uniform dispersion of metal particles (Fig. S16, ESI†). By contrary, the corresponding precursor samples (K–Fe/GA-W-0 or K–Fe/GA-C-0) show obvious differences after the utilization of high-temperature calcination (Fig. S17, ESI†). With the increase in calcination temperature, metallic Fe species appear rather than Fe3O4 in an inert N2 atmosphere. Previously, similar findings were reported for NiO supported on graphene, and this phenomenon may be ascribed to the action of graphitic carbons and hydroxyl groups.43,44 Clearly, rapid heating and slow high calcination can induce different process changes in the catalyst precursor period. It shows that the MH method is not only fast and efficient, but also more beneficial to the preparation and synthesis of specific catalysts.
The phase composition and content of surface species were also investigated by XPS (Fig. 4, Fig. S18–S20, and Tables S5–S7, ESI†). As shown in Fig. S18, the XPS spectrum of the Fe 2p region of all samples can be fitted with two spin–orbit doublets corresponding to the Fe 2p1/2 and Fe 2p3/2 peaks and the corresponding satellite peaks of Fe2+ and Fe3+, which are typical characteristic peaks of Fe3O4.45 The iron nanoparticles supported by GA and CNF have a higher surface Fe(II) content (Fig. S18, ESI†). As mentioned above, the K promoter promotes the formation of heavy hydrocarbons.
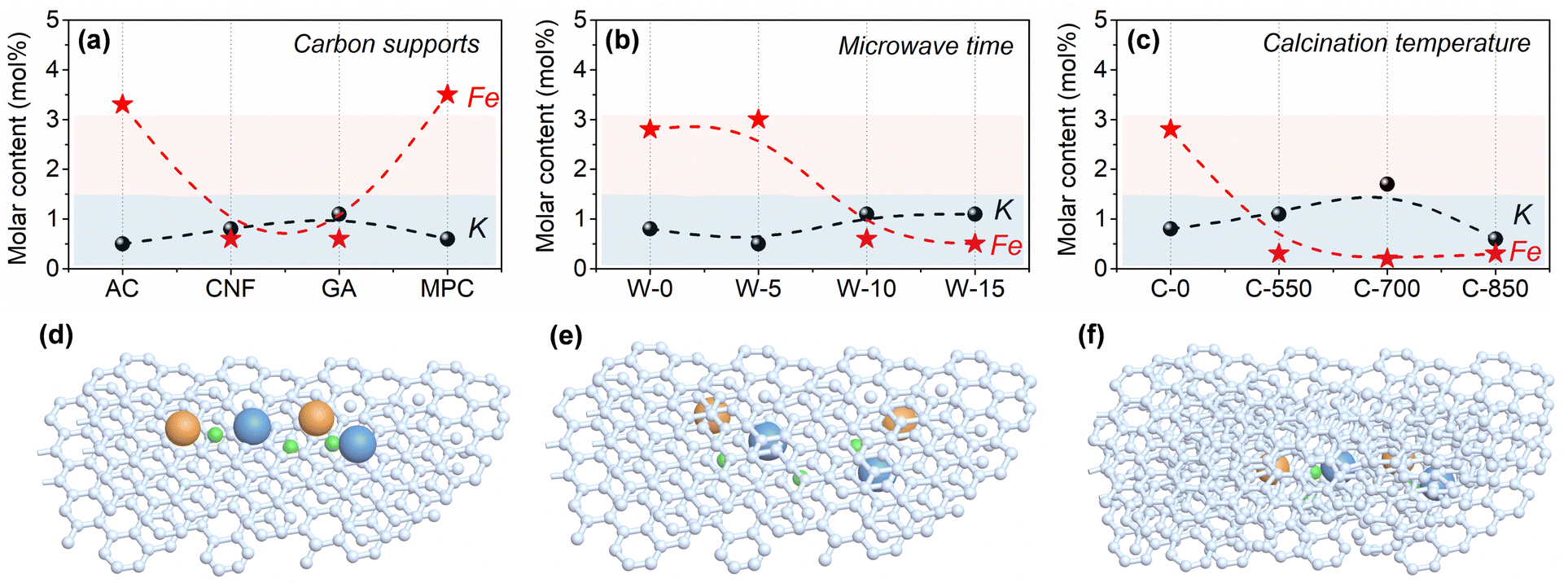 | ||
| Fig. 4 Surface element compositions of the as-prepared catalysts: (a) iron catalysts supported by various carbon materials; (b) K–Fe/GA catalysts treated by different MH times; (c) K–Fe/GA catalysts treated by different calcination temperatures using a tube furnace; (d) active metal species supported on the surface of support, e.g. AC and MPC; (e) the active metal encapsulated with appropriate graphitic carbon layers, e.g. W-10 and W-15; and (f) active metal species encapsulated with more graphitic carbon layers, e.g. C-550, C-700 and C-850. | ||
Encapsulation structure of iron nanoparticles with a carbon shell or layers
According to Fig. 4 and Table S5 (ESI†), the surface contents of iron species in K–Fe/GA-W-10 and K–Fe/CNF-W-10 samples are lower than those of K–Fe/AC-W-10 and K–Fe/MPC-W-10. In fact, the actual surface contents are much lower than the theoretical level, indicating that large amounts of iron species are encapsulated by carbon supports. In addition, with the extension of MH time, the surface content of iron species on the surface first decreases and then becomes constant, while the contents of C show an opposite trend (Table S6, ESI†). Unlike the convenient MH treatment, the surface content of iron species after calcination treatment is obviously lower than that of MH (Table S7, ESI†). Although low calcination temperatures are also conducive to the formation of uniform iron species, the inferior CO absorption and slightly large size to active Fe phase (Fig. S14 and S21; ESI† data from the difference of encapsulation structures) are unfavorable to improve the catalytic performance. These findings clearly reveal that carbon support types and heating methods can affect the surface contents of active iron species. Combined with the above-mentioned results, it can be considered that the metal iron species are encapsulated in the GA support structures or graphitic carbon layers. Similarly, You et al. reported that the hexagonal phase of Pd nanomaterials can be well encapsulated by graphite in terms of graphite templating effects and size