
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotocytotoxicity and photoinduced phosphine ligand exchange in a Ru(II) polypyridyl complex†
Sean J.
Steinke
a,
Sayak
Gupta
b,
Eric J.
Piechota
a,
Curtis E.
Moore
a,
Jeremy J.
Kodanko
*b and
Claudia
Turro
 *a
*a
aDepartment of Chemistry and Biochemistry, The Ohio State University, Columbus, OH 43210, United States. E-mail: turro.1@osu.edu
bDepartment of Chemistry, Wayne State University, Detroit, MI 48208, United States. E-mail: jkodanko@wayne.edu
First published on 1st February 2022
Abstract
Two new tris-heteroleptic Ru(II) complexes with triphenylphosphine (PPh3) coordination, cis-[Ru(phen)2(PPh3)(CH3CN)]2+ (1a, phen = 1,10-phenanthroline) and cis-[Ru(biq)(phen)(PPh3)(CH3CN)]2+ (2a, biq = 2,2′-biquinoline), were synthesized and characterized for photochemotherapeutic applications. Upon absorption of visible light, 1a exchanges a CH3CN ligand for a solvent water molecule. Surprisingly, the steady-state irradiation of 2a followed by electronic absorption and NMR spectroscopies reveals the photosubstitution of the PPh3 ligand. Phosphine photoinduced ligand exchange with visible light from a Ru(II) polypyridyl complex has not previously been reported, and calculations reveal that it results from a trans-type influence in the excited state. Complexes 1a and 2a are not toxic against the triple negative breast cancer cell line MDA-MB-231 in the dark, but upon irradiation with blue light, the activity of both complexes increases by factors of >4.2 and 5.8, respectively. Experiments with PPh3 alone show that the phototoxicity observed for 2a does not arise from the released phosphine ligand, indicating the role of the photochemically generated ruthenium aqua complex on the biological activity. These complexes represent a new design motif for the selective release of PPh3 and CH3CN for use in photochemotherapy.
Introduction
Ruthenium(II) polypyridyl complexes exhibit useful excited state properties that have been explored in photochemotherapy (PCT), photodynamic therapy (PDT), and solar energy conversion, among other applications.1–8 The spatiotemporal control possible with these complexes shows promise in alternative cancer therapies, circumventing systemic toxicity present in traditional cancer therapies, such as approved platinum drugs.9 Typically, PCT and PDT agents are activated by the absorption of visible light in the irradiated area, leading to the population of excited states that can produce cytotoxic 1O2 for PDT or induce the release of a therapeutic agent in PCT. Unlike complexes used in photochemotherapy, PDT agents rely on the presence of oxygen, which can represent a drawback in the hypoxic environments found in solid tumors,10–12 making PCT agents an important area of research to advance photoinduced treatments.13–15Coordination to the Ru(II) center through a Lewis basic site, such as a nitrile or pyridine functional group, have been explored as PCT agents and for dual PCT/PDT activity, since many drugs that can be photoreleased possess one of these groups able to coordinate to a transition metal center.16–20 A frequent challenge, however, is the ability of other strong field ligands, such as phosphines, to undergo photoinduced dissociation. Importantly, molecules with a triphenylphosphinium group and cationic compounds with a triphenylphosphine (PPh3) ligand have been shown to enhance cellular uptake,21,22 leading to an interest in the investigation of divalent ruthenium triphenylphosphine complexes for PCT.
Phosphine ligands, such as PMe3 (Me = methyl) and PPh3, have been shown to act as ancillary ligands that increase or promote the photosubstitution of other monodentate ligands in the Ru(II) coordination sphere.23–25 In particular, PR3 (R = Me, Ph) ligands are generally stronger field ligands relative to N–coordinated pyridine and acetonitrile. Strong π-backbonding to phosphine ligands has also been used to modify the electronic structure on the ruthenium center to reduce the overpotential of CO2 reduction catalysts and to tune the absorption and emission properties.26–29 Whereas CH3CN and pyridine have been previously shown to undergo photoinduced ligand exchange in Ru(II) complexes, phosphine ligands are largely inert to photosubstitution.4,24,29–34 The design of complexes that can selectively photodissociate phosphines can enable the use of drugs with phosphine motifs in PCT,35–37 as well as the synthesis of supported catalysts patterned with selective irradiation.38–40
In the present work, two new heteroleptic Ru(II) complexes containing one PPh3 and one CH3CN ligand, cis-[Ru(phen)2(PPh3)(CH3CN)]2+ (1a, phen = 1,10-phenanthroline) and cis-[Ru(biq)(phen)(PPh3)(CH3CN)]2+ (2a, biq = 2,2′-biquinoline), were synthesized and characterized, and their structures are shown in Fig. 1. The electronic absorption, electrochemistry, and photochemistry of 1a and 2a were investigated and compared to those of their bis-acetonitrile analogs, cis-[Ru(phen)2(CH3CN)2]2+ (1b) and cis-[Ru(biq)(phen)(CH3CN)2]2+ (2b). Based on the steric distortion introduced by the bulky PPh3 ligand, complexes 1a and 2a were expected to exhibit more facile CH3CN dissociation. While 1a exhibits photoinduced CH3CN exchange upon visible light excitation, 2a represents the first example of photoinduced exchange of a PPh3 ligand from a Ru(II) polypyridyl complex, a surprising departure from the commonly observed substitutional inertness of PPh3 ligands. Single-crystal X-ray structures of 1a, 2a, and the photoproduct of 2a following photolysis in CH3CN and pyridine (I), were collected and calculations were performed on 1a and 2a to gain better understanding of the origin of the unusual photoreactivity. In addition, complexes 1a and 2a were evaluated for their toxicity against the triple-negative breast cancer MDA-MB-231 cell line in the dark and upon irradiation. The present findings show enhanced activity following photoinduced ligand dissociation for both complexes and that PPh3 release from 2a results in a modest increase in toxicity as compared to CH3CN photodissociation in 1a. Importantly, both 1a and 2a exhibit significantly greater photoactivity than related complexes without PPh3 in their coordination sphere. The present work is consistent with greater cellular uptake by the PPh3-containing complexes, laying the groundwork for the design of new photoactive complexes with enhanced activity.
 | ||
| Fig. 1 Schematic representation of the molecular structures of 1a, 1b, 2a, and 2b. | ||
Experimental
Materials
All materials were used as received without further purification, including 1,10-phenanthroline, 2,2′-biquinoline, CD3CN, CD3OD, (CD3)2CO, lithium chloride, pyridine, silver tetrafluoroborate, tetrabutylammonium hexafluorophosphate, and triphenylphosphine which were purchased from Sigma-Aldrich. Ethanol (200 proof) was obtained from Decon Laboratories, acetone, acetonitrile, dichloromethane, diethyl ether, N,N-dimethylformamide, 85% H3PO4, and toluene were acquired from Fischer Scientific, and ammonium hexafluorophosphate was purchased from Oakwood Chemical. Complexes 1b and 2b,41 [Ru(phen)2Cl2],42 [Ru(p-cymene)Cl2]2,43 and triphenylphosphine oxide44 were prepared according to literature procedures.[Ru(phen)2(PPh3)(Cl)](PF6)
[Ru(phen)2Cl2] (0.16 g, 0.30 mmol), triphenylphosphine (0.14 g, 0.53 mmol), and excess LiCl were added to 10 mL ethanol/water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) mixture sparged for 15 min with N2. The reaction mixture was refluxed for 4 h under a nitrogen atmosphere, allowed to cool, concentrated by rotary evaporation, and then precipitated by adding it dropwise to a concentrated NH4PF6 solution. The product was purified by column chromatography, using a deactivated neutral alumina stationary phase and a 1:2 toluene:acetone mobile phase. The solvent was removed from the fraction containing the product via rotary evaporation, producing a dark orange solid (0.078 g, 29% yield). 1H NMR (400 MHz, (CD3)2CO, Fig. S1†): δ 9.72 (d, 1H, J = 5.4 Hz), 8.77 (m, 2H), 8.68 (d, 1H, J = 8.2 Hz), 8.55 (dd, 1H, J = 5.7, 3.9 Hz), 8.37 (s, 2H), 8.36 (d, 1H, J = 1.3 Hz), 8.30 (d, 1H, J = 8.8), 8.23 (dd, 1H, J = 20.3, 9.0 Hz), 8.13 (d, 1H, J = 8.8 Hz), 7.90 (dd, 1H, J = 8.2, 5.3 Hz), 7.86 (dd, 1H, J = 8.2, 5.3 Hz), 7.77 (dd, 2H, J = 8.3, 5.3 Hz), 7.65 (d, 1H, 5.3 Hz), 7.53 (m, 2H), 7.37 (t, 5H, J = 8.7 Hz), 7.27 (t, 2H, J = 7.6 Hz), 7.11 (m, 6H). 31P{H} NMR (400 MHz, (CD3)2CO, Fig. S2†): δ 45.1 (s, 1P).
:1, v/v) mixture sparged for 15 min with N2. The reaction mixture was refluxed for 4 h under a nitrogen atmosphere, allowed to cool, concentrated by rotary evaporation, and then precipitated by adding it dropwise to a concentrated NH4PF6 solution. The product was purified by column chromatography, using a deactivated neutral alumina stationary phase and a 1:2 toluene:acetone mobile phase. The solvent was removed from the fraction containing the product via rotary evaporation, producing a dark orange solid (0.078 g, 29% yield). 1H NMR (400 MHz, (CD3)2CO, Fig. S1†): δ 9.72 (d, 1H, J = 5.4 Hz), 8.77 (m, 2H), 8.68 (d, 1H, J = 8.2 Hz), 8.55 (dd, 1H, J = 5.7, 3.9 Hz), 8.37 (s, 2H), 8.36 (d, 1H, J = 1.3 Hz), 8.30 (d, 1H, J = 8.8), 8.23 (dd, 1H, J = 20.3, 9.0 Hz), 8.13 (d, 1H, J = 8.8 Hz), 7.90 (dd, 1H, J = 8.2, 5.3 Hz), 7.86 (dd, 1H, J = 8.2, 5.3 Hz), 7.77 (dd, 2H, J = 8.3, 5.3 Hz), 7.65 (d, 1H, 5.3 Hz), 7.53 (m, 2H), 7.37 (t, 5H, J = 8.7 Hz), 7.27 (t, 2H, J = 7.6 Hz), 7.11 (m, 6H). 31P{H} NMR (400 MHz, (CD3)2CO, Fig. S2†): δ 45.1 (s, 1P).
[Ru(phen)2(PPh3)(CH3CN)](PF6)2 (1a)
[Ru(phen)2(PPh3)Cl](PF6) (0.058 g, 0.064 mmol) was dissolved in 10 mL of acetonitrile/H2O (1:1, v/v) mixture and, under an atmosphere of nitrogen, was refluxed overnight. After cooling to room temperature, the reaction solution was added dropwise to a concentrated aqueous NH4PF6 solution. The precipitate that formed was collected by filtering over Celite and purified on a neutral alumina column eluted with a 1:2 toluene:acetone mobile phase. The purified solution was collected and the solvent was removed via rotary evaporation, affording the desired product as a yellow-orange solid (0.031 g, 53% yield). 1H NMR (400 MHz, CD3CN, Fig. S3†): δ 9.41 (d, 1H, J = 5.3 Hz), 9.09 (d, 1H, J = 5.2 Hz), 8.82 (dd, 1H, J = 8.4, 1.2 Hz), 8.59 (dd, 1H, J = 8.3, 1.2 Hz), 8.55 (dd, 1H, J = 8.2, 1.3 Hz), 8.37 (dd, 1H, J = 8.3, 1.2 Hz), 8.24 (dd, 2H, J = 28, 8.8 Hz), 8.13 (dd, 2H, J = 18, 8.8 Hz), 7.90 (dd, 1H, J = 3.0, 5.3 Hz), 7.75 (dd, 1H, J = 3.0, 5.3 Hz), 7.49 (m, 1H), 7.43 (m, 1H), 7.37 (m, 4H), 7.19 (m, 7H), 7.03 (td, 6H, J = 9.4, 1.1 Hz), 2.17 (s, 3H). 31P{H} NMR (400 MHz, CD3CN, Fig. S4†): δ 45.3 (s, 1P). ESI-MS(+): [M–PF6]+m/z = 910.193 (calc. m/z = 910.123).
