
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRadical boron migration of allylboronic esters†
Xiangzhang
Tao
,
Shengyang
Ni
,
Lingyu
Kong
,
Yi
Wang
 * and
Yi
Pan
* and
Yi
Pan
State Key Laboratory of Coordination Chemistry, Jiangsu Key Laboratory of Advanced Organic Materials, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210023, China. E-mail: yiwang@nju.edu.cn
First published on 17th January 2022
Abstract
A photocatalyzed 1,3-boron shift of allylboronic esters is reported. The boron atom migration through the allylic carbon skeleton proceeds via consecutive 1,2-boron migrations and Smiles-type rearrangement to furnish a variety of terminally functionalized alkyl boronates. Several types of migrating variations of heteronuclei radicals and dearomatization processes are also tolerated, allowing for further elaboration of highly functionalized boron-containing frameworks.
Introduction
Rearrangements have been fascinating topics to organic chemists due to their innate charm and versatile use in skeleton reconstruction.1,2 The classic ionic-type named reactions, such as Pinacol, Favorskii, Beckmann, Claisen and Smiles rearrangements, involve the re-organization of certain carbon skeletons.3–6 In contrast, the examples for heteroatom shifts are relatively rare. For example, the Brook rearrangement7,8 and Doyle–Kirmse reactions9–12 involve silyl and thiol group migrations. A series of 1,2-boryl migrations of α-boryl expoxides and carboxylic acids have been reported by Yudin and co-workers.13,14 Besides the ionic type functional group migrations (FGM), the rearrangements that involve free radicals have demonstrated versatile reactivities towards remote FGM for the construction of more complex molecular frameworks.15–19 Recent progress on aryl, cyano, alkynyl, and carbonyl group migration protocols have been well-documented for the construction of new C–C bonds via radical pathways.1,20–32 Boron shift chemistry, which represents tremendous importance for the broad utility of boronic acids and esters, has attracted much attention of organic chemists. The first example of radical type boron shift was reported by Batey and Smil.33 In 2019, Aggarwal described a radical 1,2-boron shift of diboronates under photocatalysis to access 1,2-difunctionalized boronates (Fig. 1A).34 Studer and co-workers applied the boron migration protocol to 1,3-difunctionalized trifluoromethylated alkynyl boronates (Fig. 1B).35 In contrast, the analogous 1,3-shift process has encountered inevitable difficulties because of the disfavored four-membered cyclic transition state and no example of γ-boron migration has been reported.36–42 Theoretical studies on such 1,3-boron migrating processes have been concluded to be thermodynamically infavorable and experimentally inaccessible.43 We envisioned that through consecutive boron migration on the carbon skeleton, the boronic ester moiety could switch to the remote end of the allylic backbones for the formation of more stable radical intermediates. Herein, we described a radical 1,3-boron migration process merging double 1,2-boron shifts of allylboronates and Smiles-type rearrangement of arylsulfonyl radicals to generate aryl γ-boronates (Fig. 1C). Moreover, this boron migration protocol could be further extended to other heteronuclei radicals and the formation of dearomatized frameworks.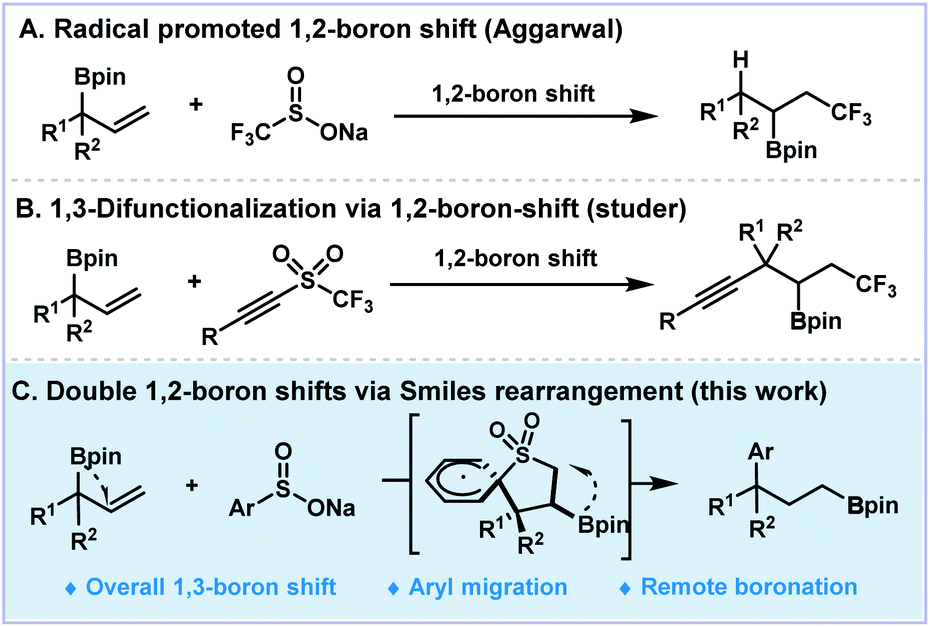 | ||