effects.46Different from the K–Fe/SiO2-C-550 reference catalyst, the content of Fe(II) is higher for carbon-supported Fe catalysts (Fig. S19, ESI†), an active phase for catalyzing CO2 molecule hydrogenation. According to the C 1s and O 1s spectra, there are more C–C species and low content of C–O/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species, indicating that more adsorption species are converted into hydrocarbon products over K–Fe/GA-W-10 (Fig. S20, ESI†). Notably, the low intensity of Fe species especially for K–Fe/GA-W-10 indicates that iron species are encapsulated into the support or carbon shells (Fig. 3 and Fig. S7, S10, ESI†). This phenomenon could also be confirmed based on the surface content analysis of iron species (Table S8, ESI†). As for the K–Fe/GA-W-10 catalyst, there is no metal species coated by a carbon shell, thus Fe nanoparticles are more likely to be encapsulated in the graphene support or graphitic carbon layers. Based on this discussion, the schematic diagram of the active iron species in/on the support materials is depicted (Fig. 4(d)–(f)). For the used samples, the data of K and Fe surface content were also compared, as shown in Fig. S21 (ESI†). Similar to the samples before the reaction, the samples after reaction showed the same trend, indicating that the packaging structure was still obvious in the reaction process, and gradually appeared for AC- and MPC-supported iron catalysts. As shown in Fig. S22 (ESI†), CO-TPD experiments were further conducted to investigate the Fe accessibility. Compared with K–Fe/GA-W-0, the CO adsorption intensity decreases with the assistance of microwave heating. By contrast, the catalyst with calcination treatment presents inferior CO adsorption at low temperatures. Moreover, the desorption temperature of CO in the high-temperature region increases further. These results further support these findings to some extent (Fig. 4). In addition, the K 2p XPS spectra of used samples were compared, as shown in Fig. S23 (ESI†). As indicated, with the increase in K content, the binding energy shifts to a high binding energy location, indicating that K donates electrons to iron species. Generally, the change in electronic density enhances the strength of the Fe–C bond. In addition, the K promoter can suppress olefin secondary hydrogenation and then promote the formation of heavy olefin-rich hydrocarbons. Meanwhile, it is reported that the contact angle of H2O on graphenes is significantly higher than that on graphite, which indicates that graphenes have the most hydrophobic surfaces with very weak interactions with the H2O molecules.47 Xu et al. also reported that nanoscale zero-valent iron prepared by a carbothermal reduction method catalyzed the amorphous carbon to form hydrophobic graphitic layers.48 Generally, the presence of H2O molecules can oxidize the active phase and thus reduce the CO2 hydrogenation performance. Quick water removal from hydrogenation processes plays a crucial role for promoting COx hydrogenation.25,49 The water contact angle test shows that the graphene-supported catalyst has benign hydrophobicity, and the hydrophobicity of K–Fe/GA after MH treatment is enhanced (Fig. S24, ESI†). However, after the reaction, the hydrophilic and hydrophobic properties will change with the occurrence of the reaction. Thus, Fe nanoparticles encapsulated in the hydrophobic graphene support or graphitic carbon layers could be an ideal sample for catalyzing CO2 hydrogenation.
