
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Isothiourea-catalyzed formal enantioselective conjugate addition of benzophenone imines to β-fluorinated α,β-unsaturated esters†
Jerson E.
Lapetaje
,
Claire M.
Young
 ,
Chang
Shu
and
Andrew D.
Smith
*
,
Chang
Shu
and
Andrew D.
Smith
*
EaStCHEM, School of Chemistry, University of St Andrews, North Haugh, St. Andrews KY16 9ST, UK. E-mail: ads10@st-andrews.ac.uk
First published on 20th May 2022
Abstract
The isothiourea-catalyzed formal enantioselective conjugate addition of 2-hydroxybenzophenone imine derivatives to α,β-unsaturated para-nitrophenyl esters has been developed. Investigations of the scope and limitations of this procedure showed that β-electron withdrawing substituents within the α,β-unsaturated ester component are required for good product yield, giving rise to a range of β-imino ester and amide derivatives in moderate to good isolated yields with excellent enantioselectivity (20 examples, up to 81% yield and 97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 er).
:3 er).
The development of methods for the enantioselective synthesis of β-amino acid derivatives1a is of widespread importance due to the prevalence of this structural motif in natural products and medicinally relevant compounds.1 Among the synthetic methods that have been developed for the preparation of β-amino acid derivatives, arguably the most simple and elegant involves the asymmetric conjugate addition of an ammonia equivalent to an α,β-unsaturated carbonyl motif. As an example of this approach, the conjugate addition of enantiomerically pure lithium amide derivatives to α,β-unsaturated esters has been developed and exploited extensively by Davies and co-workers. Conjugate addition of lithium N-benzyl-N-α-methylbenzylamide to an α,β-unsaturated ester gives the corresponding β-amino ester with high diastereoselectivity (>95
:5 dr), with N-deprotection through hydrogenolysis giving the corresponding β-amino ester derivatives (Scheme 1a).2
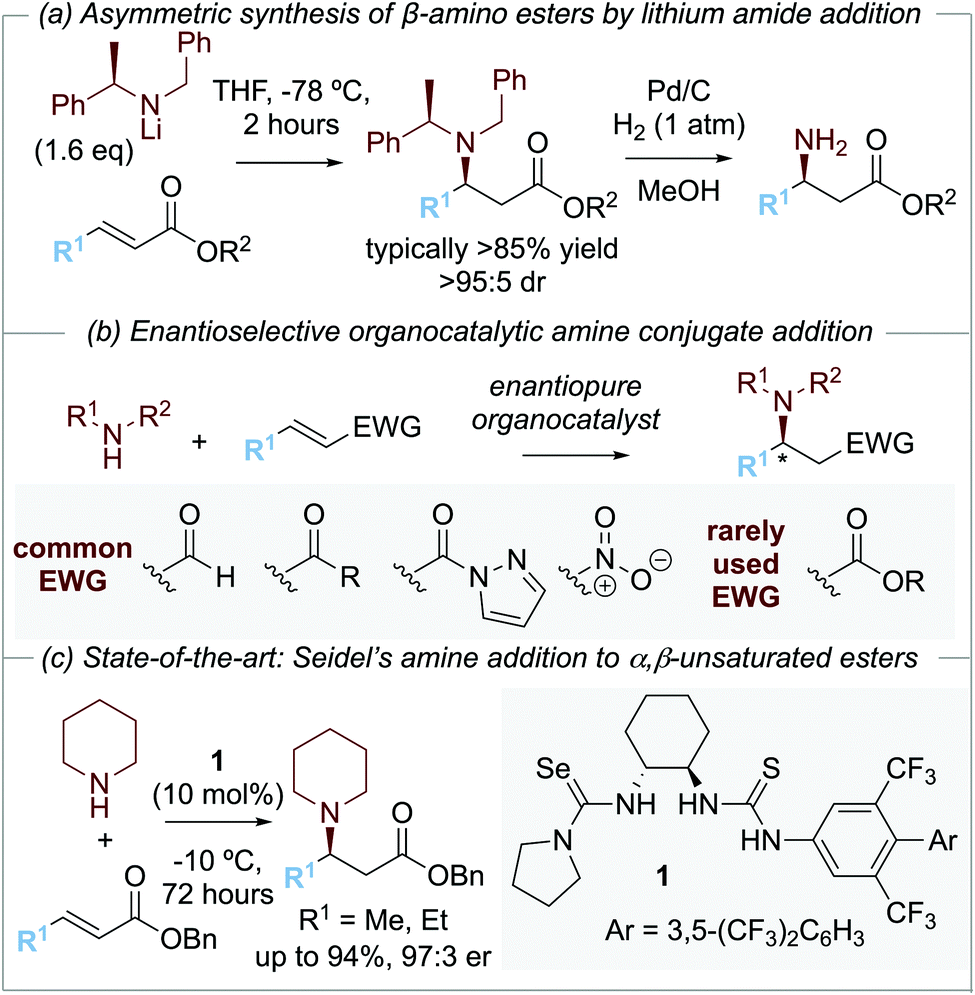 | ||
| Scheme 1 Synthesis of β-amino ester derivatives. | ||
Over the last two decades, several enantioselective organocatalytic approaches to amine conjugate addition have been introduced. To date, these successful approaches rely upon enals,3 enones,4N-acyl pyrazoles,5 and nitro-olefins6 as Michael acceptors, with the use of bifunctional thiourea4a,5b,7,8a–c,e or squaramide4,5c,8a,b,e organocatalysts, or Lewis basic pyrrolidines3,8 commonplace. Catalytic enantioselective amine conjugate additions to α,β-unsaturated esters are rare, reflecting the recognized recalcitrance of α,β-unsaturated esters as Michael acceptors (Scheme 1b). To date, the current state-of-the-art organocatalytic approach is represented by Seidel and co-workers’9 demonstration of the conjugate addition of cyclic secondary amines to β-alkyl-α,β-unsaturated benzyl esters using a selenourea-thiourea catalyst 1 (Scheme 1c). Although limited to β-alkyl substituted Michael acceptors, this impressive methodology was applicable to a range of cyclic amines and the kinetic resolution of (±)-cyclic 2-arylamines.
