
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantiopure nanohoops through racemic resolution of diketo[n]CPPs by chiral derivatization as precursors to DBP[n]CPPs†
Daniel
Wassy
 a,
Mathias
Hermann
a,
Jan S.
Wössner
a,
Lucas
Frédéric
b,
Grégory
Pieters
b and
Birgit
Esser
*acd
a,
Mathias
Hermann
a,
Jan S.
Wössner
a,
Lucas
Frédéric
b,
Grégory
Pieters
b and
Birgit
Esser
*acd
aInstitute for Organic Chemistry, University of Freiburg, Albertstraße 21, 79104 Freiburg, Germany. E-mail: besser@oc.uni-freiburg.de; Web: https://www.esser-lab.uni-freiburg.de
bUniversité Paris-Saclay, CEA, INRAE, Département Médicaments et Technologies pour la Santé (DMTS), SCBM, 91191 Gif-sur-Yvette, France
cFreiburg Materials Research Center, University of Freiburg, Stefan-Meier-Str. 21, 79104 Freiburg, Germany
dFreiburg Center for Interactive Materials and Bioinspired Technologies, University of Freiburg, Georges-Köhler-Allee 105, 79110 Freiburg, Germany
First published on 22nd June 2021
Abstract
Strained conjugated nanohoops are attractive synthetic targets due to the bending of their π-system, which leads to intriguing optoelectronic properties, among others. By incorporating non-mirror-symmetric aromatic panels, chiral nanohoops can be obtained. We herein present a strategy to enantiopure nanohoops by racemic resolution through chiral derivatization of diketone-embedded hoops. The resulting diketo[n]CPPs (n = 6, 7) contain two stereogenic carbon atoms each and possess high fluorescence quantum yields paired with circularly polarized luminescence. These are versatile precursors to chiral dibenzo[a,e]pentalene-based nanohoops DBP[n]CPPs with antiaromatic character and ambipolar electrochemical behavior. Due to their strained structures the DBP[n]CPPs do not racemize at room temperature, which is supported by high calculated isomerization barriers. X-ray crystallographic investigations on the DBP[n]CPPs and their precursors as well as DFT calculations provide insight into the build-up of strain energy during the synthetic transformations.
Introduction
Hoop-shaped π-systems have fascinated chemists since the 1950s.1–3 The effect of curvature on the π-systems has always been of prime interest, and the design of topologically unique carbon nanostructures is an on-going challenge.4,5 Since the discovery of carbon nanotubes conjugated hoops have also been considered as molecular segments of these.6,7 Their inwardly oriented π-orbitals make them attractive for supramolecular chemistry,8,9 and based on their optoelectronic properties emerging applications have been identified.10,11 Their synthesis, however, has always remained a challenge. Since 2008, several synthetic strategies to [n]cycloparaphenylenes ([n]CPPs) have been established,12–14 some of which rely on using naturally bent precursors to oligo(paraphenylene) units. These strategies have enabled access to a large variety of conjugated nanohoops11,15,16 and allowed to expand the structural variety to hoops incorporating polycyclic aromatic hydrocarbons (PAHs).15 Axially chiral nanohoops can be obtained by unsymmetrically incorporating PAHs, if their rotation through the hoop is hindered. This has been reported for a number of different PAHs.17–22 Another strategy to obtain chiral nanohoops is by topological design.23–25In most of these cases the nanohoops were synthesized as racemic or diastereomeric mixtures, and pure stereoisomers were separated by chiral HPLC.17–22 Exceptions are the use of a chiral catalyst to obtain an enantiomeric excess of a cyclophenylene26 and a rhodium-catalyzed diastereo- and enantioselective synthesis of CPPs,27 both recently reported by Tanaka and coworkers, and the stereoselective synthesis of chiral nanohoops using enantiomerically pure bent precursors reported by us.28 Preparative chiral HPLC is costly and time-intensive, since multiple runs or a recycling mode using expensive chiral columns are required, and hence only allows to isolate small quantities of enantiopure hoops. Racemic resolution by preparative synthetic chemistry, on the other hand, can easily be upscaled. However, to the best of our knowledge there is no report of a racemic resolution of nanohoops using chiral derivatization. We herein report such a strategy, using precursors to DBP[n]CPPs (1 and 2 in Fig. 1), namely