
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDual emission in purely organic materials for optoelectronic applications
Nadzeya A.
Kukhta
 * and
Martin R.
Bryce
*
* and
Martin R.
Bryce
*
Department of Chemistry, Durham University, Lower Mountjoy, Stockton Road, Durham, DH1 3LE, UK. E-mail: nadzeya.kukhta@gmail.com; m.r.bryce@durham.ac.uk
First published on 21st September 2020
Abstract
Purely organic molecules, which emit light by dual emissive (DE) pathways, have received increased attention in the last decade. These materials are now being utilized in practical optoelectronic, sensing and biomedical applications. In order to further extend the application of the DE emitters, it is crucial to gain a fundamental understanding of the links between the molecular structure and the underlying photophysical processes. This review categorizes the types of DE according to the spin multiplicity and time range of the emission, with emphasis on recent experimental advances. The design rules towards novel DE molecular candidates, the most perspective types of DE and possible future applications are outlined. These exciting developments highlight the opportunities for new materials synthesis and pave the way for accelerated future innovation and developments in this area.
1. Introduction
Purely organic light-emitting compounds with an aromatic backbone have emerged as promising materials for optoelectronic and biomedical applications. In turn, multicolour-emissive systems, in particular dual emitters (DEs), show a great potential to extend the applications to sensors,1 data encryption,2 anticounterfeiting3 and high-sensitivity bioimaging,4 as well as the production of cost-efficient white organic light emitting diodes (WOLEDs).5–7In biology, small-molecule fluorescent probes are used extensively for cellular imaging owing to their simplicity of operation, short response time and non-invasiveness.8 Fluorescent imaging reagents which give cellular images with two different colours, can distinguish sites of interest from false positive signals generated by adventitious fluorescent biomolecules in the monitoring processes.9 They can also allow for in situ fluorescence based ratiometric reporting of cell conditions such as pH, temperature, hydrophilicity/phobicity, or oxygen concentration.
In an alternate application, the construction of WOLEDs generally requires the use of two (blue and yellow) or three (blue, green, and red) different colour emitters, covering the whole visible light range from 400 to 700 nm to achieve good colour rendering. This requirement often leads to complex device architectures, demanding fabrication processes and high cost in order to accommodate multiple emissive materials.10 In contrast to systems composed of several fluorescent molecules, single-molecular systems with white emission through DE have several advantages: they can avoid the drawbacks of phase separation, colour aging, and degradation; moreover, they lead to enhanced reproducibility and stability, as well as simpler device fabrication.11,12 However, the design of purely organic molecules capable of blue/yellow DE suitable for efficient WOLEDs requires in-depth understanding of the different DE mechanisms. As an additional complication,11 ideal WOLEDs should also harvest the electro-generated excited triplet states in order to reach the highest internal quantum efficiency of 100% by utilizing thermally activated delayed fluorescence (TADF)13 or phosphorescence (PH)14 most commonly for the yellow-emitting excited state.
Finally, materials with time-dependent and/or excitation-dependant luminescence colour changes have recently attracted experimental attention for encryption and anticounterfeiting, thereby increasing the range of tools for modern data security.15
An interesting but often overlooked DE phenomenon stems simply from the existence of two (or more) emissive singlet states in a molecule.16 Originally discovered in azulene,17 the rare occurrence of simultaneous emission from the first and second singlet excited states violates Kasha's rule.18 The DE phenomenon was further explored in the intramolecular charge transfer (ICT) states in a push–pull 4-N,N-dimethylaminobenzonitrile (DMABN) compound by Lippert et al. in 1959.19 The insights gained from the myriad of subsequent studies have led to a much deeper understanding of the locally excited (LE) and charge transfer (CT) states that give rise to fluorescence, and in some cases DE.
DE remains rare and remarkable when discovered in new materials, as fluorescence is a fast relaxation process in which a molecule in the excited singlet state returns to the ground state by emitting energy in the form of photons, following the initial photoabsorption (Fig. 1(a)).13 In most materials, rapid internal conversion (IC) to the lowest excited singlet state significantly outcompetes all other radiative processes, and so emission is observed only from that state – in accordance with Kasha's rule, and precluding DE. In contrast, in ICT materials the equilibrium of a parent LE state and a twisted intramolecular charge transfer (TICT) state represents the most common case of dual fluorescence,20 although singlet DE can also originate from the existence of dual CT states,21 excimer states,22 excited state intramolecular proton transfer (ESIPT)23 and structural modification of the emitter upon absorption of UV light.24
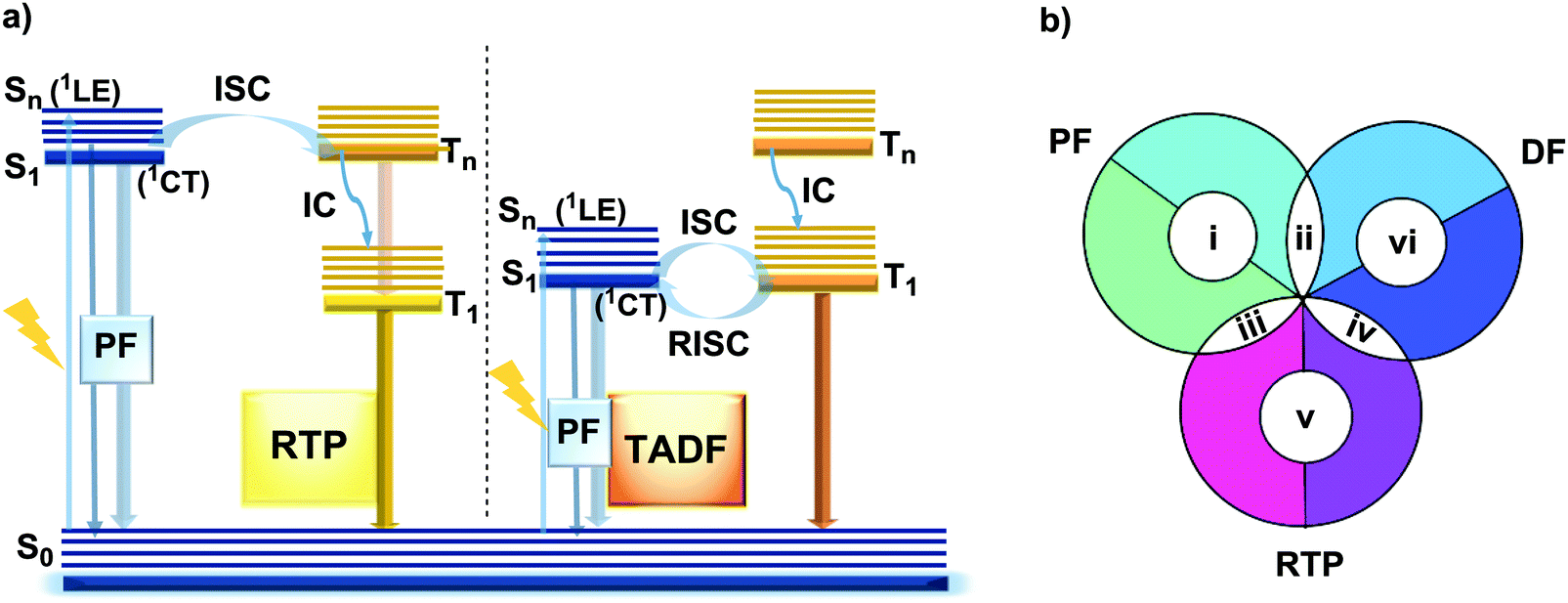 | ||
| Fig. 1 (a) Jablonski diagram, presenting an overview of photoabsorption, prompt fluorescence (PF), thermally activated delayed fluorescence (TADF) and room temperature phosphorescence (RTP). ISC – intersystem crossing; RISC – reverse intersystem crossing; IC – internal conversion; (b) combinations of DE. | ||
However, DE in a single molecule can also be achieved with the involvement of states of different multiplicity. An appropriate balance of emission and intersystem crossing (ISC) rates can generate coexisting populations of singlet and triplet states, both of which are sensitive to molecular structure and the environment.25 Importantly, for this balance to be achieved, two conditions should be satisfied: (i) sufficiently fast ISC rate to ensure that all singlet states are converted to triplets; (ii) long enough triplet lifetime to insure the occurrence of PH. While the majority of such materials require the absence of oxygen, low temperature and rigid host matrices, in some cases PH can be observed in absence of these stringent requirements.26 Hence, DE comprised of simultaneous fast fluorescence and slower phosphorescence with lifetimes in microsecond (µs) or millisecond (ms) scales can be observed at room temperature and sometimes under ambient conditions.27 In contrast, a reverse ISC (RISC) process, occurring from the lowest triplet (T1) to singlet (S1) states with assistance of a small energy gap (ΔEST ≤ 0.3 eV) at room temperature, leads to the well-known TADF phenomenon, with a lifetime in the microsecond range.28–30 TADF processes rarely lead to DE, as both the prompt fluorescence (PF) and delayed fluorescence (DF) arise from the same excited state and generate the same emission spectrum. In materials with a moderate ΔEST (0.3–0.6 eV) as well as the assistance of higher triplet states (a small S1–Tn gap),31 room temperature phosphorescence (RTP) with comparable luminescence lifetimes (µs vs. ms) but a different spectrum can sometimes be obtained, together generating DE.32 Due to the great difference in the lifetimes of TADF and RTP, tuning the balance in the population of singlet and triplet excitons via ISC and RISC processes of S1 ↔ T1 is hard to achieve.25 However, recently it was found that TADF can be promoted through the Tn–S1 channel, leading to simultaneous TADF and RTP emission.33 Apart from the nanosecond range DE arising from combined fluorescence and phosphorescence, DE though dual phosphorescence stemming from higher triplet states has also been reported.34
Given the complexity of engineering any of the DE combinations, the study of the mechanism of dual photoluminescence is particularly important and highly challenging as it needs to consider not only the molecule itself but also the surrounding environment in solution, crystalline state or thin film.20 Firstly, conformational isomerism, achieved by restricting covalent bonds at different dihedral angles, leads to different spatial arrangements of atoms in a molecule.35 Owing to the resulting changes in electron distribution and interactions, the conformers generally exhibit very distinct photophysical properties with distinct emission from each conformer.36,37 Structural equilibrium between the conformers or distributions of the conformers arising from film preparation opens an ideal pathway for the observation of dual (or multiple) emission. Furthermore, supramolecular structures and intermolecular noncovalent interactions can dominate the excited-state and emission properties in some materials.38 Thus, inter- and intramolecular hydrogen bonds were confirmed to restrict intramolecular rotations and increase molecular rigidity, contributing to the suppression of non-radiative rates.39 Furthermore, the diffusion of oxygen is slowed down in ordered media such as an organic crystal, therefore suppressing DF/phosphorescence quenching.40 As a result, the ratio between fluorescence and delayed emission can be manipulated by the degree of crystallinity of a sample.41
The diversity of fortuitously discovered DE materials has prompted us to assess and review the different reported cases of this phenomenon. This review categorizes the types of DE according to the spin multiplicity and time range of the emission. Therefore, six types of DE are considered: (i) dual prompt fluorescence (PF) (LE/CT, CT/CT); (ii) PF and delayed fluorescence (DF); (iii) PF and RTP; (iv) DF and RTP; (v) dual RTP and (vi) dual DF (Fig. 1(b)). By focusing on each type, we aim to study in detail the structure–property and environment–property relationships of the dual emitters and answer several essential questions: (i) is it possible to control DE? (ii) can efficient DE materials be purposely designed, and which types of DE are achievable based on a consistent set of design principles? (iii) which new applications could arise from the successful design of dual emitters?
2. Combinations of DE
2.1. Dual prompt photoluminescence
In the case of dual prompt fluorescence, simultaneous emission takes place from two singlet states of either locally excited (LE) or charge transfer (CT) nature. Dual PF is the most widely investigated type of dual emission, as it can be easily detected by steady-state spectroscopy in ambient conditions (as oxygen-sensitive triplet states are not involved). Closely linked to the molecular structure, dual PF can be observed in purely organic molecules with both donor–acceptor (D–A) and non-D–A architectures. Generally, prompt DE can be divided into several sub-types according to the nature of the chromophores and excited states involved, coupling between the units, and the influence of molecular aggregation: (a) LE/CT, (b) CT/CT, (c) ESIPT, (d) structural reorganization and (e) aggregation-induced dual emission (AIDE) (Fig. 2). For the observation of LE/CT and CT/CT types of DE a D–A molecular backbone featuring a rotatable single bond between the D and A units is a necessary condition. Photoexcitation initiates electron transfer (ET) from the donor to the acceptor, leading to a change in the excited singlet state geometry by either a twisted intramolecular charge transfer (TICT) or a planarized intramolecular charge transfer (PICT) mechanism. Thus, both TICT and PICT compounds require rotation around a single bond in the twisted excited state. TICT materials start from an almost planar geometry in the ground state and twist in the excited state; on the contrary, PICT compounds start from a twisted geometry in the electronic ground state, and a planarized system is formed in the excited state by similar rotation around a single bond.42,43 A change in the S1 geometry usually results in a change in the wavelength and intensity of the PF. Such distinct emission properties of TICT and PICT systems are potentially environment-dependent, making them ideal sensors for solvents, (micro)viscosity, and chemical species.43 For the observation of DE, both planarized and twisted conformations of the emitter must exist in equilibrium, emitting simultaneously with distinct spectra. For instance, intramolecular CT, followed by the combination of polarization stabilization and skeletal motion towards planarization, was observed in a series of N,N′-disubstituted-dihydrodibenzo[a,c]phenazines reported by the groups of Tian and Chou.44 The combination of the initial CT state and the final planarization state renders the full spectrum, which can be tuned upon optimization of the surrounding media. Furthermore, the elaboration of the host-enhanced intramolecular charge-transfer and host-induced restriction of intramolecular rotation in the novel host–guest complex bearing a D–A–D′ dual PF emitter, comprising binaphthol and coumarin donors, led to the generation of white-light emission with CIE coordinates (0.33, 0.34).45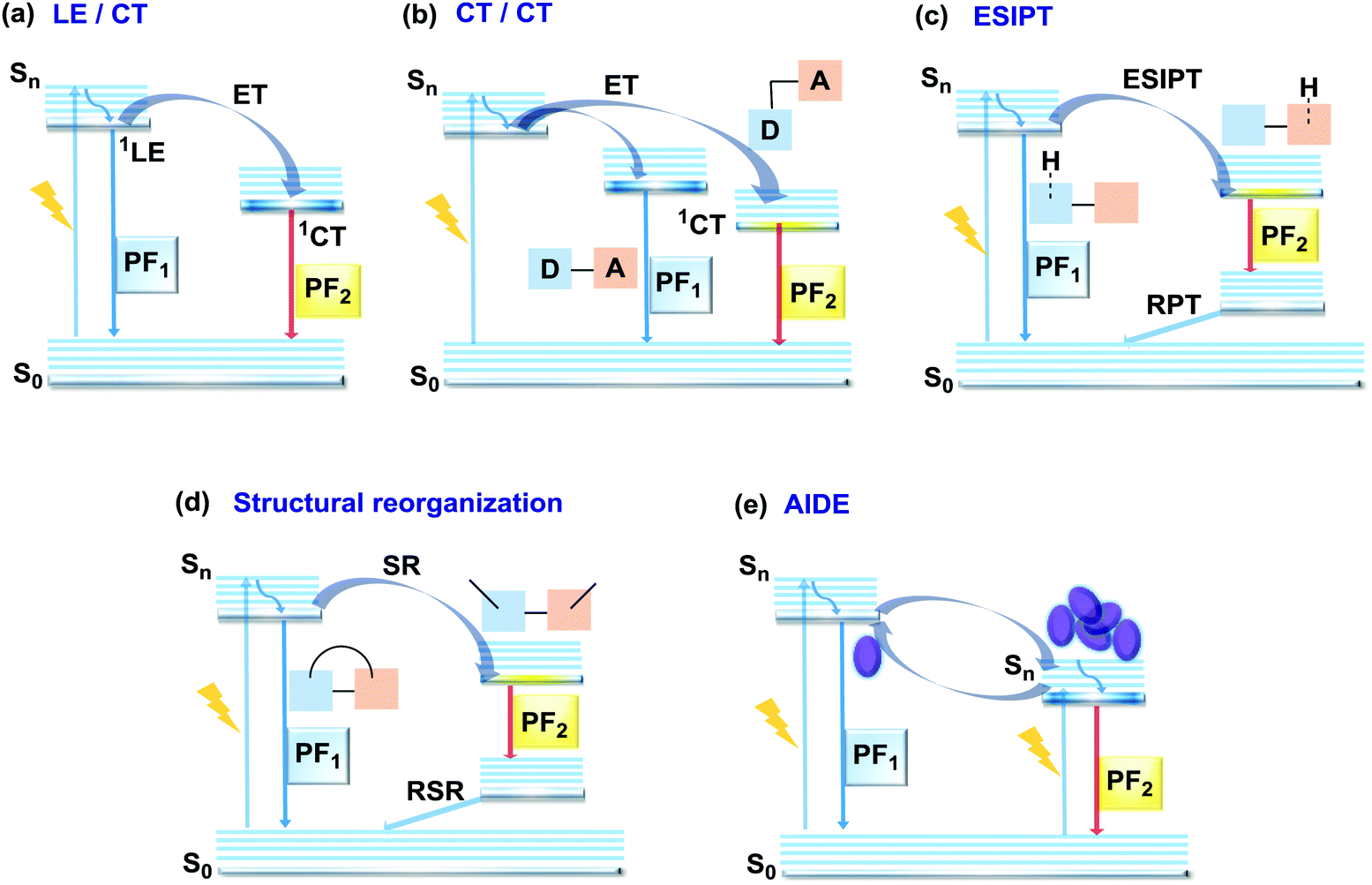 | ||