[Ru(p-cymene)(phen)Cl]
[Ru(p-cymene)Cl2]2 (0.30 g, 0.50 mmol) and 1,10-phenanthroline (0.19 g, 1.1 mmol) were dissolved in 4 mL acetonitrile and refluxed under a nitrogen atmosphere for 2 h, during which time a change from a red to orange solution was observed. A yellow-orange solid was collected by filtering over Celite (0.37 g, 82% yield). 1H NMR (400 MHz, CD3OD, Fig. S5†): δ 9.84 (d, 2H, J = 5.5 Hz), 8.84 (d, 2H, J = 8.3 Hz), 8.21 (s, 2H), 8.11 (dd, 2H, J = 8.3, 5.3 Hz), 6.23 (d, 2H, J = 6.5 Hz), 6.00 (d, 2H, J = 6.2 Hz), 2.66 (q, 1H, J = 7.0 Hz), 2.27 (s, 3H), 0.99 (d, 6H, J = 6.9 Hz).[Ru(biq)(phen)Cl2]
[Ru(p-cymene)(phen)Cl] (0.33 g, 0.68 mmol), 2,2′-biquinoline (0.18 g, 0.69 mmol), and excess LiCl were dissolved in 2 mL N,N-dimethylformamide and refluxed under a nitrogen atmosphere for 90 min. After refluxing was complete, the reaction mixture was allowed to cool to room temperature and then added dropwise to 30 mL aqueous LiCl solution, producing a dark green solution. A dark green solid was collected via vacuum filtration and was rinsed three times each with 20 mL H2O and 20 mL diethyl ether. The solid was dissolved using 1 L of a CH2Cl2/methanol (1:1, v/v) solvent mixture, which was then removed by rotary evaporation to afford the desired product as a dark green solid (0.18 g, 44% yield).
[Ru(biq)(phen)(PPh3)Cl](PF6)
[Ru(biq)(phen)Cl2] (0.048 g, 0.080 mmol), triphenylphosphine (0.039 g, 0.15 mmol), and excess LiCl were added to a 10 mL ethanol/water (1:1, v/v) mixture sparged for 15 min with N2. The reaction mixture was refluxed for 4 h under a nitrogen atmosphere, allowed to cool, concentrated by rotary evaporation, and then added dropwise to a concentrated NH4PF6 solution to produce a purple precipitate. The product was purified by column chromatography, using a neutral alumina stationary phase and an acetone mobile phase. The solvent was removed from the fraction containing the product via rotary evaporation producing a red-purple solid (0.052 g, 66% yield). 1H NMR (400 MHz, (CD3)2CO, Fig. S6†): δ 10.21 (s, 1H), 8.90 (m, 4H), 8.61 (d, 2H, J = 7.8 Hz), 8.53 (d, 1H, J = 8.1 Hz), 8.30 (t, 2H, J = 7.3 Hz), 8.12 (d, 1H, J = 8.2 Hz), 8.06 (dd, 1H, J = 8.1, 5.5 Hz), 7.85 (dd, 3H, J = 32.8, 9.0 Hz), 7.63 (m, 3H), 7.52 (t, 1H, J = 7.6 Hz), 7.28 (t, 2H, 7.5 Hz), 7.17 (s, 3H), 6.95 (m, 12H). 31P{H} NMR (400 MHz, (CD3)2CO, Fig. S7†): δ 42.5 (s, 1P).
[Ru(biq)(phen)(PPh3)(CH3CN)](PF6)2 (2a)
[Ru(biq)(phen)(PPh3)Cl](PF6) (0.044 g, 0.038 mmol) and AgBF4 (0.019 g, 0.099 mmol) were dissolved in 10 mL of a CH3CN/H2O (1:1, v/v) mixture and were refluxed overnight under a N2 atmosphere. After cooling to room temperature, the reaction mixture was added dropwise to a concentrated NH4PF6 solution, precipitate was collected by filtering over Celite, and then purified via bulk recrystallization using vapor diffusion of ether into a concentrated solution of 2a in acetonitrile, which afforded a red-orange solid (0.013 g, 35% yield). 1H NMR (400 MHz, CD3CN, Fig. S8†): δ 10.1 (d, 1H, J = 5.3 Hz), 8.96 (d, 2H, J = 8.7 Hz), 8.69 (d, 2H, J = 8.8 Hz), 8.30 (m, 5H), 8.19 (m, 5H), 8.07 (dd, 2H, J = 5.4, 2.8 Hz), 7.64 (m, 4H), 7.54 (m, 5H), 7.28 (td, 3H, J = 7.4, 1.1 Hz), 7.21 (td, 3H, J = 7.8, 1.5 Hz), 7.16 (td, 3H, J = 7.9, 1.4 Hz), 2.34 (s, 3H). 31P{H} NMR (400 MHz, CD3CN, Fig. S9†): δ 42.3 (s, 1P). ESI-MS(+): [M–PF6]+m/z = 986.252 (calc. m/z = 986.155).
Instrumentation and methods
Electronic absorption spectra were collected using a Hewlett-Packard 8454 diode array spectrophotometer in 1 cm × 1 cm quartz cuvettes. The irradiation source for photolysis experiments was a 150 W Xe arc lamp (UHSIO) in a MilliArc lamp housing unit equipped with an LPS-220 power supply and an LPS-221 igniter (PTI). Irradiation wavelengths for quantum yield determination were selected by using bandpass and long-pass filters (CVI Melles Griot). Samples for photolysis were prepared under red light, sealed in an NMR tube (NMR) or cuvette (UV-Vis), and purged with N2 for 15 min prior to irradiation.The 1H and 31P{H} NMR spectra were obtained using a Bruker 400 MHz DPX instrument in CD3CN, (CD3)2CO, or CD3OD. 1H chemical shifts were referenced to the residual protonated solvent peak and 31P{H} shifts were referenced to an external 85% H3PO4 standard (0 ppm). 1H and 31P{H} NMR photolysis experiments were performed in CD3CN. Electrospray ionization mass spectrometry (ESI-MS) was performed using a Bruker microTOF instrument. For ESI-MS experiments, samples were dissolved in CH3CN and referenced to a sodium trifluoroacetate standard.
Electrochemistry experiments were performed on a BASi model CV-50 W voltammetric analyzer (Bioanalytical Systems, Inc.) with a three-electrode cell utilizing a glassy carbon working electrode, a Pt wire auxiliary electrode, and a saturated Ag/AgCl (3 M NaCl) reference electrode. Samples were dissolved in acetonitrile containing 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) as an electrolyte, data was collected at a scan rate of 200 mV s−1, and ferrocene was added at the end of each experiment as an internal reference (+0.43 V vs. Ag/AgCl in acetonitrile).45
Crystals suitable for X-ray diffraction were obtained through vapor diffusion of diethyl ether into concentrated acetonitrile or pyridine solutions of the desired complex. Single crystal X-ray diffraction for 2a was performed using a dark red rectangular plate crystal in a nitrogen gas stream at 150 K. The diffraction pattern was collected using a Nonius Kappa APEXII CCD diffractometer and Mo Kα radiation (λ = 0.7107 Å). Data were integrated using the Bruker SAINT software program and scaled using the SADABS software program. Structures were solved and refined with the Bruker SHELXT Software Package within APEX2 and Olex2. Other single crystal X-ray diffraction measurements were performed on a Bruker Kappa Photon II CPAD diffractometer equipped with Mo Kα radiation (1a and 2b, λ = 0.71073 Å) or Cu Kα radiation (I, λ = 1.54178 Å) using a dark red crystal (1a), orange plate (2b), or red blade (I) in a nitrogen gas stream at 100(2) K. The data were integrated using the Bruker SAINT software program and scaled using the SADABS software program. Solution by direct methods (SHELXT) produced a complete phasing model consistent with the proposed structure.
Spin restricted and unrestricted density functional theory (DFT) calculations were performed using the Gaussian09 program package.46 Geometry optimizations and vibrational frequency calculations were performed with the SDD47 basis set on Ru and the TZVP48 basis set on all other atoms with the PBE exchange–correlation functional.49,50 The geometries of 1a, 2a, and 2b were fully optimized starting from X-ray crystal structures and were verified to have positive harmonic frequencies, confirming the calculated structures as electronic energy minima. Molecular orbital calculations utilized the hybrid functional B3LYP,51–53 with the SDD basis set on Ru and the TZVP basis set on all other atoms. Spin densities were calculated using Mulliken population analysis (MPA) methods. Molecular orbitals from the Gaussian calculations were plotted using the Chemcraft program,54 and the analysis of the molecular orbitals and Mayer bond order calculations were performed using AOMix-FO within the AOMix program.55,56
The cell viability of all the synthesized complexes were determined by plating MDA-MB-231 cells in a 96 well plate at a density of 7000 cells per well in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% FBS and 1000 units per mL penicillin/streptomycin. The plates were incubated overnight in a 37 °C humidified incubator ventilated with 5% CO2. The media was aspirated off and then quadruplicate wells were treated with DMEM supplemented with 10% FBS and 1000 units per mL penicillin/streptomycin containing different concentrations (30 μM to 500 nM) of the synthesized complexes in 1% DMSO. Plates containing wells with no cells were designated as blank wells whereas wells with cells that were not treated with the compound but only DMEM supplemented with 10% FBS and 1000 units per mL penicillin/streptomycin containing 1% DMSO (vehicle) were designated as control wells. The plates were then again incubated in a 37 °C humidified incubator ventilated with 5% CO2. After 1 h of incubation, the cells were either irradiated with blue light (tirr = 20 min, λirr = 460–470 nm, 56 J cm−2) or kept in the dark. After 20 minutes, the plates were placed in a 37 °C humidified incubator with 5% CO2 for 72 h, after which time, 10 μL of 3-(4,5-dimethylthiazol-2-yl)diphenyl tetrazolium bromide (MTT) reagent (5 mg mL−1 in PBS) was added to each well of the 96 well plate and incubated in a 37 °C humidified incubator ventilated with 5% CO2 for 2 h. The media was then aspirated off and 100 μL of DMSO was added. The plates were then shaken for 20 min to ensure complete dissolution of the purple formazan crystals formed. Absorbance of each well was then measured at 570 nm. The mean absorbance values of the blank wells were calculated and subtracted from absorbance values for each well treated with a certain concentration of a compound. The absorbance of the control wells was also taken and subtracted with the average of the blank wells. The mean of these corrected control absorbances were then calculated. Viability of the cells was finally determined by dividing the corrected absorbance of the compound wells by the mean corrected absorbance of the control wells and expressing the mean of the ratio as a percentage value. The % viability was plotted against the log of concentration (in molarity) of the compounds and the antilog of the concentration value at 50% viability was used to determine the EC50 value of each complex against MDA-MB-231 cells.