diketo[n]CPPs (3 and 4 in Fig. 1). The carbonyl groups in these hoops offer a convenient handle for attachment of a chiral auxiliary. We demonstrate this by transformation of the smaller diketo[6]CPP 3 into its chiral sulfoximine adducts, separation of the resulting diastereomers using conventional column chromatography, and simple thermal removal of the chiral auxiliary to furnish (S,S)- and (R,R)-3. This process can easily be upscaled and adapted to other hoop sizes n to provide access to enantiopure chiral diketo[n]CPPs. (S,S)- and (R,R)-3 displayed circularly polarized absorption and emission properties and the derived DBP[6]CPPs (M)-1 and (P)-1 circularly polarized absorption. We further investigated (racemic) strained DBP[n]CPPs 1 and 2 as well as diketo[n]CPPs 3 and 4 regarding their optoelectronic and structural properties, strain and antiaromatic character.
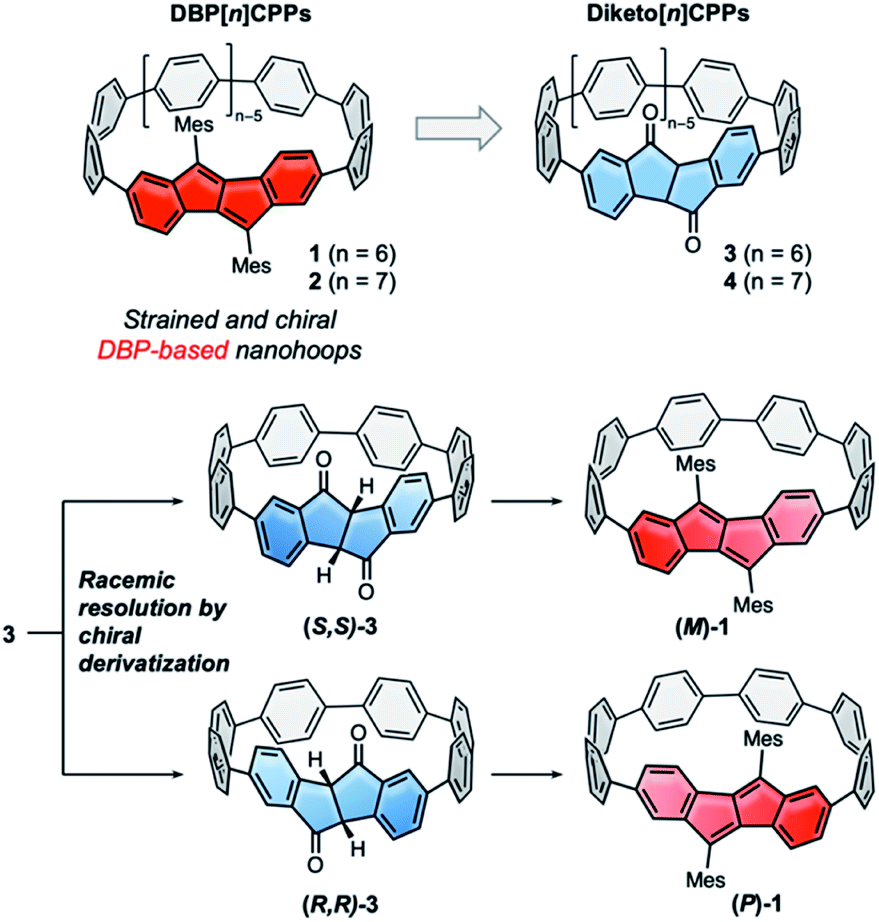 | ||
| Fig. 1 DBP[n]CPPs 1 and 2 as strained and chiral dibenzo[a,e]pentalene-based hoops, which can be accessed from diketo[n]CPPs 3 and 4. Their racemic resolution by chiral derivatization of the carbonyl groups provides access to enantiopure nanohoops. | ||
Results and discussion
Synthesis
We initiated this research by synthesizing diketo[n]CPPs 3 and 4 as well as transforming these into racemic DBP[n]CPPs 1 and 2. Our synthetic route combines the strategies introduced by Itami13 or Jasti, Bertozzi12 by using L-shaped corner units a or b as bent precursors to terphenyl groups with the bent cyclic diketone units as precursors to a DBP unit29 (Scheme 1A). Both syntheses commenced from bis-boronic ester 5 (for molecular structure in the solid state see ESI†), which was synthesized in five steps from 2-(4-bromophenyl)acetic acid (see ref. 29 and ESI†). For the smaller diketo[6]CPP 3 we tested two different routes. Using Itami's corner unit a,13 Suzuki–Miyaura coupling of bis-boronic ester 5 with 6a afforded U-shaped cyclization precursor 7a in 55% yield. Ni-mediated intramolecular cyclization furnished cycle 8a in 40% yield, which was aromatized under acidic/oxidizing conditions to yield 3. In the other route we made use of Jasti's corner unit 6b,12 which we attached to 5via Suzuki–Miyaura coupling to yield cyclization precursor 7b in 59% yield. Its intramolecular cyclization mediated by Ni proceeded in 38% yield to furnish 8b. The last aromatization step under reductive conditions led to 3 in 75% yield. Comparing all synthetic and purification steps, the latter route to diketo[6]CPP 3 was easier to reproduce in larger quantities. The structure of cyclic intermediate 8a in the solid state was resolved by X-ray diffraction (see below).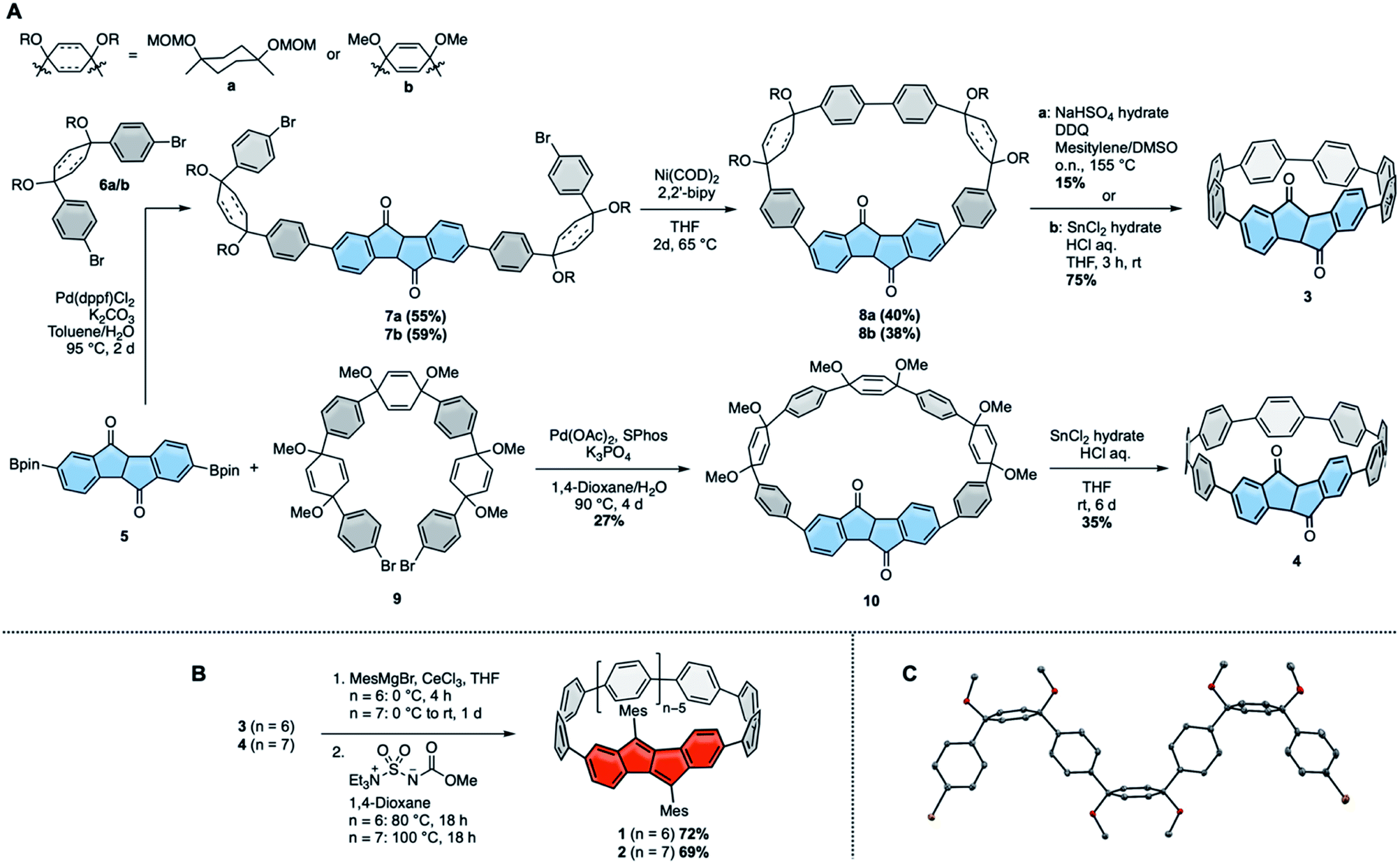 | ||
| Scheme 1 Synthesis of diketo[n]CPPs 3 and 4 (A) and DBP[n]CPPs 1 and 2 (B) (5 was used as a racemic mixture, hence all derived compounds are racemic. For simplicity, a single enantiomer is shown for 1–4); (C) molecular structure of 9 in the solid state (displacement ellipsoids are shown at the 50% probability level; hydrogen atoms are omitted for clarity). | ||
For the larger diketo[7]CPP 4 we made use of Du's C-shaped synthon 9 for a [7]paraphenylene ([7]PP) linker, containing three of Jasti's corner units.30 We optimized and slightly changed the synthetic route to 9, including a full characterization of each intermediate, so that its synthesis can now easily be reproduced on large scale (see ESI†). Crystals suitable for X-ray diffraction were obtained by slow evaporation of an ethyl acetate solution. The structure is shown in Scheme 1C. Suzuki–Miyaura coupling of bis-boronic ester 5 with C-shaped 9 furnished cycle 10 in 27% yield. Its aromatization under reductive conditions led to diketo[7]CPP 4 in 35% yield.
In the next step both diketo[n]CPPs 3 and 4 were transformed into DBP[n]CPPs 1 and 2 using a procedure initially established for strained DBP-phanes (Scheme 1B).29 We chose mesityl substituents to the DBP units to provide steric protection in case of an increased reactivity of the DBP core through bending and conjugation. Cerium trichloride-mediated31 Grignard addition of mesityl magnesium bromide was followed by two-fold water elimination with Burgess's reagent as mild dehydration method, which provided 1 and 2 in high yields of 72% and 69% over these two steps.