| Fig. 1 Radical promoted boron shift chemistry. | ||
Results and discussion
Our investigations began with the model reaction of sodium arylsulfonate 1a and allylboronic ester 2a. The desired 1,3-boron migration product 3a could be obtained in 95% yield under blue LED irradiation for 12 h with 10-phenylphenothiazine (PTH) as the photocatalyst and acetic acid as the additive in CHCl3 (entry 1). For other photocatalysts such as Ir(ppy)3 and 4CzIPN, lower yields were noted (20–35%, entries 2 and 3). Replacing the PTH photocatalyst with Acr+ClO4− led to a slightly lower yield. (entry 4). The reaction could not occur in DMF or MeOH (entry 5 and 6). In the absence of acetic acid, the reaction yield was reduced significantly (entry 7). Control experiments indicated that light and the photocatalyst are necessary for this boron shift strategy (entries 8 and 9, see the ESI†) (Table 1). | ||
| Fig. 2 Scope of the 1,3-boron shift. | ||
|
|
||
|---|---|---|
| Entry | Variation form the above conditions | Yield of 3ab |
| a Reaction conditions: la (0.3 mmol), 2a (02 mmol), 10-phenylphenothiazine (PTH, 8 mol%), HOAc (4.0 equiv.), CHCI3 (2 mL), Ar, irradiation with 60 W blue LEDs at r.t. for 12 h. b Crude yields were determined by 19F NMR using 1,4-difluorobenzene as the internal standard. c The isolated yields were determined from the corresponding oxidized product. | ||
| 1 | None | 95 (82%)c |
| 2 | Ir(ppy)3 instead of PTH | 20 |
| 3 | 4CzIPN instead of PTH | 35 |
| 4 | Acr+CIO4− instead of PTH | 85 |
| 5 | DMF instead of CHCI3 | 0 |
| 6 | MeOH instead of CHCI3 | 0 |
| 7 | w/o HOAc | 58 |
| 8 | w/o PTH | 0 |
| 9 | In darkness | 0 |

With the optimized reaction conditions in hand, we explored the generality of the protocol with various sodium arylsulfonates and allylboronic esters (Fig. 2). Arylsulfonates bearing either electron-withdrawing or electron-donating groups delivered the desired 1,3-boron shift products in moderate to good yields (3b–3l, 25–87%). The ortho-fluorinated arylsulfonate provided the product 3h in 80% yield, while the migration of para- and meta-fluorinated substrates was less efficient (30% for 3e and 27% for 3j). Multiple fluorine or chlorine substituted arylsulfonates could be tolerated (3k and 3l). Heteroarene 3m was compatible with the reaction conditions, and the low yield was due to the electronic effect of thiophene. Allylboronates with various substitutions were also investigated. For example, substrates bearing the cyclohexyl group gave the boronation product with a lower efficiency (3n, 38%). The arylboronate 3o with unsymmetrical alkyl substituents was afforded in 44% yield (R1 = Me, R2 = n-Hex). Furthermore, this boron shift was proven successful with stabilized benzyl radical precursors (R1 = Ar, R2 = H). Aryl substituted allylboronates have been subjected to the standard conditions and diarylpropylboronic products were readily accessed. The α-phenyl-substituted allylboronic esters containing phenyl, para-methyl and para-fluorine phenyl and naphthyl could deliver the corresponding products (3p–3s). The low yields of 3e, 3n and 3q were due to the loss in the purification process.