Results and discussion
Electronic absorption and electrochemistry
The electronic absorption spectra of 1a, 1b, 2a, and 2b are shown in Fig. 2 and the corresponding absorption maxima and extinction coefficients are listed in Table 1. The singlet metal-to-ligand charge transfer (1MLCT) absorption maxima of 1a in CH3CN, attributed to Ru(dπ)→phen(π*) transitions, are observed at 372 nm and 411 nm. For 2a the Ru(dπ)→phen(π*) and Ru(πp)→biq(π*) 1MLCT bands are observed at 407 nm and 477 nm, respectively, in CH3CN. The substitution of one of the phen ligands in 1a for biq in 2a results in a bathochromic shift in the 1MLCT absorption maximum, as expected from the increased conjugation and subsequently increased π-accepting character of the biq ligand as compared to phen. A similar shift is observed in the 1MLCT maxima of the bis-acetonitrile analogs, 1b and 2b, at 420 nm and 497 nm, respectively. In addition, the lower energy of the 1MLCT transitions in 1b and 2b, as compared to the corresponding peaks in 1a and 2a, are consistent with the increased π-accepting character of the phosphine ligand as compared to acetonitrile.26,31,57–59 | ||
| Fig. 2 Electronic absorption spectra of 1a (solid blue), 1b (dashed blue), 2a (solid red), and 2b (dashed red) in CH3CN. | ||
| Complex | λ abs/nm (ε/× 103 M−1 cm−1) | E 1/2/Va |
|---|---|---|
| a 0.1 M TBAPF6, vs. Ag/AgCl in CH3CN. b From ref. 41. c Irreversible. | ||
| 1a | 372 (9.1), 411 (7.4) | +1.62, −1.29, −1.51 |
| 1b | 383 (11.2), 420 (10.2) | +1.50, −1.34, −1.50 |
| 2a | 360 (18), 377 (20), 407 (4.2), 477 (5.7) | +1.67, −0.82, −1.36c |
| 2b | 356 (23), 375 (27), 406 (3.9), 497 (7.8) | +1.55, −0.86, −1.40 |
The electrochemical reduction potentials for complexes 1a and 2a obtained from cyclic voltammetry (CV) experiments are listed in Table 1 and the corresponding CVs are shown in Fig. S10,† and are compared to those previously reported for 1b and 2b.41 The first reversible reduction events of 1a and 1b are localized on one of the phen ligands, with E1/2 values at −1.29 V and −1.34 V vs. Ag/AgCl, respectively, and compare well to those reported for related complexes.60,61 In contrast, the first reduction couples of 2a and 2b observed at −0.82 V and −0.86 V vs. Ag/AgCl, respectively, are centered on the biquinoline ligand in each complex, consistent with the lower energy lowest unoccupied π* orbital in biq and similar to those measured in related complexes.60,62
The second reduction wave is localized on the phen ligand in 2a and 2b, observed at −1.36 V and −1.40 V vs. Ag/AgCl, respectively, and on the remaining phen ligand in 1a and 1b, at −1.51 V and −1.50 V vs. Ag/AgCl, respectively. The reversible oxidation events ranging from +1.50 to +1.67 V vs. Ag/AgCl are assigned to the RuIII/II redox couple (Fig. S10† and Table 1). The ∼120 mV shift of the RuIII/II couples to more positive potentials in 1a and 2a relative to those in 1b and 2b, respectively, is consistent with the greater π-accepting character of triphenylphosphine, stabilizing the highest occupied molecular orbital (HOMO). Taken together, the electrochemical data indicate that the synthetic substitution of PPh3 in 1a and 2a for CH3CN in 1b and 2b primarily affects the energy of the Ru(dπ) t2g-type orbitals.
Photochemistry
In order to explore the light-induced ligand dissociation in complexes 1a and 2a, their photoreactivity was investigated by monitoring changes in the electronic absorption and 1H and 31P{H} NMR spectra as a function of irradiation time. Irradiation of 1a in water (<5% acetone) with visible light results in a decrease in intensity of the absorption peak at 372 nm and a concomitant increase in the 430–550 nm range with a shoulder at 455 nm, along with an isosbestic point at 407 nm (Fig. 3a). The presence of the isosbestic point is indicative of the reaction proceeding from the starting material to a single product. The changes to the 1H NMR spectrum of 1a in CD3CN were also monitored as a function of irradiation time, resulting in a decrease in the resonance at 2.17 ppm associated with CH3CN bound to ruthenium and the concomitant appearance of a resonance at 1.96 ppm, corresponding to free CH3CN (Fig. 3b). These data indicate that the irradiation of 1a results in the substitution of the CH3CN ligand with a solvent molecule, in this case CD3CN, with the absence of any additional photochemical reactions. The bathochromic shift in Fig. 3a is also consistent with this conclusion, as the photolysis of 1a in water results in the formation of cis-[Ru(phen)2(PPh3)(H2O)]2+, where the bound CH3CN is substituted for the weaker-field, π-donating H2O ligand, thus raising the energy of the Ru(dp) t2g-type set and lowering the energy of the 1MLCT transition.34,63,64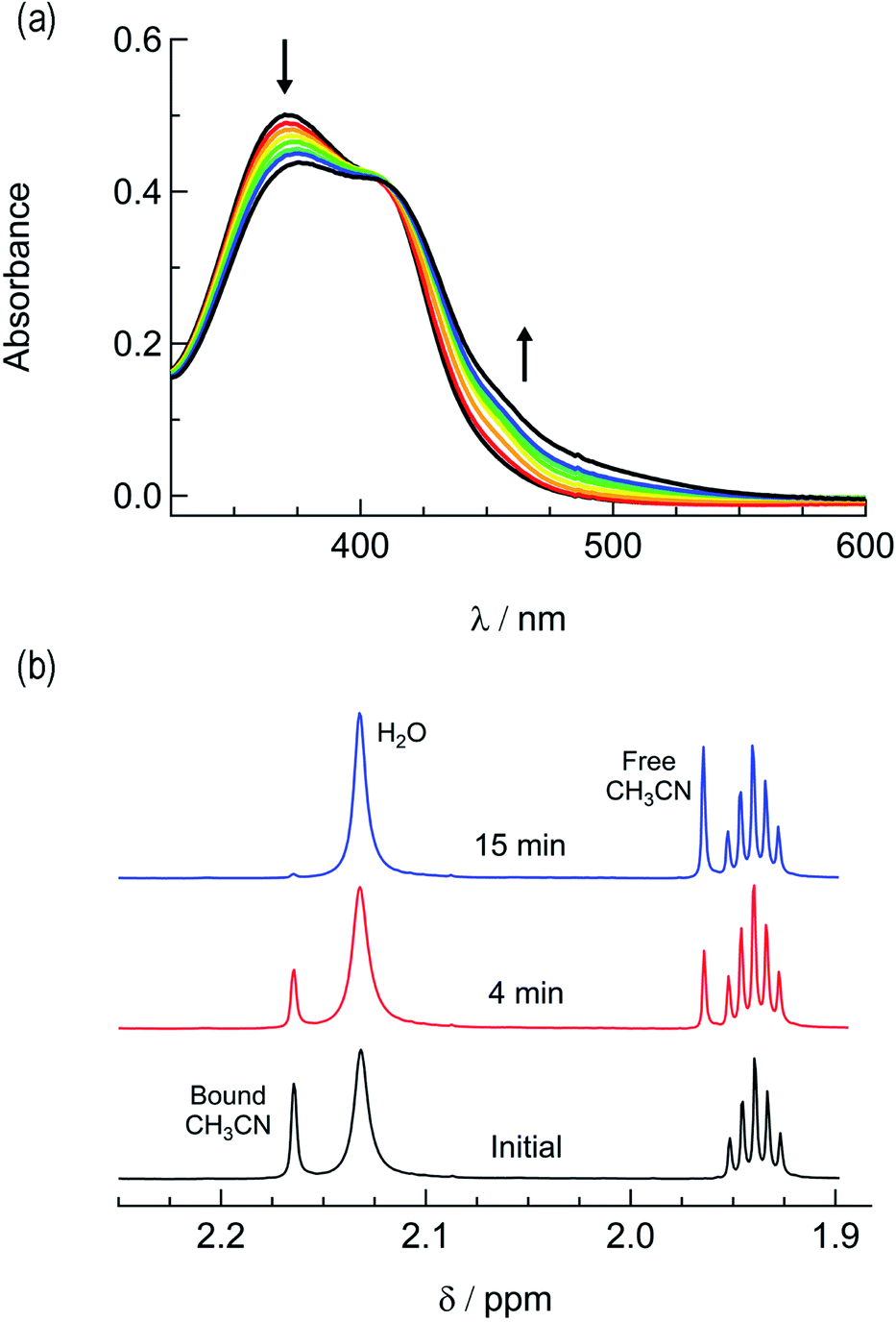 | ||