Structural properties
The structures of diketo[n]CPPs 3 and 4 as well as DBP[n]CPPs 1 and 2 were unambiguously determined using one- and two-dimensional NMR spectroscopy, mass spectrometry, X-ray crystallography (for 1–3) as well as optical methods, supported by DFT calculations. In the NMR spectra of DBP[n]CPPs 1 and 2 the para-phenylene (PP) protons resonate between 7.35–7.6 ppm, while the resonances of the DBP and aromatic mesityl protons appear between 6.4–7.1 ppm (Fig. 2). Due to the steric hindrance imposed by their ortho-methyl groups, the mesityl groups do not rotate around the CDBP–CMes bond, which leads to a splitting of the proton signals of the ortho-methyl groups in both hoops. The barrier for rotation amounts to ca. 18.9 kcal mol−1, as determined by VT NMR spectroscopy for a bis-ortho-tolyl-DBP (see ESI Section 9†). Interestingly, the methyl groups pointing inside the nanohoops are high-field shifted to 1.23 and 1.30 ppm for 1 and 2, respectively. We had observed an even stronger high-field shift in more strained DBP-phanes before.29 The methyl protons must experience a shielding effect through the diamagnetic ring current of the somewhat aromatic six-membered rings of the DBP units. This apparently outperforms a potential de-shielding effect from the paramagnetic ring currents of the antiaromatic five-membered DBP rings and leads in sum to a high-field shift.32 All other protons experience only a small shift with increasing strain from 2 to 1.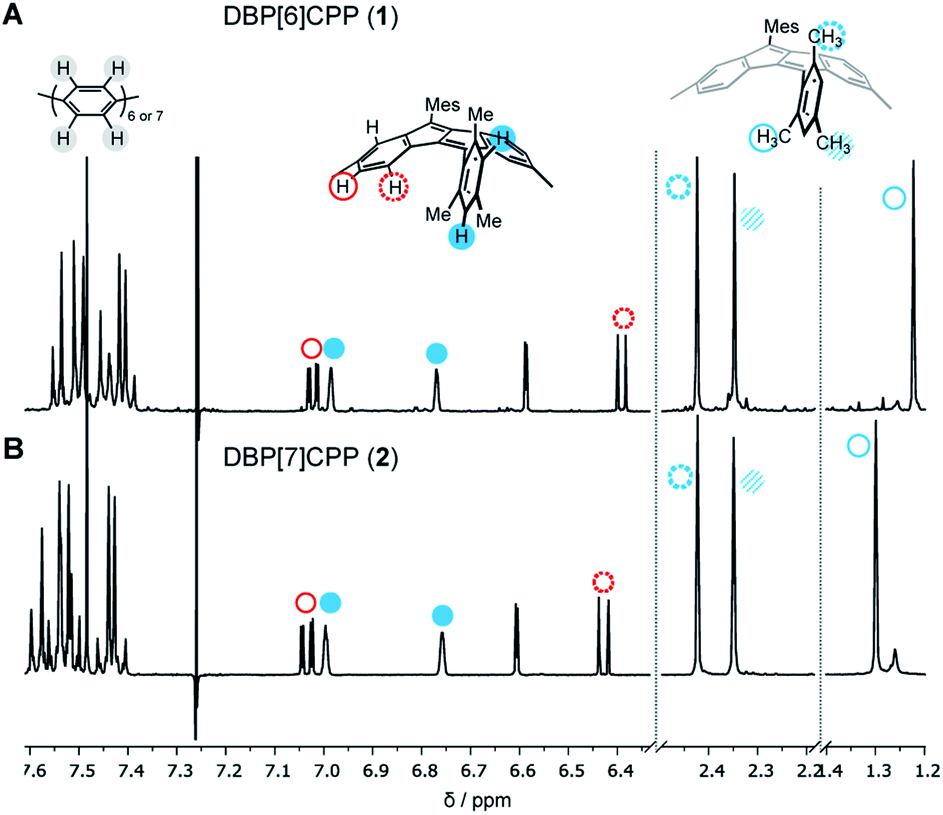 | ||
| Fig. 2 Selected regions of the 1H NMR spectra of DBP[n]CPPs 1 and 2 (CDCl3, 500 MHz, 300 K). | ||
Crystals suitable for X-ray diffraction were obtained by vapor diffusion of n-pentane into a dichloroethane solution for DBP[6]CPP 1 and by vapor diffusion of n-pentane into a CDCl3 solution or vapor diffusion of isopropanol into a chlorobenzene solution for DBP[7]CPP 2, which allowed their structural characterization in the solid state. We were also able to obtain single crystals for enantiopure diketo[6]CPP (S,S)-3, which we synthesized as described below by racemic resolution of 3, by vapor diffusion of methanol into a chlorobenzene solution, as well as crystals of cyclic intermediate 8a by vapor diffusion of n-hexane into a chlorobenzene solution.
As shown in Fig. 3A and B, the DBP moieties in 1 and 2 are distorted from planarity. The bending of the DBP units can be quantified using the so-called bend angles ΘDBP, originally defined by Bodwell and coworkers for pyrenophanes.33,34ΘDBP amounted to 79.6° for 1 and 63.4° for 2 (Fig. 3). These distortions lie slightly below those we had reported for DBP-phanes of up to 87.6°.29 The shapes of the hoops are almost circular with diameters of 11–12 Å for 1 and 12.9 Å for 2. These lie in the range of [8]- and [9]CPP (11 Å respective 12 Å).35
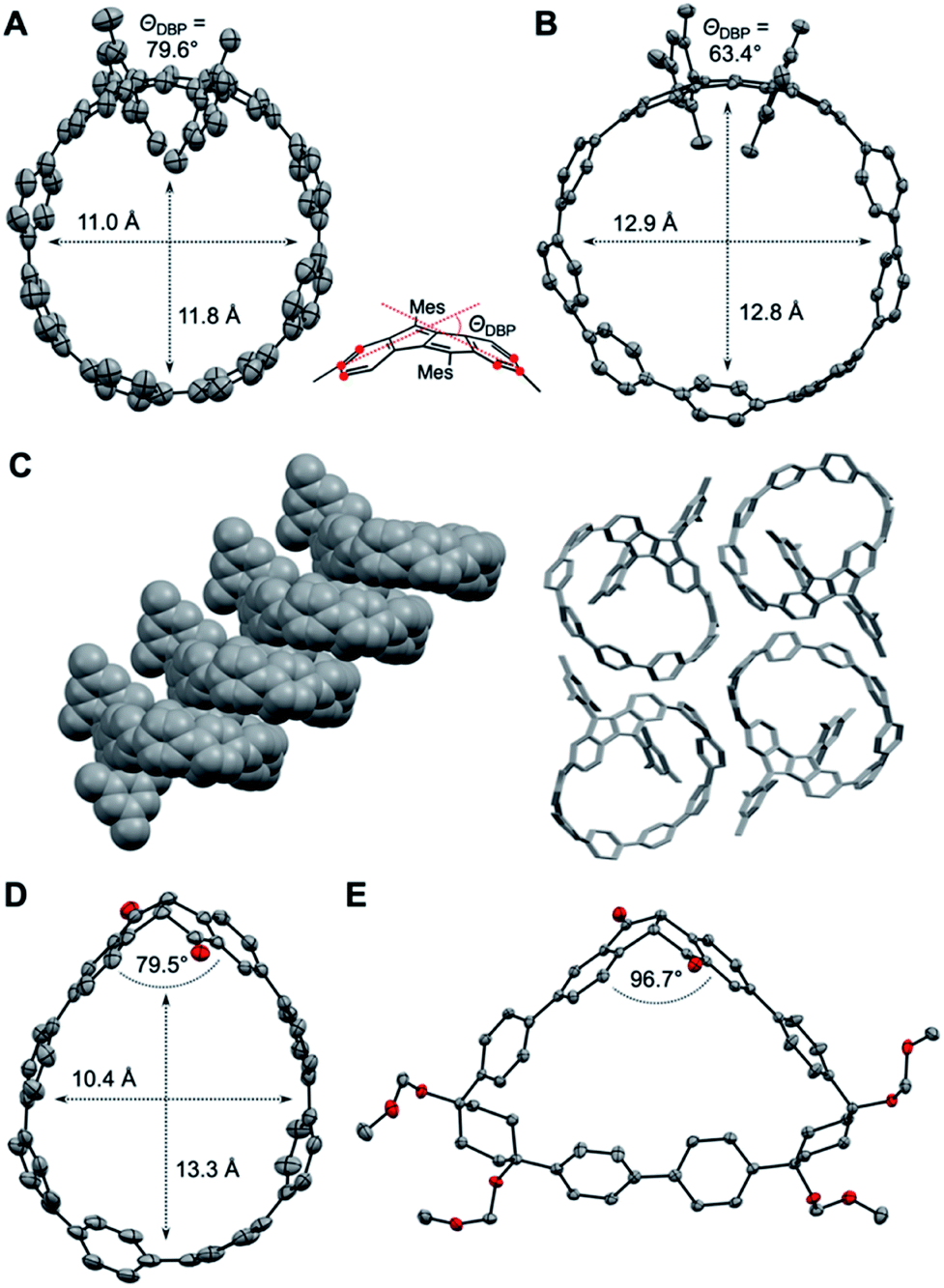 | ||
| Fig. 3 Molecular structures of 1 (A), 2 (B) with molecular packing (C), (S,S)-3 ((D), one of two independent molecules in the asymmetric unit) and 8a (E) in the solid state ((A, B, D and E) displacement ellipsoids are shown at the 50% probability level; hydrogen atoms, solvent molecules, and disordered atoms are omitted for clarity). | ||
2 forms tubular structures in the solid state (Fig. 3C). The molecules are stacked in a ladder-type fashion, where one mesityl group of each hoop points inside the cavity of the neighbouring hoop. The remaining “empty space” is filled by solvent molecules (not shown in Fig. 3C). 1, on the other hand, packs in a herringbone-type pattern (see ESI Section 10†).