| Fig. 2 Jablonski diagrams, presenting an overview of the processes in (a) LE/CT, (b) CT/CT, (c) ESIPT, (d) structural reorganization and (e) AIDE types of prompt DE. ET – electron transfer; RPT – reverse proton transfer; SR – structural reorganization; RSR – reverse structural reorganization; AIDE – aggregation induced dual emission. | ||
The degree of conjugation between the donor and acceptor fragments significantly controls the nature of the observed emission bands. Partial coupling of D and A fragments, generally achieved via the introduction of a single C–C or N–C bond, leads to polarity-independent LE emission, generally featuring a vibronic structure and peaking at shorter wavelengths when compared to the Gaussian-shaped CT bands. For example, strong D–A coupling in the boron-dipyrromethene (BODIPY) – carbazole system 1 (Fig. 3) resulted in the equilibrium of 1LE (510 nm) and strongly emissive 1CT (620 nm) states (Fig. 4(a)), which was retained at various temperatures in solvents of different polarity.46 In turn, peculiar DE from two singlet states in 2 was observed in violation of Kasha's rule.18 DE in 2 was accompanied by a negligible structural rearrangement upon electronic excitation and subsequent relaxation, and optimal energy gaps of the two optical bands (S02 and S01) with fast (200 fs) internal conversion (S2 → S1) lead to decoupled relaxation and simultaneous radiative decay from both S2 and S1. Polarity independence of the S02 band and a red shift of the S01 band, respectively, give a hint of the LE/CT origin of the DE.47
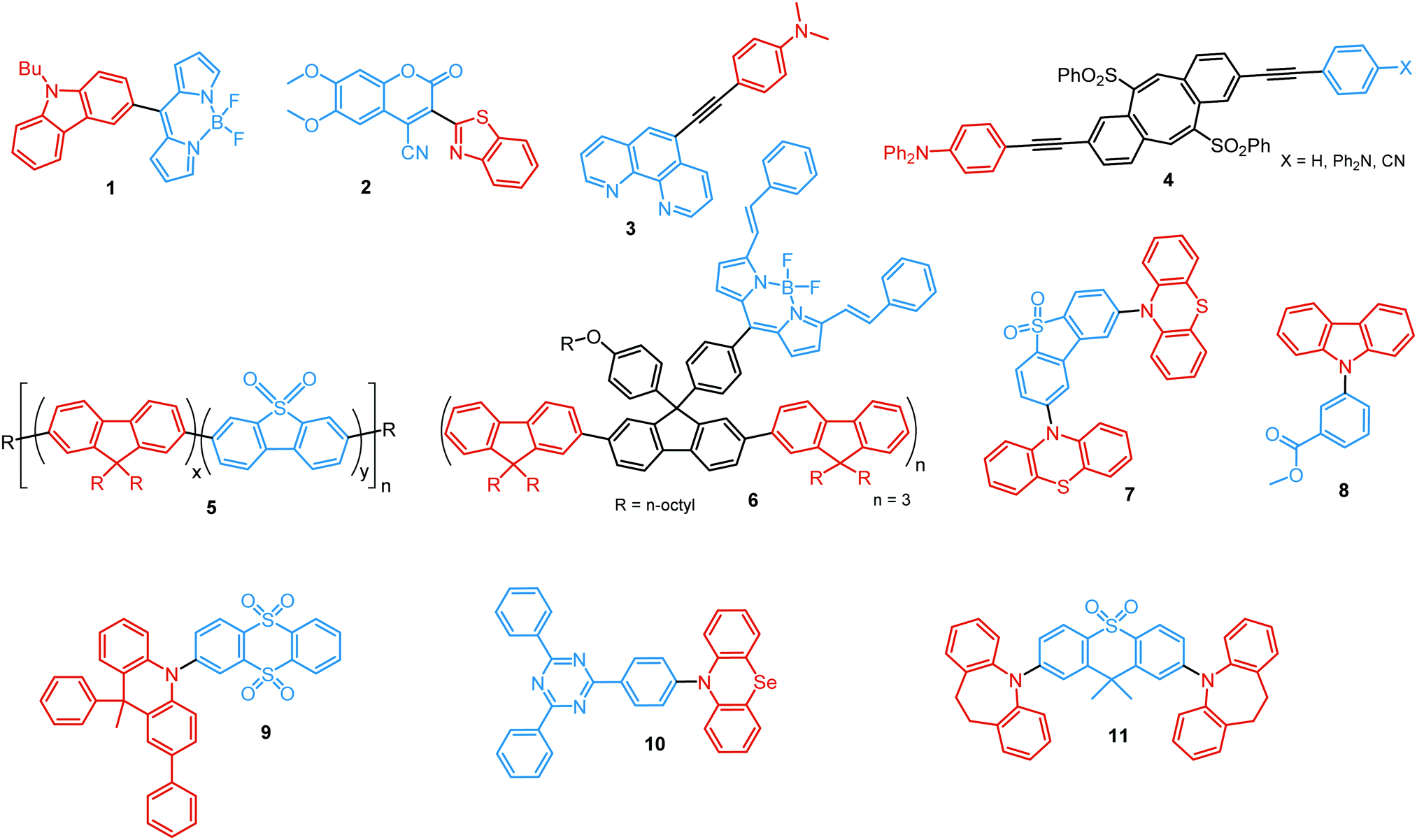 | ||
| Fig. 3 Examples of PF LE/CT and CT/CT dual emitters. Some of these compounds also possess RTP/TADF in the absence of oxygen (RT), so mixed category (i) and (ii) (Fig. 1(b)). | ||
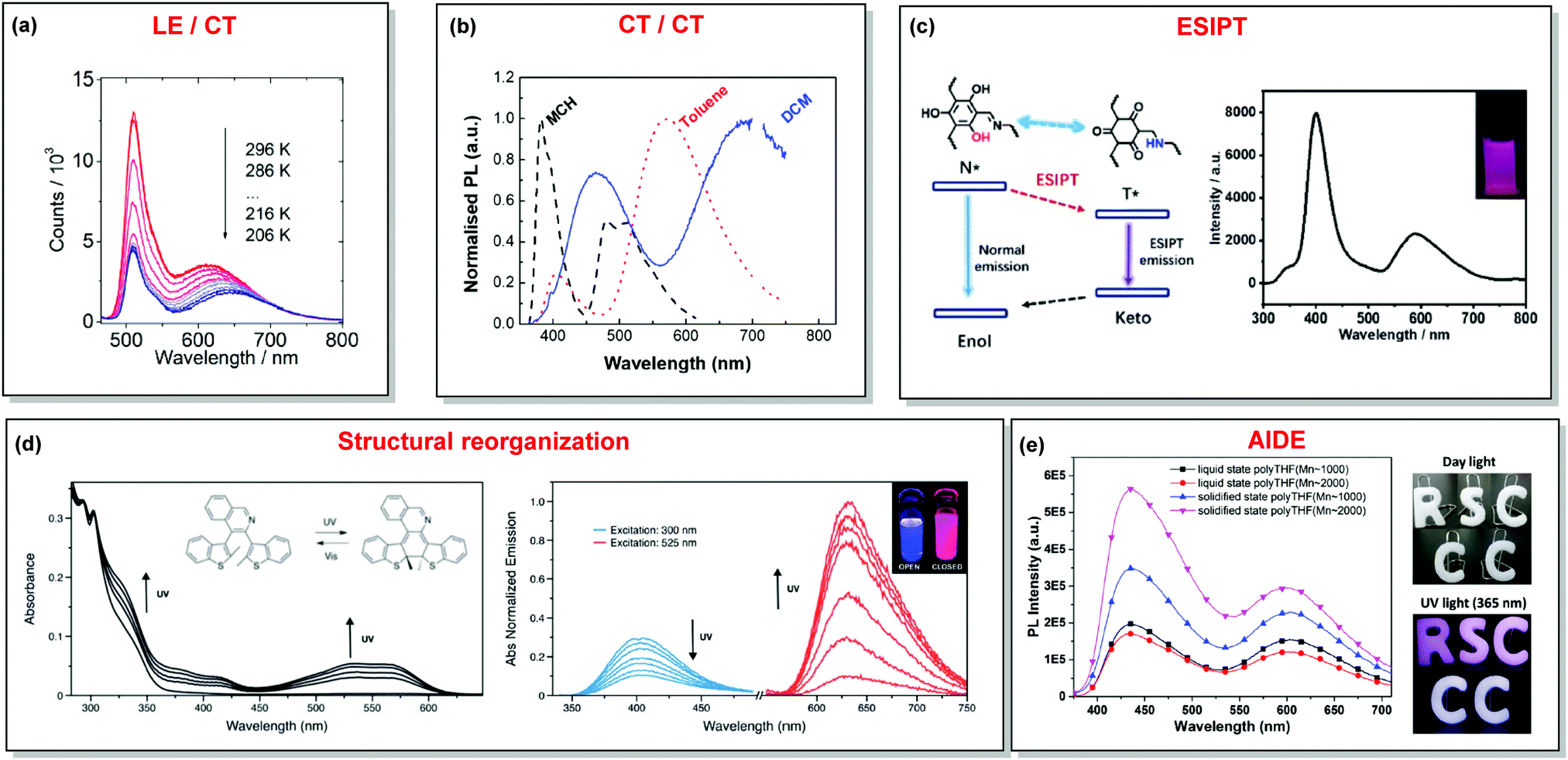 | ||
| Fig. 4 Examples of DE in (a) LE/CT (1); (b) CT/CT (10); (c) ESIPT (13); (d) structural reorganization (16); (e) AIDE (18) systems. (a) Temperature-dependent spectra of 1 in acetonitrile. Reproduced with permission.46 Copyright 2020 American Institute of Physics. (b) Normalised PL of 10 in different solvents: methylcyclohexane (MCH), toluene and dichloromethane (DCM). Excitation at 355 nm. Reproduced with permission.37 Copyright 2019 Royal Society of Chemistry. (c) The ESIPT DE mechanism of hydroxyl-containing 13; DE from 13 under 280 nm excitation; inset: a picture of 13 under 280 nm illumination. Reproduced with permission.39 Copyright 2019 Royal Society of Chemistry. (d) UV-vis spectral changes of 16 (2.5 × 10−5 M) in dichloromethane upon UV irradiation. Emission spectral changes of 16 upon UV irradiation.24 Reproduced with permission. Copyright 2020 Royal Society of Chemistry. (e) PL spectra of 18 (10−5 M) in pure liquid and solidified poly-THF at 20 °C (λex = 365 nm); photos of 18 (10−5 M) in solid letter molds under day light at 20 °C; fluorescence photos of 18 (10−5 M) in solid letter molds at 20 °C (λex = 365 nm). Reproduced with permission.59 Copyright 2019 Royal Society of Chemistry. | ||
Extension of the conjugated backbone by the insertion of triple C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bonds in the systems 3 and 4 facilitated the D–A coupling that resulted in DE from LE and CT states. Interestingly, LE emission was switched on at higher excitation energy, while the CT emission was accessible at lower excitation energy.48,49
C bonds in the systems 3 and 4 facilitated the D–A coupling that resulted in DE from LE and CT states. Interestingly, LE emission was switched on at higher excitation energy, while the CT emission was accessible at lower excitation energy.48,49
LE/CT DE has also been observed in purely organic polymers. The simultaneous observation of PL from both LE and CT states in fluorene-dibenzothiophene-S,S-dioxide random copolymer 5 was explained by the competition of the LE emission with the solvent reorganization time at low temperature in polar media. Such a competition clearly identifies the dual nature of the excited state as a function of molecular conformation (relaxation) and ratio of the monomer units.50 DE of 5 was successfully utilized in the single-polymer OLED, displaying broad greenish-white electroluminescence.
On the other hand, a fully range-separated chromophore decoupling is also responsible for unlocking the LE emission channel. Chujo et al. studied the influence of oligofluorene chain length on the electronic interaction between the chromophores in a series of oligofluorene-BODIPY dyads.51 Interestingly, only the heptamer 6 revealed supressed energy transfer resulting in deep-blue LE and red DE from the oligofluorene fragment and the ICT state to BODIPY, respectively. These results implied that the effective exciton diffusion length in oligofluorene might be from 5 to 7 fluorene units.
While simultaneous LE/CT emission is more common in C–C bonded D–A systems, it can also occur in more twisted N–C bonded D–A compounds. For instance, detailed analysis of the crystal structure of the D–A–D triad 7 revealed that conformation plays an important role in determining the fluorescence properties of the polymorphs. Thus, the band at 427 nm, assigned to the LE emission of the phenothiazine (PTZ) unit, was observed only in the polymorphs with a planarized D–A backbone, along with the CT band at 530–643 nm. The LE band completely disappeared in the crystals with orthogonal D–A orientation.20 Additionally, free rotation around the N–C bond of the D–A link is responsible for the equilibrium of pre-twisted (CT) and planar (LE) states in the push–pull carbazole-benzoate ester adduct 8. Ultrafast transient absorption spectroscopy indicated that photoexcitation into the LE state selectively accesses Frank Condon states localized on the carbazole chromophore, which react via ps electron transfer to form the emissive TICT states.52
Linking D–A units of balanced electron donating/accepting strength by an N–C bond gives rise to strong intramolecular CT states of n–π* origin, leading in turn to thermally activated delayed fluorescence (TADF).30,53,54 However, if the chromophores possess a certain degree of conformational flexibility (especially the donor unit),55 rotation around the N–C bond will inevitably lead to the equilibrium of at least two stable conformers, i.e. planar (quasi-axial) and twisted (quasi-equatorial) (Fig. 2(b)). Unlike systems consisting of partially coupled chromophores with unbalanced D–A strength (which emit from locally excited and CT states), well matched D–A combinations can exhibit CT emission from both stable conformations. For instance, chiral (R) and (S)-9-methyl-2,9-diphenyl-9,10-dihydroacridine donors in compound 9 not only switch on TADF and circularly polarized luminescence (CPL) but also stabilize planar and nearly-orthogonal conformations of the emitter. Two broad and structureless emission bands (430 and 577 nm) observed in toluene displayed a synchronous solvatochromic redshift in media of different polarity, which is a key characteristic of CT/CT DE.56 The inversion of the central six-membered ring of the D unit, leading to both quasi-axial and quasi-equatorial conformers, was discussed in detail in previous work using other examples of a flexible donor, i.e. phenothiazine and phenoselenazine.37,57 Stable conformations of the triazine-based emitter 10 reveal not only dual prompt fluorescence of CT/CT nature (Fig. 4(b)), but also the rarely observed combination of TADF and RTP. A phenothiazine-based analogue of 10, reported by Adachi et al., revealed a similar existence of two ground-state conformers with different energy gaps between the lowest singlet excited state and lowest triplet excited states.58 Increasing the size and flexibility of the donor led to the superposition of easily interchanging conformers in compound 11, featuring 10,11-dihydro-5H-dibenz[b,f]azepine (Az). As the flexibility of the Az donor unit would readily allow different conformers, dual CT emission can be ascribed to the combination of nearly-planar and nearly-orthogonal conformers. Of note, only nearly-orthogonal conformers showed TADF.21 It is worthy to mention that conformational heterogeneity in the nearly-orthogonal structures can occur not only in the systems with a flexible chromophore (e.g. phenothiazine), but also in the emitters with pseudoplanar segments. Thus, Zhang and co-workers proved that many frequently used nonaromatic planar segments, such as 9,9-dimethyl-9,10-dihydroacridine, are actually pseudoplanar segments and have two possible conformations: a planar form and a crooked form. In turn, molecules constructed from pseudoplanar segments can thus have two corresponding conformations, significantly impacting the performance of many TADF emitters.36
In addition to electron transfer upon photoexcitation, dual PF can also occur because of excited state intramolecular proton transfer (ESIPT mechanism Fig. 2(c)). First reported by Weller in 1955 for salicylic acid,60 ESIPT has been extensively studied ever since. In general, the presence of an intramolecular hydrogen bonding interaction between a hydrogen bond donor (–OH or –NH2) and a hydrogen bond acceptor (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– or –CO) is a necessary condition for the observation of ESIPT fluorescence. ESIPT is a unique four-step process, with the electronic ground state of the fluorophores typically existing in an enol form.61 Photoexcitation leads to the redistribution of the electronic charge, resulting in greater acidity for the hydrogen bond donor group and increased basicity for the hydrogen bond acceptor within the enol form. As a result, an extremely fast enol to keto phototautomerization (kESIPT ≥ 1012 s−1) takes place, with the excited state enol form rapidly converting to its excited keto form. After decaying radiatively back to its electronic ground state, a reverse proton transfer (RPT) takes place to produce the original enol form (Fig. 2(c)).62 Highly efficient ESIPT was observed in the 2-(2′-hydroxyphenyl)oxazole and triphenylamine adduct 12 (Fig. 5) and exploited in a single-component white OLED. The combination of experimental and theoretical data demonstrates that the enol form of 12 possesses hybridized local and charge-transfer (HLCT) excited state character, as opposed to the excited state keto-form, which possesses solely LE character. It was suggested that the electroluminescence in this case originated from the synergistic combination of the ESIPT equilibrium and the RISC process arising from the HLCT excited state character. The RISC of the enol form and relaxation of excitons from the triplet state to the singlet state in the electroluminescence, re-established the ESIPT equilibrium.63
N– or –CO) is a necessary condition for the observation of ESIPT fluorescence. ESIPT is a unique four-step process, with the electronic ground state of the fluorophores typically existing in an enol form.61 Photoexcitation leads to the redistribution of the electronic charge, resulting in greater acidity for the hydrogen bond donor group and increased basicity for the hydrogen bond acceptor within the enol form. As a result, an extremely fast enol to keto phototautomerization (kESIPT ≥ 1012 s−1) takes place, with the excited state enol form rapidly converting to its excited keto form. After decaying radiatively back to its electronic ground state, a reverse proton transfer (RPT) takes place to produce the original enol form (Fig. 2(c)).62 Highly efficient ESIPT was observed in the 2-(2′-hydroxyphenyl)oxazole and triphenylamine adduct 12 (Fig. 5) and exploited in a single-component white OLED. The combination of experimental and theoretical data demonstrates that the enol form of 12 possesses hybridized local and charge-transfer (HLCT) excited state character, as opposed to the excited state keto-form, which possesses solely LE character. It was suggested that the electroluminescence in this case originated from the synergistic combination of the ESIPT equilibrium and the RISC process arising from the HLCT excited state character. The RISC of the enol form and relaxation of excitons from the triplet state to the singlet state in the electroluminescence, re-established the ESIPT equilibrium.63
