DOI:
10.1039/C7QM00030H
(Research Article)
Mater. Chem. Front., 2017,
1, 1635-1640
Dithieno[3,2-a:3′,2′-j][5,6,11,12]chrysene diimides and their molecular energy level regulation†
Received
21st January 2017
, Accepted 13th April 2017
First published on 4th May 2017
Abstract
Dithieno[3,2-a:3′,2′-j][5,6,11,12]chrysene diimides (DTCDIs), a new class of organic dyes, with a twisted molecular backbone were designed and synthesized. The optical and electrochemical properties, charge transport behaviors and the single crystal structure were investigated. The cyanation of the DTCDI backbone was carried out to fine-tune the molecular energy levels. The addition of two cyano groups to the DTCDI backbone reduced the HOMO and LUMO energy levels by 0.35 and 0.30 eV, respectively. The energy levels could be further reduced with the addition of two more cyano groups leading to 0.32 and 0.22 eV reductions in HOMO and LUMO energy levels and stabilization of the energy levels. A tetracyano-substituted DTCDI derivative showed an electron mobility of 0.25 cm2 V−1 s−1 and a current on/off ratio of 106 in its organic thin film transistors.
1. Introduction
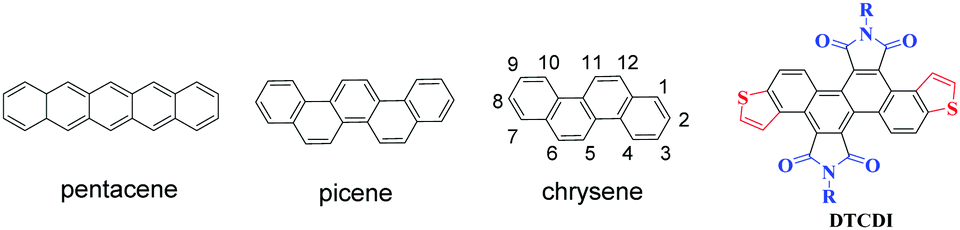
Polycyclic aromatic hydrocarbons (PAHs) have attracted ever increasing attention in the past few decades due to their potential applications in organic optoelectronic devices, such as organic field-effect transistors (OFETs), organic light-emitting diodes (OLEDs), and organic photovoltaics (OPVs).1 Among the PAH-based charge transport materials, pentacene (Fig. 1), a PAH with five fused benzene rings in a linear configuration, has become a representative material for OFETs, with hole mobilities of up to 3 cm2 V−1 s−1 in thin films and 35 cm2 V−1 s−1 in single crystals.2 However, the major drawback of pentacene is its instability on exposure to air and/or light owing to its high highest occupied molecular orbital (HOMO) energy level and high reaction activity of the 6- and 13-positions at the central benzene ring.3 Compared with the linear [n]acenes, [n]phenacenes that contain fused benzene rings with a zigzag shape (Fig. 1) have emerged as promising organic optoelectronic materials. The decreased delocalization caused by increased aromatic π-sextets brings about stabilization in the HOMO and destabilization of the lowest unoccupied molecular orbital (LUMO), both of which contribute to increased stability.4 Therefore, [n]phenacene molecules may be superior to pentacene and its analogues (acene molecules) in transistor and other applications. Recently, the zigzag phenacenes have received considerable attention, some of which showed high optoelectronic device performance.5 For example, picene is the first PAH that showed unique superconductivity through intercalation with an alkali metal,6 and a picene derivative that bears two tetradecyl groups showed a hole mobility as high as 20 cm2 V−1 s−1.7 These results demonstrate that zigzag shaped PAHs are a class of promising organic optoelectronic materials.
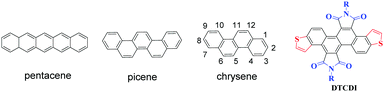 |
| | Fig. 1 Structures of pentacene, picene and chrysene, and the chemical structure of dithieno[3,2-a:3′,2′-j][5,6,11,12]chrysene diimides (DTCDIs). | |
Chrysene (Fig. 1) is a typical zigzag PAH consisting of two fused naphthalenes. However, the application of chrysene and its derivatives in organic electronics is limited.8 One reason is the lack of efficient chemical modifications of chrysene.9 Based on our previous work on 1,2,5,6-naphthalene diimides,10 we report herein the synthesis of novel laterally extended chrysene derivatives, dithieno[3,2-a:3′,2′-j][5,6,11,12]chrysene diimides (DTCDIs), which consist of a dithienochrysene skeleton and two imide groups at 5,6- and 11,12-positions of the central chrysene core (Fig. 1). The molecular design of DTCDI is based on the following considerations: (i) the extended π conjugation provided by the addition of thiophene units could enhance the intermolecular π–π overlaps in solid state and thus benefit charge carrier transport; (ii) the introduction of two imide groups on the dithienochrysene skeleton makes DTCDI a promising electron-deficient building block for constructing new donor–acceptor (D–A) conjugated small molecules and polymers; (iii) the twisted molecular π-backbone (Fig. 2 and 3) and the proper N-substituted groups could afford enough solubility and effective molecular self-assembly for solution-processable organic optoelectronic devices with high performance. Moreover, the cyanation of the molecular backbone of DTCDI was carried out to fine-tune the molecular energy levels.