The crystals of (S,S)-3 contained two independent molecules in the asymmetric unit, whose geometrical parameters are quite similar. Due to the bent shape of the diketone unit, this hoop assumes a stretched shape with a width of around 10.4 Å and a length of around 13.3 Å (Fig. 3D).
The PP units on the opposite side of the diketones experience the strongest bend (see StrainViz calculations in the ESI†). The bend angle of the diketone group (angle between its two phenylene units) amounted to 79.5° and 82.2° for the two independent molecules, which is significantly smaller than in the unsubstituted diketone (109.8°, for X-ray data see ESI, Section 10†) or in bis(boronic ester) 5 (107.0°). Hence these diketone units are able to accommodate some of the strain through a change in conformation and bend angle.29 The cyclic intermediate 8a containing the diketone as well as two Itami corner units assumes a near triangular shape (Fig. 3E), in which the diketone unit is bent with only 96.7°.
DFT calculations at the B3LYP/6-31G(d) level of theory using the StrainViz36 tool provided information on strain energies (see ESI Section 11 for details†). For DBP[n]CPPs 1 and 2, these lay at 68.4 kcal mol−1 and 63.4 kcal mol−1, respectively, which is in the range of [8]- and [9]CPP (64.2 and 70.5 kcal mol−1, respectively).36 A comparison with the strain energy for the smaller diketo[6]CPP 3 of 51.7 kcal mol−1 shows that an additional 16.7 kcal mol−1 of strain was induced in the last two synthetic steps to DBP[6]CPP 1, mostly through the two-fold water elimination to 1. The largest amount of strain, however, was built up during the aromatization reactions from 8a (strain energy: 11.9 kcal mol−1) and 8b (9.3 kcal mol−1) to furnish 3.
Bending the DBP units affects their antiaromaticity. We used NICS(1)zz (ref. 37) (nucleus-independent chemical shift38) calculations at the B3LYP/6-31G(d)-SP//PBEh-3c39 level of theory with the GIAO method to assess these changes, where dummy atoms were placed 1 Å above the 5- and 6-membered rings of the DBP units (outside the hoops). Negative NICS values indicate a diatropic ring current (associated with an aromatic character) while positive NICS values indicate a paratropic ring current (associated with an antiaromatic character). With NICS(1)zz values of 16.0 and 17.4, the 5-membered rings are antiaromatic in 1 and 2, respectively, while the 6-membered rings are aromatic with values of −13.8 and −13.2. The bending overall renders the DBP units less antiaromatic, which can be seen from comparison with unsubstituted DBP that had values of 25.7 for the 5-membered rings and −12.9 for the 6-membered rings.40 This trend is also reflected in a slightly less antiaromatic character of the DBP unit in DBP[6]CPP 1 compared to the larger DBP[7]CPP 2. A similar trend upon bending of the DBP units we had observed before in DBP-phanes29 as well as in larger hoops [2]DBP[12]CPP.41 HOMA (harmonic oscillator model of aromaticity)42,43 analyses of the bond lengths in the calculated structures (PBEh-3c) provided additional insight. For benzene as ideal aromatic system, the HOMA value is 1, antiaromatic systems show small or often negative HOMA values.44 In the six-membered rings of 1 and 2 the HOMA values are 0.953 and 0.951, respectively, and in the five-membered rings the HOMA values are −0.004 for 1 and −0.001 for 2.
Optoelectronic properties
DBP is a non-alternant, 16 π-electron system with antiaromatic character,45,46 which bestows ambipolar electrochemical character47 to DBP[n]CPPs 1 and 2.48 Its small bandgap is due to an increased HOMO and decreased LUMO energy in comparison to an alternant hydrocarbon of similar size.1 This ambipolar electrochemical character makes DBP derivatives attractive for applications such as field-effect transistors.49–51 UV/Vis absorption spectroscopy, cyclic voltammetry and TDDFT calculations provided insight into the optoelectronic properties of DBP[n]CPPs 1 and 2 as well as diketo[n]CPPs 3 and 4.The absorption spectra of 1–4 show the signature band of the PP linkers as the absorption maximum at 325–345 nm in each case (Fig. 4A, exact values in Table 1). For diketo[n]CPPs 3 and 4, these correspond to the HOMO → LUMO+1 transition, according to TDDFT calculations. The corresponding orbitals are mostly located on the PP linkers (Fig. 4B). In DBP[n]CPPs, these bands involve multiple transitions from (HOMO−4)–HOMO to LUMO–(LUMO+4). As selected orbital plots in Fig. 4B show, the frontier molecular orbitals are not solely located on either the DBP unit or the PP linker, but rather stretch out over parts of both π-systems. This stands in contrast to the larger [2]DBP[12]CPP hoops we previously reported, where a clear localization of orbital lobes was found.41
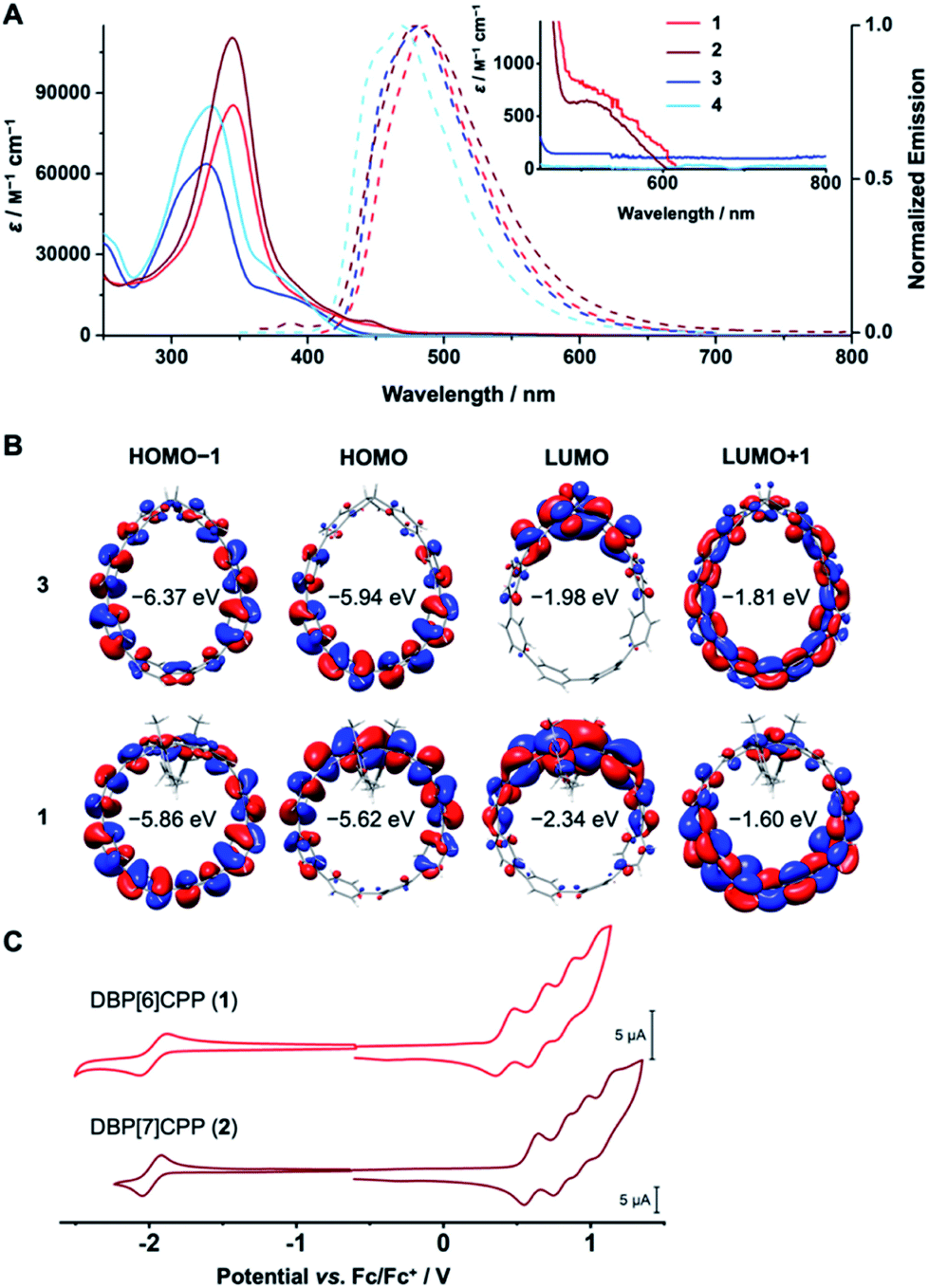 | ||
| Fig. 4 (A) Absorption (10−5 to 10−6 M) and emission spectra (10−6 to 10−7 M) of 1–4 in CH2Cl2; (B) frontier molecular orbitals of 3 and 1 (PW6B95-D3(BJ)/def2-QZVPP//PBEh-3c); (C) cyclic voltammograms of 1 and 2 (negative potential range in THF, positive in CH2Cl2, 1 mM concentration, 0.1 M n-Bu4NPF6, scan rate 0.2 V s−1, glassy carbon electrode, potentials plotted vs. Fc/Fc+ as internal standard). | ||
| Compound | λ abs-max (nm) | ε (M−1 cm−1) | λ em-max (nm) | Φ F |
|---|---|---|---|---|
| 3 | 325 | 6.4 × 104 | 481 | 0.57 |
| 4 | 329 | 8.5 × 104 | 470 | 0.66 |
| 1 | 345 | 8.5 × 104 | 487 | 0.06 |
| 2 | 345 | 1.1 × 105 | 479 | 0.01 |