O species, indicating that more adsorption species are converted into hydrocarbon products over K–Fe/GA-W-10 (Fig. S20, ESI†). Notably, the low intensity of Fe species especially for K–Fe/GA-W-10 indicates that iron species are encapsulated into the support or carbon shells (Fig. 3 and Fig. S7, S10, ESI†). This phenomenon could also be confirmed based on the surface content analysis of iron species (Table S8, ESI†). As for the K–Fe/GA-W-10 catalyst, there is no metal species coated by a carbon shell, thus Fe nanoparticles are more likely to be encapsulated in the graphene support or graphitic carbon layers. Based on this discussion, the schematic diagram of the active iron species in/on the support materials is depicted (Fig. 4(d)–(f)). For the used samples, the data of K and Fe surface content were also compared, as shown in Fig. S21 (ESI†). Similar to the samples before the reaction, the samples after reaction showed the same trend, indicating that the packaging structure was still obvious in the reaction process, and gradually appeared for AC- and MPC-supported iron catalysts. As shown in Fig. S22 (ESI†), CO-TPD experiments were further conducted to investigate the Fe accessibility. Compared with K–Fe/GA-W-0, the CO adsorption intensity decreases with the assistance of microwave heating. By contrast, the catalyst with calcination treatment presents inferior CO adsorption at low temperatures. Moreover, the desorption temperature of CO in the high-temperature region increases further. These results further support these findings to some extent (Fig. 4). In addition, the K 2p XPS spectra of used samples were compared, as shown in Fig. S23 (ESI†). As indicated, with the increase in K content, the binding energy shifts to a high binding energy location, indicating that K donates electrons to iron species. Generally, the change in electronic density enhances the strength of the Fe–C bond. In addition, the K promoter can suppress olefin secondary hydrogenation and then promote the formation of heavy olefin-rich hydrocarbons. Meanwhile, it is reported that the contact angle of H2O on graphenes is significantly higher than that on graphite, which indicates that graphenes have the most hydrophobic surfaces with very weak interactions with the H2O molecules.47 Xu et al. also reported that nanoscale zero-valent iron prepared by a carbothermal reduction method catalyzed the amorphous carbon to form hydrophobic graphitic layers.48 Generally, the presence of H2O molecules can oxidize the active phase and thus reduce the CO2 hydrogenation performance. Quick water removal from hydrogenation processes plays a crucial role for promoting COx hydrogenation.25,49 The water contact angle test shows that the graphene-supported catalyst has benign hydrophobicity, and the hydrophobicity of K–Fe/GA after MH treatment is enhanced (Fig. S24, ESI†). However, after the reaction, the hydrophilic and hydrophobic properties will change with the occurrence of the reaction. Thus, Fe nanoparticles encapsulated in the hydrophobic graphene support or graphitic carbon layers could be an ideal sample for catalyzing CO2 hydrogenation.
The differences in the structure of the as-prepared catalysts with different heating manners were further assessed by Raman spectroscopy. In terms of the Raman spectra of three catalysts, two peaks can be clearly seen at around 1580 and 1350 cm−1, which can be attributed to the G band and the D band, respectively (Fig. S25, ESI†).22,50 Thereinto, the intensity of the G band is relative to the growth of graphitic carbons, while the intensity of the D band is a feature for disordered graphite including defects and sp3-hybridized carbons; then, the relative intensity (ID/IG) of the D band and G band can reflect the defect sites or graphitization degree. Obviously, the utilization of different heating methods shows slight influence on the defect structures of supports (Table S9, ESI†), which also means that the GA-loaded iron nanoparticles have benign thermal stability during preparation steps. This finding shows that the GA-supported iron catalyst has good stability, and the difference in catalytic performance is mainly due to the formation of the package structure. In addition, the spent K–Fe/GA-W-10 catalyst obtained after 200 h of reaction was further characterized by TEM. With the prolongation of reaction time, the particle size of the catalyst increases obviously (Fig. 2 and Fig. S26, ESI†). Moreover, the crystalline Fe5C2 core is covered