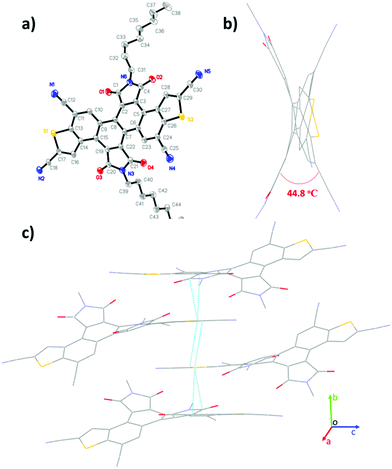 |
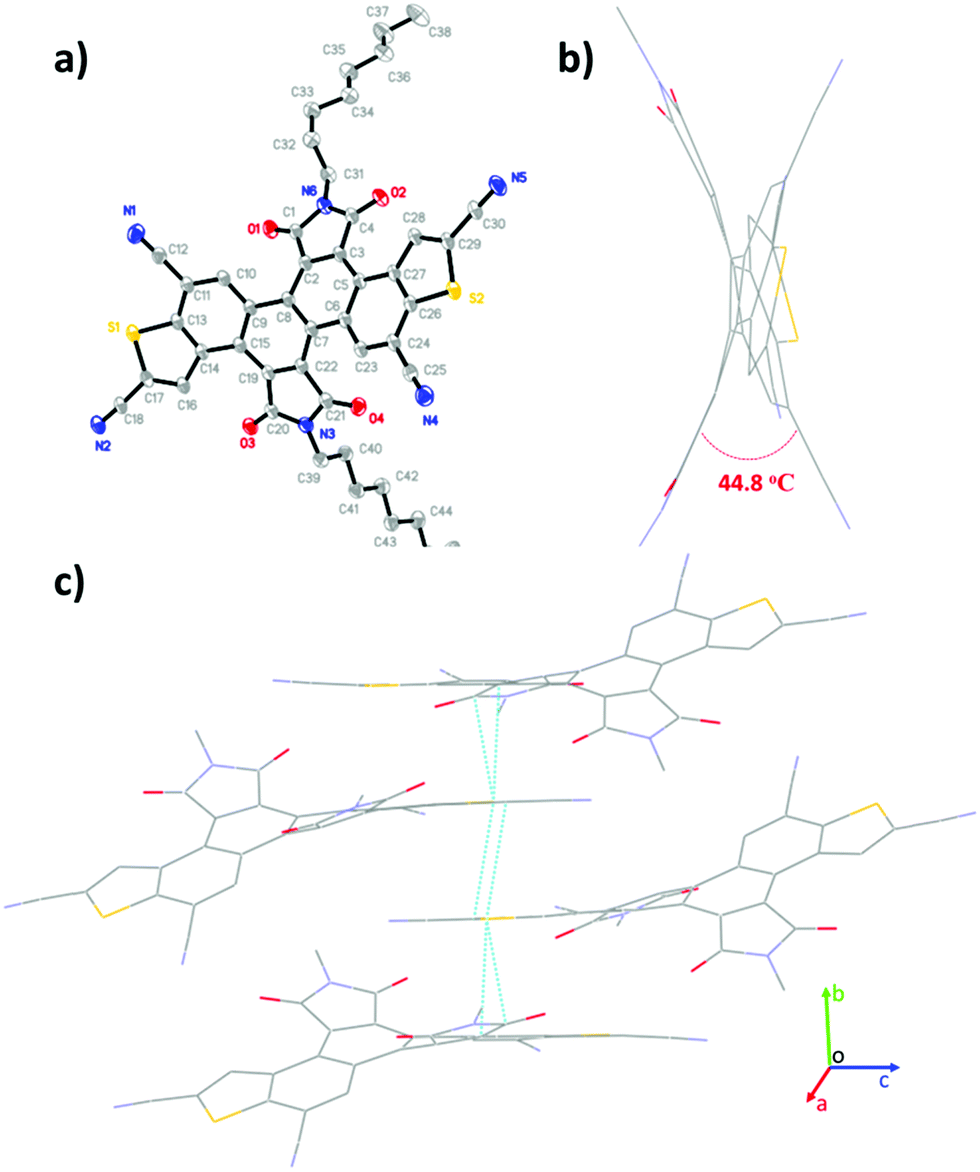
| | Fig. 2 Single-crystal structure of 3b: (a) thermal ellipsoids (probability level = 50%), (b) viewed along the c-axis, and (c) packing structure. | |
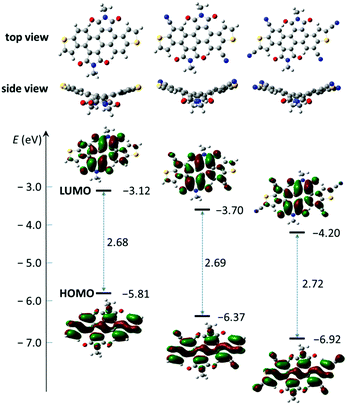 |
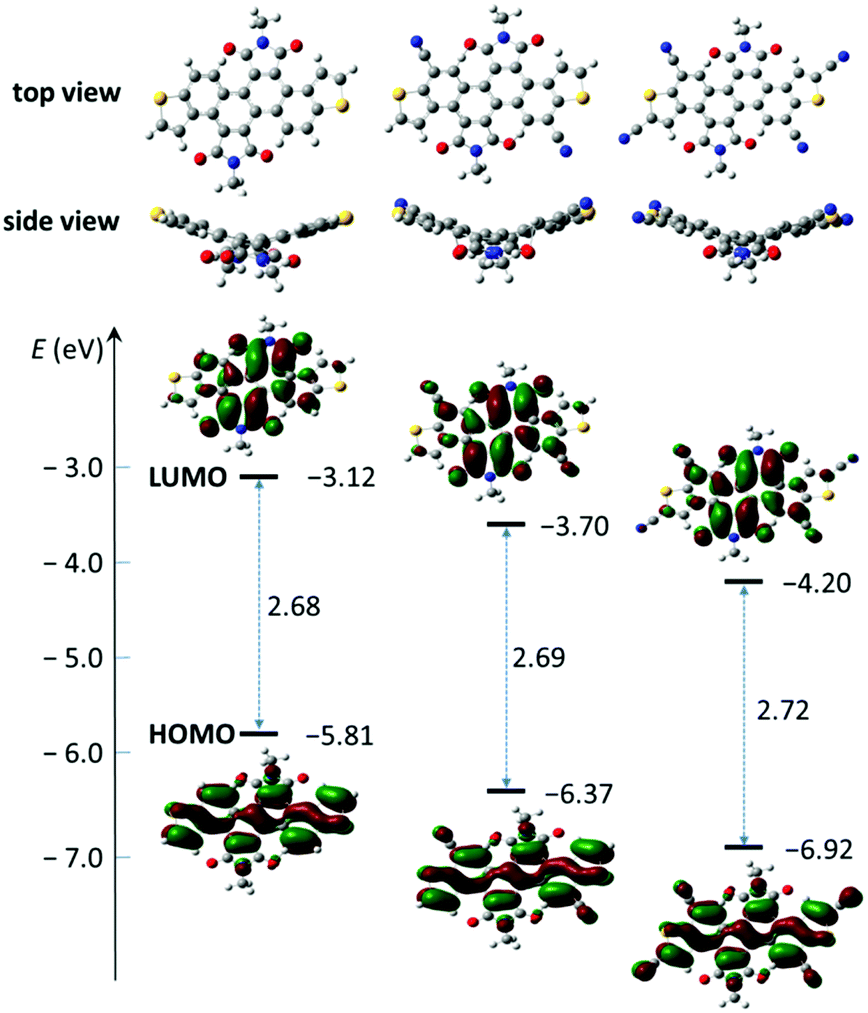
| | Fig. 3 Molecular geometries, frontier molecular orbitals, and FMO energies of model molecules for 1a, 2a and 3a obtained by DFT calculations. | |
2. Results and discussion
2.1 Synthesis
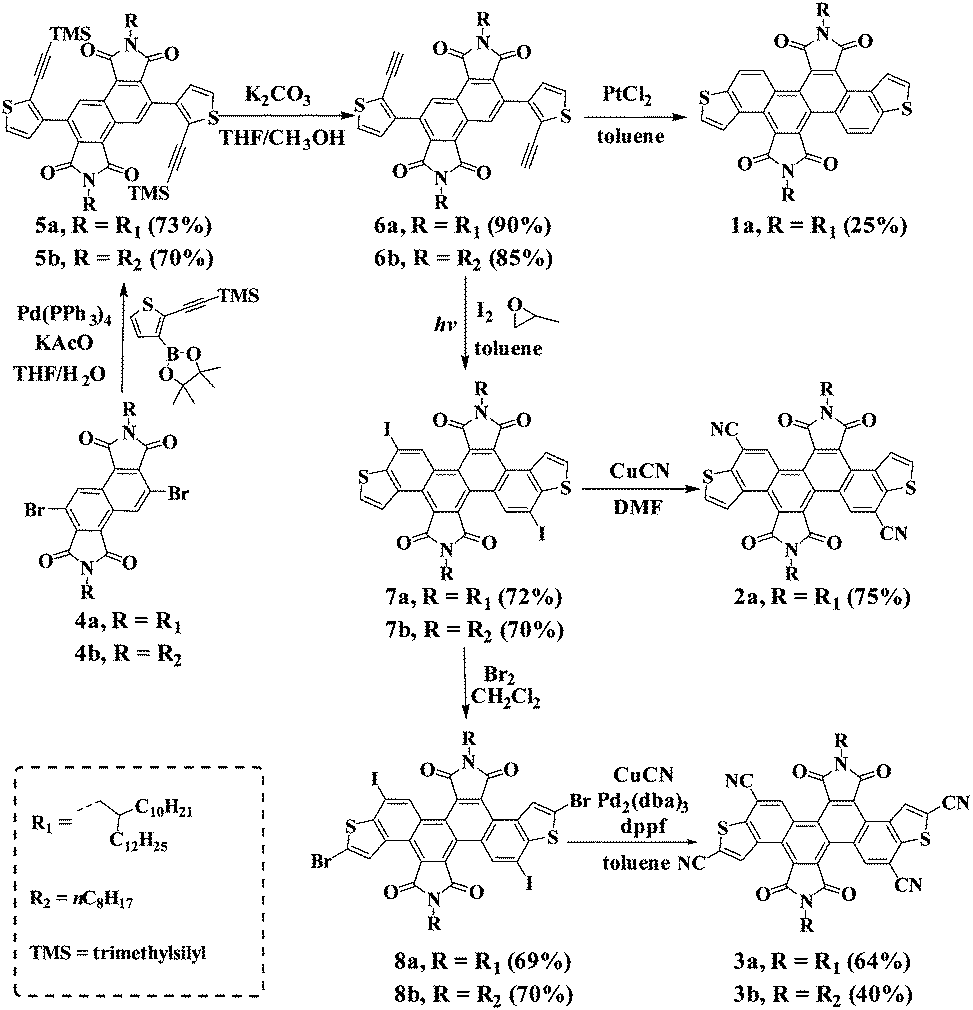
The synthetic routes for the preparation of DTCDI (1a), dicyano-substituted DTCDI (2a) and tetracyano-substituted DTCDI (3a and 3b) are shown in Scheme 1. The synthesis of 3b is similar to that of 3a. Suzuki–Miyaura couplings occurred between 3,7-dibromo-1,2,5,6-naphthalene diimides (4a) and trimethyl((5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)thiophen-2-yl)ethynyl)silane11 in the presence of Pd(PPh3)4 and KOAc, affording 5a in 73% yield. After the elimination of the trimethylsilyl-protecting groups by using K2CO3 in the mixed methanol and THF solution, compound 6a was achieved in 90% yield. Compound 1a was prepared in the presence of PtCl2 with a relatively low yield of 25%. Compound 7a was obtained in 72% yield via irradiation under a medium pressure mercury lamp in the presence of I2.12 The selective brominated intermediate compound 8a was obtained from 7a in the presence of Br2 in CH2Cl2. The cyanation of 7a took place in DMF with CuCN as the cyanating reagent, giving 2a in 75% yield. However this cyanation method failed to afford tetracyano-substituted compound 3a from 8a. A powerful Pd-catalyst in situ generated from Pd2(dba)3 and dppf in toluene was used, affording 3a with a moderate yield of 64%.13 All compounds were fully characterized by 1H- and 13C-NMR, HRMS as well as elemental analysis. 1a, 2a and 3a are soluble in common organic solvents, such as THF, chloroform and toluene, which is beneficial for solution-processable device fabrication. The thermal properties of 1a, 2a and 3a were evaluated by differential scanning calorimetry (DSC) and thermal gravimetric analysis (TGA) (Fig. S1, ESI†). Compounds 1a and 2a showed lower melting points of 29 °C and 113 °C, respectively, compared to 3a (218 °C). The TGA measurements reveal that the 5% weight loss temperatures of 1a, 2a and 3a are 442 °C, 396 °C and 427 °C, respectively, demonstrating their excellent thermal stability.