Our approach to enantioselective amine conjugate addition focused upon the use of imines as nucleophiles. The conjugate addition of (diphenylmethylene)amine to α,β-unsaturated esters, nitriles and ketones in racemic form has been demonstrated by de Meijere et al. MeOH was optimal as a solvent and a basic additive (such as NEt3) led to effective product formation (Scheme 2a).10 In 2018, Alemán and co-workers successfully demonstrated an enantioselective aza-Michael addition of nucleophilic imines to enals using secondary amine catalyst 2 (Scheme 2b).11 Trapping of the resultant β-imino aldehydes with a phosphorane gave the corresponding δ-imino esters in good yield and enantioselectivity. Notably, 2-hydroxybenzophenone imines showed increased reactivity and enantioselectivity compared with the parent benzophenone imine, attributed to an increase in acidity of the imine proton caused by intramolecular hydrogen bonding.12,13 In previous work, we and others have demonstrated a range of enantioselective Michael-addition processes of in situ generated α,β-unsaturated acyl ammonium species.14,15 Building on these precedents, we report herein the formal isothiourea-catalyzed enantioselective addition of 2-hydroxybenzophenone imines to β-fluorinated α,β-unsaturated para-nitrophenyl esters via an α,β-unsaturated acyl ammonium intermediate, giving products in up to 98:2 er (Scheme 2c).
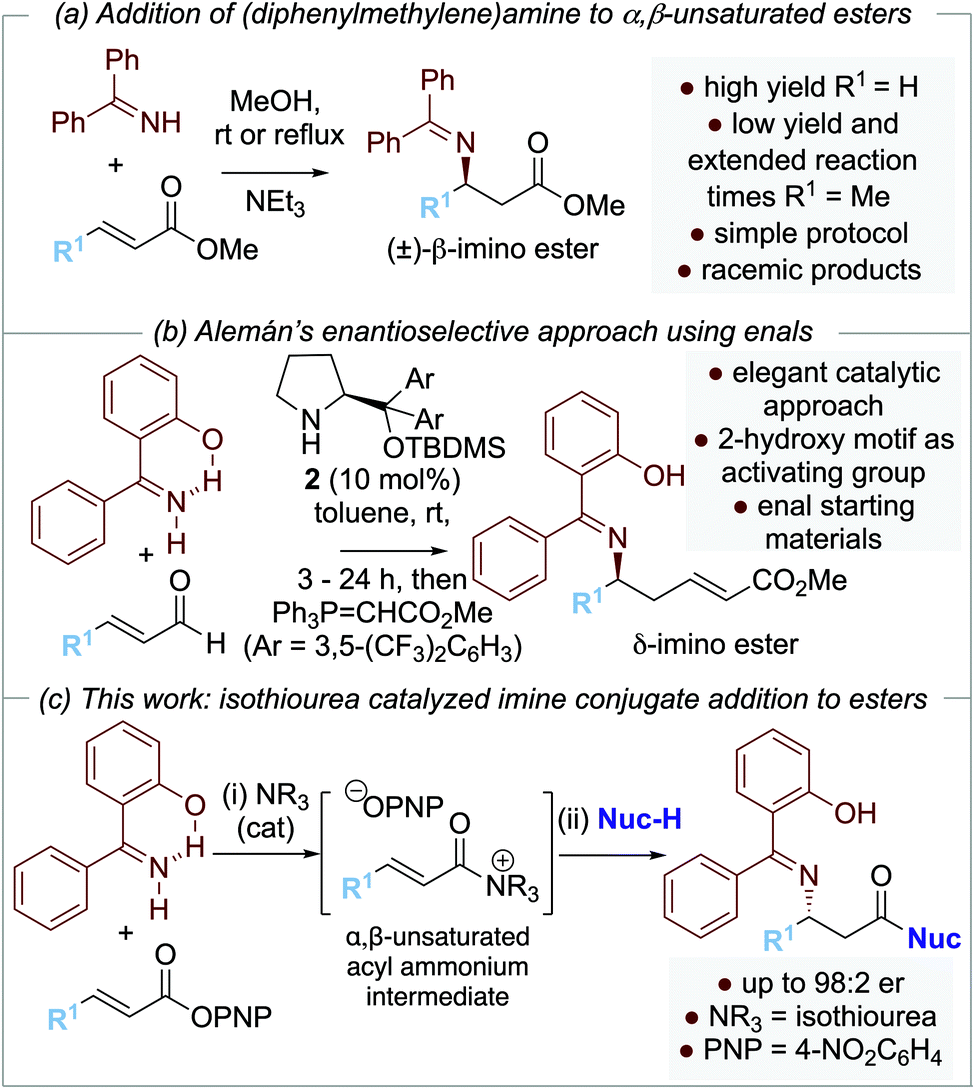 | ||
| Scheme 2 Previous imine conjugate additions and this work. | ||
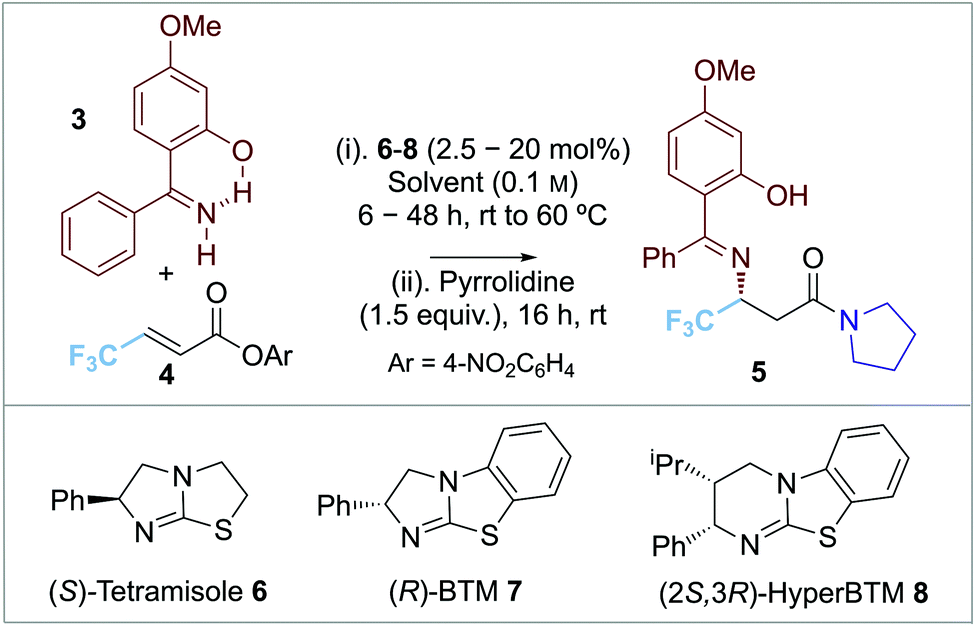