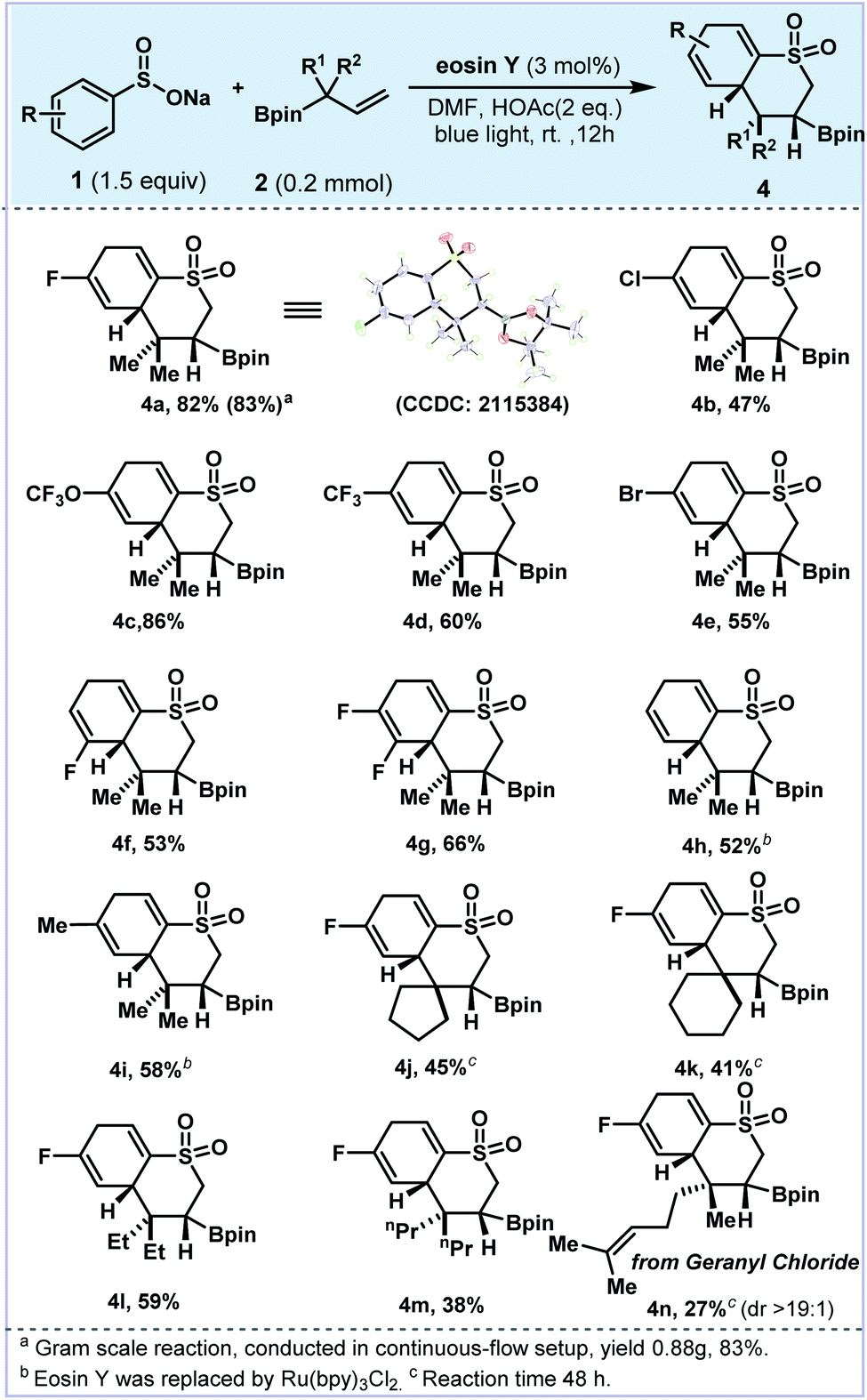 | ||
| Fig. 3 Substrate scope of dearomatized products. | ||
Photoredox-catalyzed and electrochemical dearomatization processes involving radicals have been explored with proven utility.44–50 During the investigation of this 5-exo-trig cyclization process, we also observed the 6-exo-trig cyclization dearomatizated products in the reaction mixture.51–53 As valuable pharmacophores, cyclic sulfones have been widely applied in medicinal chemistry and organic photoconducting materials due to their unique electronic and optical properties.54–56 With a slight variation of the reaction conditions (eosin Y and DMF), the dearomatized product 4 could be readily afforded via 1,2-boron shift and radical addition. As shown in Fig. 3, tetrohydrothiochromene sulfone 4a was achieved in 82% yield with excellent diastereoselectivity (>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, the absolute configuration was confirmed by XRD analysis). Similarly, electron-withdrawing group substituted 4b–4e were obtained in 47–86% yields. In the cases of 4d and 4e, small amounts of the protonated by-products were observed. Interestingly, 3-fluoro and 3,4-difluoro substituted arylsulfonate substrates generated single dearomatized regioisomers in good yields (4f and 4g), which was governed by the regio- and chemoselective nature of the cyclization step. Notably, dearomatized products 4h and 4i were furnished along with the corresponding aromatized cyclic sulfones. Replacing the photocatalyst with Ru(bpy)3Cl2, the aromatic by-products were significantly suppressed (De/Ar = 10:1 for 4i and >20:1 for 4h). Next, different allylboronic esters were explored with sodium arylsulfonate 1a under this protocol (4j–4n). Substrates bearing five- and six-membered rings also afforded the corresponding products 4j and 4k in moderate yields (41–47%). The 2,2-diethyl and 2,2-dipropyl alkenyl boronic esters delivered the dearomatized products 4l and 4m in 59% and 38% yields, respectively. The allylboronate derived from geranyl chloride gave 4n in 27% yield as a single diastereoisomer (d.r. > 19:1).