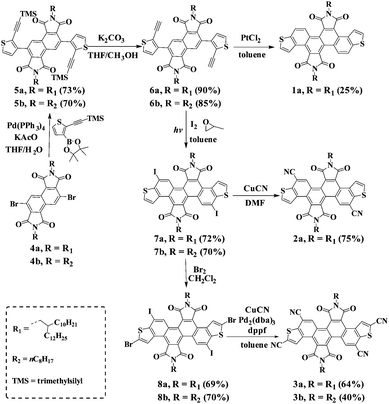 |
| | Scheme 1 Synthesis of DTCDI derivatives 1a, 2a and 3a. | |
2.2 Single crystals
Single crystals of 3b (R = n-octyl) were obtained from the slow evaporation of its methanol and dichloromethane mixed solution at room temperature. As shown in Fig. 2, the π-skeleton of 3b is twisted with a torsion angle of 44.8° between the benzothiophene units and the imide groups. This is probably due to the intense repulsive force between the O atoms of imide groups and the neighbouring C–H groups. The crystal packing of 3b displays one-dimensional π-stacking along the b-axis direction with short π–π stacking distances of 3.3–3.5 Å, which is in favor of charge carrier transport.
2.3 DFT calculations
To gain an insight into the electronic structures of 1a, 2a and 3a, DFT calculations were performed at the B3LYP/6-31G(d,p) level using Gaussian 09, and the minimum-energy conformations and the frontier molecular orbitals were obtained. Fig. 3 shows the optimized molecular geometries and the frontier molecular orbital (FMO) distribution of 1a, 2a and 3a. All alkyl chains were replaced by methyl groups to simplify the calculation. All three compounds show a similar electron density distribution, the HOMO orbital is well located along the dithienochrysene skeleton and the LUMO counterpart mainly localizes in the central 1,2,5,6-naphthalene diimide core. These calculations suggest the HOMO–LUMO transition to be an intramolecular charge transfer (ICT) in these compounds. As shown in Table 1, the calculated HOMO/LUMO energy values for 1a, 2a and 3a are −5.81/−3.12 eV, −6.37/−3.70 eV, and −6.92/−4.20 eV, respectively. The cyanation of the DTCDI core leads to a stepwise decrease of the HOMO/LUMO energies with ΔEHOMO and ΔELUMO of about 0.5–0.6 eV for every two cyano groups added (Fig. 3 and Table 1).
Table 1 Thermal, optical, and electrochemical data for compounds 1a, 2a and 3a
| Comp. |
λ
abs sol/film (nm) |
ε/M−1 cm−1 |
λ
em sol (nm) |
E
optg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (eV) a (eV) |
E
onset,red
(V) |
E
LUMO
(eV) |
E
HOMO
(eV) |
E
LUMO
(eV) |
E
HOMO
(eV) |
|
Estimated from the absorption edge.
0.1 M [Bu4N]PF6 in CH2Cl2 (vs. SCE) at a scan rate of 100 mV s−1.
Estimated from the equation14ELUMO = −(Eonset,red − E1/2(Fc/Fc+) + 5.1) (eV).
E
HOMO (eV) = ELUMO − Eoptg.