Preliminary investigations used β-CF3-substituted α,β-unsaturated para-nitrophenyl ester 4 (1.0 equiv.) in toluene as standard. Given the moderate reactivity of α,β-unsaturated acyl ammonium ions, imine 3 (2.0 equiv.) bearing an electron donor 4-OMe-substituent was postulated to enhance nucleophilicity (Table 1). Attempted isolation of the para-nitrophenyl ester product led to low and irreproducible product yields, so addition of pyrrolidine to give the isolable amide 5 was adopted. Screening of isothiourea catalysts 6–8 (10 mol%) at 1:2 substrate ratio of ester 4: imine 3 (entries 1–3) showed that tetramisole 6 and BTM 7 gave promising product yield (∼50%) whereas HyperBTM 8 showed poor catalytic activity (<10% yield). Excellent enantioselectivity (96:4 er) was observed using BTM 7. Altering the reaction stoichiometry (entries 4 and 5) led to reduced product yield. A detrimental effect on product enantioselectivity (91:9 er) was observed when the reaction temperature was increased to 40 °C or 60 °C (entries 6 and 7). Lowering the catalyst loading showed a significant decrease in product yield and enantioselectivity (entries 8 and 9), while using 20 mol% BTM 7 gave increased yield (71% yield, 96:4 er, entry 10). Screening of a alternative solvents gave high product enantioselectivity but reduced yields (entries 11–13). Further optimisation probed the effectiveness of alternative electron-deficient aryl esters. Comparison of para-nitrophenyl with 2,4,6-trichlorophenyl, pentafluorophenyl, and 3,5-bis(trifluoromethyl)phenyl esters (entries 14–16) showed that excellent enantioselectivities were observed in each case (up to 98:2 er), with the para-nitrophenyl ester leading to the best product yield (71%).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | Temp. (°C) | Solvent | 3:4 |
Yielda (%) | erb |
|
a Using 1H NMR spectroscopic analysis and 1,3,5-trimethoxybenzene as internal standard.
b Ratio of (R):(S) enantiomers determined by HPLC analysis on a chiral stationary phase.
c Ar = 4-NO2C6H4.