For diketo[n]CPPs 3 and 4 the main absorption bands appeared at wavelengths close to those of “symmetry-broken” m[n]CPPs (328 nm)52 and blue-shifted in comparison to [n]CPPs of similar size (340 nm).15 In DBP[n]CPPs 1 and 2 this band was red-shifted by 0.22 eV for 1 relative to 3 and by 0.18 eV for larger 2 relative to 4 (see values in Table 1). Concerning the absolute intensity of absorption, the molar attenuation coefficients ε were higher for the larger [7]PP-bridged hoops 4 and 2 compared to [6]PP-bridged 3 and 1 (see values in Table 1). This size-dependent trend is shared with [n]CPPs15 as well as with the “symmetry-broken” m[n]CPPs mentioned above.52 The absolute values of ε also lie in the range of these similar systems.15
The signature of the DBP units in 1 and 2 can be seen from the shoulders around 530 nm and a red-shifted long-wavelength absorption onset compared to 3 and 4. This demonstrates the low band gaps of 1.81 and 1.89 eV for DBP[6]CPP 1 and DBP[7]CPP 2, respectively, responsible for the red colour of these hoops. The shoulder or weak band at around 530 nm for both hoops (inset in Fig. 4A) corresponds to the HOMO → LUMO transitions, which have a low oscillator strength from TDDFT calculations. These orbitals are centered on the DBP units (Fig. 4B), and this transition is forbidden in planar and centrosymmetric DBP derivatives, according to Laporte's rule, when both orbitals are of au symmetry.53 Due to the bending and local symmetry-breaking of the DBP units in DBP[n]CPPs 1 and 2, however, this transition has a relatively high intensity compared to planar DBPs.31,41
All four hoops 1–4 were fluorescent with emission maxima from 470–487 nm (Fig. 4A, exact values in Table 1). These emissions stemmed from the PP linkers, which explains the similar wavelengths and shapes of the emission bands. Diketo[n]CPPs 3 and 4 had high fluorescence quantum yields ΦF of 0.57 and 0.66, respectively, which lie in a similar range as those of “symmetry-broken” m[n]CPPs.52 Also the fact that ΦF increases with increasing hoop size has been observed for these systems as well as for [n]CPPs.15 The emissions in DBP[n]CPPs 1 and 2, on the other hand, had low ΦF values of 0.06 and 0.01, respectively. This correlates well with a calculated oscillator strength of 0.04 for the S1 → S0 transition. The low fluorescence quantum yields could be due to an energy transfer from the PP linkers to the DBP units in the excited state, which is energetically feasible considering the overlap of the PP-centered emission bands with the long-wavelength absorptions of the DBP parts at 530 nm. The DBP units are non-emissive and dissipate the energy radiation-less. For the electronically similar indeno[1,2-b]fluorenes this non-emissive behaviour was rationalized through transient absorption spectroscopy measurements that showed that the excited state lifetimes were shorter than the timescale at which fluorescence is usually observed.54 Another explanation would be a photoinduced electron transfer from the excited PP part to a DBP unit, which would also lead to a fluorescence quenching.
For the more strained hoops 1 and 3 we investigated the effect of solvent polarity on the emission spectra. A slight positive solvatochromic effect was evidenced for both hoops by a bathochromic shift of the emission maximum with increasing solvent polarity (from 478 nm in cyclohexane to 487 nm in CH2Cl2 for 1 and from 476 nm (cyclohexane) to 483 nm (chloroform) for 3, see ESI Section 6† for spectra). Such a bathochromic shift indicates a higher polarity of the first excited state than the ground state.55
The cyclic voltammograms confirm the ambipolar electrochemical character of DBP[n]CPP hoops 1 and 2 (Fig. 4C). Both showed a reversible reduction in THF at a half-wave potential of E1/2 = −1.98 V vs. Fc/Fc+ as well as further, partly reversible, reductions when scanning down to −3.7 V vs. Fc/Fc+ (see ESI Section 7† for further CVs and potential values). Each DBP unit can reversibly accept up to two electrons, upon which it assumes aromatic character with 18 π-electrons.47 Hence, the first reductions stem from the DBP units, as can be rationalized by the distribution of the LUMO in 1 (Fig. 4B), while further reduction processes occur at the PP linker or the DBP unit.
In positive scan direction, both 1 and 2 displayed four reversible oxidations each at half-wave potentials of E1/2 = 0.42, 0.64, 0.83 and 1.01 V for 1 and at E1/2 = 0.60, 0.81, 0.94 and 1.10 V for 2 (all vs. Fc/Fc+). The first oxidations likely stem from the DBP units based on the HOMO distribution (Fig. 4B) and since [n]CPPs as best reference for the PP linkers are oxidized at higher potentials E1/2 = 0.59 V or 0.70 V vs. Fc/Fc+ for [8]- and [9]CPP,35 which have similar diameters and strain energies as 1 and 2. The further oxidation processes could either be based on the PP linkers or the DBP units, which can undergo two oxidations to furnish an aromatic 14π-electron system.47
In summary, the optical properties of diketo[n]CPPs 3 and 4 are dominated by the PP linkers with high fluorescent quantum yields. In DBP[n]CPPs 1 and 2, the DBP units lead to ambipolar electrochemical character with both reversible oxidations and reductions. The optical properties reflect the presence of both the PP linkers in characteristic absorption and emission bands as well as the DBP units in a red-shifted absorption onset and a partial quenching of the fluorescence intensity.