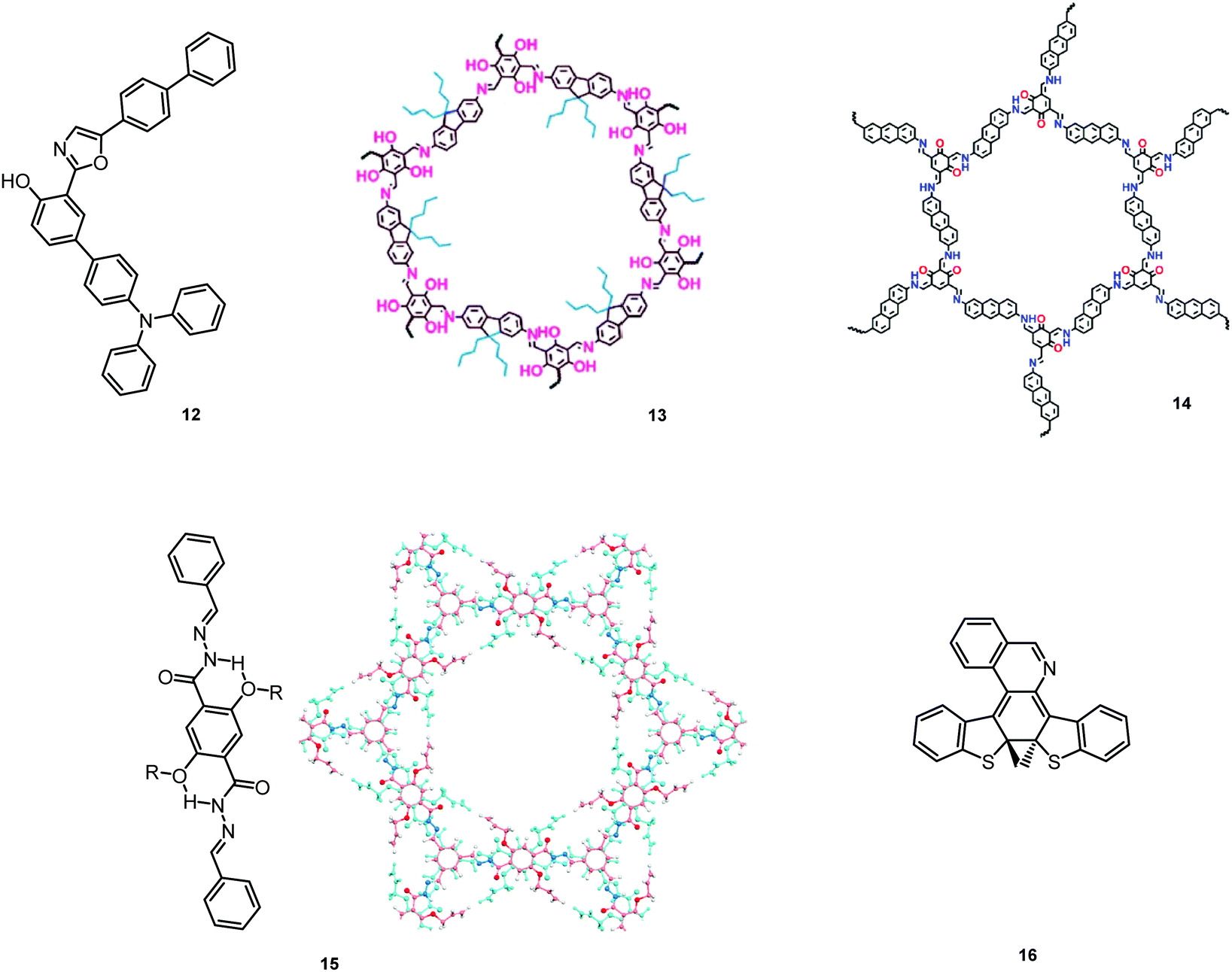 | ||
| Fig. 5 Examples of PF ESIPT and structural reorganization DE materials. 13: reproduced with permission.39 Copyright 2019 Royal Society of Chemistry. 14: reproduced with permission.65 Copyright 2018 American Chemical Society. 15: reproduced with permission.66 Copyright 2020 American Chemical Society. | ||
As opposed to a single intramolecular hydrogen bond in 12, covalent organic frameworks (COFs) are wonderful examples of employing multiple intramolecular hydrogen bonds for attaining molecular rigidity and emission enhancement.64 For example, multiple hydrogen bonds between hydroxyl and imine groups led to strong DE in 13. Fig. 4(c) illustrates the ESIPT process in 13. While blue emission at 400 nm was attributed to normal fluorescence from the enol state, the peak at 590 nm with a larger Stokes shift, was assigned to emission from the tautomeric keto form following ESIPT. Of note, both the n-butyl groups and intramolecular hydrogen bonds in 13 act as dual locks for the restricted intramolecular rotation (RIR) that enhance DE by minimizing non-radiative decay.39 Similarly, the selective incorporation of electronically active segments (anthracene and resorcinol) into the ordered structure of the COF 14 resulted in white light emission. The keto–enol tautomers give rise to DE, which can be tuned by the O-donor and N-donor matrix. For instance, a 0.32 wt% film of 14 in poly(methylmethacrylate) (PMMA) emits intense white light (CIE coordinates (0.35, 0.36)). From steady-state and time-resolved studies, the mechanism of the white light emission was unambiguously assigned to fluorescence, with the blue component originating from the π-stacked columns of anthracene, and the mixture of red and green emission from the keto–enol tautomerized resorcinol units. This study introduces COFs as a new class of readily processable, single-source white light emitters.65
While most reports on COFs are centered on the eclipsed solid state structures, Loh et al. have discovered dual PF in the two-dimensional acylhydrazone COF 15, with alkoxy side-chains solely in the antiparallel stacked structure. The intralayer and interlayer hydrogen bonding in the latter act synergistically to ensure RIR, thus turning on fluorescence. Curiously, the interlayer hydrogen bonds between the acyl oxygen and the side-chain hydrogen were found to be stronger in the DE antiparallelly stacked COF compared to those in the single emissive ones, underscoring the roles of competing inter- and intra-hydrogen bonds in the excited state conformers.66
Comparably to the ESIPT process, dual PF can also originate from structural reorganization (Fig. 2(d)) of the molecule upon external stimuli (Fig. 5, 16). Diarylethene-based compounds are a class of photochromic molecules, which can undergo photocyclization and photocycloreversion between differently coloured open and closed forms (Fig. 4(d)). Surprisingly, compound 16 is emissive in both forms: blue emission with a quantum yield of 0.04 from the open form (in dichloromethane (DCM) solution) accompanied by intense red emission with a quantum yield of 0.21 in the closed form. Due to the presence of the basic quinoline group, 16 shows pH sensitivity in DCM solution. 16 was demonstrated to be a potential fluorophore for super-resolution imaging, and also a rare example of a single-component red-blue pH switching material, reminiscent of litmus indicator but in emission rather than reflection.24
In all the above examples, the observed PF DE was the result of photoexcitation of an isolated molecule. However, some molecules emit upon aggregation (AIE)67,68 or upon the formation of an excited state complex (excimer or exciplex).69,70 Such compounds generally bear either an AIE-active unit that restricts intramolecular rotations (e.g. tetraphenylethene67,71 or 2,3,4,5-tetraphenylsilole72), or an electron rich/deficient unit that is prone to excimer/exciplex formation (e.g. pyrene73 or anthracene74). Smart molecular design incorporating such units can lead to DE from both monomeric and aggregated states in the condensed phase (Fig. 2(d)). For instance, tetraphenylethene (TPE) – anthracene-(aminomethyl)phenylboronic ester derivative 17 (Fig. 6) in tetrahydrofuran exhibits DE upon the gradual addition of water. At low water content well-resolved emission of the anthracene unit was observed in the blue spectral region, whereas upon further addition of water green fluorescence emerged due to the aggregation of the AIE-active TPE fragments. It was proposed that 17 could act as a dual-fluorescence emission sensor for detecting water in organic solvents.75
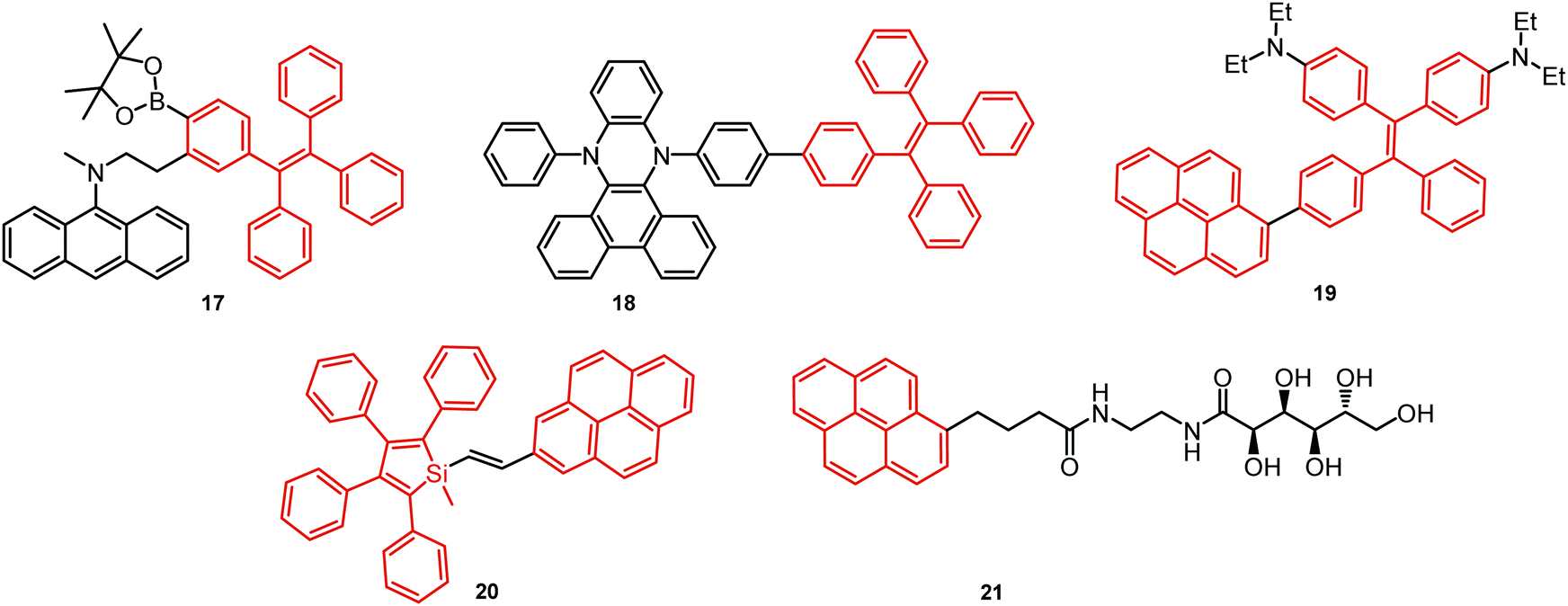 | ||
| Fig. 6 Examples of PF AIDE dual emitters. | ||
A new strategy to achieve single-component white emission was demonstrated by Mei, Su and co-workers by combining vibration-induced emission (VIE) and AIE.59 VIE-active molecules can undergo an excited-state conformational transformation from an intrinsic bent shape to a planarized shape. Bearing that in mind, a VIE-active phenazine unit and an AIE-active TPE unit were combined in compound 18. While a peak from the planarized state (610 nm) was solely observed in the solutions of low viscosity, increasing the solution viscosity unlocked the blue-green AIE emission of the TPE unit. By modulating the viscosity of toluene solutions of 18 (via changing the molar fraction of polytetrahydrofuran), single-molecular white-light emission with CIE coordinates of (0.33, 0.37) was achieved by balancing the VIE effect restricted intramolecular rotations with AIE, resulting in intense white emission (Fig. 4(e)). Another example of white emission from a single AIE-active compound was reported by Tang's group.76 A combination of blue-emissive pyrene with AIE-active TPE (forming green–yellow emissive aggregates at high water contents) yielded compound 19 with warm-white emission (CIE coordinates 0.30, 0.41) and a PLQY of 12%. Similarly to TPE, substituted siloles are also renowned AIEgens. Polymorphism-dependent AIE-DE (AIDE) of 20 was achieved by balancing the blue emission of the pyrene motif with the yellow emission of the tetraphenylsilole that restricted intramolecular rotations in the condensed state.77 As pyrenes are prone to π–π stacking and red-shifted excimer emission, DE of the organogelator 21 is engineered from the combination of monomer and excimer pyrene emissions.78
2.2. Dual prompt and delayed photoluminescence
As stated previously, free rotation around the N–C bond between a donor and acceptor, as well as steric flexibility of the units may lead to conformational heterogeneity. In this case, simultaneous radiative decay from the most stable planar (quasi-axial, ax) and twisted (quasi-equatorial, eq) forms can take place.57 Depending on the balance of donor/acceptor strengths, the planar conformer can emit either LE or CT-type prompt fluorescence. As the electron density of the planar conformer is commonly delocalized over the whole molecule (hybrid LE/CT character), the singlet–triplet energy gap (ΔEST) is often too large to harvest triplets. The twisted form with its well-decoupled D and A units is generally capable of up-converting the triplets due to the minimized ΔEST, giving rise to TADF.57 Therefore, if both planar and twisted forms can co-exist and have distinguishable emission spectra, dual PF/DF emission can occur (Fig. 7(a)).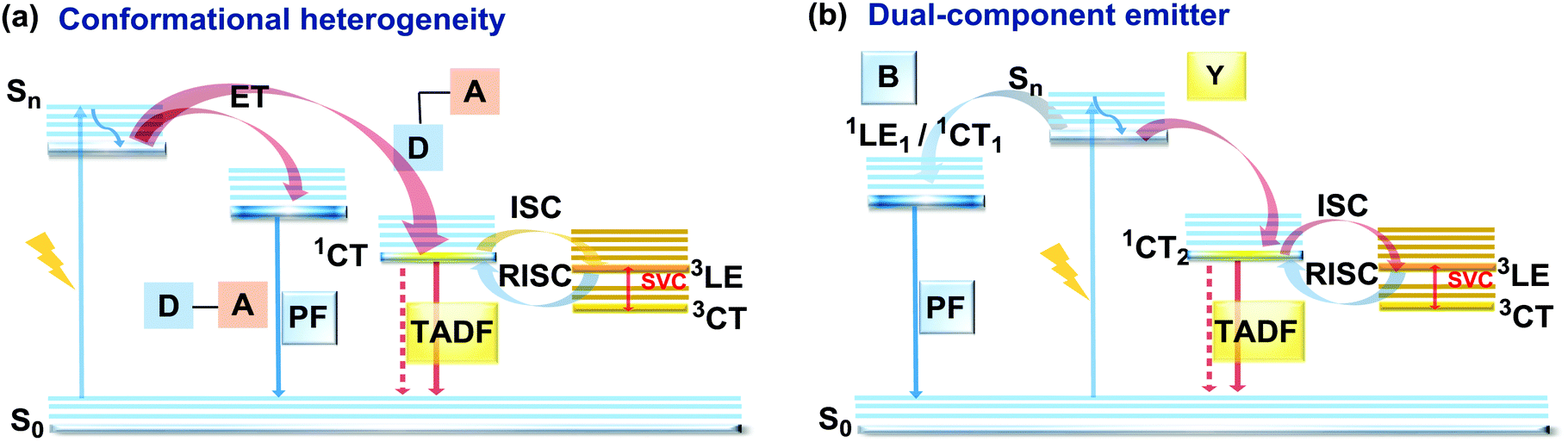 | ||
| Fig. 7 Jablonski diagrams, presenting an overview of the processes in the PF/DF dual emitters with (a) conformational heterogeneity and (b) dual (blue (B) and yellow (Y)) emissive components. SVC – spin-vibronic coupling. | ||
Conformational heterogeneity strongly depends on the rigidity of the media, as well as molecular organization and packing forces. For instance, three polymorphs with various photophysical properties were obtained for the diphenylamine-xanthone derivative 22 (Fig. 8). These polymorphs exhibited specific intermolecular non-covalent interactions (such as π⋯π stacking, hydrogen bonding, and C–H⋯π interactions) which determined their emission characteristics. In the absence of intermolecular π⋯π interactions, only LE prompt emission was observed, while strong π⋯π stacking led to only CT emission at a different wavelength. As expected, balanced weak intermolecular interactions resulted in the intermediate case, with the observation of DE. While retaining PF from the locally excited states, weak π⋯π stacking between the acceptor units facilitated the RISC process, unlocking the TADF channel.38 Coexistence of dual stable conformations in 23 was successfully exploited for the fabrication of a warm WOLED (CIE (0.41, 0.47); maximum external quantum efficiency EQEmax = 16.34%) with complementary emission colours contributed by the two conformations. Interestingly, the authors suggested that the originally wasted triplet energy of the nearly-planar conformation could be transferred to the nearly-orthogonal isomer and then harvested via TADF, fully utilizing the excitons. Furthermore, the phenyl spacer in 23 was purposely used to partially disrupt the communication between the D and A units, leading to DE of balanced intensity.6
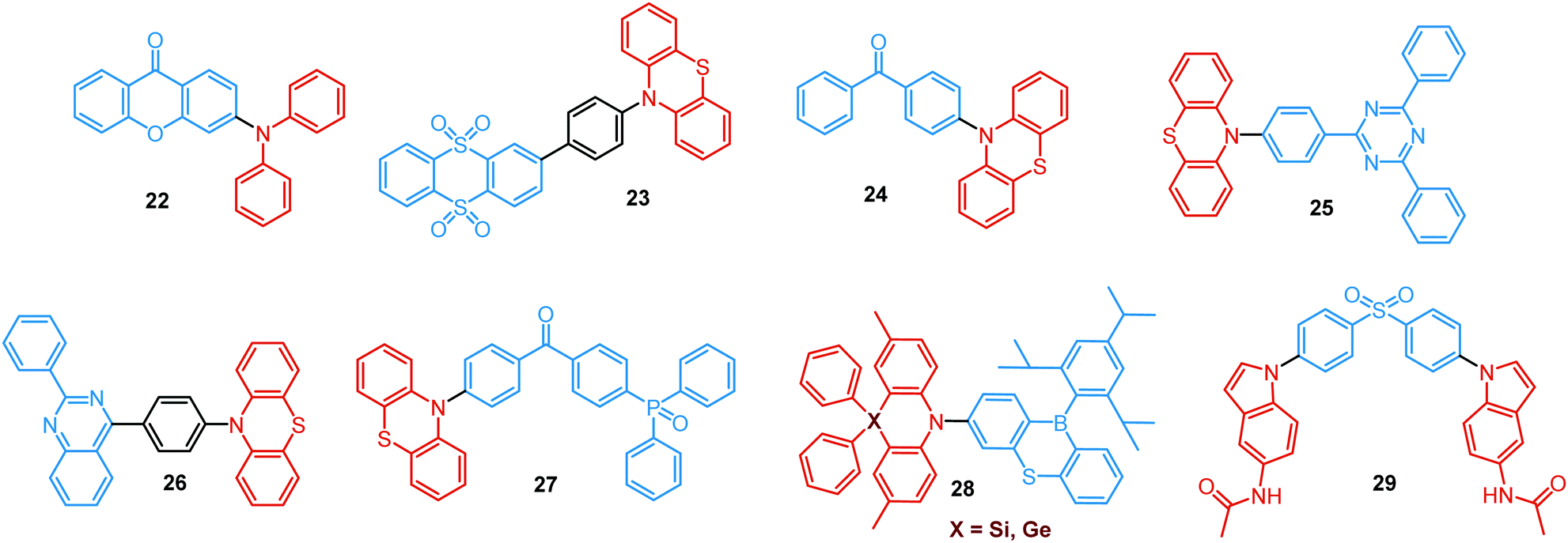 | ||
| Fig. 8 Examples of PF/DF dual emitters featuring conformational heterogeneity. | ||
Conformations of a DE molecule can also be controlled by the degree of crystallinity. For example, photoluminescence of the crystalline compound 24 is dominated by blue emission, and that of amorphous 24 by yellow TADF. This phase-dependent emission is attributed to the interchange of H-extra and H-intra molecular conformations of the phenothiazine unit in the crystalline and amorphous phases, leading to the highly twisted (eq) and planar (ax) conformations, respectively. The combination of blue and yellow emissions from a semi-crystalline film of 24 resulted in WOLEDs with a maximum EQE of 6.2% and CIE (0.34, 0.46).11 The existence of two ground-state conformers, with different singlet–triplet energy gaps (1.14 and 0.18 eV) arising from the distortion of the phenothiazine unit, was confirmed by both theoretical and experimental results for the triazine derivative 25. Interestingly, both ax and eq excited state conformers of the PTZ unit exhibited CT emission. However, only the nearly-orthogonal conformer with a small ΔEST showed TADF in toluene solution. A combination of the negligible energy barrier between the conformers and a large difference in ΔEST ensured observation of dual PF (CT)/TADF emission.58 Interestingly, further investigation of the photophysical properties of 25 by Chou et al.79 revealed that both ax and eq conformers relax to the same orthogonal structure in the excited state, accompanied by the planarization of the PTZ unit. Such simultaneous structural relaxations confirm the stability of the CT state leading to TADF. However, prior to this relaxation the ax conformer is capable of emitting PF of CT character. In turn, a combination of PF (CT) and TADF in the degassed toluene solution lead to DE.
Recently, Zhang and co-workers have outlined the essential criteria for realization of efficient single molecule WOLEDs.35 It is necessary that such emitters should (i) possess a pseudo-planar segment with non-contiguous conjugation to produce heterogeneous conformers, (ii) possess a less rigid conjugated configuration to allow the interconvertible transition between conformers, (iii) provide a low energy difference between conformers, to stabilize their co-existence, and (iv) generate complementary emissions from each conformer for white lighting. Following these design principles, 26 (Fig. 8) featuring a phenyl spacer between a PTZ donor and a quinazoline acceptor is indeed a white emitter. As expected, dual PF/DF emission of complementary blue and orange was observed in solution. Astonishingly, the single crystal X-ray diffraction analysis revealed that ax and eq conformers of 26 co-exist in a precisely 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. The co-existence of both conformers in a single crystal was reported previously,57,80 however, the balanced ratio of the conformers is hard to achieve. More importantly, WOLEDs (CIE (0.32, 0.34)) based on 26 yielded one of the highest reported EQE values (10.1%) for a purely organic single-component emitter.
:1 ratio. The co-existence of both conformers in a single crystal was reported previously,57,80 however, the balanced ratio of the conformers is hard to achieve. More importantly, WOLEDs (CIE (0.32, 0.34)) based on 26 yielded one of the highest reported EQE values (10.1%) for a purely organic single-component emitter.