d Isolated yield.
e Ar = 2,4,6-Cl3C6H2.
f Ar = C6F5.
g Ar = 3,5-(CF3)2C6H3.
|
||||||
| 1c | 6 (10) | rt | Toluene | 1:2 |
50 | 12:88 |
| 2c | 7 (10) | rt | Toluene | 1:2 |
54 | 96:4 |
| 3c | 8 (10) | rt | Toluene | 1:2 |
<10 | 68:32 |
| 4c | 7(10) | rt | Toluene | 1:1.5 |
42 | 95:5 |
| 5c | 7 (10) | rt | Toluene | 1.5:1 |
38 | 97:3 |
| 6c | 7 (10) | 40 | Toluene | 1:2 |
52 | 94:6 |
| 7c | 7 (10) | 60 | Toluene | 1:2 |
47 | 91:9 |
| 8c | 7 (2.5) | rt | Toluene | 1:2 |
<10 | 91:9 |
| 9c | 7 (5.0) | rt | Toluene | 1:2 |
18 | 96:4 |
| 10c | 7 (20) | rt | Toluene | 1:2 |
71d | 96:4 |
| 11c | 7 (20) | rt | THF | 1:2 |
31 | 96:4 |
| 12c | 7 (20) | rt | Et2O | 1:2 |
30 | 96:4 |
| 13c | 7 (20) | rt | CH2Cl2 | 1:2 |
37 | 96:4 |
| 14e | 7 (20) | rt | Toluene | 1:2 |
31 | 98:2 |
| 15f | 7 (20) | rt | Toluene | 1:2 |
42 | 96:4 |
| 16g | 7 (20) | rt | Toluene | 1:2 |
36 | 96:4 |
The scope and limitations of the developed process was explored through variation of the nucleophilic imine reaction component (Fig. 1). Variation of the electronic bias of the 4-aryl substituent within the imine component showed that decreased product yield was observed upon changing from an electron-donating 4-MeO- (5, 70% yield) to 4-Me (9, 49% yield), 4-H (11, 36% yield) and electron-withdrawing 4-Br substituent (10, 24% yield) all with >96:4 er. This is consistent with increasing electron density within the imine component leading to increased product yield. Interestingly, comparing the yield and er of products 11 and 12 indicates that the 2-hydroxy-substituent within the imine is essential for high product er, but does not affect product yield. The incorporation of an additional electron-donating 4-MeO substituent led to product 