:1, the absolute configuration was confirmed by XRD analysis). Similarly, electron-withdrawing group substituted 4b–4e were obtained in 47–86% yields. In the cases of 4d and 4e, small amounts of the protonated by-products were observed. Interestingly, 3-fluoro and 3,4-difluoro substituted arylsulfonate substrates generated single dearomatized regioisomers in good yields (4f and 4g), which was governed by the regio- and chemoselective nature of the cyclization step. Notably, dearomatized products 4h and 4i were furnished along with the corresponding aromatized cyclic sulfones. Replacing the photocatalyst with Ru(bpy)3Cl2, the aromatic by-products were significantly suppressed (De/Ar = 10:1 for 4i and >20:1 for 4h). Next, different allylboronic esters were explored with sodium arylsulfonate 1a under this protocol (4j–4n). Substrates bearing five- and six-membered rings also afforded the corresponding products 4j and 4k in moderate yields (41–47%). The 2,2-diethyl and 2,2-dipropyl alkenyl boronic esters delivered the dearomatized products 4l and 4m in 59% and 38% yields, respectively. The allylboronate derived from geranyl chloride gave 4n in 27% yield as a single diastereoisomer (d.r. > 19:1).
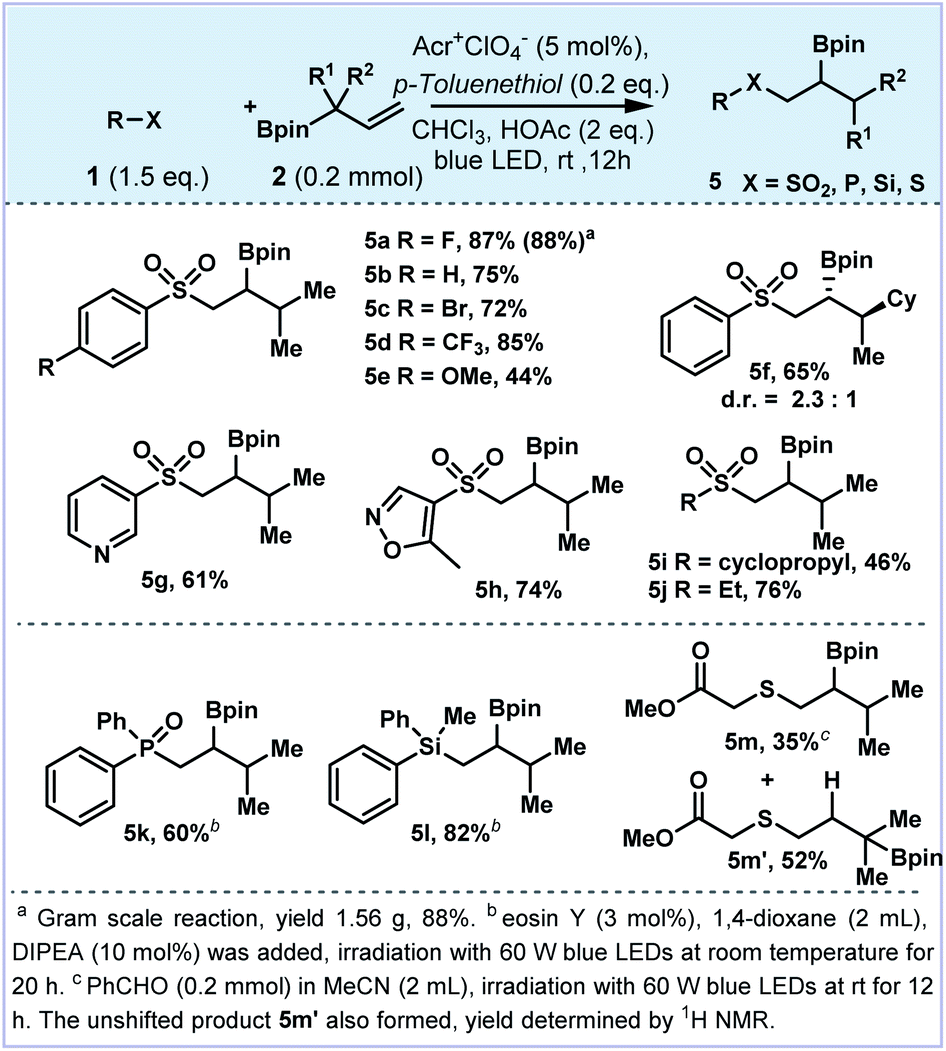 | ||
| Fig. 4 Scope of 1,2-boron shift. | ||
In order to better understand the reaction pathway, we have added a catalytic amount of thiophenol to quench the tertiary C-radical generated by 1,2-boron migration of the allylboronates (Fig. 4). A diverse set of aromatic substrates, regardless of their electronic properties were found to be compatible to produce the corresponding 1,2-difunctionalized products (5a–5e). Allylboronate containing a quaternary