TADF can occur not only as a result of intramolecular interactions, but also due to intermolecular or “exciplex” CT states.81 The combination of intra- and intermolecular CT in asymmetric 27 is related to the formation of intermolecular C–H⋯O hydrogen bonds between the D–A units (Fig. 9(a)). Surprisingly, intermolecular TADF was observed in crystals of 27, unlocking tuneable multi-emission pathways. Even though folding angles of the PTZ moieties and the molecular conformations are not the key factors for the DE properties in this case, molecular flexibility was shown to assist the formation of hydrogen bonding by adopting suitable conformations to decrease the energies of the aggregated systems.82
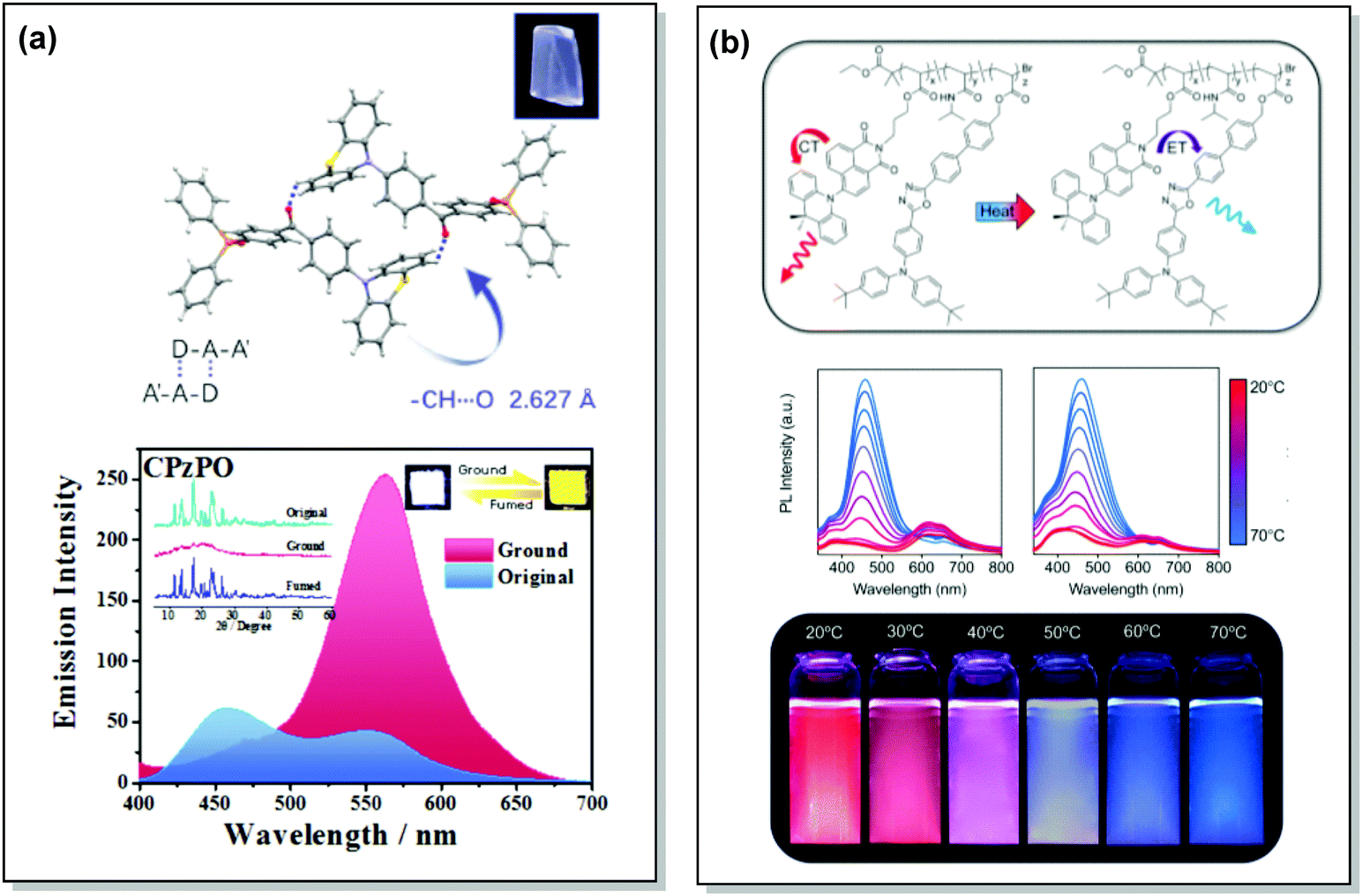 | ||
| Fig. 9 Examples of PF/DF DE in the systems with (a) conformational heterogeneity (27) and (b) multicomponent emitter (32). (a) Single-crystal structure and luminescence picture of 27; emission spectra of 27 before and after grinding (the left inset shows the pXRD spectra of the original sample, ground sample, and fumed sample; the right inset shows the luminescence photographs (under UV light) of the original and ground samples); reproduced with permission.82 Copyright 2017 Wiley-VCH. (b) Schematic representation of the thermal response for 32; temperature-dependent emission spectra of 32; visual representation of 32 at various temperatures. Reproduced with permission.85 Copyright 2020 American Chemical Society. | ||
Flexibility of the donor units is greatly enhanced by incorporation of heavy atoms (silicon, germanium), leading to conformational heterogeneity and DE in the TADF emitters 28. While both TADF molecules adopted dual conformations in solutions and thin films, efficient excited energy transfer from the ax to eq conformers took place, enhancing the TADF characteristics.83 Using 28 (X = Si) as the emitter, blue OLEDs (CIE (0.14, 0.26)) were fabricated with EQEmax 27.6% and low efficiency roll-off.
An elegant example of utilizing PF/DF dual emission for ratiometric sensing was introduced by Zhu and Ågren.84 To achieve a balanced contribution of LE (internal reference signal) and CT (sensing/reporting signal) in the excited state, a strategy was presented to regulate the electron-donating ability of the donor group by adjusting the effective conjugation length. System 29, bearing 5-acetaminoindole donors freely rotating around the D–A bond, displayed PF from the LE state in addition to TADF from the CT state. While the LE fluorescence is insensitive to the environmental polarity, the wavelength and lifetime of the TADF signal changed along with the external stimuli. This behaviour made it possible to establish a 3-D mapping method of polarity, using ratiometric comparisons of emission wavelength and lifetime. This strategy was applied for detection of changes in phospholipid microenvironmental polarity, offering convenient and accurate identification of cholesterol-releasing membrane lesions.
Another way to achieve PF/DF DE is by combining components with different emission colours within a single asymmetric D–A–D′ molecule (Fig. 7(b)). In this case, one part of the molecule is capable of only conventional PF following the initial photoexcitation, while the other part readily up-converts triplets leading to TADF. DE compound 30 was specifically designed to inherit the blue PF and yellow TADF of its symmetric carbazole (blue) and phenothiazine parents (yellow) (Fig. 10). As a result, 30 displayed both mechanochromism (Δλem,max > 100 nm) and bright white emission (CIE (0.27, 0.29)) in the solid state arising from a combination of conventional fluorescence and TADF. DE in this case was likely to originate from two different radiative decays in the molecule, arising from two independent CT transitions from carbazole or phenothiazine donors to the diphenylsulfone acceptor. The authors demonstrate that radiative decay disobeying Kasha's rule and avoiding the expected energy transfer from the blue emitting centre to the yellow emitting band occurs in this asymmetric system.86
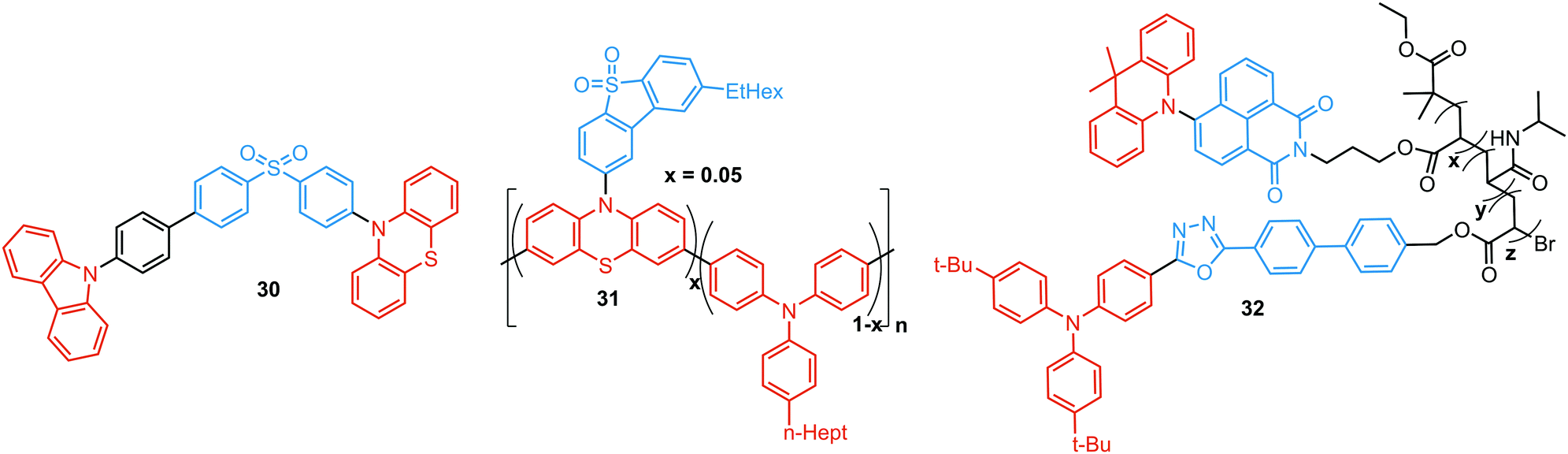 | ||
| Fig. 10 Examples of PF/DF dual-component dual emitters. | ||
Controlled copolymerization presents a wonderful opportunity to achieve white emission from blue/yellow emissive monomers, or monomers that act as donors and acceptors for through-space CT formation. Thus, by combining non-planar blue-emitting triphenylamine (TPA) host and yellow TADF dibenzothiophene-PTZ guest in the polymer backbone of 31, well-balanced white emission was produced. Interestingly, the optimized ratio of TPA monomers was found to suppress exciton concentration quenching and boost the rate of the RISC process. Therefore, a solution-processable warm WOLED (CIE coordinates (0.38, 0.35)) with EQE = 7.1% was prepared.87 Such copolymers have applications not only in WOLEDs, but also in ratiometric sensors and TADF biological imaging. For instance, Hudson's group has recently incorporated a red-emitting naphthalene-imide based TADF fluorophore and a blue-emissive Förster resonance energy transfer (FRET) oxadiazole derivative into a flexible poly(N-isopropylacrylamide) matrix, giving rise to a thermoresponsive copolymer 32 (Fig. 10).85 Owing to the sensitivity of the RISC process to both thermal energy and oxygen concentration, 32 was demonstrated to be a highly effective sensor for both temperature and molecular oxygen (Fig. 9(b)).
2.3. Dual prompt and RTP photoluminescence
Purely organic materials exhibiting room temperature phosphorescence are attracting tremendous interest due to their low toxicity, low cost, and favourable environmental impact compared to organometallic phosphors. In addition, specific optical features associated with organic phosphorescent materials such as long afterglow lifetimes have opened the way to new applications including anti-counterfeiting technologies, temperature monitoring, sensing and bio-imaging.88 However, observation of triplet emission requires efficient ISC between the singlet and triplet states. Although formally spin forbidden, El-Sayed's rule states that ISC can be promoted when the S1 → Tn transition involves a change of the molecular orbital type89 (e.g. nπ* → ππ* in case of the N–C bond).90 Similarly, the ISC rate can be further enhanced by the introduction of heavy atoms (e.g. S, Se, Br, I), which provides an alternative reservoir for conserving angular momentum through the spin–flip. As well as generating triplet states, observing RTP requires that the molecules do not suffer quenching by non-radiative processes before the slow (forbidden) emissive processes can act. This can be achieved by restriction of intramolecular rotations or vibrations, through suitable molecular packing, hydrogen/halogen bonding, host–guest effects, or (most commonly) by employing cryogenic temperatures which activate low temperature phosphorescence (Fig. 11). In most cases, molecular oxygen must also be removed, as this acts as a triplet quencher.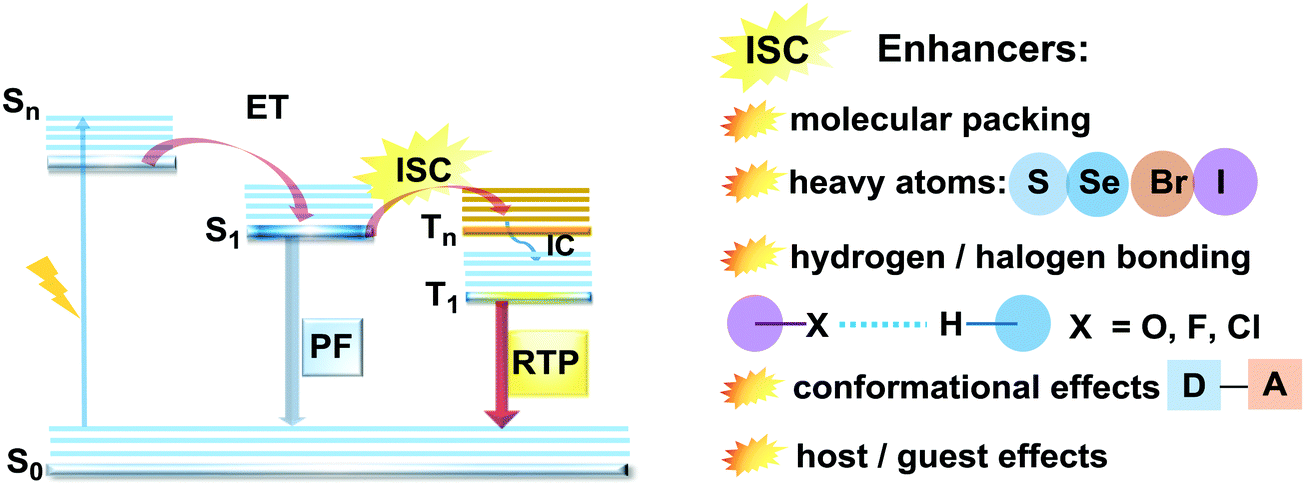 | ||
| Fig. 11 Jablonski diagram, presenting an overview of the processes in PF/RTP DE along with strategies for RTP activation. | ||
As both RISC and PH act upon the same reservoir of triplet states, the balance of RTP and TADF emission can be achieved by fine-tuning the activity of TADF though ΔEST and its sensitivity to molecular conformation. Thus, Ward et al. showed that twisting the eq conformation of the phenothiazine D units in a series of D–A–D emitters leads to TADF, while the nearly planar ax D is instead responsible for RTP.91
Bearing in mind the conditions for RTP, the essential criteria for the occurrence of dual PF/RTP can be outlined as follows: (i) strong fluorescence in the nanosecond region; (ii) strong ISC between S1 and Tn states (achieved by following El-Sayed's rule, introduction of heavy atoms, control of molecular packing, or otherwise); (iii) moderately large S1–T1 gap (for the observation of two bands); (iv) adequate suppression of triplet-quenching.
A wide range of PF/RTP dual emitters have been reported in recent years (Fig. 12), utilizing different ways to enhance ISC. For instance, PF/RTP DE along with dual mode mechanochromism was observed in the asymmetric D–A–D′ molecule 33. The presence of nπ* → ππ* transitions, in addition to the large sulphur atom and the carbonyl group, assisted in activating ISC. The carbonyl group also enhanced intermolecular interactions, highly constraining the thermal vibrations of 33 in the crystal and impeding the quenching by oxygen (Fig. 13(a)). A dimer with parallel molecular packing further promoted ISC, generating RTP with a long lifetime of ∼410 ms due to the larger transition dipole moment when compared to that of the monomer, and close energy level between S1 and Tn. Persistent RTP was attributed to the layer-by-layer stacking structure with alternating carbazole-centered or dibenzothiophene-centered columns in the crystal, impeding the non-radiative relaxation induced by oxygen and thermal vibrations.92
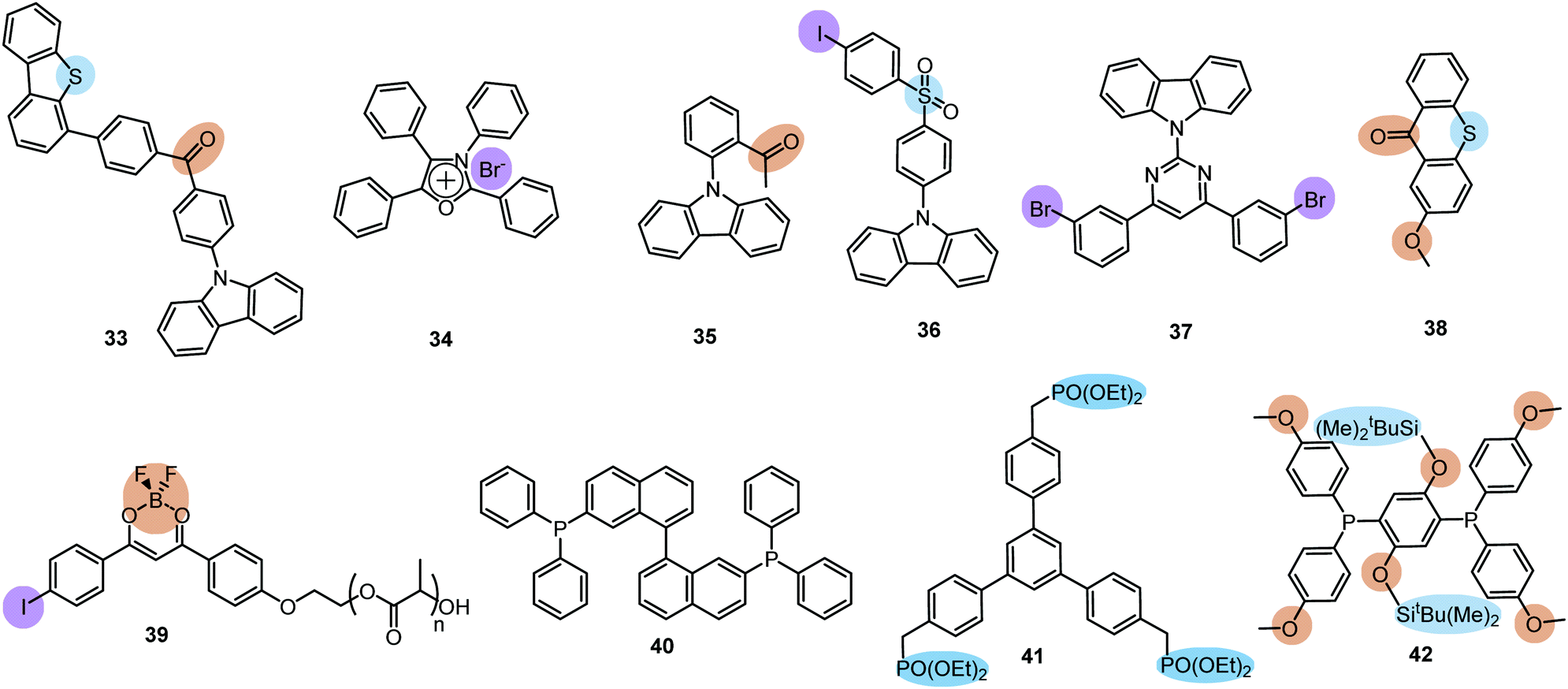 | ||
| Fig. 12 Examples of PF/RTP dual emitters. Heavy atom and hydrogen/halogen bonding features are highlighted. | ||
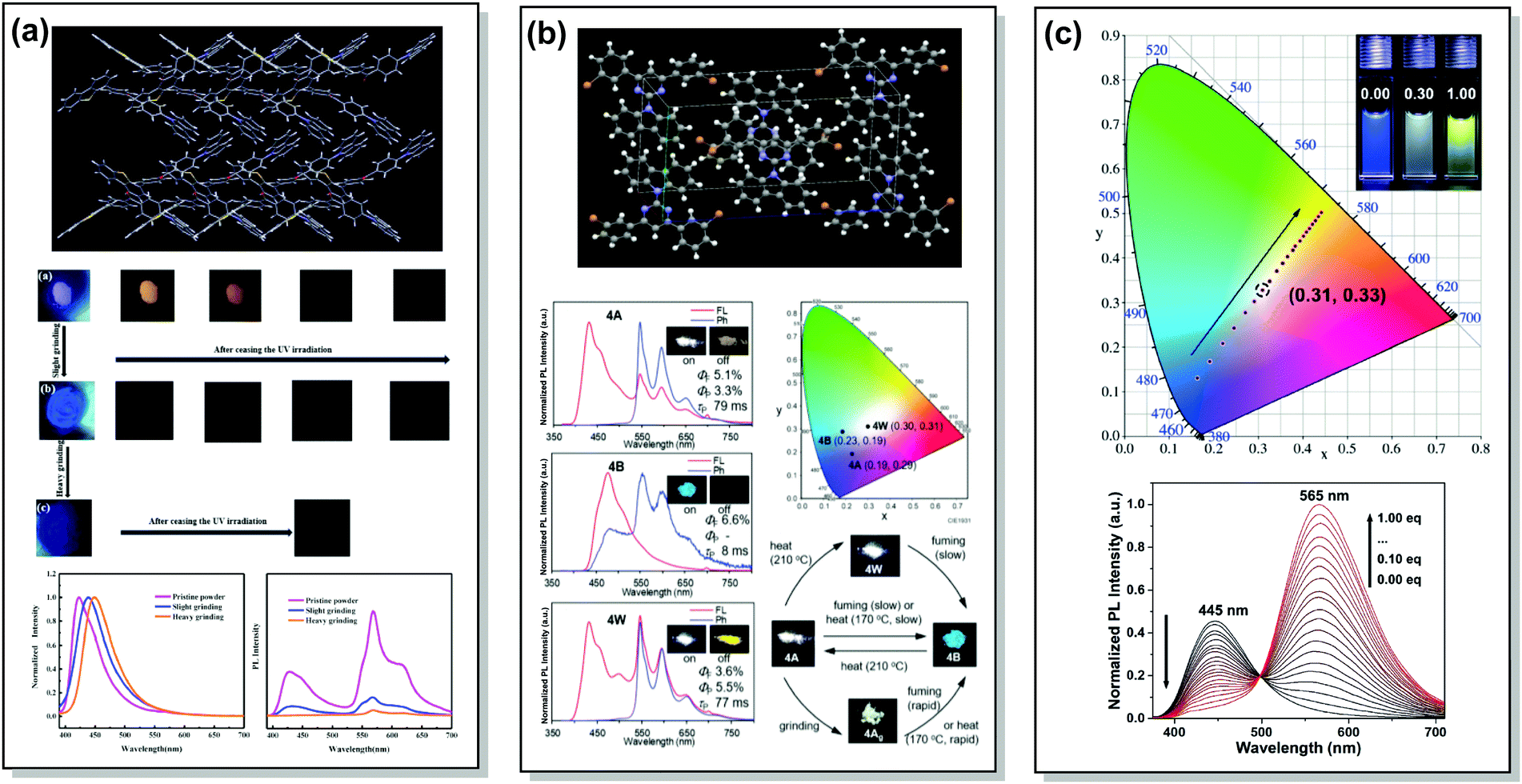 | ||
| Fig. 13 Examples of PF/RTP dual emission in systems with (a and b) heavy atom and hydrogen bonding effects (33, 37) and (c) host/guest effect (45). (a) The layer-by-layer stacking structure of 33 with alternative carbazole-centered or dibenzothiophene-centered columns; optical images of the pristine powder, slightly ground powder and heavily ground powder of 33 under 365 nm UV light and afterglow after ceasing the UV light; steady-state photoluminescence spectra and delayed emission spectra of the pristine powder, slightly ground powder and heavily ground powder of 33. Reproduced with permission.92 Copyright 2020 Elsivier. (b) X-Ray crystal packing of 37; steady-state PL and phosphorescence spectra of various polymorphs of 37 (inset photos are emission images before (on) and after (off) removal of the excitation source (black light, at 350 nm)); CIE 1931 coordinates of emission of various polymorphs of 37; emission colour switching between various polymorphs of 37 stimulated by heating, chloroform-vapor-fuming, and mechanical grinding. Reproduced with permission.12 Copyright 2020 Royal Society of Chemistry. (c) CIE coordinates of 45 with different CB[8] ratios in water at 298 K (inset: photographs of 45 with 0.00 (left), 0.30 (middle) and 1.00 (right) eq. CB[8] in water); PL spectra of 45 (50 mm) with different ratios of CB[8] (0.00–1.00 eq.) in water at 298 K (λex = 365 nm). Reproduced with permission.95 Copyright 2020 Wiley-VCH. | ||
A facile strategy for the construction of a single-molecule white emitter was reported by Tang's group by utilizing blue fluorescence and yellow RTP.41 Yellow RTP was achieved in 1,2,3,4-tetraphenyloxazolium salts with different counterions. In particular, compound 34 with a bromide counter ion exhibited outstanding RTP due to the external heavy atom effect via anion–π+ interactions, supported by single crystal X-ray diffraction analysis and theoretical calculations. As 34 also exhibited blue fluorescence in the solid state, white emission was realized from films and solids by tuning the degree of crystallinity.