Estimated from DFT calculations.
|
|
1a
|
484/489 |
6550 |
561 |
2.26 |
−0.72 |
−4.02 |
−6.28 |
−3.12 |
−5.81 |
|
2a
|
478/517 |
10340 |
550 |
2.31 |
−0.42 |
−4.32 |
−6.63 |
−3.70 |
−6.37 |
|
3a
|
461/502 |
10790 |
521 |
2.41 |
−0.20 |
−4.54 |
−6.95 |
−4.20 |
−6.92 |
2.4 Optical and electrochemical properties
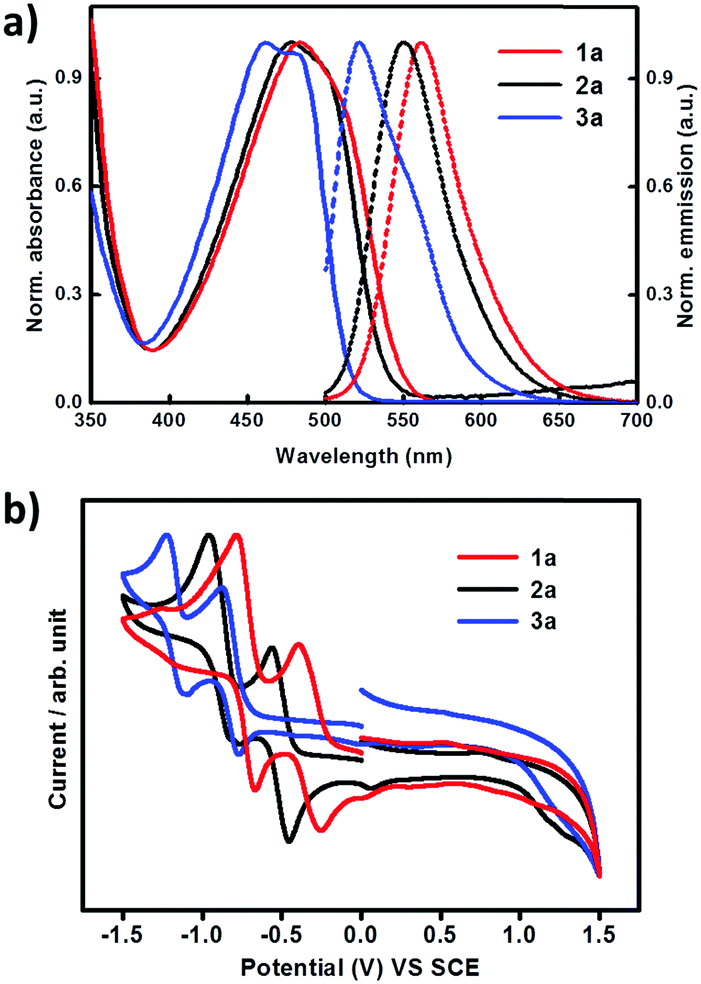
The optical properties of compounds 1a, 2a and 3a were studied by UV-vis and fluorescence spectra in CH2Cl2 solutions. As shown in Fig. 4 and Table 1, all the compounds exhibit absorption bands in the range of 380–570 nm, which have been formally assigned to intramolecular charge-transfer (ICT) transitions. For the ICT absorption, compound 3a with four cyano groups and 2a bearing two cyano groups show 23 nm and 6 nm hypochromic shifts, respectively, compared with compound 1a. The same trend is observed for their maximum emission wavelengths, with obvious blue shifts for 3a (40 nm) and 2a (11 nm) relative to 1a (Table 1). In comparison with the absorption of 1a, 2a and 3a in dichloromethane solution, their absorption edge in thin films shows obvious red shifts; the absorption edge values in solution and thin films are 549/563 nm, 538/582 nm and 514/525 nm for 1a, 2a and 3a respectively (Fig. S2, ESI†). This is probably due to the intermolecular interactions within the thin films. The electrochemical properties of 1a, 2a and 3a were analyzed by cyclic voltammetry (CV) in dichloromethane solutions with tetrabutylammonium hexafluorophosphate (Bu4NPF6) as the supporting electrolyte. As shown in Fig. 4(b) and Table 1, all compounds show two quasi-reversible reduction processes. As the number of cyano groups increases in the backbone, the reduction onset potential (Eonset,red) shifts toward more positive values. Eonset,red values of 1a, 2a and 3a were −0.72 V, −0.42 V, and −0.20 V, respectively. The LUMO energy levels of 1a, 2a and 3a estimated from the Eonset,red values are −4.02 eV, −4.32 eV and −4.54 eV, respectively.14 The results indicate an efficient electronic structure regulation of the DTCDI molecular backbone by using cyano groups. As shown in Table 1, the addition of two CN groups to the DTCDI backbone reduced the HOMO and LUMO energy levels by 0.35 and 0.30 eV, respectively. The energy levels could be further reduced with the addition of two more CN groups leading to 0.32 and 0.22 eV reductions in HOMO and LUMO energy levels and stabilization of the energy levels.
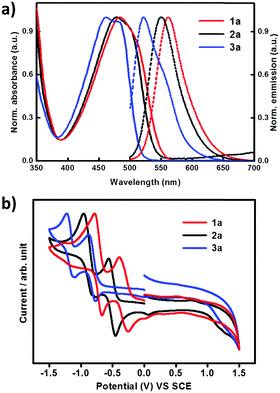 |
| | Fig. 4 (a) UV-vis absorption (solid line) and fluorescence (dashed line) spectra of 1a, 2a and 3a in CH2Cl2 solutions. (b) Cyclic voltammograms of 1a, 2a and 3a in CH2Cl2 solutions (0.1 M Bu4NPF6 as supporting electrolyte; SCE as reference electrode; scan rate 100 mV s−1). | |
2.5 Charge transport properties
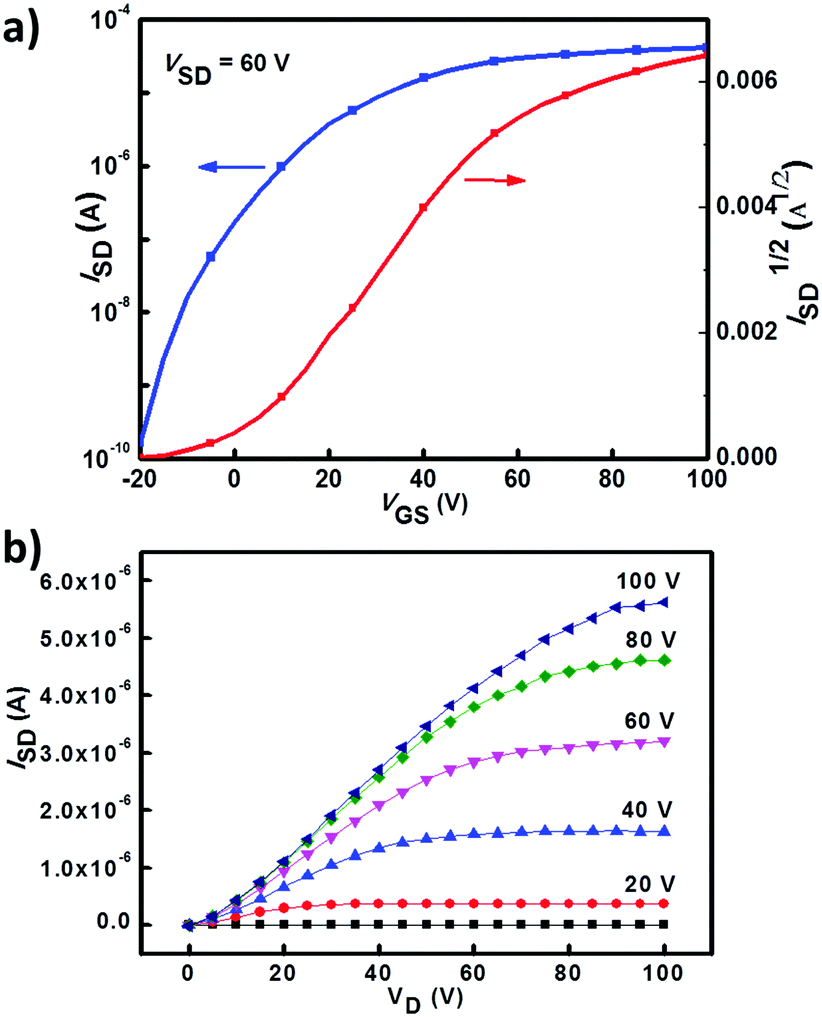
The charge transport properties of 1a, 2a and 3a were investigated by testing their spin-coated bottom-gate top-contact OFET devices (see ESI† for details). All OFET devices were measured in a N2 atmosphere. OFETs based on 2a and 3a exhibited typical electron-transporting characteristics, while 1a-based OFETs showed no obvious FET behavior. A 3a-based OFET showed an optimized electron mobility of 0.25 cm2 V−1 s−1 with a current on/off ratio of 106 (Fig. 5 shows its transfer and output characteristics), whereas 2a-based devices showed much lower electron mobilities of about 10−3–10−2 cm2 V−1 s−1. Atomic force microscopy (AFM) and X-ray diffraction (XRD) measurements were employed to study the thin film morphologies of 2a and 3a. As shown in Fig. S3 (ESI†), no obvious crystalline domains were observed for as-spun films of 2a and 3a. Their thin-film surface morphologies were changed obviously after thermal annealing, and more interconnected domains were formed (Fig. S3, ESI†). The third order diffraction peak emerged after annealing the film of 3a at 120 °C (Fig. S4, ESI†), indicating a better crystallinity of the thin films of 3a, which together with the interconnected domains in thin films of 3a could explain its relatively high device performance.