carbon center provided the corresponding product 5f with a diastereomeric ratio of 2.3:1. The heteroaryl sulfones bearing pyridine (5g) and oxazole (5h) could be tolerated. The alkyl sulfonates (5i and 5j) were also furnished in good yields. Moreover, other heteroatom radical precursors could be tolerated to construct C–P, C–Si and C–S bonds. For example, phosphate and diphenylmethylsilane delivered β-boronate products 5k and 5l in 60% and 82% yields, respectively. The mercaptan derivative afforded the thiolated boronate 5m in 35% yield in the mixture with the unshifted product 5m′.
 | ||
| Fig. 5 Plausible mechanism for 1,3- and 1,2-boron migrations. | ||
Based on the above results, a plausible mechanism was proposed (Fig. 5). First, the photocatalysts were activated to their excited state by blue light and they underwent single electron transfer (SET) with arylsulfonate to produce sulfone radical int I. The conjugate addition of int I to allylboronate 2 produced the secondary carbon radical int II and 1,2-boron shift generated the tertiary carbon radical int III. At this point, the radical int III could undergo different pathways depending on the conditions. Under condition A, 5-exo-trig cyclization followed by Smiles-type rearrangement afforded int IV which degraded to release SO2 and the second 1,2-boron shift provided the β-carbon radical int V. Next, the hydrogen atom transfer (HAT) process occurred between radical int V and CHCl3 to deliver the 1,3-boron shift products 3 and ˙CCl3 (see deuteration experiments in the ESI† for details). Finally, the trichloromethyl radical underwent SET with PC˙- to close the photoredox catalysis cycle. However under condition B, the tertiary radical int III could undergo 6-exo-trig cyclization to deliver the dearomatized product 4. Under condition C, the radical int III underwent HAT with thiophenol to form the 1,2-difunctionalized product 5.