Carbazole is a well-known building block in ultralong RTP systems, and acetophenone acts as a strong ISC facilitator. Therefore, combination of both moieties in a single D–A molecule can logically lead to DE. The simultaneous observation of fluorescence, TADF and ultralong RTP in crystals of 35 can be attributed to the RISC process from the T2 to the S1 state.25
A fascinating design strategy for pure organic PF/RTP emitters combining the benefits of a diphenylsulfone group, crystalline state, and heavy atom effects was realized in compound 36. While introduction of the iodine atom exerted only a minimal effect on the molecular energy levels, intense fluorescent (383 nm, 0.37 ns) and phosphorescent (559 nm, 19 ms) peaks were observed. Interestingly, dual emitter 36 exhibited sensitivity to mechanical stimuli, displaying a red-shift and a drop in the intensity of its prompt emission upon grinding.93
An elegant way of controlling RTP was recently realized in the polymorphs of bromine-decorated carbazole-pyrimidine derivative 37. Breaking the symmetry of the molecular structure provides three crystal polymorphs, two of which are RTP active. The fraction of phosphorescence emission was increased by heating, thereby leading to white-light emission (Fig. 13(b)).12
A complex D–A molecular architecture is not an essential condition for the observation of efficient PF/RTP DE. Multiple hydrogen bonds observed in the crystal structure of methoxy-substituted thiaxanthone 38 enhanced DE by a precise manipulation of the π–π stacking mode and intermolecular interactions compared to a non-substituted thiaxanthone. This result can be ascribed to the following factors: (i) ISC rate enhancement achieved by the introduction of the methoxy substituent; (ii) acceleration of the PF and RTP induced by the formation of M-aggregation or J-aggregation; (iii) multiple intra-and intermolecular hydrogen bonding interactions suppressing non-radiative processes. Thus, by the introduction of a simple substituent the PL efficiency of powders increased from 1.5% for the parent thioxanthone to 21.4% for 38 with pure white light emission (CIE (0.33, 0.33)).94
While the above-mentioned examples of dual PF/RTP were observed in highly-ordered crystalline states, intense dual blue PF/yellow RTP was achieved by Fraser et al. in a polymer ratiometric sensor, 39 (Fig. 12). Astonishingly, nanoparticles with balanced fluorescence and phosphorescence intensities were fabricated from a high molecular-weight polymer 39 (Mn 7.300 Da) and were successfully tested in mice as ratiometric tumour hypoxia imaging agents.96
An interesting case of phosphorescence quantum yield enhancement was observed in the racemic form of (2,2′-bis(diphenylphosphino)-1,1′-binaphthyl) (BINAP) 40, when compared to the homochiral form. Owing to the balanced intensities of PF (500 nm) and RTP emission (680 nm), compound 40 was found to be applicable in warm WOLEDs. The optimized single-molecule WOLEDs for racemic BINAP 40 achieved a EQEmax of ∼1.6% and CIE coordinates of (0.37, 0.44).97
Even though RTP efficiency often depends strongly on molecular packing, one of the first examples of halogen-free amorphous purely organic PF/DF DE was recently reported by Reineke and co-workers.98 Ultralong green RTP with lifetime >2 s was observed along with deep-blue prompt fluorescence in the amorphous solution-processed film of the terphenyl phosphonate 41. Similarly, the powder of the phosphynyl-substituted disiloxybenzene 42 revealed simultaneous fluorescence (374 nm) and phosphorescence (470 nm) under vacuum. The authors proposed that the easily detectable emission maxima of PF and RTP could enable accurate ratiometric luminescence sensing of temperature and molecular oxygen using a simple steady-state set-up.99
RTP can readily occur in compounds with a simple molecular architecture if supported by molecular packing and/or ISC-enhancing groups. Alternatively, a D–A structure with well-chosen molecular fragments allows minimization of both S1–T1 and S1–Tn gaps, which was shown to facilitate spin–orbit coupling regardless of the molecular packing.31,32 Furthermore, Ward et al. proved that manipulation of D–A conformers by introduction of substituents at the appropriate positions can switch on the RTP channel. For example, an inward-facing methyl substituent on the PTZ donors in the D–A–D derivative 43 gave a nearly planar axial conformation (Fig. 14). Locking this axial form in a solid Zeonex matrix resulted in the observation of DE from PF LE origin and PTZ-based phosphorescence.27 Similar to the incorporation of RTP emitters in a Zeonex matrix, Mei, Zhang and co-workers have recently demonstrated the use of non-luminous crystalline solvent matrices based on dimethyl sulfoxide (DMSO): [Al(DMSO)6]X3 (X− = Cl− or Br−). Encapsulating a trace amount of the aromatic-acid-based phosphor 44 as guest within the metal-solvent host matrix resulted in a single crystal complex featuring bright long-lasting RTP with a lifetime up to 1.9 s and quantum yield of 58.3%. The enhanced ISC and suppression of non-radiative pathways by the rigid matrix are responsible for the strong RTP. White light with PLQY of 29.5% was achieved utilizing the blue PF and yellow RTP emissions. The polychromatic behaviour of the complex was exploited in a proof-of-concept data encryption/decryption application.100
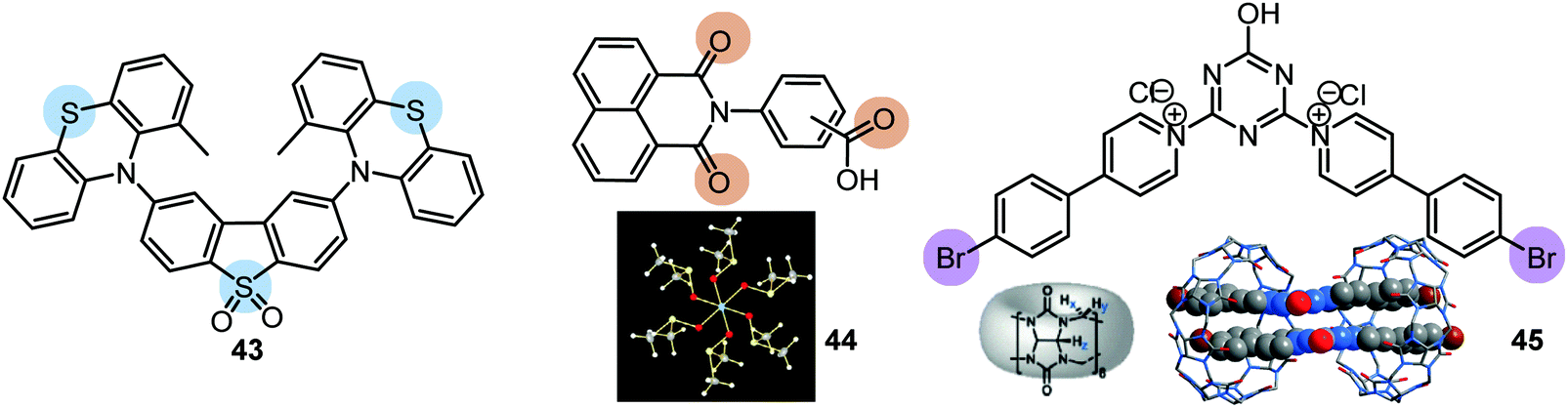 | ||
| Fig. 14 Examples of PF/RTP DE materials utilizing conformational and host/guest effects. 44: reproduced with permission.100 Copyright 2020 Wiley-VCH. 45: reproduced with permission.95 Copyright 2020 Wiley-VCH. | ||
A unique achievement, the first example of visible-light-excited pure organic RTP in aqueous solution, was recently reported in Tian's group by using a supramolecular host–guest assembly strategy.95 The circular cucurbit[8]uril (CB[8]) formed a stacked supramolecular structure with the triazine derivative 45 (Fig. 14) giving a structurally-rigid dimer with a new charge-transfer triplet state. This triplet state then produced yellow RTP emission in water under visible-light excitation. Additionally, multicolour PL from blue to white and yellow was realized by adding different molar ratios of CB[8] into the blue fluorescent solution of 45 (Fig. 13(c)). This tunable assembly-induced organic RTP emission under visible-light excitation in aqueous solution would be unthinkable in previous decades. Furthermore, RTP hydrogels and cell imaging based on the 45-CB[8] assembly systems were shown to have potential for applications such as biological sensors and time-resolved imaging.
2.4. Dual delayed and RTP photoluminescence
If the essential conditions for both RISC (D–A architecture, negligible ΔEST, spin-vibronic coupling between 3LE and 3CT) and RTP (large spin–orbit coupling, rigidified conformation) are satisfied, both delayed emission and RTP can be simultaneously observed.101 Donor–acceptor molecular architectures and the use of RTP-enhancing chromophores (containing heavy atoms, carbonyl groups) are valuable strategies for achieving dual DF/RTP emission.102 Molecular packing and the rigidity of the media are similarly important. While both RTP and TADF can arise in media with restricted intramolecular rotations/vibrations (e.g. polymer matrix), intermolecular interactions can also play a vital role in activating triplet emission. Thus, intra- and intermolecular hydrogen bonds can lead not only to rigidification of the structure (giving rise to the monomer-type RTP102) but also can promote the occurrence of intermolecular charge transfer or excited state dimers, leading to the lower energy aggregate-type RTP103 (Fig. 15). However, monomer and aggregate RTP can be hard to distinguish. Generally, computational studies may give an insight into the nature of the triplet states, as aided further by phosphorescence lifetime measurements: aggregates tend to exhibit persistent (long-lasting) RTP in the seconds time range, as opposed to conventional RTP (ms time range).4,92,102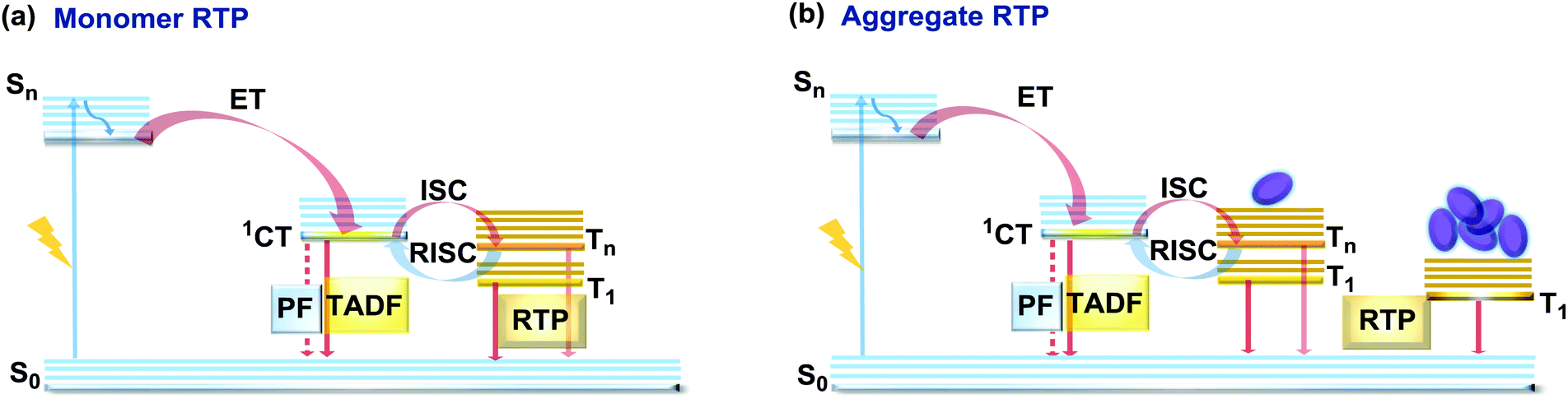 | ||
| Fig. 15 Jablonski diagram, presenting an overview of the processes in the DF/RTP DE materials with (a) monomer and (b) aggregate type RTP. | ||
For instance, Fraser et al. reported the first example of the dual DF/RTP with a monomer RTP type in 2007.104 By incorporating a classic boron dye into a common biopolymer (polylactic acid) a readily processable, single-component, multi-emissive material 46 was obtained, exhibiting intense fluorescence, delayed fluorescence, and unusual room temperature phosphorescence (Fig. 16). The time-resolved optical properties of 46 were found to be responsive to temperature, oxygen, and the polarity and rigidity of the local medium, confirming the coexistence of TADF and RTP.
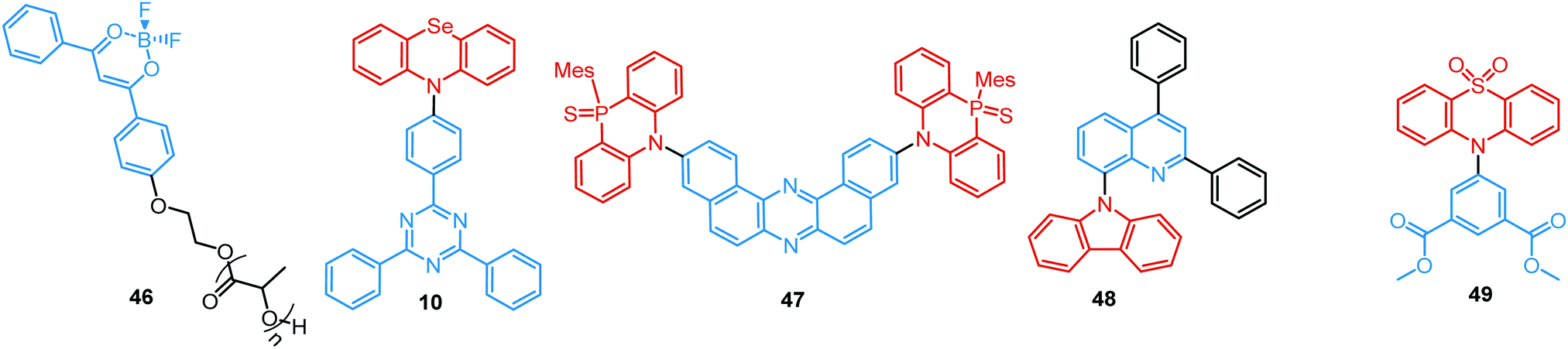 | ||
| Fig. 16 Examples of DF/RTP DE materials with monomer type RTP. | ||
As both TADF and RTP depend on the molecular conformation of the emitter,91 co-existence of eq and ax conformers combined with the heavy-atom effect resulted in DF/RTP DE in compound 10.37 Discussed previously in terms of dual prompt emission (Fig. 3 and 4(b)), 10 also possesses two different relaxation dynamics, with both excited states showing distinct frontier orbital electron distribution. Higher energy mixed HLCT orbitals in the more planar ax conformer activate RTP from the donor unit. In contrast, the nearly orthogonal eq conformer has more decoupled HOMO/LUMO orbitals, resulting in a lower energy CT state and a small ΔEST with the acceptor triplet, activating TADF. Similar conformation- and temperature-dependent interplay of TADF/RTP was detected in Zeonex films of the D–A–D triad 47, bearing the novel 10-mesityl-5H-phenophosphazinine-10-sulfide donor. In addition to the moderately large ΔEST value (0.3 eV), the host environment helped to constrain the vibrational and rotational movements and inhibit non-radiative channels from the triplet state. Together these effects boosted and balanced the TADF and RTP pathways. Of note, conformational interconversion in 47 led to remarkable reversible mechanoluminescent transformations between the visible and NIR regions in response to acid or base.105
Monomer-type RTP can occur not only in dilute solid guest–host films, but also in the crystal form. Compound 48 showed DE via RTP and TADF pathways, as well as emission enhancement at elevated temperatures. Computational and experimental studies confirmed RISC from the higher-lying triplet state (T2) due to the small energy gaps between T2 and S1 and between T1 and T2, leading to TADF, while RTP originated from the lowest triplet state (T1), (Fig. 17(a)). The torsion angle between the planes of the carbazolyl and quinolinyl moieties in 48 was suggested to be the main cause of the negligible ΔES1–T2 and ΔET1–T2, leading to the TADF/RTP DE even at the elevated temperatures.5 This torsion angle was found to be temperature-dependent (56° at room temperature vs. 105° at 70 °C), influencing the RTP and RISC rates in 48.