with an amorphous shell, which is distinctly different from the previous catalyst obtained after 6 h of reaction (Fig. S26, ESI†). The core (iron species)–shell (carbon) configuration is a typical structure for FTS Fe-based catalysts.40,51–53 Similarly, Kim et al. also reported this phenomenon for the Na–Fe3O4/CNT after 24 h of reaction.54 Chernyak et al. reported the formation of a carbide core–amorphous carbon–graphitic carbon structure.55 Zhang et al. reported that the presence of a graphene shell could inhibit the sintering and agglomeration of active phases, and reduce the negative effects of water by blocking the contact between H2O and active phases.56 It can be inferred that when the catalyst particle size is small and coated by graphitic carbon layers, the amorphous carbon is not easily formed on the surface of active phases (Fig. 2 and Fig. S10, ESI†). However, when the particle size increases significantly, the catalyst particles show a tendency to form the amorphous carbon (Fig. S26, ESI†). The presence of the graphitic carbon or amorphous carbon can provide a good stable carbon environment for active carbides, making the catalyst exhibit good catalytic performance (Fig. 5). Previously, Gupta et al. reported a carbon-coated core–shell iron nanocatalyst for efficient CO2 hydrogenation, and found that the presence of a carbon shell could prevent rapid deactivation via hindering the aggregation of particles and enhance C2+ olefin selectivity.57 Besides, porous graphene-confined Fe catalysts also presented benign activity and excellent stability for CO2 hydrogenation to C2+ olefins, which is ascribed to the confinement effect.58
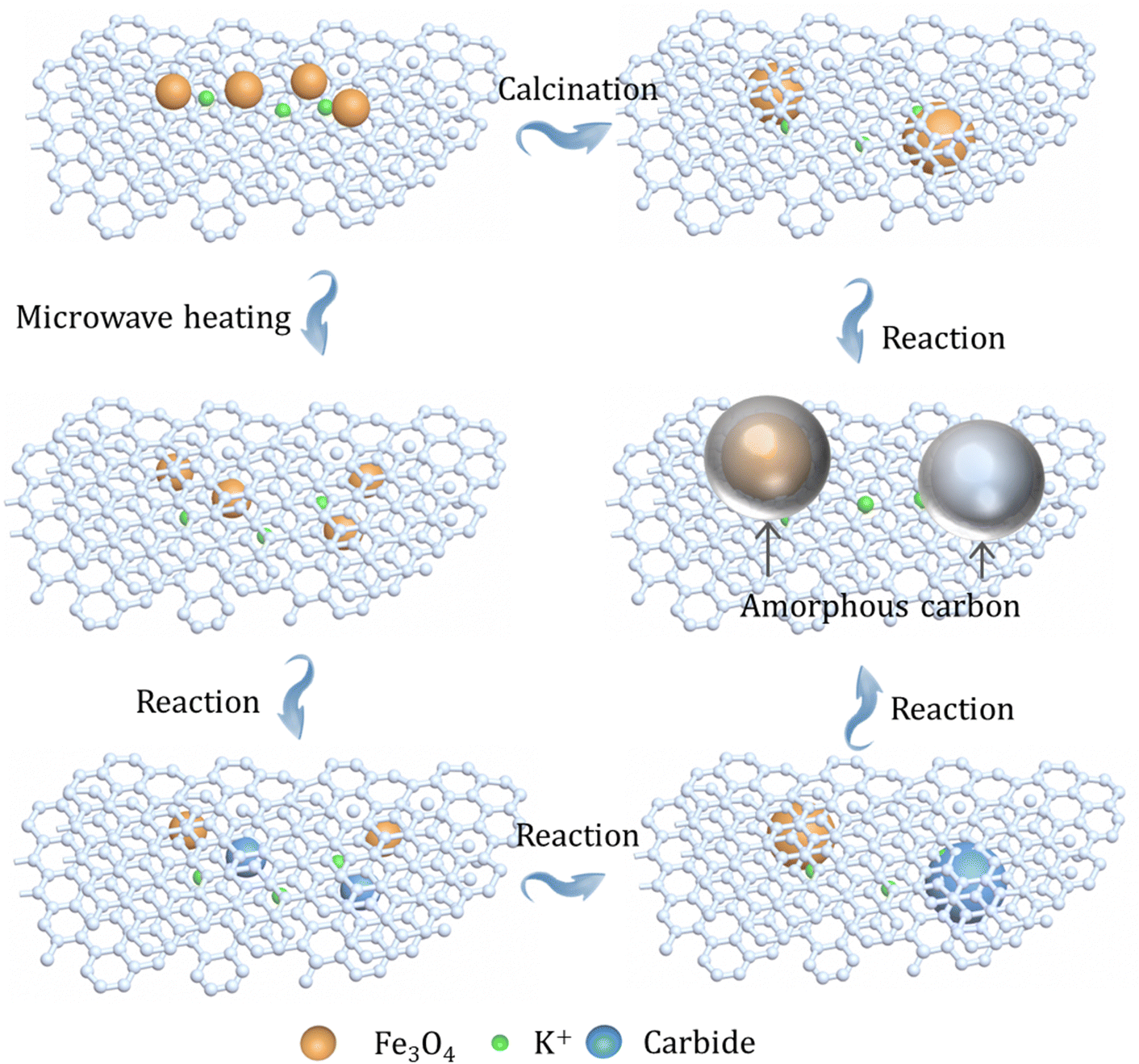 | ||
| Fig. 5 Schematic diagram of the changes in active iron species microenvironment on different catalysts after high-temperature heating treatment and reaction. | ||
Conclusions
Compared with AC-, MPC-, and CNF-supported iron catalysts, the GA-supported catalyst treated by simple MH presents a relatively small size with high exposed surface of active species and uniform dispersion. In addition, GA-supported iron also presents a benign carburization behavior. In terms of GA support, small-size active precursors are coated by graphitic carbon layers. With the introduction of reaction gases, an amorphous carbon shell is formed on the surface of active carbides for AC-, MPC-, and CNF-supported catalysts. For GA-supported iron catalysts, the presence of graphitic carbon layers can slow down the particle sintering during CO2 hydrogenation. By contrast, classic high calcination temperatures will form large particles of active precursors, which is not conducive to promote CO2 hydrogenation. With the increase in particle size, these active carbides are coated with the amorphous carbon (K–Fe/AC-W-10). In this process, the formed H2O can keep a distance with the active carbide or is quickly removed owing to the constant presence of hydrophobic carbon shells (graphenes, graphitic carbons, and amorphous carbons), which are helpful to improve the catalytic performance.Under such a space-confined carbon environment, the active iron sites are probable to express unique catalytic performance, which finally results in a favorable performance as we observed. When the MH time is 10 s, the K–Fe/GA-W-10 catalyst shows 53.2% C5+ selectivity at a CO2 conversion of 23.1%, and the test of 200 h reaction indicates good stability for CO2 hydrogenation. Carbon material-supported iron nanoparticles with the utilization of rapid MH provide a new and facile strategy for CO2 hydrogenation to produce high value-added products such as heavy hydrocarbons. Excellent absorbents of microwaves such as GA act as benign supports for catalyzing CO2 hydrogenation, and MH distinctly shortens the manufacturing cycle and production cost, which is an important factor in industrial production.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
Funds from National Natural Science Foundation of China (22102001), State Key Laboratory of Clean and Efficient Coal Utilization of Taiyuan University of Technology (SKL2022010), Key Research and Development Program of Anhui Province (202004a05020015, 006233172019), and NEDO of Japan are greatly appreciated.References
- R.-P. Ye, J. Ding, W. Gong, M. D. Argyle, Q. Zhong, Y. Wang, C. K. Russell, Z. Xu, A. G. Russell, Q. Li, M. Fan and Y.-G. Yao, CO2 hydrogenation to high-value products via heterogeneous catalysis, Nat. Commun., 2019, 10, 5698 CrossRef CAS PubMed.