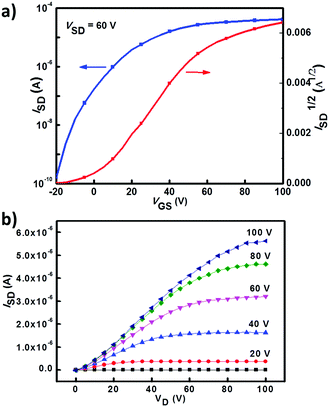 |
| | Fig. 5 Transfer and output characteristics of an OFET device based on 3a. | |
3. Conclusions
In conclusion, a new class of aromatic diimides, dithieno[3,2-a:3′,2′-j][5,6,11,12]chrysene diimides (DTCDIs), with a twisted molecular backbone were designed and synthesized. Their optical and electrochemical properties, charge transport characteristics as well as single crystal structure were investigated. The cyanation of the molecular backbone of DTCDI was carried out to tune the molecular energy levels. A tetracyano-substituted DTCDI derivative (3a) showed an electron mobility of 0.25 cm2 V−1 s−1 and a current on/off ratio of 106 in one of its OFET devices. Strong chemical derivatization, twisted molecular backbone and good electron transport property make DTCDI a promising building block for organic optoelectronic materials. Side chain engineering and polymerization of the molecular backbone of DTCDI are currently underway in our lab.
4. Experimental section
4.1 Synthesis
Chemical reagents were purchased from J&K or TCI and used as received. Solvents and other common reagents were obtained from the Sinopharm Chemical Reagent Co., Ltd. Toluene and tetrahydrofuran (THF) were distilled from sodium/benzophenone under nitrogen prior to use. 3,7-Dibromo-1,2,5,6-naphthalene diimides (4a and 4b)10,15 and ((3-bromothiophen-2-yl)ethynyl)trimethylsilane11 were synthesized according to the literature procedures.
Synthesis of trimethyl((3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)thiophen-2-yl)ethynyl)silane.
Under a nitrogen atmosphere, ((3-bromothiophen-2-yl)ethynyl)trimethylsilane (200 mg, 0.77 mmol), B2Pin2 (300 mg, 1.18 mmol), Pd(dppf)2Cl2 (45 mg, 10% mmol) and KOAc (152 mg, 1.56 mmol) were dissolved in dioxane (10 mL). The mixture solution was stirred at 80 °C for 16 h. After cooling down to rt, the mixture was extracted with CH2Cl2 and dried under reduced pressure. The crude product was purified by silica gel column chromatography (petrol ether:CH2Cl2 = 2:1) to give a viscous product (122 mg, 48% yield). 1H NMR (400 MHz, CDCl3) δ (ppm) 7.21–7.22 (d, J = 5.2, 2H), 7.18–7.19 (d, J = 5.2, 2H), 1.34 (s, 12H), 0.26 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 132.6, 126.4, 101.0, 98.3, 83.8, 77.3, 25.0, −0.2. HRMS (EI+) calcd for [C15H23O2SBSi]+ 303.1317, found: 303.1315.
Synthesis of 5a.
Under a nitrogen atmosphere, 3,7-dibromo-1,2,5,6-naphthalene diimide 4a (300 mg, 0.27 mmol), trimethyl((3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)thiophen-2-yl)ethynyl)silane (285 mg, 0.93 mmol), and Pd(PPh3)4 (20 mg, 6% mmol) were dissolved in THF (20 mL) and potassium acetate aqueous solution (2 M) (10 mL). The mixture solution was stirred at 100 °C for 12 h. After cooling down to rt, the mixture was extracted with CH2Cl2 and dried under reduced pressure. The crude product was purified by silica gel column chromatography (petrol ether:CH2Cl2 = 4:1) to give the product as a yellow viscous liquid (259 mg, 73% yield). FTIR (KBr): ν 2146.2 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C), 1712.8 (C
C), 1712.8 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) cm−1. 1H NMR (400 MHz, CDCl3) δ (ppm) 9.67 (s, 2H), 7.32–7.34 (d, J = 5.2 Hz, 2H), 7.28–7.29 (d, J = 5.2 Hz, 2H), 3.57–3.59 (d, J = 7.2 Hz, 4H), 1.90 (br, 2H), 1.23 (br, 80H), 0.85–0.87 (m, 12H), 0.09 (s, 18H). 13C NMR (100 MHz, CDCl3) δ (ppm) 168.8, 167.8, 140.3, 134.0, 132.2, 130.6, 129.9, 129.8, 128.5, 125.6, 122.5, 102.2, 97.0, 42.8, 37.3, 32.1, 31.7, 30.2, 29.8, 29.5, 26.5, 22.8, 14.2, −0.2. HRMS (MALDI-FT) calcd for [C80H122N2O4S2Si2]+ 1294.8457, found: 1295.8504 (M + 1)+.
O) cm−1. 1H NMR (400 MHz, CDCl3) δ (ppm) 9.67 (s, 2H), 7.32–7.34 (d, J = 5.2 Hz, 2H), 7.28–7.29 (d, J = 5.2 Hz, 2H), 3.57–3.59 (d, J = 7.2 Hz, 4H), 1.90 (br, 2H), 1.23 (br, 80H), 0.85–0.87 (m, 12H), 0.09 (s, 18H). 13C NMR (100 MHz, CDCl3) δ (ppm) 168.8, 167.8, 140.3, 134.0, 132.2, 130.6, 129.9, 129.8, 128.5, 125.6, 122.5, 102.2, 97.0, 42.8, 37.3, 32.1, 31.7, 30.2, 29.8, 29.5, 26.5, 22.8, 14.2, −0.2. HRMS (MALDI-FT) calcd for [C80H122N2O4S2Si2]+ 1294.8457, found: 1295.8504 (M + 1)+.