Racemic resolution to enantiopure nanohoops
For the racemic resolution of diketo[6]CPP 3 we used the chiral sulfoximine auxiliary 11 (Scheme 2).56,57 Addition of its Grignard reagent furnishes diastereomers, which can be separated by column chromatography, as we previously demonstrated for the diketone unit by itself.28 Performing the racemic resolution at the late nanohoop stage in compound 3 has several advantages compared to an early-stage racemic resolution of the diketone precursor S1 (dibromide precursor to boronic ester 5 in Scheme 1, for structure see ESI†): In an early-stage resolution (1) due to the material loss in the synthesis of diketo[6]CPP 3 from S1 (14% yield over four steps) significantly higher amounts of the chiral auxiliary would be required and (2) in order to obtain both enantiomers (S,S)- and (R,R)-3, their syntheses from S1 would have to be performed in two separate batches. Furthermore, in order to determine the enantiopurity of the chiral hoops via HPLC, a racemic sample of 3 is required in any case.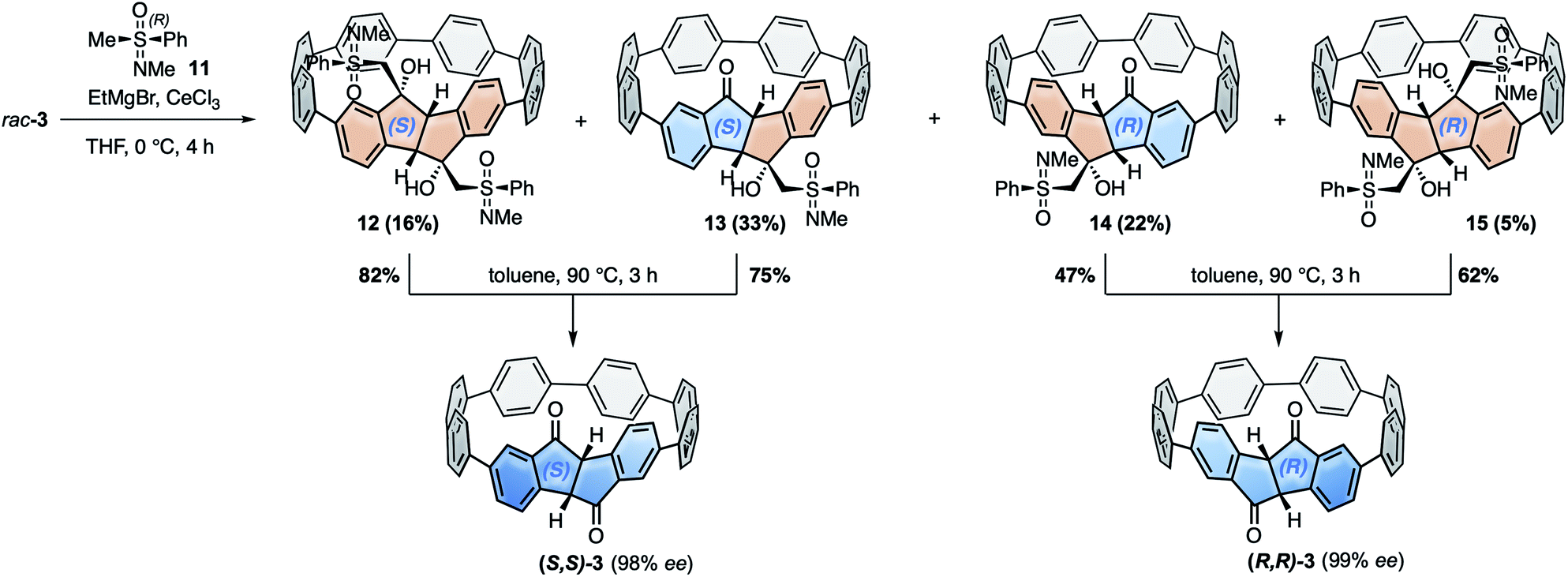 | ||
| Scheme 2 Racemic resolution by chiral derivatization of diketo[6]CPP 3 to furnish enantiopure (S,S)- and (R,R)-3. | ||
We synthesized chiral sulfoximine (R)-11 from thioanisol in three steps including racemic resolution of the intermediate sulfoximine before N-methylation.28,58–60 Its Grignard reagent diastereoselectively adds to the carbonyl groups in rac-3 from their convex side28 and furnished four products 12–15 as a result of a single or double addition in an overall yield of 76%. The double adducts 12 and 15 as well as the single adducts 13 and 14 are diastereomers. 12–15 could easily be separated by conventional column chromatography. The chiral auxiliary was removed by thermal treatment, which afforded enantiopure hoop (S,S)-3 from 12 and 13 in high yields as well as (R,R)-3 from 14 and 15. Their absolute configuration was unambiguously determined through the molecular structure of (S,S)-3 in the solid state (Fig. 3C). Analytical chiral HPLC confirmed a high enantiomeric purity of diketo[6]CPPs (S,S)- and (R,R)-3 with 98% respective 99% ee (see ESI Section 2.3† for details). To allow for CD-spectroscopic analyses we transformed small samples of (S,S)- and (R,R)-3 into enantiopure DBP[6]CPPs (M)-1 and (P)-1, respectively. The chiral descriptors M and P are based on the nomenclature of chiral molecules which resemble a helix, where P (plus) is a right-handed and M (minus) a left-handed helix (see ESI† section 2.3.1 for a drawing that illustrates our assignment).61,62 In order to assess the stability of these enantiomers towards racemization, we investigated a possible rotation of the DBP unit through the hoop. Due to the large size of the mesityl substituents compared to the hoop, calculation of a transition state for racemization was not even possible, hence rotation can be assumed to be impossible. Using methyl substituents instead resulted in a calculated rotational barrier of 36.5 kcal mol−1 for the DBP[6]CPP from the transition state energy (see ESI†). In the transition state, the DBP moiety is rotated by 90° and almost planar. Even in larger DBP[7]CPP with only methyl substituents on the DBP units the barrier still amounted to 26.9 kcal mol−1, making racemization slow at room or even higher temperatures18 and impossible with larger substituents such as mesityl.
Chiroptical properties of enantiopure nanohoops
We next turned to electronic circular dichroism (ECD) and circularly polarized luminescence (CPL) measurements to assess the chirality of the enantiopure diketo[6]CPPs and DBP[6]CPPs. The spectra can be found in Fig. 5 (in toluene), and absorption and luminescence dissymmetry factors are listed in Table 2. Additional spectra in CH2Cl2 as well as the corresponding absorption dissymmetry factor plots gabs = f(λ) can be found in the ESI Section 8.†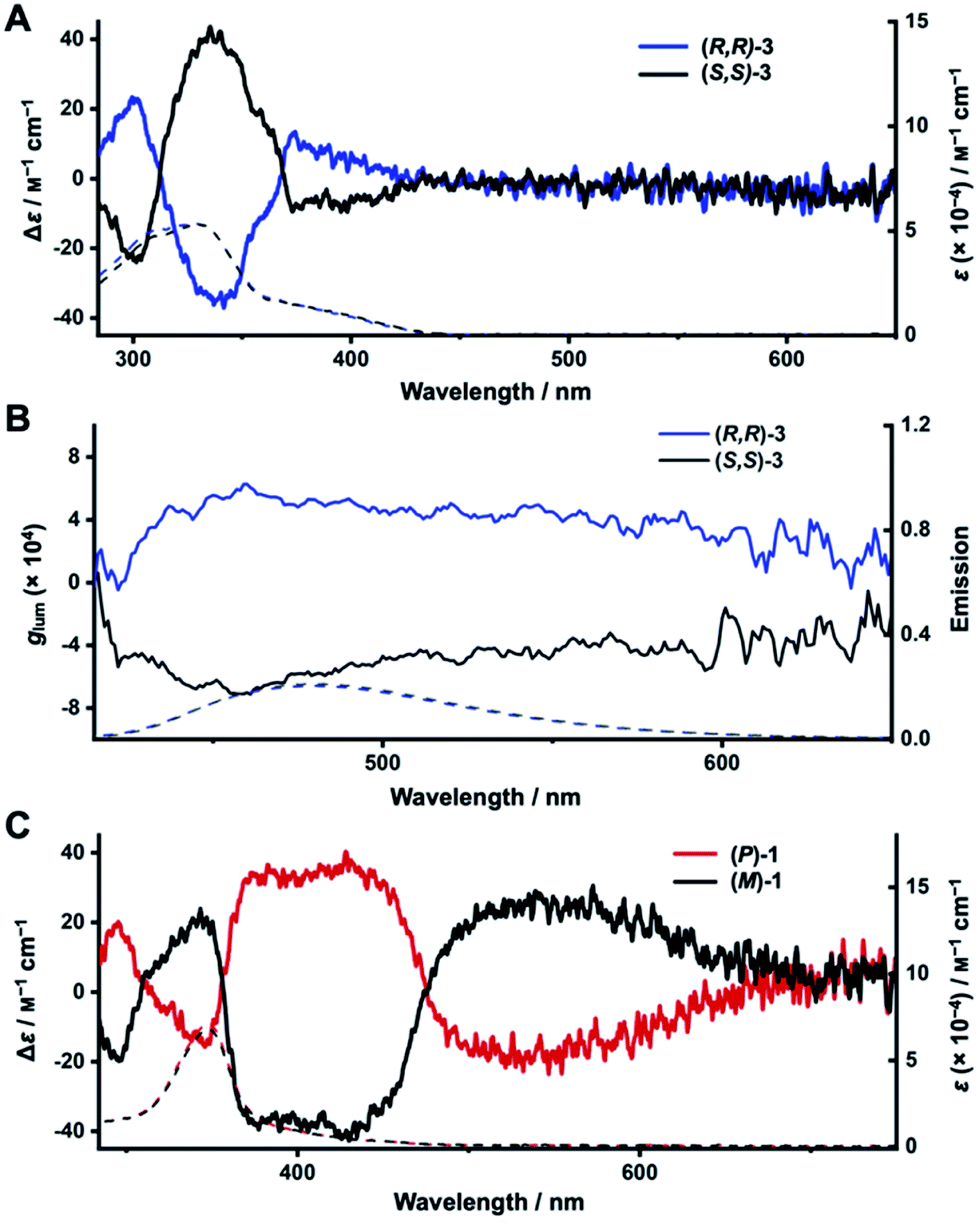 | ||
| Fig. 5 Chiroptical properties or diketo[6]CPPs and DBP[6]CPPs (5 × 10−5 M in toluene, 20 °C). (A) ECD (solid lines) and UV/Vis absorption spectra (dotted lines) of (R,R)- and (S,S)-3; (B) CPL (solid lines) and fluorescence (dotted lines) spectra of (R,R)- and (S,S)-3; (C) ECD (solid lines) and UV/Vis absorption spectra (dotted lines) of (P)- and (M)-1. | ||
The mirror image relationship between (R,R)- and (S,S)-3 as well as (P)- and (M)-1 can be seen from their ECD and CPL spectra in Fig. 5. The ECD spectrum of diketo[6]CPP (R,R)-3 shows three characteristic bands (Fig. 5A). The first one is centred at 300 nm (Δε = 23 M−1 cm−1), the second at 340 nm (Δε = −38 M−1 cm−1) and the third at 390 nm (Δε = −9 M−1 cm−1), corresponding to the HOMO → LUMO transition. The two enantiomers (R,R)- and (S,S)-3 are CPL-active, with dissymmetry factors |glum| of around 4 × 10−4 at the emission maxima (Fig. 5B), which are in the usual range of reported organic CPL emitters.63
For DBP[6]CPPs (P)- and (M)-1 the situation is quite different. The ECD signals corresponding to the absorption maximum in the UV/Vis spectra centred at 350 nm (Fig. 5C) show a moderate molar circular dichroism (|Δε| = 15 M−1 cm−1) leading to a rather small absorption dissymmetry factor of gabs = 0.23 × 10−3. These correspond to transitions mostly centred on the PP linkers, according to TDDFT calculations. ECD bands of larger molar circular dichroism were observed at higher wavelengths with two significant signals centred at 400 nm with Δε = −35 M−1 cm−1 and at 550 nm with Δε = +26 M−1 cm−1 (all values for (M)-1). These correspond to absorptions of the DBP units (see inset in the absorption spectra in Fig. 4A). Particularly noteworthy is the longest wavelength band from 730–460 nm, which stems from the HOMO → LUMO transition in (P)- and (M)-1 and is only visible as a weak shoulder in the UV/Vis absorption spectra, however, results in relatively important Δε and therefore gabs values in this region. The experimental ECD spectra agree well with the calculated ones (Fig. S93†). The intense chiroptical properties at these wavelengths (confirmed by TDDFT calculations, see ESI Section 11.4†), related to transitions mostly centred on the DBP units, can be explained by the fact that the chirality of these nanohoops depends on the orientation of the latter.63
Conclusions
We herein presented a racemic resolution strategy by chiral derivatization of conjugated nanohoops. The carbonyl groups in diketo[n]CPPs can be derivatised by reversible addition of a chiral sulfoximine auxiliary, and the resulting diastereomers separated by conventional column chromatography. This results in an easily scalable process, as exemplified for n = 6. These chiral hoops displayed circularly polarized absorption and emission properties. Two further steps transform the diketo[n]CPPs into DBP[n]CPPs with antiaromatic character of the dibenzo[a,e]pentalene units, which was demonstrated for racemic n = 6 and 7. The optoelectronic properties of the DBP[n]CPPs reflect the presence of both electrochemically ambipolar DBP units as well as the para-phenylene (PP) linkers, while those of diketo[n]CPPs are dominated by the PP linkers with high fluorescent quantum yields, similar to other symmetry-broken CPPs. X-ray crystallographic analyses of the DBP[n]CPPs as well as their precursors provided valuable insight into the build-up of strain energy during synthesis. NICS calculations indicated significant bending of the DBP units, which led to a reduction of its antiaromaticity. To the best of our knowledge, this is the first report of conjugated nanohoops, where enantiomers are separated by chiral derivatization of a functionalized nanohoop. The potential to upscale this method paves the way for an application of enantiopure nanohoops in material science or biology.Data availability
Further datasets supporting this article have been uploaded as part of the supplementary information.Author contributions
Conceptualization, supervision, resources, methodology, visualization, writing – original draft, project administration, funding acquisition B. E.; investigation, methodology, data curation, formal analysis, visualization, writing – original draft D. W.; investigation, data curation, formal analysis, writing – review and editing M. H.; investigation, data curation, formal analysis, writing – review and editing J. S. W.; investigation, data curation, formal analysis, writing – review and editing L. F.; supervision, funding acquisition, writing – review and editing G. P.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – project numbers 230408635, 434040413, INST 40/467-1 FUGG and INST 39/1081-1 FUGG – and the state of Baden-Württemberg through bwHPC. GP thanks the CEA especially the SCBM and the Labex CHARMMMAT (ANR-11-LABX-0039) for support and funding.Notes and references
- R. Gleiter and G. Haberhauer, Aromaticity and Other Conjugation Effects, Wiley-VCH, Weinheim, Germany, 2012 Search PubMed.
- K. Tahara and Y. Tobe, Chem. Rev., 2006, 106, 5274–5290 CrossRef CAS PubMed.
- R. Gleiter, B. Esser and S. C. Kornmayer, Acc. Chem. Res., 2009, 42, 1108–1116 CrossRef CAS PubMed.
- Y. Segawa, D. R. Levine and K. Itami, Acc. Chem. Res., 2019, 52, 2760–2767 CrossRef CAS PubMed.
- B. Esser and M. Hermann, Nat. Chem., 2021, 13, 209–211 CrossRef CAS PubMed.
- R. Jasti and C. R. Bertozzi, Chem. Phys. Lett., 2010, 494, 1–7 CrossRef CAS PubMed.
- Y. Segawa, A. Yagi, K. Matsui and K. Itami, Angew. Chem., Int. Ed., 2016, 55, 5136–5158 CrossRef CAS PubMed.
- Y. Xu and M. von Delius, Angew. Chem., Int. Ed., 2020, 59, 559–573 CrossRef CAS PubMed.
- D. Lu, Q. Huang, S. Wang, J. Wang, P. Huang and P. Du, Front. Chem., 2019, 7, 668 CrossRef CAS PubMed.
- E. J. Leonhardt and R. Jasti, Nat. Rev. Chem., 2019, 3, 672–686 CrossRef CAS.
- D. Wu, W. Cheng, X. Ban and J. Xia, Asian J. Org. Chem., 2018, 7, 2161–2181 CrossRef CAS.
- R. Jasti, J. Bhattacharjee, J. B. Neaton and C. R. Bertozzi, J. Am. Chem. Soc., 2008, 130, 17646–17647 CrossRef CAS PubMed.
- H. Takaba, H. Omachi, Y. Yamamoto, J. Bouffard and K. Itami, Angew. Chem., Int. Ed., 2009, 48, 6112–6116 CrossRef CAS PubMed.
- S. Yamago, Y. Watanabe and T. Iwamoto, Angew. Chem., Int. Ed., 2010, 49, 757–759 CrossRef CAS PubMed.
- M. Hermann, D. Wassy and B. Esser, Angew. Chem., Int. Ed., 2021 DOI:10.1002/anie.202007024.
- A.-F. Tran-Van and H. A. Wegner, Beilstein J. Nanotechnol., 2014, 5, 1320–1333 CrossRef PubMed.
- P. Sarkar, Z. Sun, T. Tokuhira, M. Kotani, S. Sato and H. Isobe, ACS Cent. Sci., 2016, 2, 740–747 CrossRef CAS PubMed.
- J. Wang, G. Zhuang, M. Chen, D. Lu, Z. Li, Q. Huang, H. Jia, S. Cui, X. Shao, S. Yang and P. Du, Angew. Chem., Int. Ed., 2020, 59, 1619–1626 CrossRef CAS PubMed.
- S. Hitosugi, W. Nakanishi, T. Yamasaki and H. Isobe, Nat. Commun., 2011, 2, 492 CrossRef.
- S. Hitosugi, T. Yamasaki and H. Isobe, J. Am. Chem. Soc., 2012, 134, 12442–12445 CrossRef CAS PubMed.