Experimental
General procedure for product 3 formation (condition A)
Under argon, PTH (8 mol%) and sodium benzenesulfinate 1 (0.3 mmol, 1.5 equiv.) were placed in a tube with a stirring bar, and then CHCl3 (2 mL) was added at room temperature. Subsequently, the corresponding allylboronic acid pinacol ester (0.2 mmol, 1.0 equiv.) and HOAc (0.8 mmol, 4.0 equiv.) were added dropwise via syringe. After that, the tube was exposed to 60 W blue LEDs at room temperature for about 12 h. The mixture was concentrated in vacuo. Then it was passed through a short pad of silica gel. The organic layer was concentrated under vacuum and the yellow oily residue was diluted with THF (1 mL) and water (1 mL) followed by addition of NaBO3·4H2O (61.5 mg, 0.4 mmol). The mixture was allowed to stir for 3 h at rt. The reaction mixture was washed with EtOAc (5 mL × 3) and the combined organic layers were dried over MgSO4. It was then concentrated and purified by silica gel chromatography to afford the desired product.General procedure for product 4 formation (condition B)
Under argon, Eosin Y (3 mol%) and sodium benzenesulfinate 1 (0.3 mmol, 1.5 equiv.) were placed in a tube with a stirring bar, and then CHCl3 (2 mL) was added at room temperature. Subsequently, the corresponding allylboronic acid pinacol ester (0.2 mmol, 1.0 equiv.) and HOAc (0.4 mmol, 2.0 equiv.) were added dropwise via syringe. After that, the tube was exposed to 60 W blue LEDs at room temperature for about 12–24 h. Upon completion, the solution was passed through a pad of silica gel and washed with ethyl acetate. The filtrate was concentrated under vacuum and purified by column chromatography on silica gel using 10:1–5:1 hexane:EtOAc as the eluent to give the corresponding pure product 4.
General procedure for product 5 formation (condition C)
Under argon, Acr+ClO4− (5 mol%), sodium benzenesulfinate 1 (0.3 mmol, 1.5 equiv.) and p-toluenethiol (0.04 mmol, 0.2 equiv.) were placed in a tube with a stirring bar, and then CHCl3 (2 mL) was added at room temperature. Subsequently, the corresponding allylboronic acid pinacol ester (0.2 mmol, 1.0 equiv.) and HOAc (0.4 mmol, 2.0 equiv.) were added dropwise via syringe. After that, the tube was exposed to 60 W blue LEDs at room temperature for about 12 h. The mixture was concentrated and purified by silica gel chromatography to afford the desired product 5.Conclusions
In conclusion, a photocatalytic 1,3-boron shift protocol merging radical migration and Smiles-type rearrangement is developed. The atom switch acrobatics undergoes consecutive secondary/tertiary and primary/secondary carbon radical migrations to access a variety of 1,3-difunctionalized aryl boronates. The variation of the conditions could also lead to other 1,2-boron shift products including phosphate, silyl and thiol derivatives and dearomatized sulfone-fused ring systems. This boron migration strategy has provided a new pathway for remote functional group migration. Further studies of boron shift chemistry are underway in the laboratory.Data availability
The data that support the findings of this study are available in the ESI† or on request from the corresponding author.Author contributions
X. T. conducted all experiments and characterized the novel compounds. S. N., L. K., Y. W. and Y. P. designed the experiments and wrote the manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (No. 21772085, 21971107 and 22071101).Notes and references
- X. Wu and C. Zhu, Acc. Chem. Res., 2020, 53, 1620–1636 CrossRef CAS PubMed.
- C. M. Rojas, Molecular Rearrangements in Organic Synthesis, Wiley-VCH, New York, 2015 Search PubMed.
- M. Kischkewitz, F. W. Friese and A. Studer, Adv. Synth. Catal., 2020, 362, 2077–2087 CrossRef PubMed.
- D. Leonori and V. K. Aggarwal, Acc. Chem. Res., 2014, 47, 3174–3183 CrossRef CAS PubMed.
- W. Li, W. Xu, J. Xie, S. Yu and C. Zhu, Chem. Soc. Rev., 2018, 47, 654–667 RSC.
- S. Namirembe and J. P. Morken, Chem. Soc. Rev., 2019, 48, 3464–3474 RSC.
- A. G. Brook, J. Am. Chem. Soc., 1958, 80, 1886–1889 CrossRef CAS.
- A. G. Brook, C. M. Warner and M. E. McGriskin, J. Am. Chem. Soc., 1959, 81, 981–984 CrossRef CAS.
- W. Kirmse and M. Kapps, Chem. Ber., 1968, 101, 994–1003 CrossRef CAS.
- X. Lin, Y. Tang, W. Yang, F. Tan, L. Lin, X. Liu and X. Feng, J. Am. Chem. Soc., 2018, 140, 3299–3305 CrossRef CAS PubMed.
- M. P. Doyle, W. H. Tamblyn and V. Bagheri, J. Org. Chem., 1981, 46, 5094–5102 CrossRef CAS.