13 in reduced yield but maintained high product er. Variation of the β-substituent within the α,β-unsaturated ester indicated that the incorporation of polyhalogenated or ester electron-withdrawing groups was necessary for reactivity as alkyl, aryl, ketone and amide substituted acceptors gave no significant product formation. For example, the introduction of halogenated (CF2H) and polyhalogenated substituents (CF2Cl, CF2Br, and C2F5) led to products 14–17 in up to excellent yields with high enantioselectivity (40% to 81%; >96:4 er), while the incorporation of ester substituents gave 18–19 in poor 20% product yield in up to 96:4 er. Variation of the post catalysis nucleophilic component (Nuc-H) to incorporate alcohols as well as cyclic secondary and acyclic primary amines gave a range of ester and amide products 20–24 in good yield (42% to 64%) and excellent enantioselectivity (≥96:4 er).
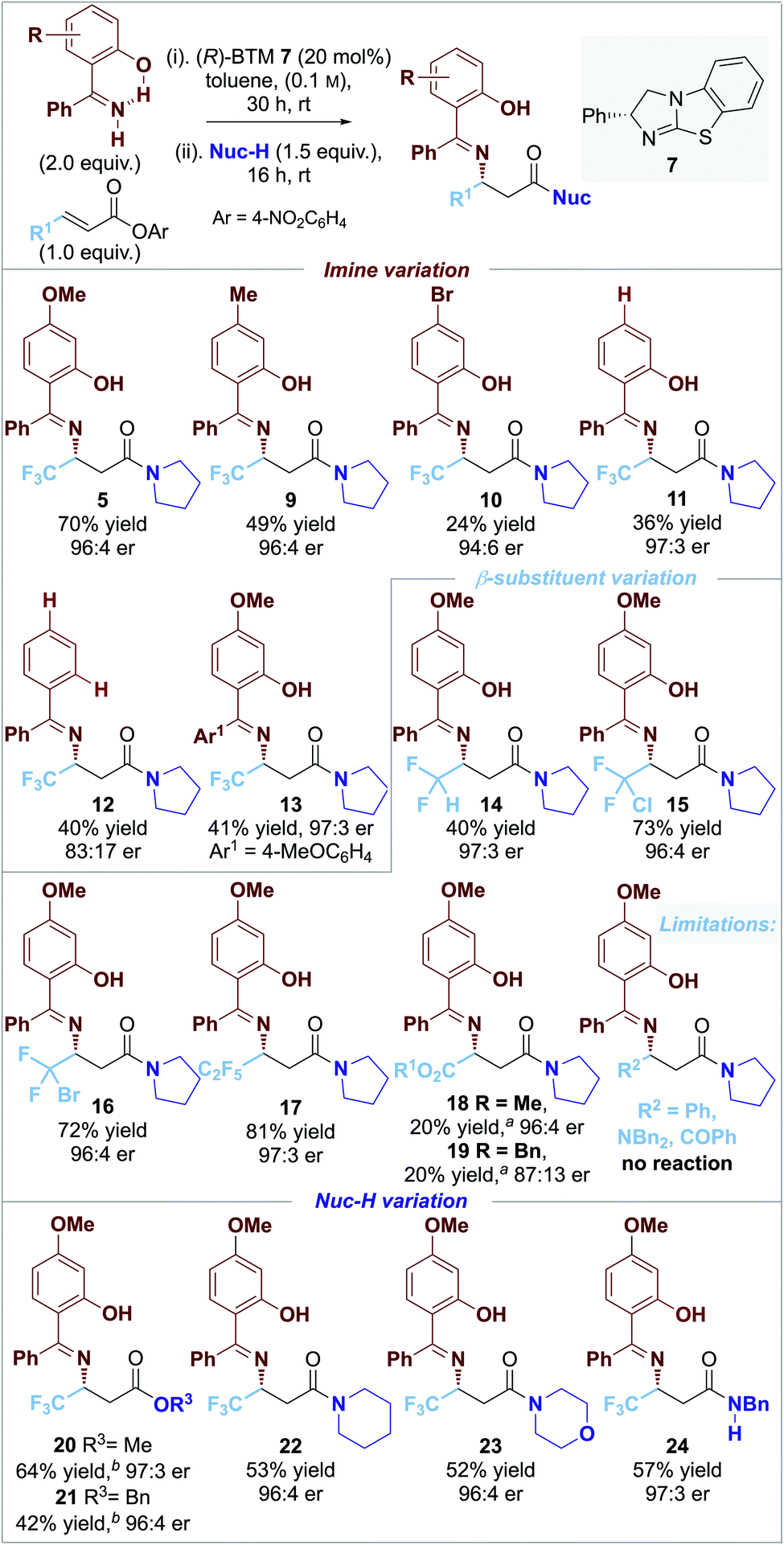 | ||
| Fig. 1 0.10 mmol scale. Isolated product yield; er determined by HPLC analysis on a chiral stationary phase; [a] 40 °C for step i; [b] DMAP 20 mol% in step ii. | ||
To further demonstrate the synthetic utility of this transformation, it was applied to the gram-scale synthesis of product 5 with consistent yield and enantioselectivity (67%, 96:4 er, Scheme 3). Hydrolysis gave the free β-amino amide product 26 in high yield and enantioselectivity (95%, 96:4 er).16
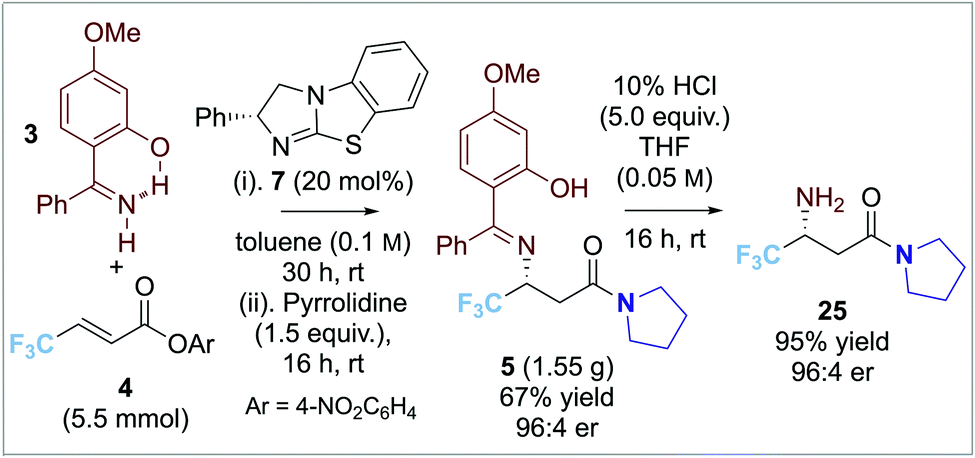 | ||
| Scheme 3 Gram scale synthesis of product 5. | ||
A proposed mechanism of this transformation is shown in Scheme 4. Reversible acylation of the isothiourea with the α,β-unsaturated ester 1a generates the key α,β-unsaturated acyl isothiouronium ion pair 26.