- T. Matsuno, S. Kamata, S. Hitosugi and H. Isobe, Chem. Sci., 2013, 4, 3179–3183 RSC.
- S. Hitosugi, S. Sato, T. Matsuno, T. Koretsune, R. Arita and H. Isobe, Angew. Chem., Int. Ed., 2017, 56, 9106–9110 CrossRef CAS PubMed.
- Y. Segawa, M. Kuwayama, Y. Hijikata, M. Fushimi, T. Nishihara, J. Pirillo, J. Shirasaki, N. Kubota and K. Itami, Science, 2019, 365, 272–276 CrossRef CAS PubMed.
- K. Senthilkumar, M. Kondratowicz, T. Lis, P. J. Chmielewski, J. Cybińska, J. L. Zafra, J. Casado, T. Vives, J. Crassous, L. Favereau and M. Stępień, J. Am. Chem. Soc., 2019, 141, 7421–7427 CrossRef CAS PubMed.
- S. Nishigaki, Y. Shibata, A. Nakajima, H. Okajima, Y. Masumoto, T. Osawa, A. Muranaka, H. Sugiyama, A. Horikawa, H. Uekusa, H. Koshino, M. Uchiyama, A. Sakamoto and K. Tanaka, J. Am. Chem. Soc., 2019, 141, 14955–14960 CrossRef CAS PubMed.
- J. Nogami, Y. Tanaka, H. Sugiyama, H. Uekusa, A. Muranaka, M. Uchiyama and K. Tanaka, J. Am. Chem. Soc., 2020, 142, 9834–9842 CAS.
- J. Nogami, Y. Nagashima, K. Miyamoto, A. Muranaka, M. Uchiyama and K. Tanaka, Chem. Sci., 2021, 12, 7858–7865 RSC.
- M. Hermann, D. Wassy, J. Kohn, P. Seitz, M. U. Betschart, S. Grimme and B. Esser, Angew. Chem., Int. Ed., 2021, 60, 10680–10689 CrossRef CAS PubMed.
- M. Hermann, D. Wassy, D. Kratzert and B. Esser, Chem. –Eur. J., 2018, 24, 7374–7387 CrossRef CAS PubMed.
- Q. Huang, G. Zhuang, H. Jia, M. Qian, S. Cui, S. Yang and P. Du, Angew. Chem., Int. Ed., 2019, 58, 6244–6249 CrossRef CAS PubMed.
- J. Wilbuer, D. C. Grenz, G. Schnakenburg and B. Esser, Org. Chem. Front., 2017, 4, 658–663 RSC.
- S. Klod and E. Kleinpeter, J. Chem. Soc. Perkin Trans., 2001, 2, 1893–1898 Search PubMed.
- G. J. Bodwell, J. J. Fleming and D. O. Miller, Tetrahedron, 2001, 57, 3577–3585 CrossRef CAS.
- B. L. Merner, K. S. Unikela, L. N. Dawe, D. W. Thompson and G. J. Bodwell, Chem. Commun., 2013, 49, 5930 RSC.
- E. R. Darzi and R. Jasti, Chem. Soc. Rev., 2015, 44, 6401–6410 RSC.
- C. E. Colwell, T. W. Price, T. Stauch and R. Jasti, Chem. Sci., 2020, 11, 3923–3930 RSC.
- C. Corminboeuf, T. Heine, G. Seifert, P. von R. Schleyer and J. Weber, Phys. Chem. Chem. Phys., 2004, 6, 273–276 RSC.
- P. von R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. van E. Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS PubMed.
- S. Grimme, J. G. Brandenburg, C. Bannwarth and A. Hansen, J. Chem. Phys., 2015, 143, 054107 CrossRef PubMed.
- M. Baranac-Stojanović and M. Stojanović, Phys. Chem. Chem. Phys., 2019, 21, 3250–3263 RSC.
- D. Wassy, M. Pfeifer and B. Esser, J. Org. Chem., 2020, 85, 34–43 CrossRef CAS PubMed.
- J. Kruszewski and T. M. Krygowski, Tetrahedron Lett., 1972, 13, 3839–3842 CrossRef.
- T. M. Krygowski and M. K. Cyrański, Chem. Rev., 2001, 101, 1385–1420 CrossRef CAS PubMed.
- P. J. Mayer, O. El Bakouri, T. Holczbauer, G. F. Samu, C. Janáky, H. Ottosson and G. London, J. Org. Chem., 2020, 85, 5158–5172 CrossRef CAS PubMed.
- H. Hopf, Angew. Chem., Int. Ed., 2013, 52, 12224–12226 CrossRef CAS PubMed.
- M. Schmidt, D. Wassy, M. Hermann, M. T. González, N. Agräit, L. A. Zotti, B. Esser and E. Leary, Chem. Commun., 2021, 57, 745–748 RSC.
- M. Hermann, T. Böttcher, M. Schorpp, S. Richert, D. Wassy, I. Krossing and B. Esser, Chem. –Eur. J., 2021, 27, 4964–4970 CrossRef CAS PubMed.
- A. Konishi and M. Yasuda, Chem. Lett., 2021, 50, 195–212 CrossRef CAS.
- T. Kawase, T. Fujiwara, C. Kitamura, A. Konishi, Y. Hirao, K. Matsumoto, H. Kurata, T. Kubo, S. Shinamura, H. Mori, E. Miyazaki and K. Takimiya, Angew. Chem., Int. Ed., 2010, 49, 7728–7732 CrossRef CAS PubMed.
- M. Hermann, R. Wu, D. C. Grenz, D. Kratzert, H. Li and B. Esser, J. Mater. Chem. C, 2018, 6, 5420–5426 RSC.
- D. C. Grenz, M. Schmidt, D. Kratzert and B. Esser, J. Org. Chem., 2018, 83, 656–663 CrossRef CAS PubMed.
- T. C. Lovell, C. E. Colwell, L. N. Zakharov and R. Jasti, Chem. Sci., 2019, 10, 3786–3790 RSC.
- C. G. Wermuth, C. R. Ganellin, P. Lindberg and L. A. Mitscher, Pure Appl. Chem., 1979, 51, 1129–1143 Search PubMed.
- B. D. Rose, L. E. Shoer, M. R. Wasielewski and M. M. Haley, Chem. Phys. Lett., 2014, 616–617, 137–141 CrossRef CAS.
- J. S. Wössner and B. Esser, J. Org. Chem., 2020, 85, 5048–5057 CrossRef PubMed.
- C. R. Johnson and J. R. Zeller, J. Am. Chem. Soc., 1982, 104, 4021–4023 CrossRef CAS.
- C. R. Johnson and J. R. Zeller, Tetrahedron, 1984, 40, 1225–1233 CrossRef CAS.
- A. Tota, M. Zenzola, S. J. Chawner, S. S. John-Campbell, C. Carlucci, G. Romanazzi, L. Degennaro, J. A. Bull and R. Luisi, Chem. Commun., 2017, 53, 348–351 RSC.
- J. Brandt and H.-J. Gais, Tetrahedron: Asymmetry, 1997, 8, 909–912 CrossRef CAS.
- C. S. Shiner and A. H. Berks, J. Org. Chem., 1988, 53, 5542–5545 CrossRef CAS.
- L. C. Cross and W. Klyne, Pure Appl. Chem., 1976, 45, 11–30 Search PubMed.
- G. P. Moss, Pure Appl. Chem., 1996, 68, 2193–2222 CAS.
- E. M. Sánchez-Carnerero, A. R. Agarrabeitia, F. Moreno, B. L. Maroto, G. Muller, M. J. Ortiz and S. de la Moya, Chem. –Eur. J., 2015, 21, 13488–13500 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, synthetic manipulations, NMR spectra, mass spectra, additional UV/Vis absorption spectra, cyclic voltammograms, ECD and CPL spectra, information on the conformational isomerism in bis-(ortho-tolyl)-DBP, single-crystal X-ray diffraction data, details on (TD) DFT calculations. CCDC 2057460 (1·(CyH) (pentane)), 2055867 (2·(pentane)), 2057160 (2·(PhCl)2(iPrOH)0.5), 2057251 ((S,S)-3·(PhCl)), 2043958 (9), 1965552 (8a), 2036550 (5), 2064603 (S6) and 1479274 (S7) contain the supplementary crystallographic data for this paper. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc02718b |
| This journal is © The Royal Society of Chemistry 2021 |