| Fig. 3 Changes following the irradiation of 1a (λirr ≥ 395 nm) to the (a) electronic absorption spectrum in H2O, tirr = 0–30 min, and (b) 1H NMR spectrum in CD3CN, tirr = 0, 4, and 14 min. | ||
The irradiation of 2a in CH3CN results in a decrease in the absorption at 407 nm and an increase a peak at 497 nm, with two isosbestic points at 396 nm and 478 nm (Fig. 4a). As shown in Fig. 4b, the spectrum of the photoproduct is nearly identical to that of 2b, providing evidence that the irradiation of 2a results in the photoinduced dissociation of the PPh3 ligand generating cis-[Ru(biq)(phen)(CH3CN)2]2+, compound 2b. The changes in the 1H NMR spectra of 2a in CD3CN recorded as a function of irradiation time are also consistent with the exchange of the phosphine ligand following visible light irradiation (Fig. 5a). For example, the resonance at 2.34 ppm, associated with the ruthenium-bound CH3CN ligand, decreases in intensity upon irradiation, with the concomitant growth of a peak at 2.47 ppm, associated with the photoproduct cis-[Ru(biq)(phen)(CH3CN)(CD3CN)]2+, similar to the resonances observed for the coordinated CH3CN ligands in 2b.41
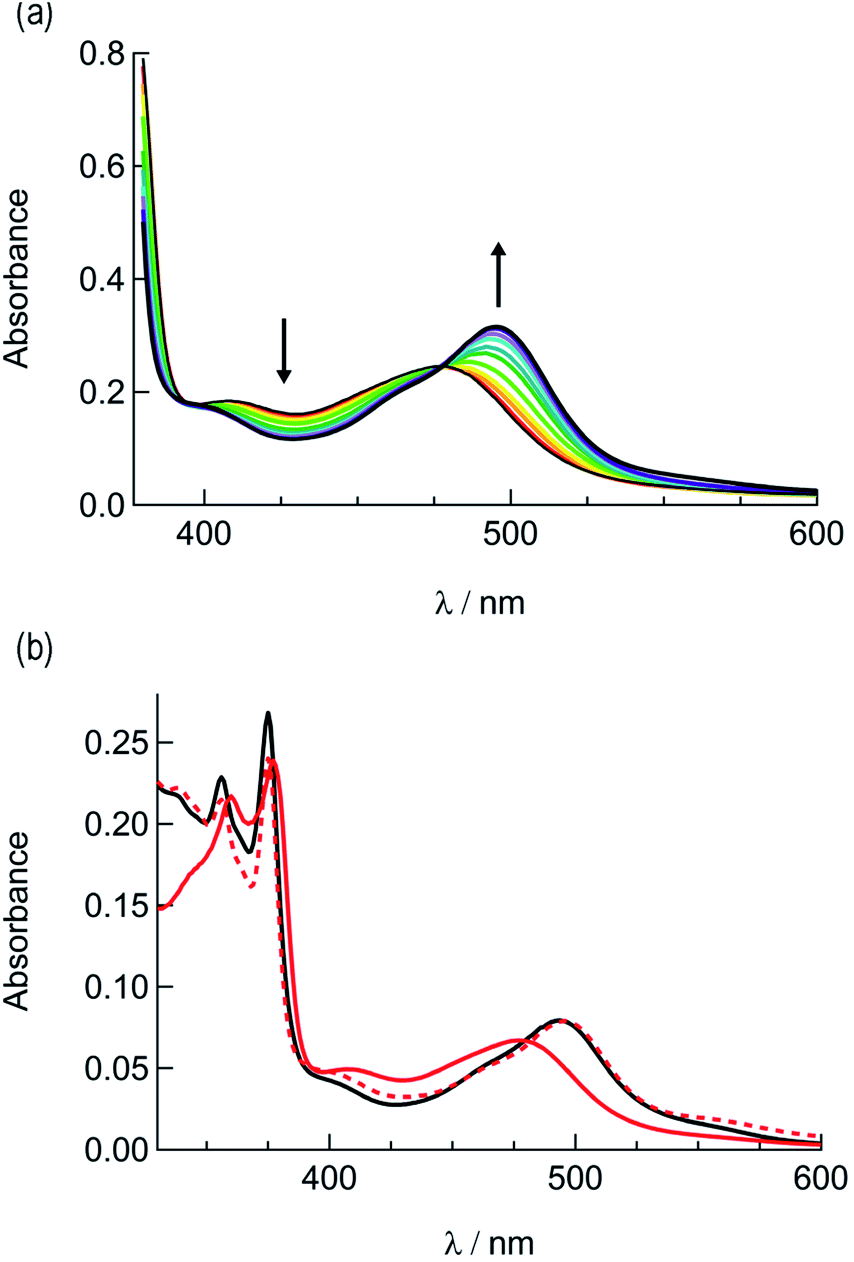 | ||
| Fig. 4 (a) Changes in the electronic absorption spectrum of 2a in CH3CN following irradiation, tirr = 0–5 min and (b) electronic absorption spectra of 2a before irradiation (solid red), following 30 min irradiation (dashed red), and 2b (black). | ||
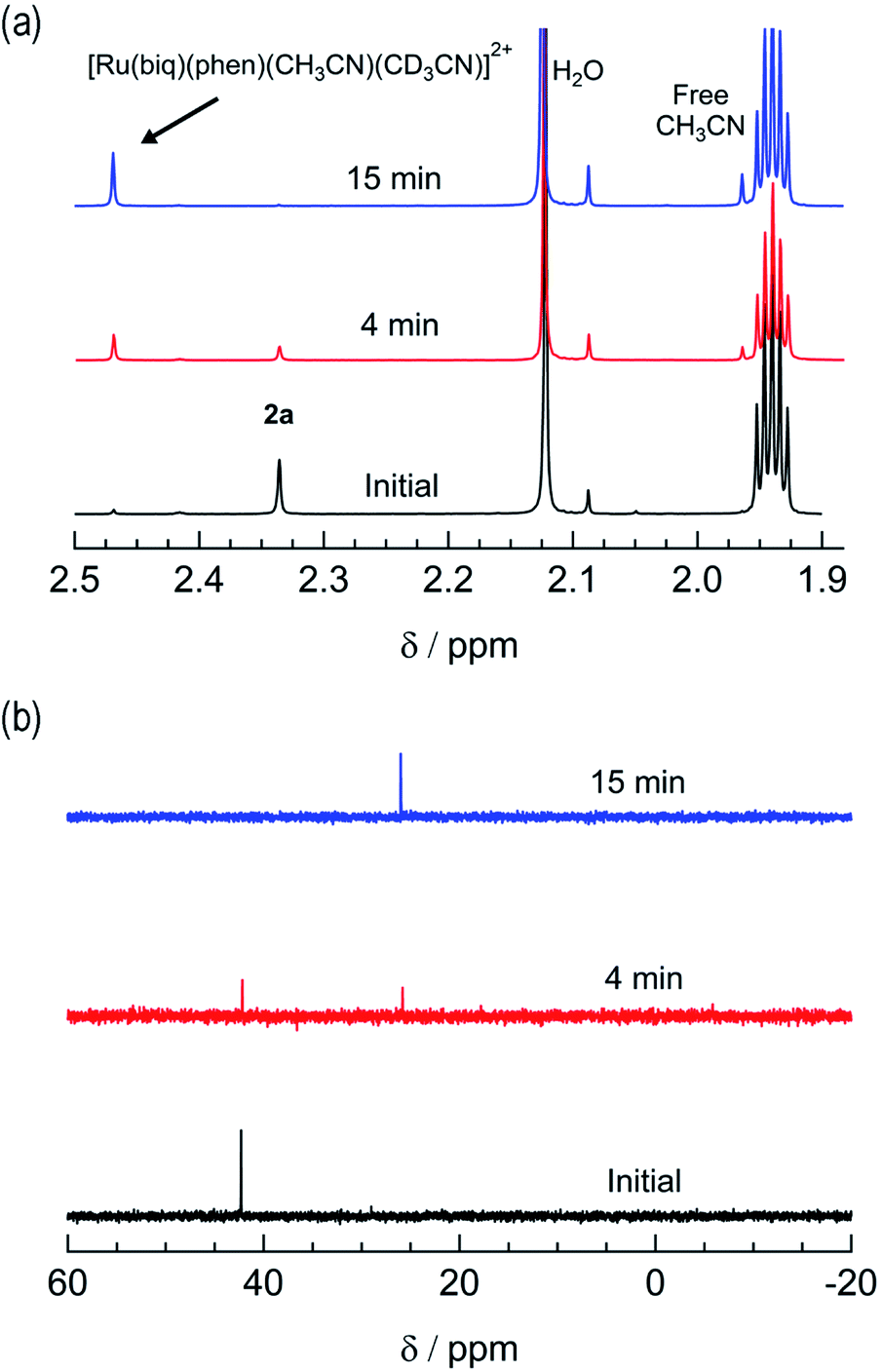 | ||
| Fig. 5 Changes in the (a) 1H and (b) 31P{H} NMR spectra of 2a in CD3CN at tirr = 0, 4, and 15 min (λirr ≥ 395 nm). | ||
The changes to the 31P{H} NMR spectra of 2a upon irradiation provide additional evidence for PPh3 exchange, where a decrease in intensity of the 31P{H} resonance at 42.3 ppm, associated with coordinated PPh3 is observed during the photolysis (Fig. 5b). Concurrently, a 31P{H} resonance corresponding to triphenylphosphine oxide centered at 26.0 ppm appeared as a function of irradiation time (Fig. 4d and S11†). Importantly, following 15 minutes of irradiation of 2a, the 31P{H} resonance associated with bound PPh3 completely disappears, while the 1H peaks of the photoproduct cis-[Ru(biq)(phen)(CH3CN)(CD3CN)]2+ persisted. In addition, the resonance at 1.96 ppm associated with free CH3CN appears concomitantly with the peak at 2.47 ppm, indicating CH3CN is also photodissociated albeit not completely on the timescale of the NMR photolysis experiment.
Based on these results, the question remains whether the irradiation of 2a results in dissociation of both PPh3 and CH3CN from the starting material, or if CH3CN exchanges only from the intermediate photoproduct cis-[Ru(biq)(phen)(CH3CN)(CD3CN)]2+ after the initial dissociation of PPh3 from the starting complex. In an effort to address this point and trap the product of the first ligand exchange step, photolysis experiments were performed in the coordinating solvent pyridine under identical illumination conditions to those previously discussed in acetonitrile, and the results are shown in Fig. 6. Inspection of Fig. 6a reveals one set of isosbestic points at early irradiation times, up to ∼2.5 min, observed at 316 nm, 448 nm, and 500 nm, and the decrease of the peak associated with 2a at 490 nm with the appearance of a band at 540 nm. A second set of isosbestic points is evident at later times (Fig. 6b), from ∼3.5 min to 20 min, at 338 nm, 361 nm, and 550 nm, with a loss of the species with absorption at 540 nm and the formation of the final product with maximum at 590 nm. These results point at the formation of solely one initial intermediate, I, with maximum at 540 nm, which then goes on to exchange a second ligand to generate the final product with a peak at 590 nm, assigned to cis-[Ru(biq)(phen)(py)2]2+ (3). As expected from the ability of CH3CN to π-backbond with the Ru(dπ) t2g-type orbitals that is not present in pyridine, the 1MLCT maximum of cis-[Ru(biq)(phen)(py)2]2+, 3, is red-shifted compared to that of the product 2b, cis-[Ru(biq)(phen)(CH3CN)2]2+, in Fig. 4a.