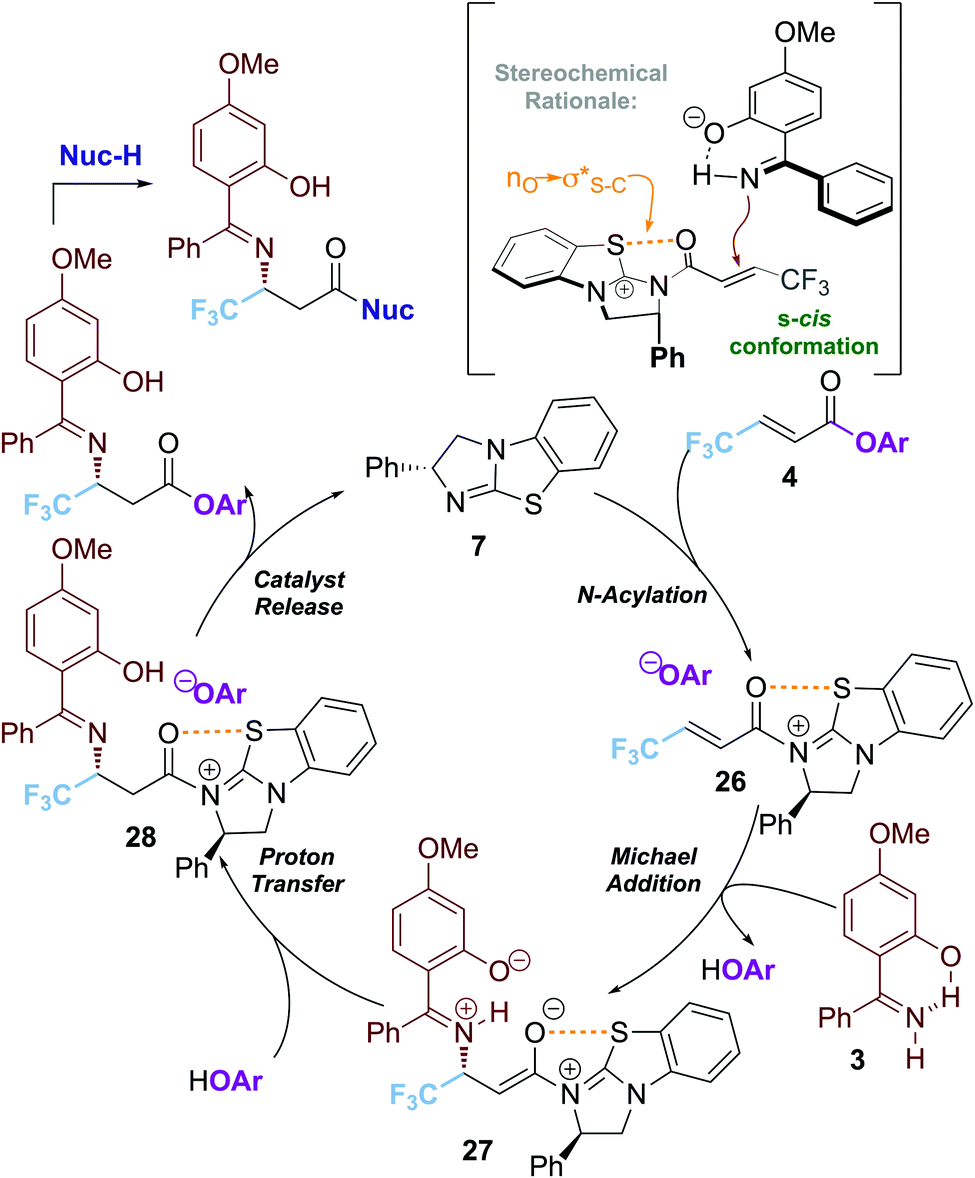 | ||
| Scheme 4 Proposed reaction mechanism. | ||
An intramolecular chalcogen 1,5-S⋯O interaction (nO → σ*S–C)17 provides a plausible stabilising effect and conformational lock. Hydrogen bonding between the 2-hydroxy-substituent and the imine N serves to conformationally restrict this functionality and facilitate deprotonation.11–13 Subsequent conjugate addition to the s-cis conformation of the α,β-unsaturated acyl isothiouronium 26anti- to the stereodirecting phenyl substituent of the isothiourea catalyst generates the ammonium enolate intermediate 27. Proton transfer generates the β-imino acyl isothiouronium intermediate 28, with catalyst turnover facilitated by the aryloxide counterion to form the product and release the isothiourea catalyst BTM 7.18
In summary, enantioselective organocatalytic conjugate addition of 2-hydroxybenzophenone imines to α,β-unsaturated esters using the isothiourea BTM as an organocatalyst gives enantioenriched β-imino amides in modest to good yield (20–81%) and excellent enantioselectivity (typically >95:5 er).19
The DOST-SEI and DOST-Foreign Graduate Scholarship Program of the Philippines are thanked for a PhD scholarship (J. E. L) and the Royal Society for a Newton Fellowship (C. S.).
Conflicts of interest
There are no conflicts of interests to declare.Notes and references
- For selected discussions of β-amino acids: (a) G. Cardillo and C. Tomasini, Chem. Soc. Rev., 1996, 25, 117–128 RSC; (b) C. Shih, L. S. Gossett, J. M. Gruber, C. S. Grossman, S. L. Andis, R. M. Schultz, J. F. Worzalla, T. H. Corbett and J. T. Metz, Bioorg. Med. Chem. Lett., 1999, 9, 69–74 CrossRef CAS PubMed; (c) W. J. Hoekstra and B. L. Poulter, Curr. Med. Chem., 1998, 5, 195–204 CrossRef CAS PubMed; (d) S. Kosemura, T. Ogawa and K. Totsuka, Tetrahedron Lett., 1993, 34, 1291–1294 CrossRef CAS.