Synthesis of 5b.
A similar procedure to that used for 5a afforded 5b as a yellow solid (135 mg, 70% yield). Mp: 154–155 °C. FTIR (KBr): ν 2143.7 (CC), 1709.1 (CO) cm−1. 1H NMR (400 MHz, CDCl3) δ (ppm) 9.65 (s, 2H), 7.33–7.35 (d, J = 5.2 Hz, 2H), 7.30–7.31 (d, J = 5.2 Hz, 2H), 3.68–3.71 (m, 4H), 1.69 (br, 4H), 1.26 (br, 24H), 0.85–0.87 (m, 6H), 0.08 (s, 18H). 13C NMR (100 MHz, CDCl3) δ 168.5, 167.5, 140.3, 133.8, 132.2, 130.7, 129.9, 129.7, 128.5, 125.7, 122.6, 102.1, 96.9, 38.5, 31.9, 29.3, 28.8, 27.1, 22.7, 14.2, −0.2. HRMS (MALDI-FT) calcd for [C48H59O4N2S2Si2]+ 847.3449, found: 847.3445 (M+).
Synthesis of 6a.
To a solution of 5a (400 mg, 0.31 mmol) in THF (20 mL) and CH3OH (10 mL), K2CO3 (254 mg, 1.86 mmol) was added. After stirring for 0.5 h at room temperature, the mixture was extracted with CH2Cl2 and dried under reduced pressure. The crude product was purified by silica gel column chromatography (petrol ether:CH2Cl2 = 3:1) to give the product as a yellow solid (320 mg, 90% yield). Mp: 120–121 °C. FTIR (KBr): ν 3294.6 (CC–H), 1715.8 (CO) cm−1. 1H NMR (400 MHz, CDCl3) δ (ppm) 9.59 (s, 2H), 7.38–7.39 (d, J = 5.2 Hz, 2H), 7.27–7.29 (d, J = 5.2 Hz, 2H), 3.60–3.61 (d, J = 7.2 Hz, 4H), 3.29 (s, 2H), 1.90 (br, 2H), 1.26–1.30 (br, 80H), 0.85–0.87 (m, 12H). 13C NMR (100 MHz, CDCl3) δ (ppm) 168.9, 167.7, 140.9, 133.7, 132.1, 130.6, 130.1, 129.5, 128.6, 126.2, 121.4, 84.2, 76.5, 42.7, 37.3, 30.2, 31.7, 30.2, 29.8, 29.5, 26.5, 22.8, 14.2. HRMS (MALDI-FT) calcd for [C74H107N2O4S2]+ 1151.7667, found: 1151.7711 (M + 1)+.
Synthesis of 6b.
A similar procedure to that used for 6a afforded 6b as a yellow solid (110 mg, 85% yield). Mp: 160–161 °C. FTIR (KBr): ν 3106.2 (CC–H), 1706.3 (CO) cm−1. 1H NMR (400 MHz, CDCl3) δ (ppm) 9.58 (s, 2H), 7.37–7.39 (d, J = 5.2 Hz, 2H), 7.28–7.29 (d, J = 5.2 Hz, 2H), 3.69–3.72 (m, 4H), 3.31 (s, 2H), 1.68–1.69 (br, 4H), 1.25 (br, 24H), 0.84–0.87 (m, 6H). 13C NMR (100 MHz, CDCl3) δ 168.6, 167.5, 140.8, 133.6, 132.0, 130.7, 129.6, 129.40, 128.7, 126.2, 121.3, 84.2, 76.4, 38.4, 31.9, 29.3, 28.8, 27.0, 22.7, 14.2. HRMS (MALDI-FT) calcd for [C42H43O4N2S2]+ 703.2659, found: 703.2663 (M + 1)+.
Synthesis of 7a.
To a solution of 6a (100 mg, 0.086 mmol) in toluene (60 mL), I2 (108 mg, 0.43 mmol) and propylene oxide (0.06 mL, 0.86 mmol) were added. After stirring for 3 h under hν conditions, the mixture was extracted with CH2Cl2 and dried under reduced pressure. The crude product was purified by silica gel column chromatography (petrol ether:CH2Cl2 = 3:1) to give the product as a red solid (87 mg, 72% yield). Mp: 124–125 °C. FTIR (KBr): ν 1686.0 (CO) cm−1. 1H NMR (400 MHz, CDCl3) δ (ppm) 9.40–9.42 (d, J = 5.5 Hz, 2H), 9.02 (s, 2H), 7.80–7.81 (d, J = 5.5 Hz, 2H), 3.63–3.64 (d, J = 7.0 Hz, 4H), 1.84 (br, 2H), 1.20 (br, 80H), 0.85–0.86 (m, 12H). 13C NMR (100 MHz, CDCl3) δ 167.6, 167.5, 148.7, 135.8, 132.5, 131.1, 130.1, 128.2, 127.3, 127.0, 121.9, 90.3, 43.5, 37.2, 32.1, 32.0, 31.7, 30.2, 29.8, 29.5, 26.5, 22.8, 14.2. HRMS (MALDI-FT) calcd for [C74H105O4N2I2S2]+ 1403.5600, found: 1403.5639 (M + 1)+. Anal. calcd for C74H104O4N2I2S2: C 63.32; H 7.47; N 2.00. Found: C 63.33; H 7.46; N 1.99.
Synthesis of 7b.
A similar procedure to that used for 7a afforded 7b as a yellow solid (130 mg, 70% yield). Mp: 151–152 °C. FTIR (KBr): ν 1701.4 (CO) cm−1. 1H NMR (400 MHz, CDCl3) δ (ppm) 9.26–9.28 (d, J = 8 Hz 2H), 9.02 (s, 2H), 7.84–7.86 (d, J = 8 Hz, 2H), 3.69–3.72 (m, 4H), 1.52 (br, 4H), 1.18 (br, 24H), 0.79–0.82 (m, 6H). 13C NMR (100 MHz, CDCl3) δ 166.9, 166.6, 148.7, 135.3, 132.3, 131.7, 129.4, 127.5, 127.0, 126.9, 121.6, 90.1, 39.4, 31.8, 29.3, 28.5, 27.0, 22.7, 14.2. HRMS (MALDI-FT) calcd for [C42H41O4N2I2S2]+ 955.0592, found: 955.0593 (M + 1)+.
Synthesis of 8a.
To a solution of 7a (188 mg, 0.134 mmol) in DCM (30 mL), Br2 was added slowly. After stirring for 10 h at 30 °C, the mixture was extracted with CH2Cl2 and dried under reduced pressure. The crude product was purified by silica gel column chromatography (petrol ether:CH2Cl2 = 2:1) to give the product as a red solid (144 mg, 69% yield). Mp: 152–153 °C. 1H NMR (400 MHz, CDCl3) δ 9.56 (s, 2H), 9.05 (s, 2H), 3.71–3.73 (d, J = 8 Hz, 4H), 1.93 (br, 2H), 1.21–1.25 (br, 80H), 0.84–0.86 (m, 12 H). 13C NMR (100 MHz, CDCl3) δ 167.2, 166.9, 149.9, 135.4, 133.8, 132.5, 129.7, 127.9, 127.1, 120.6, 116.9, 88.4, 43.7, 37.0, 32.1, 32.0, 31.6, 30.1, 29.8, 29.5, 26.4, 22.8, 14.3. MS (MALDI-TOF) m/z: 1559.5 (M + H)+. Anal. calcd for [C74H102O4N2Br2I2S2]: C 56.93; H 6.58; N 1.79. Found: C 57.33; H 6.61; N 1.77.
Synthesis of 8b.
A similar procedure to that used for 8a afforded 8b as a yellow solid (40 mg, 70% yield). Mp: 225–226 °C. FTIR (KBr): ν 1706.5 (CO) cm−1. 1H NMR (400 MHz, CDCl3) δ 9.48 (s, 2H), 9.04 (s, 2H), 3.78–3.80 (t, J = 7.2 Hz, 4H), 1.21–1.25 (br, 24H), 0.81–0.82 (m, 6H). 13C NMR (100 MHz, CDCl3) δ 166.8, 166.5, 150.0, 135.2, 134.0, 132.6, 129.5, 127.6, 127.1, 120.5, 117.0, 88.3, 39.6, 31.8, 29.2, 28.5, 27.0, 22.7, 14.2. HRMS (MALDI-FT) calcd for [C42H38O4N2Br2I2S2]+ 1109.8724, found: 1109.8745 (M + 1)+.
Synthesis of 1a.
To a solution of 6a (80 mg, 0.07 mmol) in anhydrous toluene (20 mL), PtCl2 (40 mg, 0.14 mmol) was added. After stirring for 2 h at 100 °C, the reaction mixture was cooled down to room temperature and the mixture was extracted with CH2Cl2 and dried under reduced pressure. The crude product was purified by silica gel column chromatography (petrol ether:CH2Cl2 = 5:1) to give the product as a red solid (20 mg, 25% yield). Mp: 29–30 °C. FTIR (KBr): ν 1698.5 (CO) cm−1. 1H NMR (400 MHz, CDCl3) δ (ppm) 9.23–9.25 (d, J = 5.6 Hz, 2H), 8.61–8.64 (d, J = 9.0 Hz, 2H), 8.19 (d, J = 9.0 Hz, 2H), 7.75–7.76 (d, J = 5.6 Hz, 2H), 3.66–3.68 (d, J = 7.1 Hz, 4H), 1.95 (br, 2H), 1.22 (m, 80H), 0.85 (m, 12H). 13C NMR (100 MHz, CDCl3) δ (ppm) 168.4, 168.3, 142.4, 133.7, 130.5, 130.4, 129.6, 129.0, 127.3, 126.8, 126.6, 123.6, 122.5, 43.2, 37.2, 32.0, 31.7, 30.2, 29.8, 29.5, 26.5, 22.8, 14.3. HRMS (MALDI-FT) calcd for [C74H106N2O4S2]+ 1150.7589, found: 1150.7606 (M)+.
Synthesis of 2a.
Under a nitrogen atmosphere, 7a (100 mg, 0.07 mmol) and CuCN (25 mg, 0.28 mmol) were dissolved in anhydrous DMF (20 mL). After stirring for 10 h at 160 °C, the reaction mixture was cooled down to room temperature and washed with ammonium hydroxide (30 mL). The mixture was extracted with CH2Cl2 and dried under reduced pressure. The crude product was purified by silica gel column chromatography (petrol ether:CH2Cl2 = 4:1) to give the product as a red solid (64 mg, 75% yield). Mp: 113–114 °C. FTIR (KBr): ν 2225.6 (CN), 1704.9 (CO) cm−1. 1H NMR (400 MHz, CDCl3) δ 9.35–9.36 (d, J = 5.6 Hz, 2H), 9.14 (s, 2H), 7.91–7.93 (d, J = 5.6 Hz, 2H), 3.765–3.76 (d, J = 6.8 Hz, 4H), 1.99 (br, 2H), 1.27 (br, 80H), 0.85–0.87 (m, 12H). 13C NMR (100 MHz, CDCl3) δ 167.6, 167.5, 142.7, 134.8, 132.2, 130.8, 130.0, 129.6, 129.4, 128.6, 124.6, 116.8, 108.1, 43.6, 37.3, 32.0, 31.8, 30.2, 29.8, 29.5, 26.5, 22.8, 14.2. MS (MALDI-TOF) m/z: 1201.8 (M + H)+. Anal. calcd for [C76H104O4N4I2S2]: C 75.95; H 8.72; N 4.66. Found: C 75.94; H 8.80; N 4.66.
Synthesis of 3a.
To a carefully degassed solution of compound 8a (40 mg, 0.027 mmol) in toluene, CuCN (15 mg, 0.154 mmol), Pd2(dba)3 (2.5 mg, 10% mmol) and 1,1′-bis(diphenylphosphino)ferrocene (3.0 mg, 20% mmol) were added. After stirring for 16 h at 120 °C, the reaction mixture was cooled down to room temperature and washed with ammonium hydroxide (20 mL). The solvent was removed under reduced pressure by rotary evaporation. The crude product was purified by silica gel column chromatography (petrol ether:CH2Cl2 = 2:1) to give the product as a red solid (21 mg, 64% yield). Mp: 218–219 °C. FTIR (KBr): ν 2223.5 (CN), 1703.9 (CO) cm−1. 1H NMR (400 MHz, CDCl3) δ 10.04 (s, 2H), 9.33 (s, 2H), 3.79–3.81 (d, J = 7.2 Hz, 4H), 1.98 (br, 2H), 1.25–1.27 (br, 80H), 0.85–0.86 (m, 24H). 13C NMR (100 MHz, CDCl3) δ 167.2, 167.0, 144.7, 140.3, 135.5, 132.8, 130.8, 130.5, 130.2, 129.9, 125.3, 115.6, 113.4, 112.3, 107.8, 44.0, 37.3, 32.0, 31.7, 30.2, 29.8, 29.5, 26.4, 22.8, 14.2. MS (MALDI-TOF) m/z: 1251.8 (M + H)+. Anal. calcd for [C78H102N6O4S2]: C 74.84; H 8.21; N 6.71. Found: C 74.72; H 8.40; N 6.95.
Synthesis of 3b.
A similar procedure to that used for 3a afforded 3b as a red solid (16 mg, 40% yield). Mp: 295–296 °C. FTIR (KBr): ν 2221.8 (CN), 1704.8 (CO) cm−1. 1H NMR (400 MHz, C2D2Cl4) δ 8.21 (s, 2H), 7.68 (s, 2H), 3.30–3.33 (m, 4H), 1.62–1.64 (br, 4H), 1.22 (br, 24H), 0.88–0.90 (m, 6H). 13C NMR (100 MHz, C2D2Cl4) δ 145.6, 145.4, 127.8, 124.3, 120.5, 118.4, 116.8, 116.6, 116.3, 116.0, 112.3, 104.5, 102.7, 102.0, 98.4, 44.0, 37.6, 35.5, 34.9, 33.7, 30.2, 23.4. HRMS (MALDI-FT) calcd for [C46H38O4N6S2]+ 802.2390, found: 802.2393 (M)+.
Acknowledgements
This research was financially supported by the National Natural Science Foundation (21674126 and 21522209), the “Strategic Priority Research Program” (XDB12010100) and the Shanghai Science and Technology Committee (16JC1400603).
References
-
(a) H. Usta, A. Facchetti and T. J. Marks, Acc. Chem. Res., 2011, 44, 501 CrossRef CAS PubMed;
(b) A. Facchetti, Chem. Mater., 2011, 23, 733 CrossRef CAS;
(c) M. E. Roberts, A. N. Sokolov and Z. Bao, J. Mater. Chem., 2009, 19, 3351 RSC;
(d) V. Subramanian, J. M. Fréchet, P. Chang, D. Huang, J. Lee, S. E. Molesa, A. R. Murphy, D. R. Redinger and S. K. Volkman, Proc. IEEE, 2005, 93, 1330 CrossRef CAS;
(e) C. R. Newman, C. D. Frisbie, D. A. da Silva Filho, J. L. Brédas, P. C. Ewbank and K. R. Mann, Chem. Mater., 2004, 16, 4436 CrossRef CAS;
(f) G. H. Gelinck, H. E. Huitema, E. Van Veenendaal, E. Cantatore, L. Schrijnemakers, J. B. van der Putten, T. C. Geuns, M. Beenhakkers, J. B. Giesbers, B. H. Huisman, E. J. Meijer, E. M. Benito, F. J. Touwslager, A. W. Marsman, B. J. van Rens and D. M. de Leeuw, Nat. Mater., 2004, 3, 106 CrossRef CAS PubMed.
-
(a) T. W. Kelley, D. V. Muyres, P. F. Baude, T. P. Smith and T. D. Jones, Mater. Res. Soc. Symp. Proc., 2003, 771, L6.5.1 CrossRef;
(b) O. D. Jurchescu, J. Baas and T. T. M. Palstra, Appl. Phys. Lett., 2004, 84, 3061 CrossRef CAS.
-
(a) D. Biermann and W. Schmidt, J. Am. Chem. Soc., 1980, 102, 3163 CrossRef CAS;
(b)
R. G. Havey, Polycyclic Aromatic hydrocarbons, Wiley-VCH, New York, 1997 Search PubMed;
(c) P. V. R. Schleyer, M. Manoharan, H. Jiao and F. Stahl, Org. Lett., 2001, 3, 3643 CrossRef CAS;
(d) L. Zhang, Y. Cao, N. S. Colella, Y. Liang, J. L. Brédas, K. N. Houk and A. L. Briseno, Acc. Chem. Res., 2015, 48, 500 CrossRef CAS PubMed.
-
(a) M. Randić, Chem. Rev., 2003, 103, 3449 CrossRef PubMed;
(b) Y. Kubozono, H. Mitamura, X. Lee, X. He, Y. Yamanari, Y. Takahashi, Y. Suzuki, Y. Kaji, R. Eguchi, K. Akaike, T. Kambe, H. Okamoto, A. Fujiwara, T. Kato, T. Kosugi and H. Aoki, Phys. Chem. Chem. Phys., 2011, 13, 16476 RSC;
(c)
E. Clar, The Aromatic Sextet, John Wiley & Sons, London, 1972 Search PubMed;
(d) M. Solà, Front. Chem., 2013, 1, 22 CrossRef PubMed.
-
(a) H. Okamoto, N. Kawasaki, Y. Kaji, Y. Kubozono, A. Fujiwara and M. Yamaji, J. Am. Chem. Soc., 2008, 130, 10470 CrossRef CAS PubMed;
(b) N. Kawasaki, Y. Kubozono, H. Okamoto, A. Fujiwara and M. Yamaji, Appl. Phys. Lett., 2009, 94, 043310 CrossRef;
(c) R. Eguchi, X. He, S. Hamao, H. Goto, H. Okamoto, S. Gohda, K. Sato and Y. Kubozono, Phys. Chem. Chem. Phys., 2013, 15, 20611 RSC;
(d) H. Okamoto, R. Eguchi, S. Hamao, H. Goto, K. Gotoh, Y. Sakai, M. Izumi, Y. Takaguchi, S. Gohda and Y. Kubozono, Sci. Rep., 2014, 4, 5330 CrossRef CAS PubMed.
-
(a) R. Mitsuhashi, Y. Suzuki, Y. Yamanari, H. Mitamura, T. Kambe, N. Ikeda, H. Okamoto, A. Fujiwara, M. Yamaji, N. Kawasaki, Y. Maniwa and Y. Kubozono, Nature, 2010, 464, 76 CrossRef CAS PubMed;
(b) X. Wang, R. Liu, Z. Gui, Y. Xie, Y. Yan, J. Ying, X. Luo and X. Chen, Nat. Commun., 2011, 2, 507 CrossRef CAS PubMed.
- H. Okamoto, S. Hamao, H. Goto, Y. Sakai, M. Izumi, S. Gohda, Y. Kubozono and R. Eguchi, Sci. Rep., 2014, 4, 5048 CrossRef CAS PubMed.
-
(a) A. S. Ionkin, W. J. Marshall, B. M. Fish, L. M. Bryman and Y. Wang, Chem. Commun., 2008, 2319 RSC;
(b) Y. H. Chung, L. Sheng, X. Xing, L. Zheng, M. Bian, Z. Chen, L. Xiao and Q. Gong, J. Mater. Chem. C, 2015, 3, 1794 RSC.
-
(a) O. Khorev, C. D. Bösch, M. Probst and R. Häner, Chem. Sci., 2014, 5, 1506 RSC;
(b) T. Wu, H. Chou, P. Huang, C. Cheng and R. Liu, J. Org. Chem., 2014, 79, 267 CrossRef CAS PubMed.
-
(a) Z. Zhao, F. Zhang, X. Zhang, X. Yang, H. Li, X. Gao, C. Di and D. Zhu, Macromolecules, 2013, 46, 7705 CrossRef CAS;
(b) Z. Zhao, Z. Wang, X. Zhang, S. Gao, X. Yang, Z. Duan and X. Gao, ChemPlusChem, 2015, 80, 57 CrossRef CAS;
(c) Z. Zhao, Z. Wang, C. Ge, X. Zhang, X. Yang and X. Gao, Polym. Chem., 2016, 7, 573 RSC.
- M. J. Marsella, Z. Wang, R. J. Reid and K. Yoon, Org. Lett., 2001, 3, 885 CrossRef CAS PubMed.
-
(a) S. Matsumoto, K. Takase and K. Ogura, J. Org. Chem., 2008, 73, 1726 CrossRef CAS PubMed;
(b) M. Goldfinger and T. Swager, J. Am. Chem. Soc., 1994, 116, 7895 CrossRef CAS.
- H. Qu, W. Cui, J. Li, J. Shao and C. Chi, Org. Lett., 2011, 13, 924 CrossRef CAS PubMed.
- C. M. Cardona, W. Li, A. E. Kaifer, D. Stockdale and G. C. Bazan, Adv. Mater., 2011, 23, 2367 CrossRef CAS PubMed.
- S. Chen, Q. Zhang, Q. Zheng, C. Tang and C. Lu, Chem. Commun., 2012, 48, 1254 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: TGA, DSC, AFM, XRD, NMR, MS IR spectra and crystal data. CCDC 1528872. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7qm00030h |
|
| This journal is © the Partner Organisations 2017 |