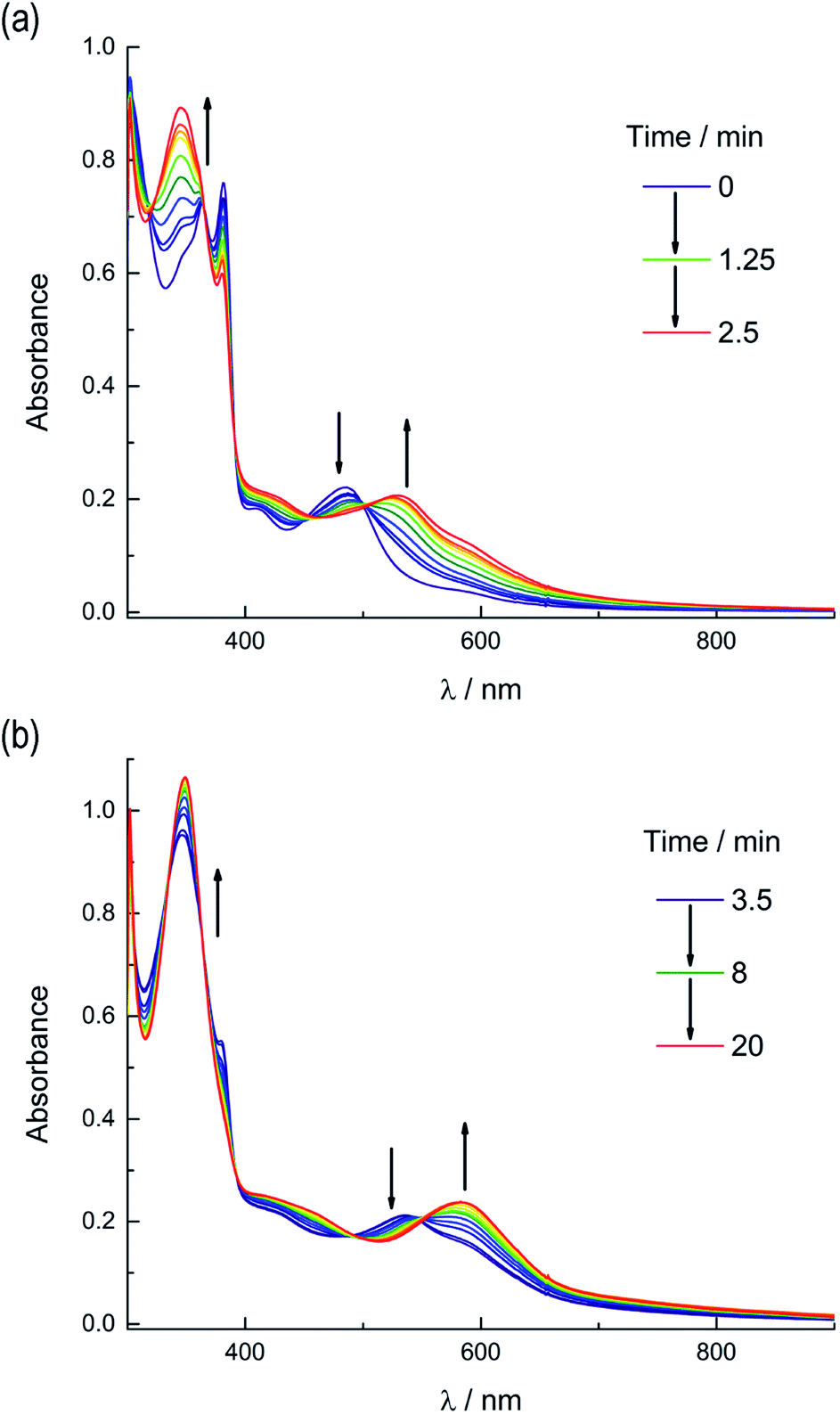 | ||
| Fig. 6 Changes following irradiation (λirr ≥ 395 nm) in the electronic absorption spectrum of 2a in pyridine from (a) tirr = 0–2.5 min and (b) tirr = 3.5–20 min. | ||
The intermediate I was identified as cis-[Ru(biq)(phen)(py)(CH3CN)]2+, generated by absorption of a single photon by 2a and photosubstitution of the triphenylphosphine ligand for a solvent pyridine molecule. Identification was supported, in conjunction with 31P{H} NMR spectra (Fig. 5b), by obtaining a single-crystal X-ray diffraction structure of the photoproduct generated by irradiating a solution of 2a in pyridine with ≥ 395 nm light for 90 s (Fig. S12 and Table S1†). No further spectral changes took place after the conclusion of irradiation, indicating I is stable in the dark. This conclusion further supported by the persistence of the complex in a pyridine solution as the compound recrystallized via diethyl ether diffusion.
The presence of the two sets of isosbestic points permits determination of time-dependent concentrations of the three individual species in solution: 2a, I, and 3, during the photolysis, where I represents an intermediate species. The deconvolution of the associated spectra is possible from the known absorption spectra and molar extinction coefficients of 2a and 3, and the details of the analysis are presented in the ESI, Table S2, and Fig. S13.† The spectra of 2a and 3, along with that of the intermediate, I, are shown in Fig. S13,† and the time dependent mole fractions of each species were calculated over the course of the photolysis and are displayed in Fig. 7. Fig. 7 shows that the loss of 2a occurs rapidly, whereby at 70 s, 50% of the starting material remains and no amount is appreciable beyond 300 s. The formation of the intermediate I begins as early as 50 s of irradiation and reaches 99% conversion to the final product, 3, at ∼900 s. A maximum fraction of ∼55% of the intermediate I is apparent at ∼150 s (Fig. 7). From the known proportions of the three species the extinction coefficient for the intermediate was estimated and compared to initial and final product values in Fig. S13.†
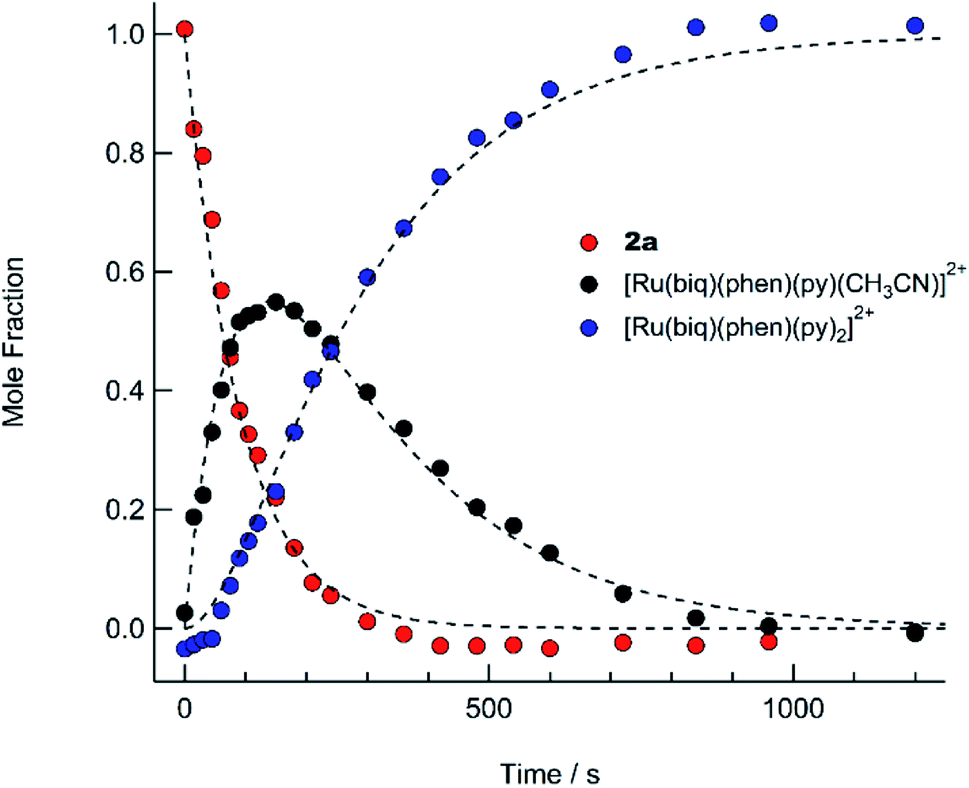 | ||
| Fig. 7 Time dependent concentrations of 2a (red circles), cis-[Ru(biq)(phen)(py)(CH3CN)]2+ (I, black circles), and cis-[Ru(biq)(phen)(py)2]2+ following irradiation (λirr ≥ 395 nm) in pyridine from t = 0 to t = 1200 s. The dashed lines are least-squares fits to a consecutive reaction model with a system of equations describing the time-dependent concentration of each compound (see text). | ||
The PPh3 ligand photodissociation from 2a apparent from the sequential formation of photoproducts in Fig. 6 and 7, as well as in the crystal structure of I, shows that irradiation of 2a does not result in the photodissociation of the CH3CN ligand, such that there is no evidence of the formation of [Ru(biq)(phen)(PPh3)(py)]2+ following irradiation. Instead CH3CN substitution must occur from further irradiation of the intermediate, cis-[Ru(biq)(phen)(CH3CN)(S)]2+ where S = coordinating solvent molecule. It is also important to note that the growth of the 1H NMR peak corresponding to free CH3CN at 1.96 ppm for the irradiation of 2a in CD3CN shown in Fig. 4c does not begin until t ∼ 4 min, which is consistent with the dissociation of CH3CN taking place from the intermediate cis-[Ru(biq)(phen)(CH3CN)(CD3CN)]2+ and not directly from 2a.
In contrast to the results for 2a, the 31P{H} NMR of 1a did not change as a function of irradiation time (Fig. S14†) in CD3CN, indicating that the PPh3 ligand is photostable in this complex and does not photodissociate. Together, these results demonstrate the photoinduced ligand exchange of PPh3 from 2a upon irradiation with λirr ≥ 395 nm, while 1a undergoes only CH3CN ligand substitution. Steric strain around the Ru(II) center due to the bulky biq ligand is known to influence the exchange of ligands from Ru(II) complexes following irradiation and likely plays a role in the photoinduced PPh3 exchange in 2a.41,65,66 The observed trends highlight a need to further investigate the geometry around the ruthenium center in phosphine complexes to identify the origin of the unusual dissociation of PPh3 in 2a.
Structural comparisons
The generally accepted model for photoinduced ligand exchange in Ru(II) polypyridyl complexes is thermal population of a 3LF (ligand field) state from a lower energy 3MLCT (metal-to-ligand charge transfer) state, which places electron density on Ru-L orbital(s) with σ* anti-bonding character, leading to ligand dissociation.67–70 Distortion in the pseudo-octahedral geometry around ruthenium metal lowers the energy of the eg-type σ* set, and consequently of the dissociative 3LF state, leading to a decrease in the activation energy required to thermally populate it from the lowest energy 3MLCT state and increasing the efficiency of ligand exchange. The introduction of steric bulk via ligands containing methyl, phenyl, or quinoline moieties has been shown to sufficiently distort the octahedral geometry, resulting in an increase the quantum yield of ligand exchange.71–74In order to better understand if steric effects account for the differences in the photoreactivity of 1a and 2a, their solid-state single-crystal X-ray structures were determined and are shown in Fig. 8, along with relevant bond lengths and angles listed in Table 2 with additional X-ray data available in Tables S3 and S4.† The Ru–N bond lengths from the ruthenium center to the bidentate ligand trans to the PPh3 ligand are 0.05 Å longer on average in 2a (biq) as compared to 1a (phen), respectively, indicating greater steric strain in the former. The greater steric hindrance in 2a relative to 1a is further supported by the Ru1–P1 bond length, which is 0.024 Å longer in 2a, consistent with a weaker Ru–P bonding interaction in the biquinoline complex. The crystal structure of 2b (Fig. 8), obtained by irradiating a solution of 2a in CH3CN with visible light overnight, possesses Ru–N3 and Ru–N4 bond lengths that are 0.075 and 0.028 Å shorter than in 2a, respectively, where N3 and N4 are the nitrogen atoms in the biquinoline ligand. Such a decrease in Ru–N bond lengths demonstrates that the photodissociation of PPh3 relieves steric strain.
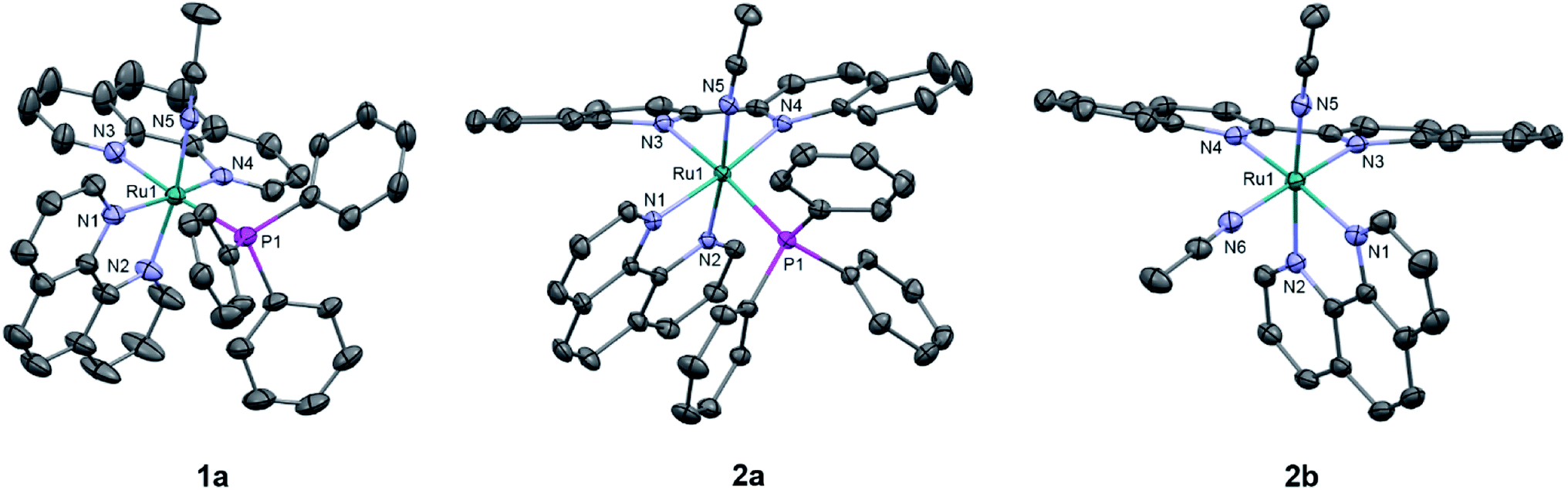 | ||
| Fig. 8 ORTEP plots of 1a, 2a, and 2b (thermal ellipsoids have been drawn at 50% probability and hydrogen atoms, PF6− molecules, and co-crystallized solvent molecules have been omitted for clarity); Ru: cyan, N: light purple, C: grey, and P: magenta. | ||
| 1a | 2a | 2b | |
|---|---|---|---|
| Bond lengths (Å) | |||
| Ru1–N3 | 2.106(9) | 2.148(1) | 2.073(2) |
| Ru1–N4 | 2.06(1) | 2.112(2) | 2.084(2) |
| Ru1–N5 | 2.030(2) | 2.036(2) | 2.046(2) |
| Ru1–P1/N6 | 2.343(1) | 2.3669(5) | 2.034(2) |
|
|||
| Torsion angles (°) | |||
| N1–C–C–N2 | 1.1(4) | 2.1(2) | 0.6(3) |
| N3–C–C–N4 | 2(1) | 10.5(2) | 3.1(3) |
|
|||
| Bond angles (°) | |||
| N1–Ru1–N3 | 87.7(2) | 94.09(9) | 100.22(7) |
| N1–Ru1–P1/N6 | 93.68(8) | 85.26(7) | 82.24(7) |
| N2–Ru1–N3 | 91.2(2) | 81.95(9) | 87.52(6) |
| N3–Ru1–N5 | 82.1(2) | 93.13(9) | 93.52(7) |
| N4–Ru1–P1/N6 | 98.8(4) | 103.63(7) | 99.31(7) |
Evidence for distortion of the bidentate ligand trans to PPh3 is apparent in the torsion angle, the angle between the two N–C–C planes formed by N3 of the bidentate ligand and the two carbon atoms bridging it to N4, expected to be 0° in an ideal octahedral geometry. In the case of 1a, the N3–C–C–N4 torsion angle in the phenanthroline ligand is 2(1)°. In contrast, a torsion angle of 10.5(2)° is measured in the biquinoline ligand in 2a. Further, geometric planes defined by N1–Ru1–N2 and N3–Ru1–N4 would be at 90° angles in an ideal octahedral geometry and deviations from this angle reveal additional steric distortion around the metal center.4 The angle between these two planes in 1a was determined to be 88.69°, which is reduced to 81.37° in 2a. Importantly, in 2b this angle is 86.93° and the N3–C–C–N4 torsion angle in the biquinoline ligand is 3.1(3)°, showing the substitution of PPh3 for CH3CN allows the complex to adopt a geometry closer to the ideal octahedral. It should be noted that the Ru–N5 bond to the CH3CN ligand does not significantly differ in length (0.006 Å) in 1a and 2a.
The bond angles provided in Table 2 demonstrate additional differences in the steric distortion in 1a, 2a, and 2b. For example, the N1–Ru1–N3 and N2–Ru1–N3 angles show the extent of distortion in the phenanthroline ligand in each complex. Substitution of biq for phen in 2a pushes N1 towards the phosphine ligand while N2 moves away from P1 to accommodate the large PPh3 unit, such that the N2–Ru1–N3 angle is nearly 10° greater in 1a than 2a. These same angles in 2b demonstrate that the dissociation of PPh3 relieves the steric strain between the polypyridyl bidentate ligands by allowing the phenanthroline ligand to move away from biq.
The angles N1–Ru1–P1 and N4–Ru1–P1 show the bulkier biquinoline ligand pushes the phosphine away from biq and toward phen in 2a, to a significantly greater extent than in the analogous phen in 1a. Similar to the bond angles, the CH3CN ligand does not display great distortion. The only notable angle change is that for N3–Ru1–N5, which is 11° smaller in 1a than in 2a, indicating that PPh3 pushes the acetonitrile ligand towards the trans bidentate ligand in 1a but the presence of the larger biq ligand prevents this displacement in 2a. In summary, the X-ray crystal structures show that there are significantly greater deviations from octahedral geometry in 2a as compared to 1a and 2b.
Calculations
Density functional theory (DFT) calculations were performed to determine if the bonding and electronic structure in the complexes could further explain the differences in the photoreactivity of 1a and 2a. Geometry optimizations in the singlet ground state (1GS) of 1a, 2a, and 2b resulted in structures in good agreement with experimental crystallographic data (Table S4). Table S4† shows that the calculated bond lengths, angles, and torsional angles are in good agreement to those obtained experimentally from the crystal structures of each complex. The 1GS highest occupied molecular orbitals (HOMOs) in 1a and 2a exhibit primarily Ru-d orbital character, as is typical for Ru(II) polypyridyl complexes.63,75–77 The lowest unoccupied molecular orbital (LUMO) in each complex is primarily localized on the ligand trans to PPh3, 1,10-phenanthroline in 1a and 2,2′-biquinoline in 2a; the latter agrees with the findings from electrochemistry (Fig. S15, S16 and Table S5†).Geometry optimizations and vibrational frequency calculations were also performed in the triplet excited states (3ES) of 1a and 2a. In the 3ES, longer Ru–NCCH3 and Ru–P bond distances are calculated in both complexes as compared to the corresponding 1GS (Table 3). The calculated Ru–P bonds in the 3ES are similar, 2.475 Å in 1a and 2.473 Å in 2a, increasing from 2.411 Å and 2.441 Å in the 1GS, respectively. However, the Ru–NCCH3 bond is 0.04 Å longer in the 3ES of 1a, 2.055 Å, as compared to that in 2a, 2.015 Å, which may indicate that the Ru–nitrile bond is weaker in the excited state of 1a relative to that in 2a.
| MBOs | Bond Lengths/Å | |||||||
|---|---|---|---|---|---|---|---|---|
| 1a | 2a | 1a | 2a | |||||
| Bond | 1GS | 3ES | 1GS | 3ES | 1GS | 3ES | 1GS | 3ES |
| a Bond to N atom of bidentate ligand trans to CH3CN. b Bond to N atom of CH3CN. c Bond to N atom of bidentate ligand trans to PPh3. | ||||||||
| Ru1–N3a | 0.370 | 0.656 | 0.223 | 0.223 | 2.080 | 2.022 | 2.096 | 2.095 |
| Ru1–N5b | 0.601 | 0.441 | 0.653 | 0.576 | 2.008 | 2.055 | 2.000 | 2.015 |
| Ru1–N1c | 0.260 | 0.408 | 0.281 | 0.368 | 2.136 | 2.103 | 2.170 | 2.146 |
| Ru1–P1 | 0.760 | 0.719 | 0.701 | 0.659 | 2.411 | 2.475 | 2.441 | 2.473 |
The differences in the bonds of 1a and 2a in the 3ES were further investigated by calculating the Mayer bond orders (MBOs) of the bonds involving ruthenium (Table 3). MBOs are an extension of Wiberg bond orders and can provide insight into the relative strengths of bonds in transition metal complexes. In the 3ES of 1a, the MBO of the Ru–NCCH3 bond (Ru1–N5) exhibits a 26.6% decrease as compared to the 1GS and the Ru–N(phen) bond trans to CH3CN (Ru1–N3) displays a dramatic 74.0% increase. These results indicate that the reduced phenanthroline ligand in the excited state exerts a trans-type influence on the CH3CN ligand and weakens the Ru1–N3 bond, likely contributing to its dissociation in the excited state as has been observed in other Ru(II) complexes.33,78,79 In contrast, the MBO of the bond to CH3CN only decreases by 11.8% in the triplet state of 2a and the order of the Ru–N bond trans to CH3CN does not change in the triplet state of 2a. In addition, the Ru–NCCH3 bond itself is significantly stronger in the 3ES of 2a as compared to 1a, 0.576 and 0.441, respectively.
Both 1a and 2a displayed a ∼6% increase in the MBO of the Ru–PPh3 bond, Ru1–P1 in Table 3, in the 3ES, although the bond is weaker in the excited state of 2a, MBO = 0.659, than in 1a, MOB = 0.719. The order of the bond trans to the phosphine ligand, Ru1–N1, is calculated to increase by 31.0% in 2a and 56.9% in 1a, indicating a trans-type influence in both complexes. However, the phenomenon is significantly stronger in 1a for the bond positioned trans to the CH3CN ligand, Ru1–N3, which may explain why the CH3CN ligand preferentially photodissociates in this complex.
Mulliken spin density (MSD) calculations were also performed on the lowest energy 3ES of 1a and 2a. These calculations determine the unpaired electron density on each atom in the 3ES and can provide information on the nature of the excited state. In complexes with 3LF as the lowest energy triplet excited state, the spin density on the Ru(II) metal center would theoretically equal two. If the lowest energy 3ES is MLCT in nature, the spin density on ruthenium is expected to be one, and any deviation from these whole numbers indicates metal/ligand mixing. The MSD on ruthenium and the summed density on each ligand in the lowest energy 3ES of 1a and 2a is shown in Fig. 9. The calculated spin densities on Ru(II) indicate the lowest energy triplet excited state is MLCT in nature with notable ligand character in 1a and significant mixing from a ligand-centered state in 2a. The summed spin density on the phenanthroline ligand trans to PPh3 in 1a is 0.447, lower than the 0.763 sum on the phen ligand trans to CH3CN, further indicating stronger trans-type influence on the nitrile ligand in the excited state. In the 3MLCT of 2a the sum of the spin density on the phen ligand, which is trans to CH3CN, is 0.074 and a sum of 1.433 is calculated on the biquinoline ligand, indicating a significant trans-type influence on the PPh3 ligand. Taken together with the calculated bond lengths and MBOs in the 3MLCT state, it can be concluded that while 1a exhibits a trans-type influence on both monodentate ligands, it is stronger in the case of the CH3CN ligand which can explain the preferential dissociation of CH3CN upon irradiation. In the 3MLCT state of 2a there is no evidence of trans-type influence on the nitrile ligand, but a significant degree of trans-type influence is calculated for the phosphine ligand. These results, in conjunction with the steric strain evident from the crystal structure, can explain the unexpected photodissociation of the PPh3 ligand in 2a.
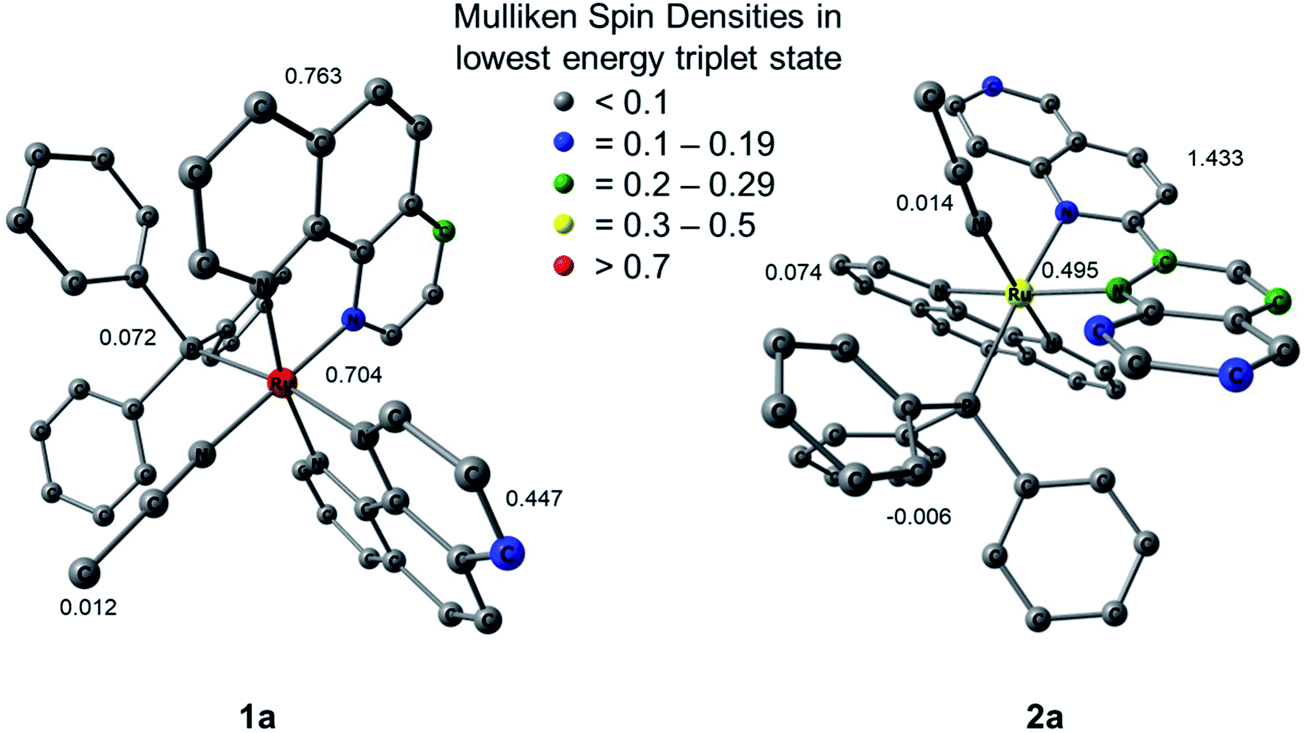 | ||
| Fig. 9 Mulliken spin densities (MSDs) on ruthenium and the summed densities on each ligand in the calculated lowest energy triplet excited states of 1a and 2a. | ||
Cell viability
The effect of photochemical reactivity on the biological behavior of 1a and 2a was investigated on the MDA-MB-231 triple-negative breast cancer cell line in the dark and upon irradiation and the results were compared to those of related complexes previously reported.18,80,81 The half effective concentration, EC50, was determined, which is defined as the concentration of the compound when the viability of the experimental cells is 50% compared to those in the absence of active compound. Complexes 1a and 2a were incubated with MDA-MB-231 cells for 1 h and were then either left in the dark or irradiated with blue light for 20 min (λirr = 460–470 nm, 56 J cm−2). Cellular viability was assessed using the MTT assay after 72 h, where the viability upon treatment with only the vehicle (1% DMSO) only in DMEM (Dulbecco's Modified Eagle Medium) was considered as 100%, and the results are listed in Table 3. Both 1a and 2a are non-toxic against MDA-MB-231 cells in the dark with ECD50 values of 26.6 ± 1.5 μM and >30 μM, respectively; the latter is above the maximum concentration allowed by the solubility in growth media (30 μM). However, irradiation with blue light significantly increased the toxicities of both complexes, resulting in ECL50 = 4.6 ± 0.6 μM for 1a and ECL50 = 7.1 ± 0.2 μM for 2a (Fig. S17 and S18†). These values of the phototherapeutic index, PI, values defined as ECD50/ECL50, were calculated to be 5.8 and >4.2 for 1a and 2a, respectively (Table 4). While complex 1a exchanges CH3CN for a solvent water molecule, 2a releases PPh3 upon irradiation. The finding that PPh3 by itself is not toxic against MDA-MB-231 cells both in the dark and when irradiated with blue light (tirr = 20 min, λirr = 460–470 nm, 56 J cm−2) in the concentrations used for complexes 1a and 2a (Fig. S19†) led to the conclusion that the corresponding aqua complexes are the major cause of toxicity upon irradiation.| Complex | ECD50/μM a | ECL50/μMa | PIb |
|---|---|---|---|
| a Data are an average of three independent experiments. b PI = ECD50/ECL50. c From ref. 82; bpy = 2,2′-bipyridine. d From ref. 83; IC50 values against HeLa cells. | |||
| 1a | 26.6 ± 1.5 | 4.6 ± 0.6 | 5.8 |
| 2a | >30 | 7.1 ± 0.2 | >4.2 |
| [Ru(bpy)2(PPh3)(CH3CN)]2+c | >30 | 7.0 ± 1.4 | >4.3 |
| cis-[Ru(bpy)2(CH3CN)2]2+d | 244 ± 23 | 223 ± 94 | 1.1 ± 0.4 |
Comparison of the cell toxicity data of cis-[Ru(bpy)2(PPh3)(CH3CN)]2+ and cis-[Ru(bpy)2(CH3CN)2]2+ in Table 3 indicates that the substitution of a CH3CN ligand for PPh3 increases the PI value of the ruthenium complex. Complexes 1a and 2a exhibit PI values similar to or exceeding that of cis-[Ru(bpy)2(PPh3)(CH3CN)]2+. These results indicate that the phosphine ligand may positively influence the activity against cancer cells of these ruthenium complexes, a conclusion supported by previous examples of phosphines improving cellular uptake and localizing complexes to the mitochondria, increasing selectivity for cancerous cells over healthy cells,22 and increasing cytotoxic activity against breast and colon cancer cells of phosphine containing cis-configured Pt(II) complexes over that of cisplatin.21,84–86
Conclusion
Two new triphenylphosphine-containing complexes, 1a and 2a, were synthesized and their ground state spectroscopic and electrochemical properties were characterized, along with their photoinduced ligand exchange and cytotoxicity against a triple-negative breast cancer cell line. Changes in the electronic absorption and NMR spectra of complex 1a revealed the substitution of a CH3CN ligand for a solvent molecule following visible light irradiation and a substitutionally inert PPh3. In contrast, the photolysis of complex 2a results in the initial exchange of the PPh3 ligand generating a solvated intermediate, and the latter goes on to absorb a second photon which then undergoes CH3CN substitution. A comparison of the single crystal X-ray structures reveals that 2a exhibits greater steric distortion around the metal center than 1a, which is subsequently relieved upon the photoinduced exchange of PPh3 for a less sterically-demanding solvent molecule. To our knowledge, this represents the first report of the photodissociation of a phosphine ligand from a Ru(II) polypyridyl complex. In addition, the ability of phosphine ligands to enhance cellular uptake was shown to enhance the photocytoxicity of 1a and 2a against a triple-negative breast cancer cell line relative to related complexes without PPh3 in their coordination sphere. This work shows that a coordinated PPh3 ligand can serve as a new architecture for potential therapeutics for use in PCT.Author contributions
Sean J. Steinke synthesized the complexes and conducted photophysical and photochemical measurements. Eric J. Piechota aided with data analysis and interpretation, Sayak Gupta performed the cell studies, and Curtis E. Moore collected and solved the crystal structures. Jeremy J. Kodanko and Claudia Turro advised their respective group members.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors thank the support from the National Science Foundation (CHE-2102508), as well as Dr J. C. Gallucci for assistance with the collection of the crystal structure data of 2a.References
- N. A. Smith and P. J. Sadler, Philos. Trans. R. Soc., A, 2013, 371, 20120519 CrossRef
.
-
(a) S. B. Vittardi, R. T. Magar, D. J. Breen and J. J. Rack, J. Am. Chem. Soc., 2021, 143, 526–537 CrossRef PubMed
-
(a) J. Karges, S. Kuang, F. Maschietto, O. Blacque, I. Ciofini, H. Chao and G. Gasser, Nat. Commun., 2020, 11, 1–13 Search PubMed
-
(a) J. K. White, R. H. Schmehl and C. Turro, Inorg. Chim. Acta, 2017, 454, 7–20 CrossRef CAS PubMed
- S. Monro, K. L. Colon, H. Yin, J. Roque, P. Konda, S. Gujar, R. P. Thummel, L. Lilge, C. G. Cameron and S. A. McFarland, Chem. Rev., 2019, 119, 797–828 CrossRef CAS PubMed
- L. Hammarström, Acc. Chem. Res., 2015, 48, 840–850 CrossRef PubMed
- M. R. Gill and J. A. Thomas, Chem. Soc. Rev., 2012, 41, 3179–3192 RSC
- L. Marcelis, J. Ghesquiere, K. Garnir, A. Kirsch-De Mesmaeker and C. Moucheron, Coord. Chem. Rev., 2012, 256, 1569–1582 CrossRef CAS
- R. Oun, Y. E. Moussa and N. J. Wheate, Dalton Trans., 2018, 47, 6645–6653 RSC
- R. Liu, L. Zhang, J. Zhao, Z. Luo, Y. Huang and S. Zhao, Adv. Thermoelectr., 2018, 1, 1800041 Search PubMed
- J. Dang, H. He, D. Chen, L. Yin and L. Biomater, Sci, 2017, 5, 1500–1511 CAS
- Y. Mir, J. E. van Lier, B. Paquette and D. Houde, Photochem. Photobiol., 2008, 84, 1182–1186 CrossRef CAS PubMed
- D. Havrylyuk, D. K. Heidary, Y. Sun, S. Parkin and E. C. Glazer, ACS Omega, 2020, 5, 18894–18906 CrossRef CAS PubMed
- H. Shi, C. Imberti and P. J. Sadler, Inorg. Chem. Front., 2019, 6, 1623–1638 RSC
- V. H. S. van Rixel, V. Ramu, A. B. Auyeung, N. Beztsinna, D. Y. Leger, L. N. Lameijer, S. T. Hilt, S. E. Le Devedec, T. Yildiz, T. Betancourt, M. B. Gildner, T. W. Hudnall, V. Sol, B. Liagre, A. Kornienko and S. Bonnet, J. Am. Chem. Soc., 2019, 141, 18444–18454 CrossRef CAS PubMed
- M. K. Herroon, R. Sharma, E. Rajagurubandara, C. Turro, J. J. Kodanko and I. Podgorski, Biol. Chem., 2016, 397, 571–582 CAS
- M. Huisman, J. K. White, V. G. Lewalski, I. Podgorski, C. Turro and J. J. Kodanko, Chem. Commun., 2016, 52, 12590–12593 RSC
- K. Arora, M. Herroon, M. H. Al-Afyouni, N. P. Toupin, T. N. Rohrabaugh, L. M. Loftus, I. Podgorski, C. Turro and J. J. Kodanko, J. Am. Chem. Soc., 2018, 140, 14367–14380 CrossRef CAS PubMed
- T. N. Rohrabaugh, A. M. Rohrabaugh, J. J. Kodanko, J. K. White and C. Turro, Chem. Commun., 2018, 54, 5193–5196 RSC
- A. Li, R. Yadav, J. K. White, M. K. Herroon, B. P. Callahan, I. Podgorski, C. Turro, E. E. Scott and J. J. Kodanko, Chem. Commun., 2017, 53, 3673–3676 RSC
-
(a) A. A. Khan, K. S. Allemailen, A. Almatroudi, S. A. Almatroodi, M. A. Alsahli and A. H. Rahmani, J. Drug Delivery Sci. Technol., 2021, 61, 102315 CrossRef CAS
- R. S. Correa, L. M. Bomfim, K. M. Oliveira, D. R. M. Moreira, M. B. P. Soares, J. Ellena, D. P. Bezerra and A. A. Batista, J. Biol. Inorg Chem., 2019, 198, 110751 CAS
- Y. R. Pérez and R. Etchenique, Photochem. Photobiol. Sci., 2019, 18, 208–212 CrossRef PubMed
- S. M. Veronica, M. Alvarez, O. Filevich, R. Etchenique and A. del Campo, Langmuir, 2012, 28, 1217–1221 CrossRef PubMed
- L. Zayat, M. G. Noval, J. Campi, C. I. Calero, D. J. Calvo and R. Etchenique, Chembiochem, 2007, 8, 2035–2038 CrossRef CAS PubMed
- S. K. Lee, M. Kondo, G. Nakamura, M. Okamura and S. Masaoka, Chem. Commun., 2018, 54, 6915–6918 RSC
- I. M. Dixon, E. Lebon, G. Loustau, P. Sutra, L. Vendier and A. Juris, Dalton Trans., 2008, 5627–5635 RSC
- E. Lebon, I. M. Dixon, L. Vendier, A. Igau and P. Sutra, Inorg. Chim. Acta, 2007, 360, 1235–1239 CrossRef CAS
- I. M. Dixon, E. Lebon, P. Sutra and A. Igau, Chem. Soc. Rev., 2009, 38, 1621–1634 RSC
- D. V. Pinnick and B. Durham, Inorg. Chem., 1984, 23, 1440–1445 CrossRef CAS
- L. Zayat, O. Filevich, L. Baraldo and R. Etchenique, Philos. Trans. R. Soc., A, 2013, 371, 20120330 CrossRef PubMed
- J. D. Knoll, B. A. Albani and C. Turro, Chem. Commun., 2015, 51, 8777–8780 RSC
- L. M. Loftus, A. Li, K. L. Fillman, P. D. Martin, J. J. Kodanko and C. Turro, J. Am. Chem. Soc., 2017, 139, 18295–18306 CrossRef CAS PubMed
- T. N. Rohrabaugh, Jr., K. A. Collins, C. Xue, J. K. White, J. J. Kodanko and C. Turro, Dalton Trans., 2018, 47, 11851–11858 RSC
- W.-S. Huang, S. Liu, D. Zou, M. Thomas, Y. Wang, T. Zhou, J. Romero, A. Kohlmann, F. Li, J. Qi, L. Cai, T. A. Dwight, Y. Xu, R. Xu, R. Dodd, A. Toms, L. Parillon, X. Lu, R. Anjum, S. Zhang, F. Wang, J. Keats, S. D. Wardwell, Y. Ning, Q. Xu, L. E. Moran, Q. K. Mohemmad, H. G. Jang, T. Clackson, N. I. Narasimhan, V. M. Rivera, X. Zhu, D. Dalgarno and W. C. Shakespeare, J. Med. Chem., 2016, 59, 4938–4964 Search PubMed
- R. Wang, Y. Chen, X. Zhao, S. Yu, B. Yang, T. Wu, J. Guo, C. Hao, D. Zhao and M. Cheng, Eur. J. Med. Chem., 2019, 183, 111716 CrossRef CAS PubMed
- J. Tu, L. T. Song, H. L. Zhai, J. Wang and X. Y. Zhang, Int. J. Biol. Macromol., 2018, 118, 1149–1156 CrossRef CAS PubMed
- S. Bischoff and M. Kant, Catal. Today, 2001, 66, 183–189 CrossRef CAS
- A. Riisager, K. M. Eriksen, P. Wasserscheid and R. Fehrmann, Catal. Lett., 2003, 90, 149–153 CrossRef CAS
- B. A. Harper, D. A. Knight, C. George, S. L. Brandow, W. J. Dressick and C. S. Dalcey, Inorg. Chem., 2003, 42, 516–524 CrossRef CAS PubMed
- B. A. Albani, C. B. Durr and C. Turro, J. Phys. Chem. A, 2013, 117, 13885–13892 CrossRef CAS PubMed
- Z. Assefa and D. M. Stanbury, J. Am. Chem. Soc., 1997, 119, 521–530 CrossRef CAS
- M. A. Bennett and A. K. Smith, J. Chem. Soc., Dalton Trans., 1974, 233–241 RSC
- A. Alberti, P. Astolfi, P. Carloni, L. Greci, C. Rizzoli and P. Stipa, New J. Chem., 2015, 39, 8964–8970 RSC
- V. V. Pavlishchuk and A. W. Addison, Inorg. Chim. Acta, 2000, 298, 97–102 CrossRef CAS
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayahsi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, revision E.01. Gaussian, Inc.: Wallingford, CT, 2016 Search PubMed
- D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 1396 CrossRef PubMed
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed
- B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200–206 CrossRef CAS
-
Chemcraft; Graphical Software for Visualization of Quantum Chemistry Computations. https://www.chemcraftprog.com Search PubMed
- S. I. Gorelsky and A. B. P. Lever, J. Organomet. Chem., 2001, 635, 187–196 CrossRef CAS
-
S. I. Gorelsky, AOMix: Program for Molecular Orbital Analysis, 2015 Search PubMed
- A. G. Orpen and N. G. Connelly, J. Chem. Soc., Chem. Commun., 1985, 19, 1310–1311 RSC
- D. M. Klassen, Chem. Phys. Lett., 1982, 93, 383–386 CrossRef CAS
- M. Mitoraj and A. Michalak, Organometallics, 2007, 26, 6576–6580 CrossRef CAS
- B. A. Albani, B. Peña, K. R. Dunbar and C. Turro, Photochem. Photobiol. Sci., 2014, 13, 272–280 CrossRef CAS PubMed
- P. Bonneson, J. L. Walsh, W. T. Pennington, A. W. Cordes and B. Durham, Inorg. Chem., 1983, 22, 1761–1765 CrossRef CAS
- A. Juris, S. Campagna, V. Balzani, G. Gremaud and A. von Zelewsky, Inorg. Chem., 1988, 27, 3652–3655 CrossRef CAS
- L. M. Loftus, K. F. Al-Afyouni, T. N. Rohrabaugh Jr, J. C. Gallucci, C. E. Moore, J. J. Rack and C. Turro, J. Phys. Chem. C, 2019, 123, 10291–10299 CrossRef CAS
- L. M. Loftus, K. F. Al-Afyouni and C. Turro, Chem. - Eur. J., 2018, 24, 11550–11553 CrossRef CAS PubMed
- E. Wachter, D. K. Heidary, B. S. Howerton, S. Parkin and E. C. Glazer, Chem. Commun., 2012, 48, 9649–9651 RSC
- E. Baranoff, J.-P. Collin, J. Furusho, Y. Furusho, A.-C. Laemmel and J.-P. Sauvage, Inorg. Chem., 2002, 41, 1215–1222 CrossRef CAS PubMed
- J. V. Caspar and T. J. Meyer, Inorg. Chem., 1983, 22, 2444–2453 CrossRef CAS
- B. Durham, J. V. Caspar, J. K. Nagle and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 4803–4810 CrossRef CAS
- B. Durham, J. L. Walsh, C. L. Carter and T. J. Meyer, Inorg. Chem., 1980, 19, 860–865 CrossRef CAS
- G. H. Allen, R. P. White, D. P. Rillema and T. J. Meyer, J. Am. Chem. Soc., 1984, 106, 2613–2620 CrossRef CAS
- J. D. Knoll, B. A. Albani, C. B. Durr and C. Turro, J. Phys. Chem. A, 2014, 118, 10603–10610 CrossRef CAS PubMed
- S. Bonnet, J. P. Collin, J. P. Sauvage and E. Schofield, Inorg. Chem., 2004, 43, 8346–8354 CrossRef CAS PubMed
- A.-C. Laemmel, J.-P. Collin and J.-P. Sauvage, Eur. J. Inorg. Chem., 1999, 383–386 CrossRef CAS
- B. S. Howerton, D. K. Heidary and E. C. Glazer, J. Am. Chem. Soc., 2012, 134, 8324–8327 CrossRef CAS PubMed
- M. H. Al-Afyouni, T. N. Rohrabaugh, K. F. Al-Afyouni and C. Turro, Chem. Sci., 2018, 9, 6711–6720 RSC
- M. Abrahamsson, M. Jäger, R. J. Kumar, T. Österman, P. Persson, H. C. Becker, O. Johansson and L. Hammarström, J. Am. Chem. Soc., 2008, 130, 15533–15542 CrossRef CAS PubMed
- J. Romanova, Y. Sadik, M. R. Ranga Prabhath, J. D. Carey and P. D. Jarowski, J. Phys. Chem. C, 2017, 121, 2333–2343 CrossRef CAS
- E. Galardon, P. Le Maux, L. Toupet and G. Simonneaux, Organometallics, 1998, 17, 565–569 CrossRef CAS
- A. C. H. Da Silva, J. L. F. Da Silva and D. W. Franco, Dalton Trans., 2016, 45, 4907–4915 RSC
- S. D. Ramalho, R. Sharma, J. K. White, N. Aggarwal, A. Chalasani, M. Sameni, K. Moin, P. C. Vieira, C. Turro, J. J. Kodanko and B. F. Sloane, PLoS One, 2015, 10, e0142527/1–e0142527/17 CAS
- N. P. Toupin, S. Nadella, S. J. Steinke, C. Turro and J. J. Kodanko, Inorg. Chem., 2020, 59, 3919–3933 CrossRef CAS PubMed
- A. P. Lanquist, S. Gupta, M. Al-Afyouni, J. J. Kodanko and C. Turro, Chem. Sci., 2021, 12, 12056–12067 RSC
- B. A. Albani, B. Peña, N. A. Leed, N. A. B. G. de Paula, C. Pavani, M. S. Baptista, K. R. Dunbar and C. Turro, J. Am. Chem. Soc., 2014, 136, 17095–17101 CrossRef CAS PubMed
- H. Scheffler, Y. You and I. Ott, Polyhedron, 2010, 29, 66–69 CrossRef CAS
- V. Gandin, A. P. Frenandes, M. P. Rigobello, B. Dani, F. Sorrentino, F. Tisato, A. Björnstedt, A. Bindoli, A. Sturaro, R. Rella and C. Marzano, Biochem. Pharmacol., 2010, 79, 90–101 CrossRef CAS PubMed
- C. Icsel, V. T. Yilmaz, B. Cevatemre, M. Aygun and E. Ulukaya, J. Biol. Inorg Chem., 2020, 25, 75–87 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 31P{H} NMR spectra, electrochemical and X-ray crystallographic data, and DFT information (PDF). CCDC 2108338, 2109146, 2108340 and 2108925. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc05647f |
| This journal is © The Royal Society of Chemistry 2022 |