- (a) S. G. Davies and O. Ichihara, Tetrahedron: Asymmetry, 1991, 2, 183–186 CrossRef CAS for reviews see; (b) S. G. Davies, A. D. Smith and P. D. Price, Tetrahedron: Asymmetry, 2005, 17, 2883–2891 Search PubMed; (c) S. G. Davies, A. M. Fletcher, P. M. Roberts and J. E. Thomson, Tetrahedron: Asymmetry, 2012, 23, 1111–1153 CrossRef CAS.
- Selected examples for aza-Michael additions to enals: (a) Y. K. Chen, M. Yoshida and D. W.-C. MacMillan, J. Am. Chem. Soc., 2006, 128, 9328–9329 CrossRef CAS PubMed; (b) J. Vesely, I. Ibrahem, R. Rios, G.-L. Zhao, Y. Xu and A. Córdova, Tetrahedron Lett., 2007, 48, 2193–2198 CrossRef CAS; (c) H. Jiang, J. B. Nielsen, M. Nielsen and K. A. Jørgensen, Chem. – Eur. J., 2007, 13, 9068–9075 CrossRef CAS PubMed; (d) P. Dinér, M. Nielsen, M. Marigo and K. A. Jørgensen, Angew. Chem., Int. Ed., 2007, 46, 1983–1987 CrossRef PubMed.
- Selected examples for aza-Michael additions to enones: (a) D. Pettersen, F. Piana, L. Bernardi, F. Fini, M. Fochi, V. Sgarzani and A. Ricci, Tetrahedron Lett., 2007, 48, 7805–7808 CrossRef CAS; (b) X. Lu and L. Deng, Angew. Chem., Int. Ed., 2008, 120, 7824–7827 CrossRef; (c) S. Ma, L. Wu, M. Liu, X. Xu, Y. Huang and Y. Wang, RSC Adv., 2013, 3, 11498–11501 RSC.
- Selected examples for aza-Michael additions to N-acylpyrazoles: (a) M. P. Sibi, J. J. Shay, M. Liu and C. P. Jasperse, J. Am. Chem. Soc., 1998, 120, 6615–6616 CrossRef CAS; (b) L. Simón and J. M. Goodman, Org. Biomol. Chem., 2009, 7, 483–487 RSC; (c) M. Sánchez-Roselló, C. Mulet, M. Guerola, C. del Pozo and S. Fustero, Chem. – Eur. J., 2014, 20, 15697–15701 CrossRef PubMed.
- Selected examples for aza-Michael additions to nitroolefins: (a) L. Lykke, D. Monge, M. Nielsen and K. A. Jørgensen, Chem. – Eur. J., 2010, 16, 13330–13334 CrossRef CAS PubMed; (b) S. Ma, L. Wu, M. Liu, Y. Huang and Y. Wang, Tetrahedron, 2013, 69, 2613–2618 CrossRef CAS; (c) B. L. Zhao, Y. Lin, H. H. Yan and D. M. Du, Org. Biomol. Chem., 2015, 13, 11351–11361 RSC; (d) M. Moczulski, P. Drelich and L. Albrecht, Org. Biomol. Chem., 2018, 16, 376–379 RSC.
- (a) Y. Sohtome, Y. Hashimoto and K. Nagasawa, Adv. Synth. Catal., 2005, 347, 1643–1648 CrossRef CAS; (b) J. Wang, L. Zu, H. Li, H. Xie and W. Wang, Synthesis, 2007, 2576–2580 CrossRef CAS.
- Reviews on organocatalytic aza-Michael addition reactions highlighting the use of thiourea, squaramide, and Lewis base pyrrolidine catalysts: (a) D. Enders, C. Wang and J. X. Liebich, Chem. – Eur. J., 2009, 15, 11058–11076 CrossRef CAS PubMed; (b) J. Wang, P. Li, P. Y. Choy, A. S. C. Chan and F. Y. Kwong, ChemCatChem, 2012, 4, 917–925 CrossRef CAS; (c) C. Bhanja, S. Jena, S. Nayak and S. Mohapatra, Beilstein J. Org. Chem., 2012, 8, 1668–1694 CrossRef CAS PubMed; (d) S. D. Pasuparthy and B. Maiti, ChemistrySelect, 2022, 7, e202104261 CrossRef CAS; (e) Y. X. Song and D. M. Du, Adv. Synth. Catal., 2021, 363, 4667–4694 CrossRef CAS.
- Y. Lin, W. J. Hirschi, A. Kunadia, A. Paul, I. Ghiviriga, K. A. Abboud, R. W. Karugu, M. J. Vetticatt, J. S. Hirschi and D. Seidel, J. Am. Chem. Soc., 2020, 142, 5627–5635 CrossRef CAS PubMed.
- (a) L. Wessjohann, G. Mcgaffin and A. de Meijere, Synthesis, 1989, 359–363 CrossRef CAS; (b) T. Meiresonne, S. Mangelinckx and N. De Kimpe, Org. Biomol. Chem., 2011, 9, 7085–7091 RSC.
- H. Choubane, A. F. Garrido-Castro, C. Alvarado, A. Martín-Somer, A. Guerrero-Corella, M. Daaou, S. Díaz-Tendero, M. Carmen Maestro, A. Fraile and J. Alemán, Chem. Commun., 2018, 54, 3399–3402 RSC.
- (a) A. Guerrero-Corella, M. A. Valle-Amores, A. Fraile and J. Alemán, Adv. Synth. Catal., 2021, 363, 3845–3851 CrossRef CAS; (b) A. Guerrero-Corella, F. Esteban, M. Iniesta, A. Martín-Somer, M. Parra, S. Díaz-Tendero, A. Fraile and J. Alemán, Angew. Chem., Int. Ed., 2018, 57, 5350–5354 CrossRef CAS PubMed.
- For a review: A. Guerrero-Corella, A. Fraile and J. Alemán, ACS Org. Inorg. Au, 2022 DOI:10.1021/acsorginorgau.1c00053.
- (a) A. Matviitsuk, M. D. Greenhalgh, D. J.-B. Antúnez, A. M.-Z. Slawin and A. D. Smith, Angew. Chem., Int. Ed., 2017, 56, 12282–12287 CrossRef CAS PubMed; (b) C. Shu, H. Liu, A. M.-Z. Slawin, C. Carpenter-Warren and A. D. Smith, Chem. Sci., 2020, 11, 241–247 RSC; (c) M. D. Greenhalgh, S. Qu, A. M.-Z. Slawin and A. D. Smith, Chem. Sci., 2018, 9, 4909–4918 RSC.
- For a review see: J. Bitai, M. Westwood and A. D. Smith, Org. Biomol. Chem., 2021, 19, 2366–2384 RSC.
- The absolute configuration was confirmed by hydrolysis of 25 to obtain the free β-amino acid (exp: [α]20D = +17.9; lit: [α]20D = +24.4): N. Shibata, T. Nishimine, N. Shibata, E. Tokunaga, K. Kawada, T. Kagawa, J. L. Aceña, A. E. Sorochinsky and V. A. Soloshonok, Org. Biomol. Chem., 2014, 12, 1454–1462 RSC.
- S⋯O interactions in isothiourea catalysis: (a) V. B. Birman, X. Li and Z. Han, Org. Lett., 2007, 9, 37–40 CrossRef CAS PubMed; (b) P. Liu, X. Yang, V. B. Birman and K. N. Houk, Org. Lett., 2012, 14, 3288–3291 CrossRef CAS PubMed; (c) M. E. Abbasov, B. M. Hudson, D. J. Tantillo and D. Romo, J. Am. Chem. Soc., 2014, 136, 4492–4495 CrossRef CAS PubMed; (d) E. R.-T. Robinson, D. M. Walden, C. Fallan, M. D. Greenhalgh, P. H.-Y. Cheong and A. D. Smith, Chem. Sci., 2016, 7, 6919–6927 RSC; (e) M. D. Greenhalgh, S. M. Smith, D. M. Walden, J. E. Taylor, Z. Brice, E. R.-T. Robinson, C. Fallan, D. B. Cordes, A. M.-Z. Slawin, H. C. Richardson, M. A. Grove, P. H.-Y. Cheong and A. D. Smith, Angew. Chem., Int. Ed., 2018, 57, 3200–3206 CrossRef CAS PubMed; (f) C. M. Young, A. Elmi, D. J. Pascoe, R. K. Morris, C. McLaughlin, A. M. Woods, A. B. Frost, A. de la Houpliere, K. B. Ling, T. K. Smith, A. M.-Z. Slawin, P. H. Willoughby, S. L. Cockroft and A. D. Smith, Angew. Chem., Int. Ed., 2020, 59, 3705–3710 CrossRef CAS PubMed In medicinal chemistry: ; (g) B. R. Beno, K.-S. Yeung, M. D. Bartberger, L. D. Pennington and N. A. Meanwell, J. Med. Chem., 2015, 58, 4383–4438 CrossRef CAS PubMed.
- For discussion on aryloxide facilitated catalyst turnover: (a) A. Matviitsuk, M. D. Greenhalgh, D. J.-B. Antúnez, A. M.-Z. Slawin and A. D. Smith, Angew. Chem., Int. Ed., 2017, 56, 12282–12287 CrossRef CAS PubMed; (b) W. C. Hartley, T. J.-C. O’Riordan and A. D. Smith, Synthesis, 2017, 49, 3303–3310 CrossRef CAS; (c) T. H. West, D. S.-B. Daniels, A. M.-Z. Slawin and A. D. Smith, J. Am. Chem. Soc., 2014, 136, 4476–4479 CrossRef CAS PubMed.
- Research data supporting publication can be accessed at https://doi.org/10.17630/fb29b2d4-41d4-4143-a19b-5f59cb71447a.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc01936a |
| This journal is © The Royal Society of Chemistry 2022 |