- Z. Zhang, Z. Sheng, W. Yu, G. Wu, R. Zhang, W. D. Chu, Y. Zhang and J. Wang, Nat. Chem., 2017, 9, 970–976 CrossRef CAS PubMed.
- Z. He, A. Zajdlik, J. D. St Denis, N. Assem and A. K. Yudin, J. Am. Chem. Soc., 2012, 134, 9926–9929 CrossRef CAS PubMed.
- C. F. Lee, D. B. Diaz, A. Holownia, S. J. Kaldas, S. K. Liew, G. E. Garrett, T. Dudding and A. K. Yudin, Nat. Chem., 2018, 10, 1062–1070 CrossRef CAS PubMed.
- Z. Li, M. Wang and Z. Shi, Angew. Chem., Int. Ed., 2021, 60, 186–190 CrossRef CAS PubMed.
- A. A. Tabolin and S. L. Ioffe, Chem. Rev., 2014, 114, 5426–5476 CrossRef CAS PubMed.
- T. H. West, S. S. M. Spoehrle, K. Kasten, J. E. Taylor and A. D. Smith, ACS Catal., 2015, 5, 7446–7479 CrossRef CAS.
- J. J. Zhang, L. Yang, F. Liu, Y. Fu, J. Liu, A. A. Popov, J. Ma and X. Feng, Angew. Chem., Int. Ed., 2021, 69, 25695–25700 CrossRef PubMed.
- Y. Zhu, L. Sun, P. Lu and Y. Wang, ACS Catal., 2014, 4, 1911–1925 CrossRef CAS.
- W. H. Urry and M. S. Kharasch, J. Am. Chem. Soc., 1944, 66, 1438–1440 CrossRef CAS.
- N. Fuentes, W. Kong, L. Fernandez-Sanchez, E. Merino and C. Nevado, J. Am. Chem. Soc., 2015, 137, 964–973 CrossRef CAS PubMed.
- W. Kong, M. Casimiro, N. Fuentes, E. Merino and C. Nevado, Angew. Chem., Int. Ed., 2013, 52, 13086–13090 CrossRef CAS PubMed.
- W. Kong, E. Merino and C. Nevado, Angew. Chem., Int. Ed., 2014, 53, 5078–5082 CrossRef CAS PubMed.
- C. Hervieu, M. S. Kirillova, T. Suarez, M. Muller, E. Merino and C. Nevado, Nat. Chem., 2021, 13, 327–334 CrossRef CAS PubMed.
- W. Kong, N. Fuentes, A. Garcia-Dominguez, E. Merino and C. Nevado, Angew. Chem., Int. Ed., 2015, 54, 2487–2491 CrossRef CAS PubMed.
- W. Kong, M. Casimiro, E. Merino and C. Nevado, J. Am. Chem. Soc., 2013, 135, 14480–14483 CrossRef CAS PubMed.
- J. J. Douglas, H. Albright, M. J. Sevrin, K. P. Cole and C. R. Stephenson, Angew. Chem., Int. Ed., 2015, 54, 14898–14902 CrossRef CAS PubMed.
- D. Alpers, K. P. Cole and C. R. J. Stephenson, Angew. Chem., Int. Ed., 2018, 57, 12167–12170 CrossRef CAS PubMed.
- Y. Xia and A. Studer, Angew. Chem., Int. Ed., 2019, 58, 9836–9840 CrossRef CAS PubMed.
- N. Radhoff and A. Studer, Angew. Chem., Int. Ed., 2021, 60, 3561–3565 CrossRef CAS PubMed.
- Y. Xia, L. Wang and A. Studer, Angew. Chem., Int. Ed., 2018, 57, 12940–12944 CrossRef CAS PubMed.
- F. W. Friese, C. Mück-Lichtenfeld and A. Studer, Nat. Commun., 2018, 9, 2808 CrossRef PubMed.
- R. A. Batey and D. V. Smil, Angew. Chem., Int. Ed., 1999, 38, 1798–1800 CrossRef CAS PubMed.
- D. Kaiser, A. Noble, V. Fasano and V. K. Aggarwal, J. Am. Chem. Soc., 2019, 141, 14104–14109 CrossRef CAS PubMed.
- K. Jana, A. Bhunia and A. Studer, Chem, 2020, 6, 512–522 CAS.