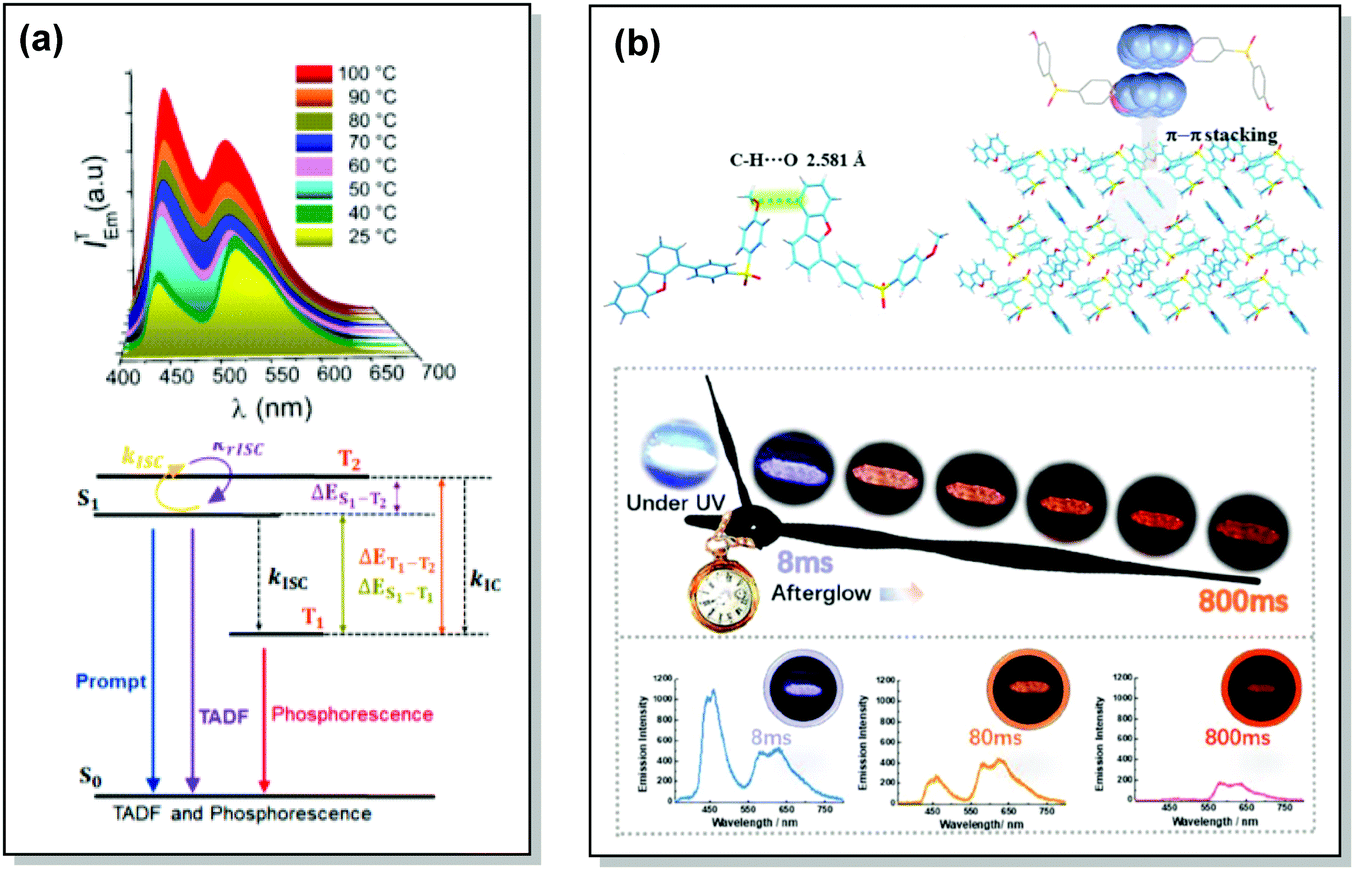 | ||
| Fig. 17 Examples of DF/RTP dual emission in the systems with (a) monomer-type RTP (48) and (b) aggregate-type RTP (51). (a) Steady-state PL of 48 spectra upon heating from 25 to 100 °C; Jablonski diagram for DE via TADF and phosphorescence in the proposed molecular design (48). Reproduced with permission.5 Copyright 2018 American Chemical Society. (b) Single crystal structure of 51 and π–π stacking properties of 51 in the single-crystal structure; emission photos of 51 during the afterglow emission processes; afterglow emission spectra of 51 at 8, 80, and 800 ms along with emission photos in ambient conditions. Reproduced with permission.107 Copyright 2019 Wiley-VCH. | ||
The critical role of π–π interactions in the origin of RTP was discussed by Tang and co-workers.102 By employing RTP-inducing donor and acceptor units (namely phenothiazine-S,S-dioxide and dimethyl isophthalate, respectively) compound 49 revealed the origin of several PL mechanisms, including persistent RTP, singlet excimer and TADF. Upon photoexcitation, both singlet excitons and excimers displayed DF emission with different wavelengths and lifetimes. However, natural transition orbital (NTO) calculations106 proved that only one molecule was involved in the triplet state for the dimer despite strong π–π interactions. Therefore, the persistent RTP was attributed to the monomer-dominated emission species, with the π–π interactions simply weakening the phosphorescence radiative decay. This study provided clear insight into the excited state electronic transition associated with the different emission processes, and demonstrated that π–π interactions are not a prerequisite for persistent RTP.
As opposed to monomer-type RTP, aggregation-induced phosphorescence is activated by molecular packing and intermolecular effects. For instance, Dias et al. performed a comprehensive photophysical study on crystals of carbazolyl and thianthrene derivative 50 (Fig. 18). The rigid environment within organic crystals is known to slow down the rate of diffusion of oxygen and molecular vibrations.40 Both these factors facilitate the observation of simultaneous fluorescence and phosphorescence in air, giving rise to strong white luminescence. Triplet–triplet annihilation (TTA) can also be active in crystalline systems due to triplet exciton diffusion through adjacent chromophores. While the results suggested that rigidity in ordered aggregates (such as crystals) promoted RTP by suppressing large-amplitude vibrations, the nature of DF was also examined. As indicated by the power-dependence experiment, the DF in 50 is of mixed TADF/TTA nature. An active TTA channel suggests that delayed emission (DF and RTP) in this system may originate from molecular assemblies in the crystal.
 | ||
| Fig. 18 Examples of DF/RTP dual emitters with aggregate-type RTP. | ||
Recently, Chi and co-workers have proven that incorporation of the appropriate substituent into the aromatic backbone can lead to strong hydrogen bonds, enhancing both RTP and DF. Thus, the presence of the methoxy-group in the dibenzofuran-containing sulfonyldibenzene 51 aided the interaction of neighbouring molecules through intermolecular hydrogen bonds, promoting CT emission with ultralong lifetimes (627 ms) when compared to the unsubstituted analogue (afterglow lifetime 107 ms). In addition, the intermolecular hydrogen bonding could immobilize the molecular conformations and enhance the RTP emission. Compound 51 displayed strong DE afterglow, with the colour gradually changing from cold white to orange during the decay process (Fig. 17(b)). In neighbouring molecules the dibenzofuran and methoxy groups are arranged at short distances within the crystal through C–H⋯O intermolecular hydrogen bonding. Therefore, alongside the persistent dibenzofuran-based RTP, a new afterglow emission band with higher energy stemming from the ICT is also observed. Thus, intermolecular hydrogen bonding capable of promoting ICT plays a key role in the DE afterglow properties of 51.107 In turn, CT character enhancement by intramolecular charge transfer in 52 appeared to be a facile way to minimize ΔEST and maximize ISC/RISC. The resulting small energy gap (ΔEST 0.17 eV) boosted dual-channel triplet harvesting via ultralong TADF and H-aggregation phosphorescence, both assisted by suppressed triplet quenching. H-aggregation granted 52 RTP with an ultralong lifetime of 0.84 s.108
Finally, by incorporating an AIE unit into the molecular backbone, efficient intramolecular TADF and intermolecular TADF/RTP DE was switched on in single crystals of 53 (Fig. 18). Theoretical calculations confirmed strong intermolecular electronic coupling in dimers of 53, responsible for the AIE channels. Of note, in this work the novel dual TADF/RTP emission was exploited to obtain a WOLED (CIE (0.35, 0.35)) with high colour rendering index (88.8).109
2.5. Dual RTP photoluminescence
One of the promising pathways towards combining triplet-harvesting and white emission is the rarely observed dual RTP. In principle, if chromophores can decay radiatively from both a higher triplet state (e.g. T2) and the lowest triplet state (T1), dual phosphorescence and even white light emission could be observed34 (Fig. 19). To achieve this, several conditions should be satisfied: (i) sufficiently large energy separation between S1 and Tn to prevent triplet up-conversion by TADF; (ii) energetic proximity and strong vibronic mixing of T1 and Tn; (iii) different nature (ππ*, nπ*) of T1 and Tn to allow alternatively fast or slow radiative decay; (iv) sufficiently slow triplet relaxation time compared to radiative decay rates.110–112 When these conditions are satisfied, dual RTP with various radiative decay lifetimes can be observed in a violation of Kasha's rule.18 Dual RTP does not depend solely on the molecular packing and medium rigidity, and can be engineered by careful molecular design. For the occurrence of dual RTP, ISC-enhancing units with similar triplet state energy levels, but different triplet state nature (ππ*, nπ*) should be exploited, and the junction pattern also plays a decisive role in endowing T1 and Tn with the required molecular orbital configurations.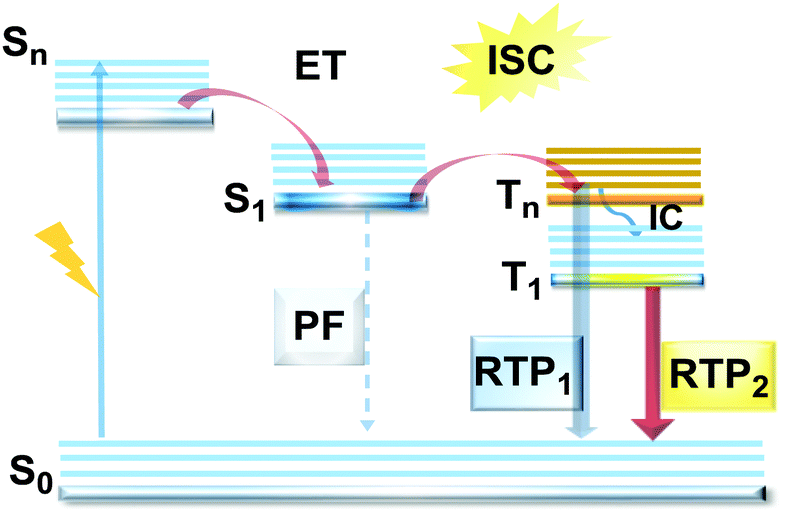 | ||
| Fig. 19 Jablonski diagram, presenting an overview of the processes in the dual RTP emitters. | ||
While most dual RTP emitters have a donor–acceptor molecular architecture, this is not an essential criterion. For instance, Yang, Ma, et al. successfully achieved cold white emission with CIE coordinates of (0.28, 0.33) from dibenzo[a,c]phenazine 54 (Fig. 20) as a result of simultaneous triple emission processes: fluorescence, T1-RTP and T2-RTP. Theoretical calculations and photophysical measurements suggested that T1-RTP and T2-RTP have relatively independent origins, namely ππ* and nπ* states, which is beneficial for achieving white emission (Fig. 21(a)). As the internal conversion of the T2 → T1 transition is slowed down due to the different orbital natures of T2 and T1, the radiative T2 → S0 transition can compete with the IC, unlocking the faster triplet emission from T2 along with the slower phosphorescence from T1. Comparison of 54 with the purely hydrocarbon counterpart proved the importance of heteroatoms in the spin–orbit coupling and ISC.16
 | ||
| Fig. 20 Examples of dual RTP emitters. | ||
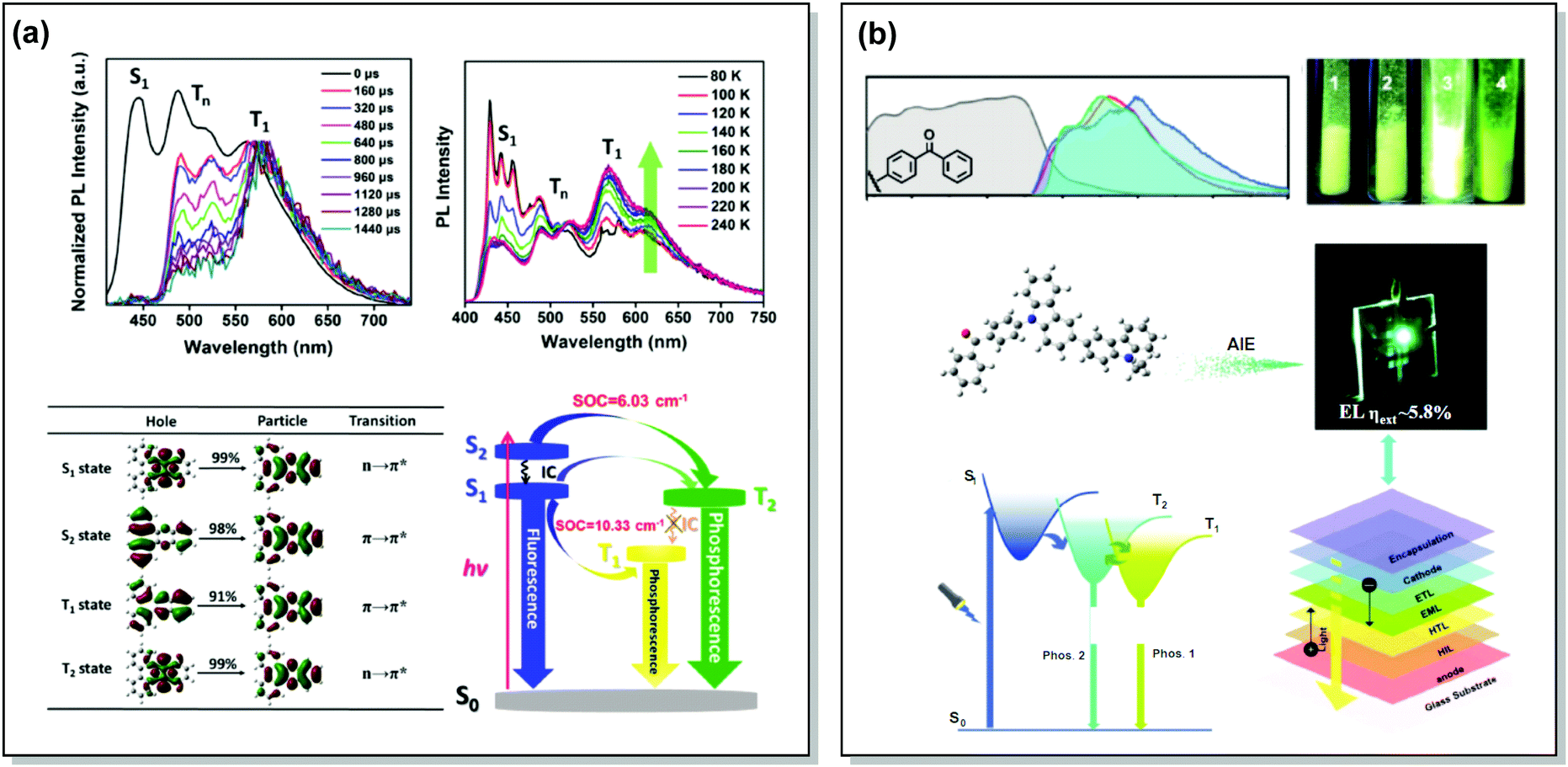 | ||
| Fig. 21 Examples dual RTP emission in the systems (a) 54 and (b) 55. (a) Normalized time-resolved PL spectra of 54 neat powder (the spectra are normalized to the T1 emission for comparison); temperature-dependent PL spectra of 54 from 80 to 240 K; natural transition orbitals (NTO) images of the S1, S2, T1, and T2 excited states of 54; schematic Jablonski diagram with SOC matrix elements of RTP-related ISC processes in 54. Reproduced with permission.16 Copyright 2018 Wiley-VCH. (b) Absorption spectra (gray lines), steady-state PL emission spectra in air at 298 K (red lines) and at 77 K (blue lines) and delayed emission spectra at 77 K (green lines, delayed time: 1 ms) of 55; PL photos of 55 taken in air at 298 K; an energy diagram showing the two main emitting triplet states of 55 and the fabricated OLED based on 55. Reproduced with permission.113 Copyright 2019 Wiley-VCH. | ||
Aggregation-induced emission, and in particular aggregation-induced phosphorescence (AIP), was shown to be a beneficial design strategy for white light emitters. By coupling a bicarbazolyl donor and benzophenone acceptor an efficient white emitter 55 was obtained. Theoretical calculations suggested hybrid nπ* and ππ* character for S1, T1, and T3 due to the contribution of the lone pairs from the carbonyl group and delocalized π electrons from the donor. Provided that S1 and T1 are dominated by nπ* and ππ* transitions, the spin–orbit coupling between S1 and T1 is favourable by El-Sayed's rule. In contrast, a relatively large T1–T2 gap decouples these states, slowing IC and allowing T2 excitons to decay radiatively, finally resulting in the observed dual phosphorescence (Fig. 21(b)). Taking advantage of the AIP, a white OLED with a small roll-off and an EQEmax of 5.8% was fabricated based on 55.113
Tang's group reported another example of white emission stemming from dual RTP. Thus, purely phosphorescent white light was observed in the organic crystals of 56. The white emission was attributed to the mixing of two RTP bands with different wavelengths and lifetimes, which originate from two excited triplet states with different excitation energies and transition orbital features. The presence of two ISC-enhancing chromophores in 56 with different triplet emission origins, i.e.3ππ* for dibenzothiophene and 3nπ* for benzophenone, is responsible for DE. Additionally, the junction mode with a phenyl group shared between the chromophores, lead to appropriate mixing in the molecular orbitals. The presence of the chlorine atom was shown to tune the ratio of T1 and T2 states, thereby balancing the emission from both states, leading to warm-white light (CIE (0.33, 0.35)).34 Of note, DE was also observed in the analogues of 56, featuring hydrogen, fluorine or bromine atoms, at the same position. However, only chlorinated derivative 56 displayed balanced DE with comparable intensity of the bands.
DE can be established by exploitation of the pertinent chromophores and by fine-tuning the molecular conformation. For example, the methyl-substituted compound 57 exhibited dual RTP, whereas the analogues bearing bulkier iso-propyl and tert-butyl donor substituents showed singular RTP instead.114 The fluorescence in 57 was accompanied by the phosphorescence from the accepting dibenzothiophene unit (450 nm), as well as phosphorescence from the donating 1-methyl-phenothiazine (540 nm). The observation of dual phosphorescence is an apparent violation of Kasha's rule,18 which strongly indicates the presence of two conformers in 57, separated by a considerable energy barrier. While only the axial form of the PTZ units was identified in the ground state, both ax and eq forms were found to exist in the excited-state based on the combination of theoretical and photophysical data. Unlike short-lived fluorescence which does not allow conformational reorganization, structural relaxation to the low energy triplet is possible during the long triplet state lifetime. Therefore, at high temperatures a subset of 57 molecules can cross the conformational barrier and emit dual phosphorescence. This work proved that relaxation between the two triplet states involves an energy barrier, which is likely to increase with the bulkiness of the substituents. Hence, by the introduction of suitable substituents the triplet emission can be manipulated.