- W. Wang, S. Wang, X. Ma and J. Gong, Recent advances in catalytic hydrogenation of carbon dioxide, Chem. Soc. Rev., 2011, 40(7), 3703–3727 RSC.
- E. V. Kondratenko, G. Mul, J. Baltrusaitis, G. O. Larrazábalc and J. Pérez-Ramírez, Status and perspectives of CO2 conversion into fuels and chemicals by catalytic, photocatalytic and electrocatalytic processes, Energy Environ. Sci., 2013, 6, 3112–3135 RSC.
- W. Zhou, K. Cheng, J. Kang, C. Zhou, V. Subramanian, Q. Zhang and Y. Wang, New horizon in C1 chemistry: breaking the selectivity limitation in transformation of syngas and hydrogenation of CO2 into hydrocarbon chemicals and fuels, Chem. Soc. Rev., 2019, 48, 3193–3228 RSC.
- J. Zhu, G. Zhang, W. Li, X. Zhang, F. Ding, C. Song and X. Guo, Deconvolution of the Particle Size Effect on CO2 Hydrogenation over Iron-Based Catalysts, ACS Catal., 2020, 10(13), 7424–7433 CrossRef CAS.
- L. Guo, J. Sun, Q. Ge and N. Tsubaki, Recent Advances in Direct Catalytic Hydrogenation of Carbon Dioxide to Valuable C2+ Hydrocarbons, J. Mater. Chem. A, 2018, 6, 23244–23262 RSC.
- Y. Ni, Z. Chen, Y. Fu, Y. Liu, W. Zhu and Z. Liu, Selective Conversion of CO2 and H2 into Aromatics, Nat. Commun., 2018, 9, 3457 CrossRef PubMed.
- H. Li, C. Qiu, S. Ren, Q. Dong, S. Zhang, F. Zhou, X. Liang, J. Wang, S. Li and M. Yu, Na+-gated water-conducting nanochannels for boosting CO2 conversion to liquid fuels, Science, 2020, 367, 667–671 CrossRef CAS PubMed.
- J. Graciani, K. Mudiyanselage, F. Xu, A. E. Baber, J. Evans, S. D. Senanayake, D. J. Stacchiola, P. Liu, J. Hrbek, J. F. Sanz and J. A. Rodriguez, Highly active copper-ceria and copper-ceria-titania catalysts for methanol synthesis from CO2, Nature, 2014, 345, 6196 Search PubMed.
- J. Wei, Q. Ge, R. Yao, Z. Wen, C. Fang, L. Guo, H. Xu and J. Sun, Directly converting CO2 into a gasoline fuel, Nat. Commun., 2017, 8, 15174–15181 CrossRef PubMed.
- C. Zhou, J. Shi, W. Zhou, K. Cheng, Q. Zhang, J. Kang and Y. Wang, Highly Active ZnO–ZrO2 Aerogels Integrated with H-ZSM-5 for Aromatics Synthesis from Carbon Dioxide, ACS Catal., 2020, 10, 302–310 CrossRef CAS.
- P. Gao, S. Li, X. Bu, S. Dang, Z. Liu, H. Wang, L. Zhong, M. Qiu, C. Yang, J. Cai, W. Wei and Y. Sun, Direct conversion of CO2 into liquid fuels with high selectivity over a bifunctional catalyst, Nat. Chem., 2017, 9, 1019–1024 CrossRef CAS PubMed.
- Z. He, M. Cui, Q. Qian, J. Zhang, H. Liu and B. Han, Synthesis of liquid fuel via direct hydrogenation of CO2, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 12654–12659 CrossRef CAS PubMed.
- R. McGinnis, CO2-to-Fuels Renewable Gasoline and Jet Fuel Can Soon Be Price Competitive with Fossil Fuels, Joule, 2020, 4, 1–3 CrossRef.
- L. Zhang, Y. Dang, X. Zhou, P. Gao, A. Petrus van Bavel, H. Wang, S. Li, L. Shi, Y. Yang, E. I. Vovk, Y. Gao and Y. Sun, Direct conversion of CO2 to a jet fuel over CoFe alloy catalysts, Innovation, 2021, 2(4), 100170 CAS.
- G. Song, M. Li, P. Yan, M. A. Nawaz and D. Liu, High Conversion to Aromatics via CO2-FT over a CO-Reduced Cu–Fe2O3 Catalyst Integrated with HZSM-5, ACS Catal., 2020, 10(19), 11268–11279 CrossRef CAS.
- X. Dong, F. Li, N. Zhao, F. Xiao, J. Wang and Y. Tan, CO2 hydrogenation to methanol over Cu/ZnO/ZrO2 catalysts prepared by precipitation-reduction method, Appl. Catal., B, 2016, 191, 8–17 CrossRef CAS.
- L. Tan, P. Zhang, Y. Cui, Y. Suzuki, H. Li, L. Guo, G. Yang and N. Tsubaki, Direct CO2 hydrogenation to light olefins by suppressing CO by-product formation, Fuel Process. Technol., 2019, 196, 106174 CrossRef CAS.
- M. Amoyal, R. Vidruk-Nehemya, M. V. Landau and M. Herskowit, Effect of potassium on the active phases of Fe catalysts for carbon dioxide conversion to liquid fuels through hydrogenation, J. Catal., 2017, 348, 29–39 CrossRef CAS.
- P. Zhai, C. Xu, R. Gao, X. Liu, M. Li, W. Li, X. Fu, C. Jia, J. Xie, M. Zhao, X. Wang, Y.-W. Li, Q. Zhang, X.-D. Wen and D. Ma, Highly tunable selectivity for syngas-derived alkenes over Zinc and Sodium-modulated Fe5C2 catalyst, Angew. Chem., Int. Ed., 2016, 55, 9902 CrossRef CAS PubMed.
- R. Yao, J. Wei, Q. Ge, J. Xu, Y. Han, Q. Ma, H. Xu and J. Sun, Monometallic iron catalysts with synergistic Na and S for higher alcohols synthesis via CO2 hydrogenation, Appl. Catal., B, 2021, 298, 120556 CrossRef CAS.
- L. Guo, P. Zhang, Y. Cui, G. Liu, J. Wu, G. Yang, Y. Yoneyama and N. Tsubaki, One-Pot Hydrothermal Synthesis of Nitrogen Functionalized Carbonaceous Material Catalysts with Embedded Iron Nanoparticles for CO2 Hydrogenation, ACS Sustainable Chem. Eng., 2019, 7, 8331–8339 CrossRef CAS.
- Y. H. Choi, E. C. Ra, E. H. Kim, K. Y. Kim, Y. J. Jang, K.-N. Kang, S. H. Choi, J.-H. Jang and J. S. Lee, Sodium-Containing Spinel Zinc Ferrite as a Catalyst Precursor for the Selective Synthesis of Liquid Hydrocarbon Fuels, ChemSusChem, 2017, 10, 4764–4770 CrossRef CAS PubMed.
- L. Guo, J. Sun, X. Ji, J. Wei, Z. Wen, R. Yao, H. Xu and Q. Ge, Directly converting carbon dioxide to linear α-olefins on bio-promoted catalysts, Commun. Chem., 2018, 1, 11 CrossRef.
- L. Guo, J. Li, Y. Cui, R. Kosol, Y. Zeng, G. Liu, J. Wu, T. Zhao, G. Yang, L. Shao, P. Zhan, J. Chen and N. Tsubaki, Spinel-structure catalyst catalyzing CO2 hydrogenation to full spectrum alkenes with an ultra-high yield, Chem. Commun., 2020, 56, 9372–9375 RSC.
- J. Wei, J. Sun, Z. Wen, C. Fang, Q. Ge and H. Xu, New insights into the effect of sodium on Fe3O4-based nanocatalysts for CO2 hydrogenation to light olefins, Catal. Sci. Technol., 2016, 6(13), 4786–4793 RSC.
- S. Wang, T. Wu, J. Lin, Y. Ji, S. Yan, Y. Pei, S. Xie, B. Zong and M. Qiao, Iron–Potassium on Single-Walled Carbon Nanotubes as Efficient Catalyst for CO2 Hydrogenation to Heavy Olefins, ACS Catal., 2020, 10(11), 6389–6401 CrossRef CAS.
- M. Zhu, P. Tian, R. Kurtz, T. Lunkenbein, J. Xu, R. Schlögl, I. E. Wachs and Y.-F. Han, Strong Metal–Support Interactions between Copper and Iron Oxide during the High-Temperature Water-Gas Shift Reaction, Angew. Chem., Int. Ed., 2019, 58(27), 9083–9087 CrossRef CAS PubMed.
- S. Li, Y. Xu, Y. Chen, W. Li, L. Lin, M. Li, Y. Deng, X. Wang, B. Ge, Y. Ce, S. Yao, J. Xie, Y. Li, X. Liu and D. Ma, Tuning the Selectivity of Catalytic Carbon Dioxide Hydrogenation over Iridium/Cerium Oxide Catalysts with a Strong Metal–Support Interaction, Angew. Chem., Int. Ed., 2017, 56(36), 10761–10765 CrossRef CAS PubMed.
- S.-M. Hwang, C. Zhang, S. J. Han, H.-G. Park, Y. T. Kim, S. Yang, K.-W. Jun and S. K. Kim, Mesoporous carbon as an effective support for Fe catalyst for CO2 hydrogenation to liquid hydrocarbons, J. CO2 Util., 2020, 37, 65–73 CrossRef CAS.
- P. Kangvansura, L. M. Chew, C. Kongmark, P. Santawaja, H. Ruland, W. Xia, H. Schulz, A. Worayingyong and M. Muhler, Effects of Potassium and Manganese Promoters on Nitrogen-Doped Carbon Nanotube-Supported Iron Catalysts for CO2 Hydrogenation, Engineering, 2017, 3(3), 385–392 CrossRef.
- Y. Cheng, J. Lin, K. Xu, H. Wang, X. Yao, Y. Pei, S. Yan, M. Qiao and B. Zong, Fischer-Tropsch Synthesis to Lower Olefins over Potassium-Promoted Reduced Graphene Oxide Supported Iron Catalysts, ACS Catal., 2016, 6(1), 389–399 CrossRef CAS.
- H. Xiong, L. L. Jewell and N. J. Coville, Shaped Carbons As Supports for the Catalytic Conversion of Syngas to Clean Fuels, ACS Catal., 2015, 5(4), 2640–2658 CrossRef CAS.
- L. Guo, Z. Guo, J. Liang, X. Yong, S. Sun, W. Zhang, J. Sun, T. Zhao, J. Li, Y. Cui, B. Zhang, G. Yang and N. Tsubaki, Quick Microwave Assembling Nitrogen-Regulated Graphene Supported Iron Nanoparticles for Fischer-Tropsch Synthesis, Chem. Eng. J., 2022, 429, 132063 CrossRef CAS.
- F. Qin and C. Brosseau, A review and analysis of microwave absorption in polymer composites filled with carbonaceous particles, J. Appl. Phys., 2012, 111(6), 061301 CrossRef.
- R. Kumar, S. Sahoo, E. Joanni and R. K. Singh, A review on the current research on microwave processing techniques applied to graphene-based supercapacitor electrodes: An emerging approach beyond conventional heating, J. Energy Chem., 2022, 74, 252–282 CrossRef CAS.
- R. Jakhar, J. E. Yap and R. Joshi, Microwave reduction of graphene oxide, Carbon, 2020, 170, 277–293 CrossRef CAS.
- J. A. Menéndez, A. Arenillas, B. Fidalgo, Y. Fernández, L. Zubizarreta, E. G. Calvo and J. M. Bermúdez, Microwave heating processes involving carbon materials, Fuel Process. Technol., 2010, 91(1), 1–8 CrossRef.
- L. Guo, X. Gao, W. Gao, H. Wu, X. Wang, S. Sun, Y. Wei, Y. Kugue, X. Guo, J. Sun and N. Tsubaki, High-yield production of liquid fuels in CO2 hydrogenation on a zeolite-free Fe-based catalyst, Chem. Sci., 2023, 14(1), 171–178 RSC.
- Y. Han, C. Fang, X. Ji, J. Wei, Q. Ge and J. Sun, Interfacing with Carbonaceous Potassium Promoters Boosts Catalytic CO2 Hydrogenation of Iron, ACS Catal., 2020, 10(20), 12098–12108 CrossRef CAS.
- R. Kosol, L. Guo, N. Kodama, P. Zhang, P. Reubroycharoen, T. Vitidsant, A. Taguchi, T. Abe, J. Cheng, G. Yang, Y. Yoneyama and N. Tsubaki, Iron Catalysts Supported on Nitrogen Functionalized Carbon for Improved CO2 Hydrogenation Performance, Catal. Commun., 2021, 149, 106216 CrossRef CAS.
- X.-P. Fu, W.-Z. Yu, C. Ma, J. Lin, S.-Q. Sun, S.-Q. Li, P.-N. Ren, F.-Y. Jia, M.-Y. Li, W.-W. Wang, X. Wang, C.-J. Jia, K. Wu, R. Si and C.-H. Yan, Supported Fe2C catalysts originated from Fe2N phase and active for Fischer-Tropsch synthesis, Appl. Catal., B, 2021, 284, 119702 CrossRef CAS.
- Y. J. Mai, J. P. Tu, C. D. Gu and X. L. Wang, Graphene anchored with nickel nanoparticles as a high-performance anode material for lithium ion batteries, J. Power Sources, 2012, 209, 1–6 CrossRef CAS.
- L. Wang, Y. Jiao, S. Yao, P. Li, R. Wang and G. Chen, MOF-derived NiO/Ni architecture encapsulated into N-doped carbon nanotubes for advanced asymmetric supercapacitors, Inorg. Chem. Front., 2019, 6(6), 1553–1560 RSC.