- Y. Ishida, I. Nakamura and M. Terada, J. Am. Chem. Soc., 2018, 140, 8629–8633 CrossRef CAS PubMed.
- Y. Chen, Y. Liu, Z. Li, S. Dong, X. Liu and X. Feng, Angew. Chem., Int. Ed., 2020, 59, 8052–8056 CrossRef CAS PubMed.
- M. N. Alam, S. R. Dash, A. Mukherjee, S. Pandole, U. K. Marelli, K. Vanka and P. Maity, Org. Lett., 2021, 23, 890–895 CrossRef CAS PubMed.
- I. Nakamura and M. Terada, Tetrahedron Lett., 2019, 60, 689–698 CrossRef CAS.
- K. Wlodarczyk, P. Borowski and M. Stankevic, Beilstein J. Org. Chem., 2020, 16, 88–105 CrossRef CAS PubMed.
- M. E. Gurskii, P. A. Belyakov, K. A. Lyssenko, A. L. Semenova and Y. N. Bubnov, Russ. Chem. Bull., 2014, 63, 480–486 CrossRef CAS.
- D. Wang, K. Jana and A. Studer, Org. Lett., 2021, 23, 5876–5879 CrossRef CAS PubMed.
- D. Wang, C. Muck-Lichtenfeld and A. Studer, J. Am. Chem. Soc., 2020, 142, 9119–9123 CrossRef CAS PubMed.
- B. K. Peters, K. X. Rodriguez, S. H. Reisberg, S. B. Beil, D. P. Hickey, Y. Kawamata, M. Collins, J. Starr, L. Chen, S. Udyavara, K. Klunder, T. J. Gorey, S. L. Anderson, M. Neurock, S. D. Minteer and P. S. Baran, Science, 2019, 363, 838–845 CrossRef CAS PubMed.
- A. Chatterjee and B. Konig, Angew. Chem., Int. Ed., 2019, 58, 14289–14294 CrossRef CAS PubMed.
- Y. Z. Cheng, X. L. Huang, W. H. Zhuang, Q. R. Zhao, X. Zhang, T. S. Mei and S. L. You, Angew. Chem., Int. Ed., 2020, 59, 18062–18067 CrossRef CAS PubMed.
- Y. Z. Cheng, Q. R. Zhao, X. Zhang and S. L. You, Angew. Chem., Int. Ed., 2019, 58, 18069–18074 CrossRef CAS PubMed.
- A. R. Flynn, K. A. McDaniel, M. E. Hughes, D. B. Vogt and N. T. Jui, J. Am. Chem. Soc., 2020, 142, 9163–9168 CrossRef CAS PubMed.
- M. J. James, J. L. Schwarz, F. Strieth-Kalthoff, B. Wibbeling and F. Glorius, J. Am. Chem. Soc., 2018, 140, 8624–8628 CrossRef CAS PubMed.
- E. H. Southgate, J. Pospech, J. Fu, D. R. Holycross and D. Sarlah, Nat. Chem., 2016, 8, 922–928 CrossRef CAS PubMed.
- T. M. Monos, R. C. McAtee and C. R. J. Stephenson, Science, 2018, 361, 1369–1373 CrossRef CAS PubMed.
- J. Shi, L. Li, C. Shan, Z. Chen, L. Dai, M. Tan, Y. Lan and Y. Li, J. Am. Chem. Soc., 2021, 143, 10530–10536 CrossRef CAS PubMed.
- R. C. McAtee, E. A. Noten and C. R. J. Stephenson, Nat. Commun., 2020, 11, 2528 CrossRef CAS PubMed.
- M. E. Cinar and T. Ozturk, Chem. Rev., 2015, 115, 3036–3140 CrossRef CAS PubMed.
- L. Duan, J. Qiao, Y. Sun and Y. Qiu, Adv. Mater., 2011, 23, 1137–1144 CrossRef CAS PubMed.
- F. Velazquez, M. Sannigrahi, F. Bennett, R. G. Lovey, A. Arasappan, S. Bogen, L. Nair, S. Venkatraman, M. Blackman, S. Hendrata, Y. Huang, R. Huelgas, P. Pinto, K. C. Cheng, X. Tong, A. T. McPhail and F. G. Njoroge, J. Med. Chem., 2010, 53, 3075–3085 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2115384. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc06760e |
| This journal is © The Royal Society of Chemistry 2022 |