Finally, yet importantly, energy transfer, such as Förster Resonance Energy Transfer (FRET), between the chromophores can lead to dual RTP. In recent work of Kubo et al., Rhodamine B fluorophores were grafted onto the surface of 3-benzo-2,4,8,10-tetraoxa-3,9-diboraspiro[5.5]-undecanes (BP) particles to obtain the compound 58. Grafting enabled FRET from the triplet excited state of the polymeric form of BP to the singlet of the fluorophore due to spectral overlap between the BP's phosphorescence and the absorption band of the grafted dye, resulting in the long-lived DE bands peaking at 500 and 600 nm. While presented as an example of dual RTP, in this case the longer wavelength band is more accurately an example of phosphorescence sensitised delayed/long-lived fluorescence from the rhodamine fluorophore. A similar case of sensitising orange fluorescence by green RTP was reported by Reineke and coworkers.115 This sophisticated interplay between the phosphor and fluorophore in a self-assembled system offers new methodology in the development of colour-variable afterglow chemosensors for the detection of various analytes.116
2.6. Dual delayed fluorescence
The last, but not least, known type of DE is dual DF. There are two ways in which triplet-singlet up-conversion can occur. Firstly, delayed fluorescence can occur via a monomolecular (involving a single triplet) thermally activated up-conversion process to give TADF emission (discussed in detail in sections 2.2 and 2.4). The essential criteria for TADF are: an electron donor–acceptor structure, negligible ΔEST, decoupled HOMO/LUMO, chromophore rigidity and presumably conformational stability.117,118 In turn, if the singlet–triplet energy gap is moderately high (1 eV ≥ ΔEST ≥ 0.3 eV), and the singlet energy is higher than double the triplet energy, two triplets can instead collide and generate sufficient energy to up-convert to a single emissive singlet state. This type of delayed emission is bimolecular triplet–triplet annihilation (TTA).119 TTA is very often regarded as an undesirable process, leading to enhanced efficiency roll-off in OLEDs. However if TTA is controlled it can be utilized to achieve efficient deep-blue emission that has so far eluded TADF research.120 In terms of the design and synthesis, dual DF emitters often require the construction of the diverse sterically congested sp3-rich skeletal ring systems. For instance, an excellent way towards construction of tricyclic products via a reductive diversion of the Minisci reaction was recently reported by Duarte and Dixon.121 Interestingly, if the emitter can generate two or more TADF-active states (Fig. 22(a)), or if the conditions for both TADF and TTA are satisfied (Fig. 22(b)), dual DF can be observed. However, unlike other types of DE, dual DF is often hard to distinguish and unambiguously identify, as the emission spectra for each process are often very similar. Different types of DF can be identified by analysis of time-resolved photoluminescence decay kinetics and laser fluence experiments (dependence of DF intensity on the excitation pulse power)21,90,120,122,123 However, even though TADF and TTA can often coexist in a single molecule,90 not many studies thoroughly analyse the dual DF case. Therefore, in this section we focus on the handful of well-documented cases of dual DF.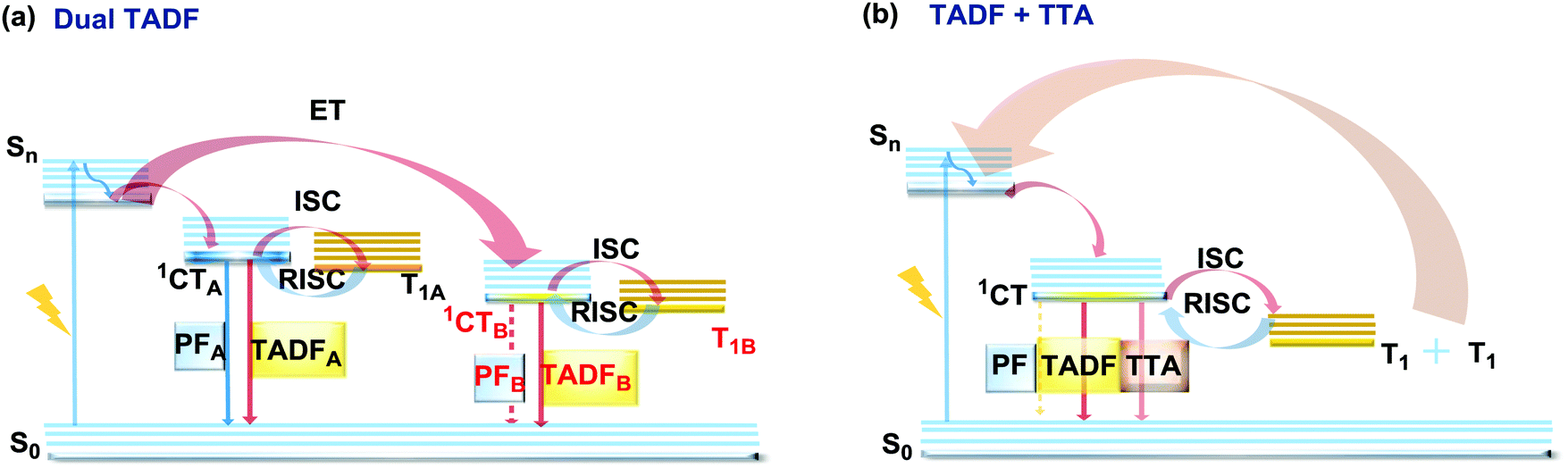 | ||
| Fig. 22 Jablonski diagram, presenting an overview of the processes in (a) dual TADF and (b) dual TADF and TTA DF emitters. | ||
By careful investigation of the PL decay kinetics of films of 1,3-bis(N-carbazolyl)benzene (mCP) doped with emitter 59 (Fig. 23), Wang et al. distinguished two TADF processes with characteristic lifetimes of around 1.1 and 9.1 µs, respectively. The presence of dual DF was attributed to the conformational heterogeneity, resulting from the slight rotation around the N–C bond between the A and D units, leading to the formation of slightly different 1CT and T1 states (Fig. 24(a)). Conformational heterogeneity might have originated from the differences in packing and polarity of the aggregates of 59 in mCP. Utilization of the 59:mCP emissive layer in an OLED manifested enhanced efficiency and reduced roll-off, which was assigned to the suppression of TTA in this material.124
 | ||
| Fig. 23 Examples of dual DF emitters. | ||
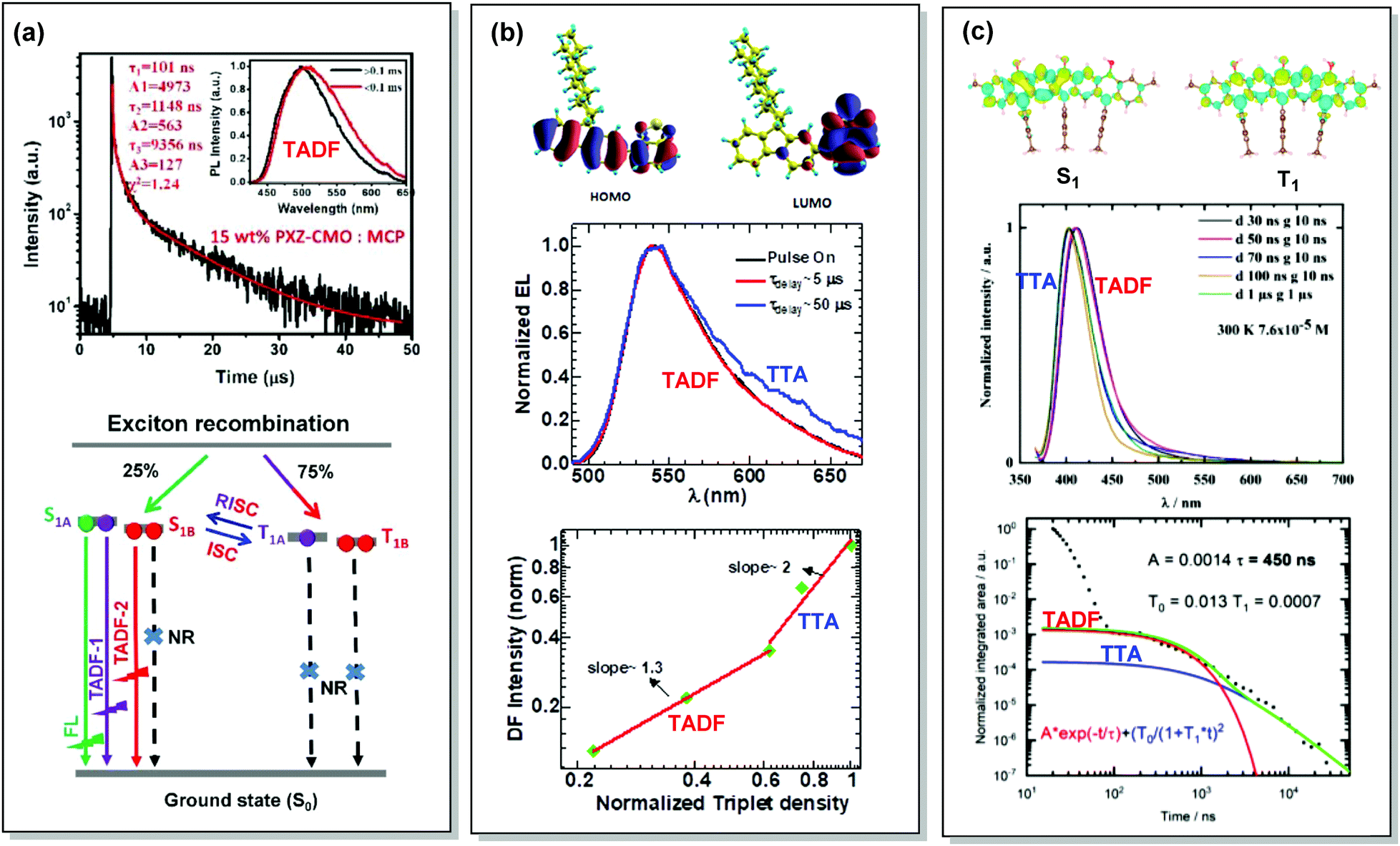 | ||
| Fig. 24 Examples dual DF emission in the systems (a) 59, (b) 60 and (c) 61. (a) Transient decay spectra of the 15 wt% 59:MCP film (inset: emission spectra of TADF components); schematic representation of RISC processes of emitters with dual delayed excited states. Reproduced with permission.124 Copyrights 2019 Frontiers. (b) Calculated HOMO and LUMO spatial distribution in 60, with isosurface value ±0.02 a.u. at the TD-B3LYP/6-311+G(d,p) level of theory; electroluminescence spectra during electrical pulse ON time and delayed emission with a time delay of 5 and 50 µs in 60 PLED; the time-integrated DF intensity vs. laser fluence plots for 60 solid film, linearly fitted with dual slopes. Reproduced with permission.126 Copyright 2018 Springer Nature. (c) Different density plots for lowest singlet and triplet excited states for 61 calculated in the gas phase by using SCS-CC2; emission spectra of 61 in THF at 7.6 × 10−5 M at 300 K; PL transients of 61 taken in 7.6 × 10−6 M solution, excited at 355 nm, integrated between 380 and 420 nm at 300 K. Reproduced with permission.127 Copyright 2020 American Chemical Society. | ||
Interestingly, the origin of DF in the OLEDs based on poly-(9,9-dioctylfluorene-alt-benzothiadiazole) polymer 60 had initially been assigned to TTA.125 However, in depth investigation of the triplet transfer processes as a function of solid-state packing revealed a different picture. To clarify the role of the DF in enhancing OLED efficiency (either by TTA or TADF), Kabra and co-workers recorded the electrically excited DF kinetics after the end of the electrical pulse applied to a working OLED.126 At low applied bias voltage (∼8 V) the transient EL followed exponential decay, which successively changed to a power law decay at higher applied bias (∼13 V). In the case of the higher bias voltage, at early times a normalized triplet density with a slope of 1.3 was observed, which at longer time delays changed to slope 2 (Fig. 24(b)). The close spectral match of PF and DF suggested a similar origin for both processes in electrically excited polymer OLEDs of 60. The slight spectral broadening in the DF was attributed to spatial diffusion of the triplet excitons towards lower energy sites in conjugated polymer systems. Based on the lifetimes of the time resolved EL, presence of monomolecular DF at the low laser fluency, and bimolecular-induced DF at the high laser fluency, is a clear indication of the mixed TADF and TTA DF. Revealing the character of the DF finally helped to explain the reported EQE value of 4.6%, falling beyond the limits of the spin statistics.125 Diffusive triplet transport in the polymer system 60 provides inspiration for molecular engineering, where efficiency can be tuned significantly by facilitating the TTA process with optimised film thickness or packing of chains.
Finally, Zysman-Colman et al. have recently reported an interesting deep-blue multi-resonance TADF emitter 61 with dominant TTA at low-temperatures.127 The low-temperature TTA in this case stemmed from the presence of aggregates of 61 in THF solution upon cooling. TADF replaces TTA at elevated temperatures due to the dissolution of the aggregates. Careful investigation of the decay kinetics revealed dual DF with an exceptionally short TADF lifetime (450 ns) (Fig. 24(c)). Such a TADF/TTA combination in a single material is envisaged to enable long-lived and highly efficient OLEDs in future.
Conclusions and outlook
Herein we have given an overview of the various types of dual emitters known to date. Overall, DE involving purely prompt fluorescence has been studied the most thoroughly over the last two decades. More recently, combinations of PF with delayed emission and room temperature phosphorescence have become fairly common due to the parallel development of TADF and RTP emitters. However, examples of dual delayed fluorescence, dual RTP or simultaneous DF/RTP remain quite rare.In general, DE is dependent on several factors: (i) molecular structure: conformation, molecular bonding pattern, donor–acceptor balance of the chromophores, the presence of ISC/RISC-promoting molecular fragments; (ii) molecular organization and packing; (iii) inter- and intramolecular interactions and (iv) external media and host effects. These factors impose certain restrictions on the practical applications of dual emitters. Therefore, even though promising applications such as efficient WOLEDs, ratiometric sensing, time-resolved bio-imaging, encryption/decryption were reported, in the majority of cases successful identification of DE can be attributed to accidental observations, rather than deliberate molecular design. Nonetheless, utilization of DE provides valuable optoelectronic material properties such as reduced cost, simplified fabrication and usage, and superior response to multidimensional stimuli, compared to mono-emissive materials. These attractive properties can be best exploited using single-component dual emitters. The combination of delayed emission with room temperature phosphorescence tends to be the most perspective DE type, as it (i) additionally includes prompt fluorescence; (ii) provides well-separated emission bands; (iii) offers targeted stimuli-response ability; (iv) includes time as an additional response parameter, as the emission covers the timeframe from ns to s.
However, the question remains: is it possible to control DE and design the materials with targeted properties? We optimistically claim that by careful molecular design, control over DE can indeed be achieved. We have outlined the following rules to guide the preparation of efficient DF/RTP dual emitters: (i) exploiting D–A molecular architectures; (ii) the use of AIE-active chromophores: (iii) introduction of ISC-enhancing molecular fragments; (iv) ensuring the presence of rigidifying intramolecular interactions (e.g. hydrogen/halogen bonding); (v) incorporation of self-hosting moieties/structures to eliminate the dependence on external host media and ensure dominance of a singular conformation. These conditions have already been realized in covalent organic frameworks, cyclophanes and grafted polymers, and could be developed further in small molecule DE materials.
We note that a key challenge is to achieve DE without requiring assistance from molecular packing (i.e. DE present various polymorphic forms). In the above mentioned COFs, polymers, and cyclophanes, such control can indeed be achieved regardless of the molecular organization (crystalline vs. amorphous). Independence from molecular packing is an essential feature in simplifying the fabrication process towards target applications of DE materials. As crystals with the desirable properties are difficult to prepare, using amorphous powders and gels can offer a more reliable and simplified approach. WOLEDs with a gel-based emissive layer, 2D and 3D stimuli-responsive coatings, security tags with multidimensional encryptions, and combinations of sensing and bio-imaging128 are just a few of the possible applications of versatile DE emitters.
In conclusion, in this review we have sought to consolidate recent new insights for the design and applications of the efficient novel DE materials. It is anticipated that this knowledge will lead to accelerated future innovation and developments in this area.
Conflicts of interest
There are no conflicts to declare.References
- R. Gui, H. Jin, X. Bu, Y. Fu, Z. Wang and Q. Liu, Coord. Chem. Rev., 2019, 383, 82–103 CrossRef CAS.
- X. Wang, H. Ma, M. Gu, C. Lin, N. Gan, Z. Xie, H. Wang, L. Bian, L. Fu, S. Cai, Z. Chi, W. Yao, Z. An, H. Shi and W. Huang, Chem. Mater., 2019, 31, 5584–5591 CrossRef CAS.
- B. Zhou and D. Yan, Adv. Funct. Mater., 2019, 29, 1970023 CrossRef.
- S. M. A. Fateminia, Z. Mao, S. Xu, Z. Yang, Z. Chi and B. Liu, Angew. Chem., Int. Ed., 2017, 56, 12160–12164 CrossRef CAS.
- I. Bhattacharjee, N. Acharya, H. Bhatia and D. Ray, J. Phys. Chem. Lett., 2018, 9, 2733–2738 CrossRef CAS.
- K. Wang, Y. Shi, C.-J. Zheng, W. Liu, K. Liang, X. Li, M. Zhang, H. Lin, S. Tao, C.-S. Lee, X.-M. Ou and X. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 31515–31525 CrossRef CAS.
- H. L. Lee, H. J. Jang and J. Y. Lee, J. Mater. Chem. C, 2020, 8, 10302–10308 RSC.
- S. V. Mulay, Y. Kim, M. Choi, D. Y. Lee, J. Choi, Y. Lee, S. Jon and D. G. Churchill, Anal. Chem., 2018, 90, 2648–2654 CrossRef CAS.
- X. Guan, B. Lu, Q. Jin, Z. Li, L. Wang, K. Wang, S. Lai and Z. Lei, Ind. Eng. Chem. Res., 2018, 57, 14889–14898 CrossRef CAS.
- B. P. Yan, C. C. C. Cheung, S. C. F. Kui, H. F. Xiang, V. A. L. Roy, S. J. Xu and C. M. Che, Adv. Mater., 2007, 19, 3599–3603 CrossRef CAS.
- C. Li, J. Liang, B. Liang, Z. Li, Z. Cheng, G. Yang and Y. Wang, Adv. Opt. Mater., 2019, 7, 1801667 CrossRef.
- T. Ishi-I, H. Tanaka, I. S. Park, T. Yasuda, S. I. Kato, M. Ito, H. Hiyoshi and T. Matsumoto, Chem. Commun., 2020, 56, 4051–4054 RSC.
- P. Data and Y. Takeda, Chem. – Asian J., 2019, 14, 1613–1636 CrossRef CAS.
- W.-Y. Hung, L.-C. Chi, W.-J. Chen, Y.-M. Chen, S.-H. Chou and K.-T. Wong, J. Mater. Chem., 2010, 20, 10113 RSC.
- J.-X. Wang, Y.-G. Fang, C.-X. Li, L.-Y. Niu, W.-H. Fang, G. Cui and Q.-Z. Yang, Angew. Chem., Int. Ed., 2020, 59, 10032–10036 CrossRef CAS.
- C. Zhou, S. Zhang, Y. Gao, H. Liu, T. Shan, X. Liang, B. Yang and Y. Ma, Adv. Funct. Mater., 2018, 28, 1802407 CrossRef.
- G. Eber, F. Grüneis, S. Schneider and F. Dörr, Chem. Phys. Lett., 1974, 29, 397–404 CrossRef CAS.
- M. Kasha, Discuss. Faraday Soc., 1950, 9, 14–19 RSC.
- A. Mangini, Advances in molecular spectroscopy. VOlume 2: proceedings of the IVth International Meeting on Molecular spectroscopy, Symposium Publications Division, Pergamon Press, Oxford, 1962 Search PubMed.
- Z. Zhang, Y. Gao, H. Liu, Q. Bai, J. Li, L. Liu, C. Wu, B. Yang, K. Wang, B. Zou, Y. Wang and P. Lu, Dyes Pigm., 2017, 145, 294–300 CrossRef CAS.
- N. A. Kukhta, A. S. Batsanov, M. R. Bryce and A. P. Monkman, J. Phys. Chem. C, 2018, 122, 28564–28575 CrossRef CAS.
- M. Rohini, M. Baral and B. K. Kanungo, RSC Adv., 2016, 6, 108017–108027 RSC.
- C. Azarias, Š. Budzák, A. D. Laurent, G. Ulrich and D. Jacquemin, Chem. Sci., 2016, 7, 3763–3774 RSC.
- Z. Xu, Q. T. Liu, X. Wang, Q. Liu, D. Hean, K. C. Chou and M. O. Wolf, Chem. Sci., 2020, 11, 2729–2734 RSC.
- K. Ling, H. Shi, H. Wang, L. Fu, A. Lv, K. Huang, W. Ye, M. Gu, C. Ma, X. Yao, W. Jia, J. Zhi, W. Yao, Z. An, H. Ma and W. Huang, Adv. Opt. Mater., 2019, 7, 1901076 CrossRef CAS.
- M. Louis, H. Thomas, M. Gmelch, F. Fries, A. Haft, J. Lindenthal and S. Reineke, Adv. Opt. Mater., 2020, 2000427 CrossRef CAS.
- J. S. Ward, R. S. Nobuyasu, M. A. Fox, A. S. Batsanov, J. Santos, F. B. Dias and M. R. Bryce, J. Org. Chem., 2018, 83, 14431–14442 CrossRef CAS.
- J. Gibson, A. P. Monkman and T. Penfold, ChemPhysChem, 2016, 17, 2956–2961 CrossRef CAS.
- F. B. Dias, J. Santos, D. R. Graves, P. Data, R. S. Nobuyasu, M. A. Fox, A. S. Batsanov, T. Palmeira, M. N. Berberan-Santos, M. R. Bryce and A. P. Monkman, Adv. Sci., 2016, 3, 1600080 CrossRef.