- Y. Chen, L. Ma, R. Zhang, R. Ye, W. Liu, J. Wei, V. V. Ordomsky and J. Liu, Carbon-supported Fe catalysts with well-defined active sites for highly selective alcohol production from Fischer-Tropsch synthesis, Appl. Catal., B, 2022, 312, 121393 CrossRef CAS.
- R. You, Z. Wu, J. Yu, F. Wang, S. Chen, Z.-K. Han, W. Yuan, H. Yang and Y. Wang, Revealing Surface Restraint-Induced Hexagonal Pd Nanocrystals via In Situ Transmission Electron Microscopy, Nano Lett., 2022, 22(11), 4333–4339 CrossRef CAS PubMed.
- S. O. Moussa, L. S. Panchakarla, M. Q. Ho and M. S. El-Shall, Graphene-Supported, Iron-Based Nanoparticles for Catalytic Production of Liquid Hydrocarbons from Synthesis Gas: The Role of the Graphene Support in Comparison with Carbon Nanotubes, ACS Catal., 2014, 4, 535–545 CrossRef CAS.
- Q. Xu, X. Liu, D. Lai, Z. Xing, P. Ndagijimana, Z. Li and Y. Wang, One-step synthesis of nanoscale zero-valent iron modified hydrophobic mesoporous activated carbon for efficient removal of bulky organic pollutants, J. Cleaner Prod., 2022, 356, 131854 CrossRef CAS.
- W. Fang, C. Wang, Z. Liu, L. Wang, L. Liu, H. Li, S. Xu, A. Zheng, X. Qin, L. Liu and F.-S. Xiao, Physical mixing of a catalyst and a hydrophobic polymer promotes CO hydrogenation through dehydration, Science, 2022, 377, 406–410 CrossRef CAS PubMed.
- H. Xiong, M. Moyo, M. A. Motchelaho, Z. N. Tetana, S. M. A. Dube, L. L. Jewell and N. J. Coville, Fischer-Tropsch synthesis: Iron catalysts supported on N-doped carbon spheres prepared by chemical vapor deposition and hydrothermal approaches, J. Catal., 2014, 311, 80–87 CrossRef CAS.
- C. Ma, W. Zhang, Q. Chang, X. Wang, H. Wang, H. Chen, Y. Wei, C. Zhang, H. Xiang, Y. Yang and Y. Li, θ-Fe3C dominated Fe@C core–shell catalysts for Fischer-Tropsch synthesis: Roles of θ-Fe3C and carbon shell, J. Catal., 2021, 393, 238–246 CrossRef CAS.
- C. Wei, W. Tu, L. Jia, Y. Liu, H. Lian, P. Wang and Z. Zhang, The evolutions of carbon and iron species modified by Na and their tuning effect on the hydrogenation of CO2 to olefins, Appl. Surf. Sci., 2020, 525, 146622 CrossRef CAS.
- S. Lyu, C. Liu, G. Wang, Y. Zhang, J. Li and L. Wang, Structural evolution of carbon in an Fe@C catalyst during the Fischer–Tropsch synthesis reaction, Catal. Sci. Technol., 2019, 9(4), 1013–1020 RSC.
- K. Y. Kim, H. Lee, W. Y. Noh, J. Shin, S. J. Han, S. K. Kim, K. An and J. S. Lee, Cobalt Ferrite Nanoparticles to Form a Catalytic Co–Fe Alloy Carbide Phase for Selective CO2 Hydrogenation to Light Olefins, ACS Catal., 2020, 10(15), 8660–8671 CrossRef CAS.
- S. A. Chernyak, A. S. Ivanov, S. V. Maksimov, K. I. Maslakov, O. Y. Isaikina, P. A. Chernavskii, R. V. Kazantsev, O. L. Eliseev and S. S. Savilov, Fischer-Tropsch synthesis over carbon-encapsulated cobalt and iron nanoparticles embedded in 3D-framework of carbon nanotubes, J. Catal., 2020, 389, 270–284 CrossRef CAS.
- P. Zhang, F. Han, J. Yan, X. Qiao, Q. Guan and W. Li, N-doped ordered mesoporous carbon (N-OMC) confined Fe3O4–FeCx heterojunction for efficient conversion of CO2 to light olefins, Appl. Catal., B, 2021, 299, 120639 CrossRef CAS.
- S. Gupta, V. K. Jain and D. Jagadeesan, Fine Tuning the Composition and Nanostructure of Fe-Based Core–Shell Nanocatalyst for Efficient CO2 Hydrogenation, ChemNanoMat, 2016, 2(10), 989–996 CrossRef CAS.
- T. Wu, J. Lin, Y. Cheng, J. Tian, S. Wang, S. Xie, Y. Pei, S. Yan, M. Qiao, H. Xu and B. Zong, Porous Graphene-Confined Fe–K as Highly Efficient Catalyst for CO2 Direct Hydrogenation to Light Olefins, ACS Appl. Mater. Interfaces, 2018, 10(28), 23439–23443 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ey00071k |
| This journal is © The Royal Society of Chemistry 2023 |