- H. Tanaka, K. Shizu, H. Miyazaki and C. Adachi, Chem. Commun., 2012, 48, 11392–11394 RSC.
- W. Zhao, Z. He, J. W. Y. Lam, Q. Peng, H. Ma, Z. Shuai, G. Bai, J. Hao and B. Z. Tang, Chem, 2016, 1, 592–602 CAS.
- N. A. Kukhta, R. Huang, A. S. Batsanov, M. R. Bryce and F. B. Dias, J. Phys. Chem. C, 2019, 123, 26536–26546 CrossRef CAS.
- R. Huang, J. Avó, T. Northey, E. Chaning-Pearce, P. L. Dos Santos, J. S. Ward, P. Data, M. K. Etherington, M. A. Fox, T. J. Penfold, M. N. Berberan-Santos, J. C. Lima, M. R. Bryce and F. B. Dias, J. Mater. Chem. C, 2017, 5, 6269–6280 RSC.
- Z. He, W. Zhao, J. W. Y. Lam, Q. Peng, H. Ma, G. Liang, Z. Shuai and B. Z. Tang, Nat. Commun., 2017, 8, 416 CrossRef.
- B. Li, Z. Li, F. Guo, J. Song, X. Jiang, Y. Wang, S. Gao, J. Wang, X. Pang, L. Zhao and Y. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 14233–14243 CrossRef CAS.
- K. Wang, C.-J. Zheng, W. Liu, K. Liang, Y.-Z. Shi, S.-L. Tao, C.-S. Lee, X.-M. Ou and X.-H. Zhang, Adv. Mater., 2017, 29, 1701476 CrossRef.
- D. de Sa Pereira, D. R. Lee, N. A. Kukhta, K. H. Lee, C. L. Kim, A. S. Batsanov, J. Y. Lee and A. P. Monkman, J. Mater. Chem. C, 2019, 7, 10481–10490 RSC.
- Y. Zhang, Y. Miao, X. Song, Y. Gao, Z. Zhang, K. Ye and Y. Wang, J. Phys. Chem. Lett., 2017, 8, 4808–4813 CrossRef CAS.
- H. Q. Yin, F. Yin and X. B. Yin, Chem. Sci., 2019, 10, 11103–11109 RSC.
- P. Pander, A. Swist, R. Turczyn, S. Pouget, D. Djurado, A. Lazauskas, R. Pashazadeh, J. V. Grazulevicius, R. Motyka, A. Klimash, P. J. Skabara, P. Data, J. Soloducho and F. B. Dias, J. Phys. Chem. C, 2018, 122, 24958–24966 CrossRef CAS.
- J. Wang, X. Gu, H. Ma, Q. Peng, X. Huang, X. Zheng, S. H. P. Sung, G. Shan, J. W. Y. Lam, Z. Shuai and B. Z. Tang, Nat. Commun., 2018, 9, 2963 CrossRef.
- G. Haberhauer, Chem. – Eur. J., 2017, 23, 9288–9296 CrossRef CAS.
- S. Sasaki, G. P. C. Drummen and G. I. Konishi, J. Mater. Chem. C, 2016, 4, 2731–2743 RSC.
- Z. Zhang, Y. S. Wu, K. C. Tang, C. L. Chen, J. W. Ho, J. Su, H. Tian and P. T. Chou, J. Am. Chem. Soc., 2015, 137, 8509–8520 CrossRef CAS.
- Q. W. Zhang, D. Li, X. Li, P. B. White, J. Mecinović, X. Ma, H. Ågren, R. J. M. Nolte and H. Tian, J. Am. Chem. Soc., 2016, 138, 13541–13550 CrossRef CAS.
- Y. Hou, I. Kurganskii, A. Elmali, H. Zhang, Y. Gao, L. Lv, J. Zhao, A. Karatay, L. Luo and M. Fedin, J. Chem. Phys., 2020, 152, 114701 CrossRef CAS.
- G. Brancato, G. Signore, P. Neyroz, D. Polli, G. Cerullo, G. Abbandonato, L. Nucara, V. Barone, F. Beltram and R. Bizzarri, J. Phys. Chem. B, 2015, 119, 6144–6154 CrossRef CAS.
- S. Chevreux, C. Allain, L. Wilbraham, K. Nakatani, P. Jacques, I. Ciofini and G. Lemercier, Faraday Discuss., 2015, 185, 285–297 RSC.
- T. Nishida, S. Ohta, F. Xu, K. Shinohara, T. Kamada, H. Akashi, M. Takezaki, K. Wakamatsu and A. Orita, Org. Lett., 2016, 18, 3988–3991 CrossRef CAS.
- S. M. King, I. I. Perepichka, I. F. Perepichka, F. B. Dias, M. R. Bryce and A. P. Monkman, Adv. Funct. Mater., 2009, 19, 586–591 CrossRef CAS.
- H. Yeo, K. Tanaka and Y. Chujo, Macromolecules, 2016, 49, 8899–8904 CrossRef CAS.
- L. M. Lifshits, D. S. Budkina, V. Singh, S. M. Matveev, A. N. Tarnovsky and J. K. Klosterman, Phys. Chem. Chem. Phys., 2016, 18, 27671–27683 RSC.
- P. L. dos Santos, J. S. Ward, P. Data, A. Batsanov, M. R. Bryce, F. Dias and A. P. Monkman, J. Mater. Chem. C, 2016, 4, 3815–3824 RSC.
- J. Gibson, A. P. Monkman and T. J. Penfold, ChemPhysChem, 2016, 17, 2956–2961 CrossRef CAS.
- N. A. Kukhta, H. F. Higginbotham, T. Matulaitis, A. Danos, A. N. Bismillah, N. Haase, M. K. Etherington, D. S. Yufit, P. R. McGonigal, J. V. Gražulevičius and A. P. Monkman, J. Mater. Chem. C, 2019, 7, 9184–9194 RSC.
- F. Y. Hao, Y. Z. Shi, K. Wang, S. Y. Xiong, X. C. Fan, L. Wu, C. J. Zheng, Y. Q. Li, X. M. Ou and X. H. Zhang, Dyes Pigm., 2020, 178, 108336 CrossRef CAS.
- M. K. Etherington, F. Franchello, J. Gibson, T. Northey, J. Santos, J. S. Ward, H. F. Higginbotham, P. Data, A. Kurowska, P. L. Dos Santos, D. R. Graves, A. S. Batsanov, F. B. Dias, M. R. Bryce, T. J. Penfold and A. P. Monkman, Nat. Commun., 2017, 8, 14987 CrossRef CAS.
- H. Tanaka, K. Shizu, H. Nakanotani and C. Adachi, J. Phys. Chem. C, 2014, 118, 15985–15994 CrossRef CAS.
- H. Wang, Y. Li, Y. Zhang, J. Mei and J. Su, Chem. Commun., 2019, 55, 1879–1882 RSC.
- A. Weller, Naturwissenschaften, 1955, 42, 175–176 CrossRef CAS.
- Y. Long, M. Mamada, C. Li, P. L. dos Santos, M. Colella, A. Danos, C. Adachi and A. Monkman, J. Phys. Chem. Lett., 2020, 11, 3305–3312 CrossRef CAS.
- A. C. Sedgwick, L. Wu, H. H. Han, S. D. Bull, X. P. He, T. D. James, J. L. Sessler, B. Z. Tang, H. Tian and J. Yoon, Chem. Soc. Rev., 2018, 47, 8842–8880 RSC.
- B. Li, L. Zhou, H. Cheng, Q. Huang, J. Lan, L. Zhou and J. You, Chem. Sci., 2018, 9, 1213–1220 RSC.
- D. Cui, D. F. Perepichka, J. M. MacLeod and F. Rosei, Chem. Soc. Rev., 2020, 49, 2020–2038 RSC.
- S. Haldar, D. Chakraborty, B. Roy, G. Banappanavar, K. Rinku, D. Mullangi, P. Hazra, D. Kabra and R. Vaidhyanathan, J. Am. Chem. Soc., 2018, 140, 13367–13374 CrossRef CAS.
- X. Li, J. Qiao, S. Y. Quek and K. P. Loh, ACS Mater. Lett., 2020, 654–657 CrossRef CAS.
- Z. Zhao, J. W. Y. Lam and B. Z. Tang, J. Mater. Chem., 2012, 22, 23726 RSC.
- R. Hu, N. L. C. Leung and B. Z. Tang, Chem. Soc. Rev., 2014, 43, 4494–4562 RSC.
- Y. Kim, J. Bouffard, S. E. Kooi and T. M. Swager, J. Am. Chem. Soc., 2005, 127, 13726–13731 CrossRef CAS.
- C. Duan, C. Han, R. Du, Y. Wei and H. Xu, Adv. Opt. Mater., 2018, 6, 1800437 CrossRef.
- D. A. Shultz and M. A. Fox, J. Am. Chem. Soc., 1989, 111, 6311–6320 CrossRef CAS.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, B. Z. Tang, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu and D. Zhu, Chem. Commun., 2001, 1740–1741 RSC.
- G. K. Bains, S. H. Kim, E. J. Sorin and V. Narayanaswami, Biochemistry, 2012, 51, 6207–6219 CrossRef CAS.
- M. Sugino, Y. Araki, K. Hatanaka, I. Hisaki, M. Miyata and N. Tohnai, Cryst. Growth Des., 2013, 13, 4986–4992 CrossRef CAS.
- Y. Ooyama, R. Nomura, T. Enoki, R. Sagisaka, N. Tsunoji and J. Ohshita, ChemistrySelect, 2017, 2, 7765–7770 CrossRef CAS.
- X. Feng, C. Qi, H. T. Feng, Z. Zhao, H. H. Y. Sung, I. D. Williams, R. T. K. Kwok, J. W. Y. Lam, A. Qin and B. Z. Tang, Chem. Sci., 2018, 9, 5679–5687 RSC.
- R. Cao, L. Ai, H. Yang, S. Li and C. Xu, New J. Chem., 2020, 44, 5049–5055 RSC.
- Z. Liu, Y. Jiang, J. Jiang, C. Yuan, D. Wang and M. Liu, RSC Adv., 2020, 10, 6772–6776 RSC.
- D. G. Chen, T. C. Lin, Y. A. Chen, Y. H. Chen, T. C. Lin, Y. T. Chen and P. T. Chou, J. Phys. Chem. C, 2018, 122, 12215–12221 CrossRef CAS.
- M. Okazaki, Y. Takeda, P. Data, P. Pander, H. Higginbotham, A. P. Monkman and S. Minakata, Chem. Sci., 2017, 8, 2677–2686 RSC.
- M. Colella, A. Danos and A. P. Monkman, J. Phys. Chem. Lett., 2019, 10, 793–798 CrossRef CAS.
- Z. Xie, Q. Huang, T. Yu, L. Wang, Z. Mao, W. Li, Z. Yang, Y. Zhang, S. Liu, J. Xu, Z. Chi and M. P. Aldred, Adv. Funct. Mater., 2017, 27, 1703918 CrossRef.
- K. Matsuo and T. Yasuda, Chem. Sci., 2019, 10, 10687–10697 RSC.
- X. Li, G. Baryshnikov, C. Deng, X. Bao, B. Wu, Y. Zhou, H. Ågren and L. Zhu, Nat. Commun., 2019, 10, 731 CrossRef CAS.
- C. J. Christopherson, D. M. Mayder, J. Poisson, N. R. Paisley, C. M. Tonge and Z. M. Hudson, ACS Appl. Mater. Interfaces, 2020, 12, 20000–20011 CrossRef CAS.
- B. Xu, Y. Mu, Z. Mao, Z. Xie, H. Wu, Y. Zhang, C. Jin, Z. Chi, S. Liu, J. Xu, Y. C. Wu, P. Y. Lu, A. Lien and M. R. Bryce, Chem. Sci., 2016, 7, 2201–2206 RSC.
- Y. Liu, G. Xie, Z. Ren and S. Yan, ACS Appl. Polym. Mater., 2019, 1, 2204–2212 CrossRef CAS.
- A. Forni, E. Lucenti, C. Botta and E. Cariati, J. Mater. Chem. C, 2018, 6, 4603–4626 RSC.
- M. A. El-Sayed, Acc. Chem. Res., 1968, 1, 8–16 CrossRef CAS.
- F. B. Dias, K. N. Bourdakos, V. Jankus, K. C. Moss, K. T. Kamtekar, V. Bhalla, J. Santos, M. R. Bryce and A. P. Monkman, Adv. Mater., 2013, 25, 3707–3714 CrossRef CAS.
- J. S. Ward, R. S. Nobuyasu, A. S. Batsanov, P. Data, A. P. Monkman, F. B. Dias and M. R. Bryce, Chem. Commun., 2016, 52, 2612–2615 RSC.
- L. Huang, J. Liu, L. Liu, Q. Yang, Z. Ma and X. Jia, Dyes Pigm., 2020, 173, 107963 CrossRef CAS.
- Z. Mao, Z. Yang, Y. Mu, Y. Zhang, Y. F. Wang, Z. Chi, C. C. Lo, S. Liu, A. Lien and J. Xu, Angew. Chem., Int. Ed., 2015, 54, 6270–6273 CrossRef CAS.
- Y. Wen, H. Liu, S. Zhang, J. Cao, J. De and B. Yang, Adv. Opt. Mater., 2020, 8, 1901995 CrossRef CAS.
- J. Wang, Z. Huang, X. Ma and H. Tian, Angew. Chem., Int. Ed., 2020, 59, 9928–9933 CrossRef CAS.
- G. Zhang, G. M. Palmer, M. W. Dewhirst and C. L. Fraser, Nat. Mater., 2009, 8, 747–751 CrossRef CAS.
- X. Wu, C. Y. Huang, D. G. Chen, D. Liu, C. Wu, K. J. Chou, B. Zhang, Y. Wang, Y. Liu, E. Y. Li, W. Zhu and P. T. Chou, Nat. Commun., 2020, 11, 2145 CrossRef CAS.
- H. Thomas, D. L. Pastoetter, M. Gmelch, T. Achenbach, A. Schlögl, M. Louis, X. Feng and S. Reineke, Adv. Mater., 2020, 32, 2000880 CrossRef CAS.
- M. Shimizu, S. Nagano and T. Kinoshita, Chem. – Eur. J., 2020, 26, 5162–5167 CrossRef CAS.
- W. Fang, J. Zhang, H. Zhao, J. Ni, S. Liu, Z. Liu, A. Ni, P. Zhang and H. Wei, Adv. Opt. Mater., 2020, 2000482 CrossRef CAS.
- R. Huang, N. A. Kukhta, J. S. Ward, A. Danos, A. S. Batsanov, M. R. Bryce and F. B. Dias, J. Mater. Chem. C, 2019, 7, 13224–13234 RSC.
- Y. Wang, J. Yang, Y. Tian, M. Fang, Q. Liao, L. Wang, W. Hu, B. Z. Tang and Z. Li, Chem. Sci., 2020, 11, 833–838 RSC.
- Z. Mao, Z. Yang, C. Xu, Z. Xie, L. Jiang, F. L. Gu, J. Zhao, Y. Zhang, M. P. Aldred and Z. Chi, Chem. Sci., 2019, 10, 7352–7357 RSC.
- G. Zhang, J. Chen, S. J. Payne, S. E. Kooi, J. N. Demas and C. L. Fraser, J. Am. Chem. Soc., 2007, 129, 8942–8943 CrossRef CAS.
- Y. Takeda, T. Kaihara, M. Okazaki, H. Higginbotham, P. Data, N. Tohnai and S. Minakata, Chem. Commun., 2018, 54, 6847–6850 RSC.
- R. L. Martin, J. Chem. Phys., 2003, 118, 4775–4777 CrossRef CAS.
- J. Chen, T. Yu, E. Ubba, Z. Xie, Z. Yang, Y. Zhang, S. Liu, J. Xu, M. P. Aldred and Z. Chi, Adv. Opt. Mater., 2019, 7, 1801593 CrossRef.
- Z. Mao, Z. Yang, C. Xu, Z. Xie, L. Jiang, F. L. Gu, J. Zhao, Y. Zhang, M. P. Aldred and Z. Chi, Chem. Sci., 2019, 10, 179–184 RSC.
- J.-H. Tan, W.-C. Chen, S.-F. Ni, Z. Qiu, Y. Zhan, Z. Yang, J. Xiong, C. Cao, Y. Huo and C.-S. Lee, J. Mater. Chem. C, 2020, 8, 8061–8068 RSC.
- T. Itoh, J. Lumin., 2004, 109, 221–225 CrossRef CAS.
- M. F. R. Prieto, B. Nickel, K. H. Grellmann and A. Mordziński, Chem. Phys. Lett., 1988, 146, 387–392 CrossRef.
- S. Y. Chu and L. Goodman, Chem. Phys. Lett., 1975, 32, 24–27 CrossRef CAS.
- T. Wang, X. Su, X. Zhang, X. Nie, L. Huang, X. Zhang, X. Sun, Y. Luo and G. Zhang, Adv. Mater., 2019, 31, 1904273 CrossRef CAS.
- R. Huang, J. S. Ward, N. A. Kukhta, J. Avó, J. Gibson, T. Penfold, J. C. Lima, A. S. Batsanov, M. N. Berberan-Santos, M. R. Bryce and F. B. Dias, J. Mater. Chem. C, 2018, 6, 9238–9247 RSC.
- A. Kirch, M. Gmelch and S. Reineke, J. Phys. Chem. Lett., 2019, 10, 310–315 CrossRef CAS.
- M. Hoshi, R. Nishiyabu, Y. Hayashi, S. Yagi and Y. Kubo, Chem. – Asian J., 2020, 15, 787–795 CrossRef CAS.
- Q. Zhang, J. Li, K. Shizu, S. Huang, S. Hirata, H. Miyazaki and C. Adachi, J. Am. Chem. Soc., 2012, 134, 14706–14709 CrossRef CAS.
- V. Jankus, P. Data, D. Graves, C. McGuinness, J. Santos, M. R. Bryce, F. B. Dias and A. P. Monkman, Adv. Funct. Mater., 2014, 24, 6178–6186 CrossRef CAS.
- A. P. Monkman, ISRN Mater. Sci., 2013, 670130 Search PubMed.
- N. A. Kukhta, T. Matulaitis, D. Volyniuk, K. Ivaniuk, P. Turyk, P. Stakhira, J. V. Grazulevicius and A. P. Monkman, J. Phys. Chem. Lett., 2017, 8, 6199–6205 CrossRef CAS.
- J. A. Leitch, T. Rogova, F. Duarte and D. J. Dixon, Angew. Chem., Int. Ed., 2020, 59, 4121–4130 CrossRef CAS.
- P. L. dos Santos, J. S. Ward, P. Data, A. S. Batsanov, M. R. Bryce, F. B. Dias and A. P. Monkman, J. Mater. Chem. C, 2016, 4, 3815–3824 RSC.
- C. Chen, R. Huang, A. S. Batsanov, P. Pander, Y.-T. Hsu, Z. Chi, F. B. Dias and M. R. Bryce, Angew. Chem., Int. Ed., 2018, 130, 16407–16411 CrossRef.
- Y. Zhang, Z. Li, C. Li and Y. Wang, Front. Chem., 2019, 7, 302 CrossRef CAS.
- B. H. Wallikewitz, D. Kabra, S. Gélinas and R. H. Friend, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 045209 CrossRef.
- A. Dey, N. Chandrasekaran, D. Chakraborty, P. Johari, C. R. McNeill, A. Rao and D. Kabra, npj Flexible Electron., 2018, 2, 28 CrossRef.
- S. M. Suresh, E. Duda, D. Hall, Z. Yao, S. Bagnich, A. M. Z. Slawin, H. Bässler, D. Beljonne, M. Buck, Y. Olivier, A. Köhler and E. Zysman-Colman, J. Am. Chem. Soc., 2020, 142, 6588–6599 CrossRef CAS.
- M. Luo, X. Li, L. Ding, G. Baryshnikov, S. Shen, M. Zhu, L. Zhou, M. Zhang, J. Lu, H. Ågren, X.-D. Wang and L. Zhu, Angew. Chem., Int. Ed., 2020, 59, 17018–17025 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |