
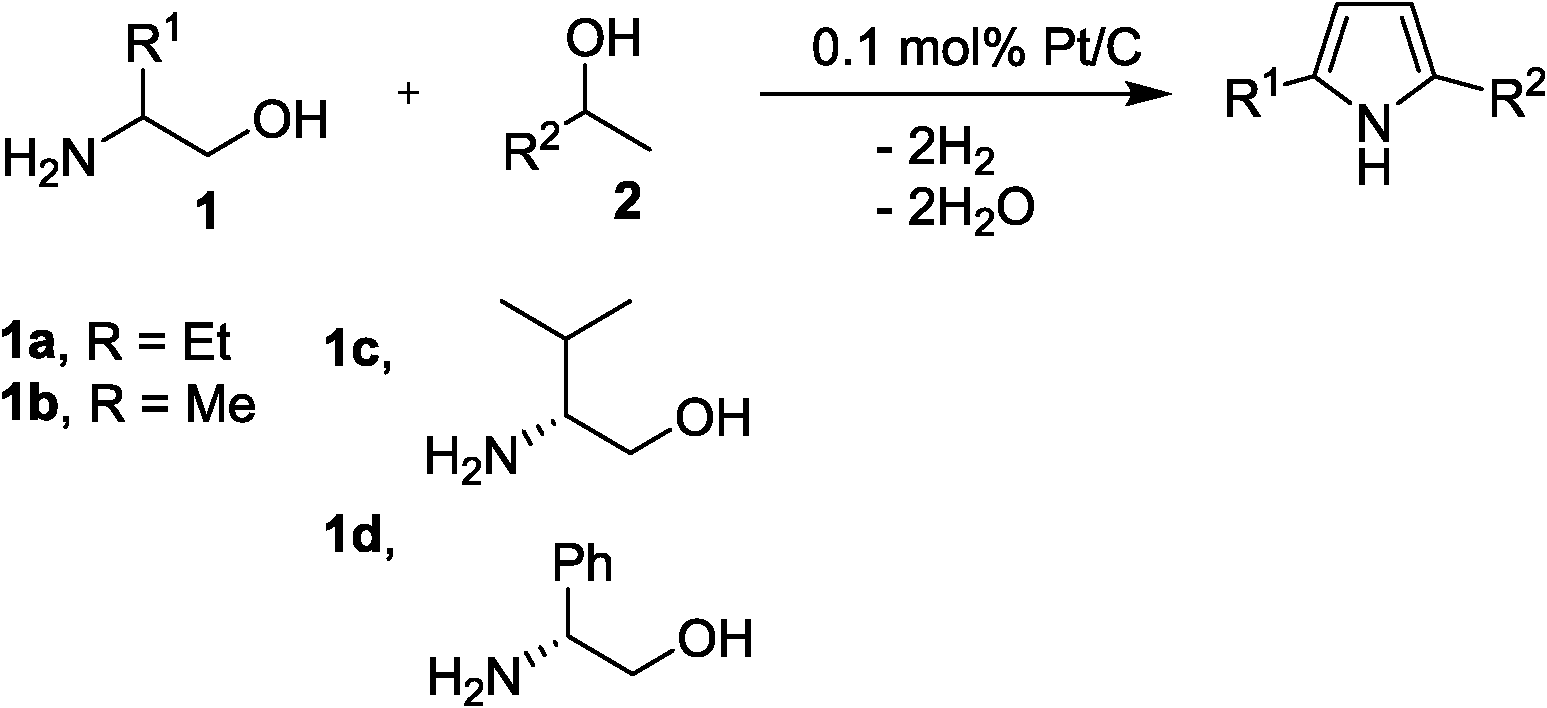
Synthesis of 2,5-disubstituted pyrroles via dehydrogenative condensation of secondary alcohols and 1,2-amino alcohols by supported platinum catalysts†
S. M. A. H.
Siddiki
a,
Abeda S.
Touchy
a,
Chandan
Chaudhari
a,
Kenichi
Kon
a,
Takashi
Toyao
ab and
Ken-ichi
Shimizu
*ab
aInstitute for Catalysis, Hokkaido University, N-21, W-10, Sapporo 001-0021, Japan. E-mail: kshimizu@cat.hokudai.ac.jp
bElements Strategy Initiative for Catalysts and Batteries, Kyoto University, Katsura, Kyoto 615-8520, Japan
First published on 11th May 2016
Abstract
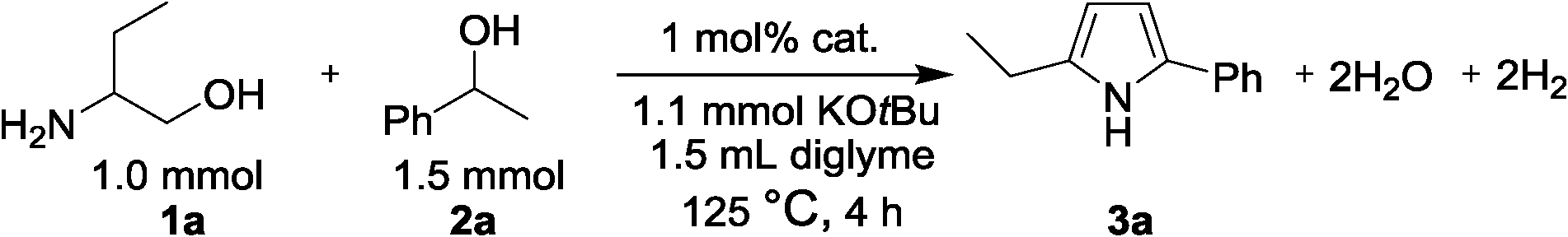
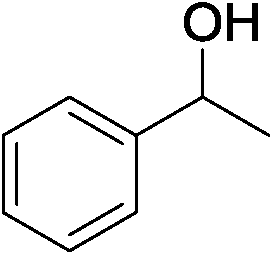
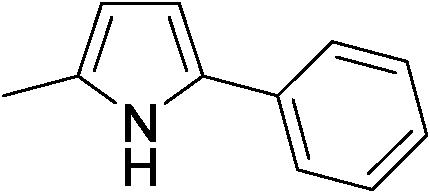
Direct synthesis of 2,5-disubstituted pyrroles has been achieved via acceptorless dehydrogenative heterocyclization of 1,2-aminoalcohols and secondary alcohols by utilizing a heterogeneous carbon-supported Pt catalyst (Pt/C). The optimized method gave 92% yield of 2-ethyl-5-phenyl-1H-pyrrole as a desired product from 2-amino-1-butanol and 1-phenylethanol in the presence of 0.1 mol% of Pt/C and 1.1 equiv. of KOtBu. It has been revealed that Pt/C demonstrates superior catalytic activity to a number of catalysts tested in this study including other transition metal-loaded carbon and various metal-oxide-supported Pt catalysts. In addition, the turnover number (TON) obtained in the present system was found to be higher than those obtained in previously-reported catalytic systems. It is significant that the Pt/C catalyst could be recycled as a heterogeneous catalyst without significant loss in the activity and showed a wide substrate scope for the 2,5-disubstituted pyrrole forming process.
Introduction
Pyrroles are key compounds in organic synthesis1 and industrial production of pharmaceutical compounds with various biological activities1 as well as functional materials used for solar cells and batteries.2 Classical synthetic methods of pyrroles such as Hantzsch and Paal–Knorr reactions1 and recent catalytic methods1,3a–h suffer from several problems including the multistep procedure or the difficult availability of the starting compounds. Following the pioneering studies,3i,j several groups4–8 have recently reported new synthetic protocols to obtain pyrroles from easily available starting materials. The protocols employ the acceptorless dehydrogenative coupling methodology.9 Three groups (Kempe,4 Saito,5 Milstein6) have discovered new catalytic methods for the direct synthesis of pyrroles through dehydrogenative condensation of 1,2-amino alcohols with secondary alcohols (or ketones) using homogeneous Ir or Ru catalysts with a basic additive (KOtBu). Beller et al.7 also developed the direct dehydrogenative synthesis of pyrroles from benzylic ketones, vicinal diols, and amines by utilizing a homogeneous Ru catalyst. However, these systems require elaborate homogeneous catalysts and have difficulties in catalyst recycling. From the viewpoint of sustainable chemistry and for industrial applications, these processes should be carried out by a recyclable heterogeneous catalyst which is advantageous for easy product separation and recycling of the catalysts. In this sense, Kempe's group developed a new heterogeneous catalyst, Ir@SiCN, that was effective for acceptorless dehydrogenative synthesis of pyrroles from 1,2-amino alcohols and secondary alcohols with KOtBu.8 Although the heterogeneous system was successfully developed, the catalyst preparation could be complicated since they made a precursor by themselves and the preparation procedure requires several steps. Therefore, the development of versatile heterogeneous catalytic systems is desirable for easy access to this organic synthesis and its practical applications. We have previously reported a series of studies on the acceptorless dehydrogenation of alcohols10a and the acceptorless dehydrogenative coupling reactions of alcohols10b–e promoted by heterogeneous Pt catalysts. The Pt catalysts are synthesized by a facile impregnation method by employing commercially-available Pt precursors and carbon supports. In addition, these catalytic systems show high versatility toward various dehydrogenation reactions with a wide substrate scope, and thus, it is expected that our Pt/C-catalyzed system could be utilized for the acceptorless dehydrogenative synthesis of pyrroles. The observations made in this effort show that this expectation is correct. Specifically, we report herein the first example of Pt-catalyzed acceptorless dehydrogenative synthesis of 2,5-substituted pyrroles from 1,2-amino alcohols and secondary alcohols. This new method shows good catalyst reusability and a higher turnover number (TON) than previously-reported catalysts.4,6,8Results and discussion
The catalysts were screened by employing the reaction of 2-amino-1-butanol (1a) and 1.5 equiv. of 1-phenylethanol (2a) with 1.1 equiv. of KOtBu in diglyme at 125 °C for 4 h under N2 as a model system. Table 1 shows the conversion of 1a and the yield of 2-ethyl-5-phenyl-1H-pyrrole (3a) based on 1a. Under the present conditions, no reaction took place without catalysts (entry 1). Active carbon itself (entry 2) and platinum oxide-loaded carbon (PtOx/C in entry 3) were also inactive for the reaction. In contrast, metallic Pt-loaded carbon (Pt/C), pre-reduced in H2 at 300 °C, gave 92% yield (entry 5). Once the pre-reduced Pt/C was exposed to air at room temperature, the air-exposed Pt/C (Pt/C-air in entry 4) showed lower yield (60%) than Pt/C. Entries 5–13 compare the results for various transition metal-loaded carbon catalysts, pre-reduced in H2 at 300 °C, containing 0.01 mmol of metals (1 mol% with respect to 1a). Among the catalysts tested (Pt, Re, Ir, Rh, Pd, Ru, Ni, Co, Cu), Pt-loaded carbon (Pt/C) showed the highest yield of the desired product 3a. Other precious metals (Re, Ir, Rh, Pd, Ru) gave moderate to low yields of 48–18%, and base metals (Ni, Co, Cu) gave only 1% yield. Next, the catalytic properties of Pt/C were compared with various metal oxide-supported Pt catalysts (entries 14–23). Pt/C showed a higher yield (92%) than the other Pt catalysts. For example, Pt-loaded Al2O3, CeO2, TiO2, HBEA zeolite, Nb2O5, ZrO2 and SiO2 gave moderate to low yields of 61–20%. Entries 5 and 24–26 compare the results for different Pt-loaded carbon catalysts. The Pt-loaded different carbon materials (Ketjenblack EC-600JD in entry 24; Vulcan-XC72 in entry 25) and a commercial Pt catalyst from Sigma-Aldrich (Pt/CSA, entry 26) showed slightly lower yields (83–90%) than the Pt/C catalyst developed in this study (entry 5; the carbon support was supplied by Kishida Chemical). This result indicates that the activity of Pt-loaded carbon catalysts does not markedly depend on the type of the carbon and preparation methods. Hereafter the Pt/C catalyst in entry 5 is used as the standard Pt/C catalyst. Recently, we have reported a detailed study on the structure of the Pt/C.10e The results demonstrated that Pt nanoparticles in the Pt/C catalyst exist in the metallic state and have an average particle size of 4.4 nm.|
|
|||
|---|---|---|---|
| Entry | Catalysts | 1a conv.a (%) | 3a yielda (%) |
| a GC yields. b 0.039 g. c Pre-reduced Pt/C was exposed to air at room temperature for 0.5 h. | |||
| 1 | Blank | 0 | 0 |
| 2 | Cb | 3 | 0 |
| 3 | PtOx/C | 4 | 0 |
| 4 | Pt/C-airc | 72 | 60 |
| 5 | Pt/C | 100 | 92 |
| 6 | Re/C | 56 | 48 |
| 7 | Ir/C | 44 | 35 |
| 8 | Rh/C | 33 | 26 |
| 9 | Pd/C | 29 | 21 |
| 10 | Ru/C | 25 | 18 |
| 11 | Ni/C | 4 | 1 |
| 12 | Co/C | 3 | 1 |
| 13 | Cu/C | 3 | 1 |
| 14 | Pt/Al2O3 | 72 | 61 |
| 15 | Pt/CeO2 | 59 | 51 |
| 16 | Pt/TiO2 | 43 | 35 |
| 17 | Pt/HBEA | 40 | 34 |
| 18 | Pt/Nb2O5 | 28 | 22 |
| 19 | Pt/ZrO2 | 27 | 20 |
| 20 | Pt/SiO2 | 29 | 20 |
| 21 | Pt/SiO2-Al2O3 | 19 | 14 |
| 22 | Pt/MgO | 11 | 6 |
| 23 | Pt/SnO2 | 3 | 1 |
| 24 | Pt/CKB | 100 | 83 |
| 25 | Pt/CVX | 100 | 90 |
| 26 | Pt/CSA | 100 | 85 |
In order to optimize the reaction conditions such as the amount of reactants, effects of bases and solvents for the pyrrole synthesis, various reactions were carried out over Pt/C for 4 h. The results are given in Table 2. We tested the reaction of 1 mmol of 1a with 1 mmol of 2a in diglyme at 125 °C with different amounts of KOtBu (entries 1–5). The product 3a was not obtained in the absence of KOtBu (entry 1). This indicates that the base is indispensible for the reaction. The addition of 1.1 equiv. of KOtBu gave the highest yield (entry 4). The reaction at different temperatures (results not shown) showed that the lower (105 °C) and higher (162 °C) temperatures gave lower yields (49% and 64%) than the standard temperature (75% yield at 125 °C). In the presence of the optimal amount (1.1 mmol) of KOtBu, the reactions with different amounts (0.5–2.0 equiv.) of 1-phenylethanol (2a) were compared in entries 4 and 6–9. It was found that 1.5 equiv. of 2a (entry 8) gave the highest yield of 3a based on 1a. For the reaction of 1 mmol of 1a and 1.5 equiv. of 2a in the presence of 1.1 equiv. of KOtBu, we tested the solvent effect (entries 8 and 10–13). The result shows that diglyme (entry 8) would serve as the best solvent. The reaction with various basic additives (entries 8, 14–18) showed that KOtBu (entry 8) was more effective than other bases such as K2CO3, Cs2CO3, NaOH, NaOMe and KOH.
|
|
|||||
|---|---|---|---|---|---|
| Entry | 1a (mmol) | 2a (mmol) | Base (x mmol) | Solvent | 3a yielda (%) |
| a GC yields. b Reflux conditions. | |||||
| 1 | 1.0 | 1.0 | — | Diglyme | 0 |
| 2 | 1.0 | 1.0 | KOtBu (0.5) | Diglyme | 41 |
| 3 | 1.0 | 1.0 | KOtBu (1.0) | Diglyme | 67 |
| 4 | 1.0 | 1.0 | KOtBu (1.1) | Diglyme | 75 |
| 5 | 1.0 | 1.0 | KOtBu (1.5) | Diglyme | 71 |
| 6 | 1.0 | 0.5 | KOtBu (1.1) | Diglyme | 39 |
| 7 | 1.0 | 1.25 | KOtBu (1.1) | Diglyme | 83 |
| 8 | 1.0 | 1.5 | KOtBu (1.1) | Diglyme | 92 |
| 9 | 1.0 | 2.0 | KOtBu (1.1) | Diglyme | 84 |
| 10b | 1.0 | 1.5 | KOtBu (1.1) | Toluene | 81 |
| 11b | 1.0 | 1.5 | KOtBu (1.1) | 1,4-Dioxane | 73 |
| 12 | 1.0 | 1.5 | KOtBu (1.1) | o-Xylene | 78 |
| 13 | 1.0 | 1.5 | KOtBu (1.1) | Mesitylene | 75 |
| 14 | 1.0 | 1.5 | K2CO3 (1.1) | Diglyme | 13 |
| 15 | 1.0 | 1.5 | Cs2CO3 (1.1) | Diglyme | 21 |
| 16 | 1.0 | 1.5 | NaOH (1.1) | Diglyme | 47 |
| 17 | 1.0 | 1.5 | NaOMe (1.1) | Diglyme | 39 |
| 18 | 1.0 | 1.5 | KOH (1.1) | Diglyme | 51 |
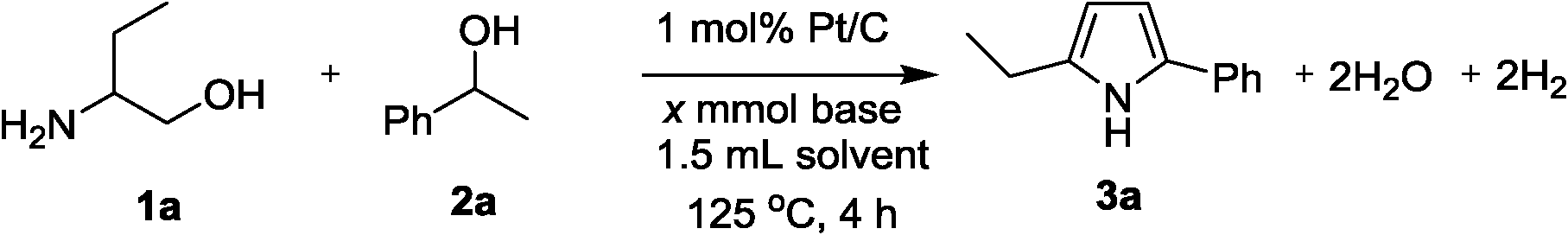
The detailed catalytic properties of the Pt/C-catalyzed system were further investigated under the optimized conditions. Fig. 1 shows the results of the catalyst recycling experiments. After completion of the first run, 2-propanol (3 mL) was added to the reaction mixture and the catalyst was separated by centrifugation, followed by washing the catalyst with water (1 × 3 mL) and acetone (3 × 3 mL), drying at 100 °C for 3 h and by H2-reduction at 300 °C for 0.5 h. The recovered Pt/C catalyst showed high yield (88–92%) for the next three cycles. ICP-AES analysis of the filtrate after the first cycle showed that the content of Pt in the solution was below the detection limit. These results demonstrate that the present catalytic system is able to be recycled and offers advantages as a heterogeneous catalytic process even under basic reaction conditions. Furthermore, the TON of this system was investigated in a gram scale synthesis of 3a. The reaction of 10 mmol of 1a and 15 mmol of 2a was carried out using a small amount of the Pt/C catalyst in this attempt. As shown in eqn. (1), the reactions with 0.03 mol% of Pt/C for 96 h resulted in 83% yield, corresponding to a TON of 2767. This TON is larger than those for the previous homogeneous Ir (TON = 1860)4 and Ru (TON = 150)6 catalysts and a heterogeneous Ir catalyst (TON = 260)8 for the same reaction.
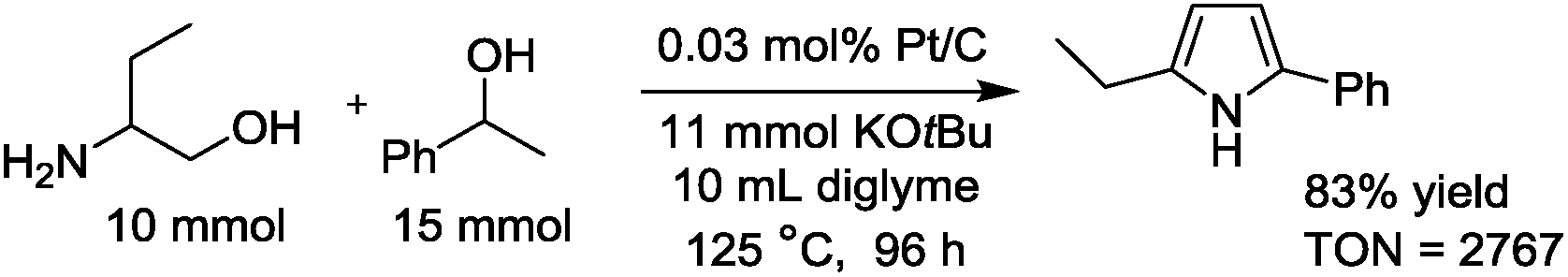 | (1) |
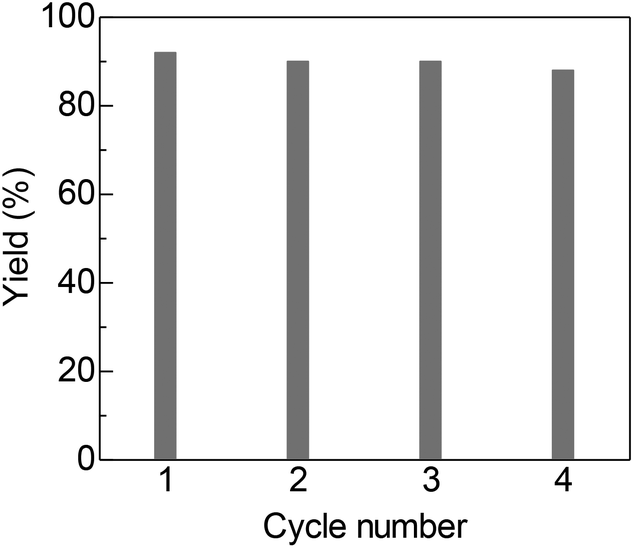 | ||
| Fig. 1 Catalyst reuse for synthesis of 3a from 1a and 2a by Pt/C under the conditions for entry 8 in Table 2. | ||
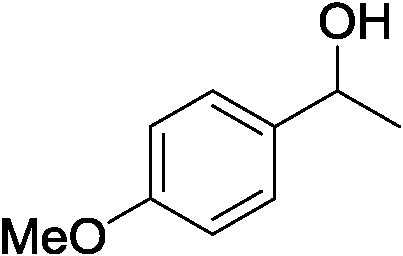
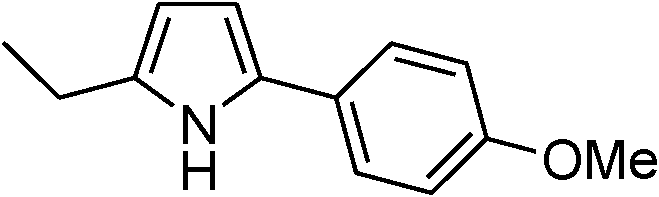
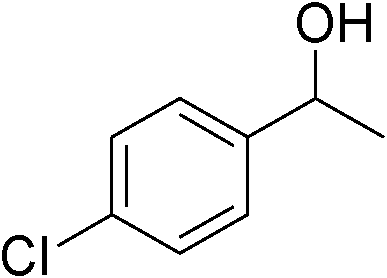
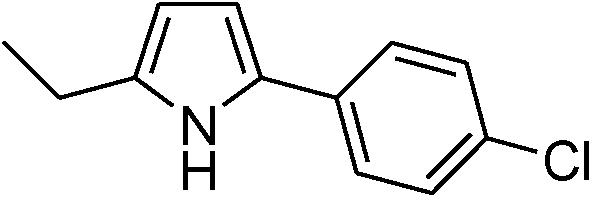
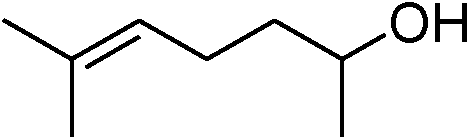
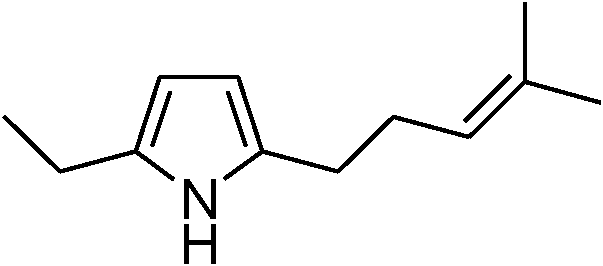
Table 3 demonstrates the substrate scope of the dehydrogenative synthesis of 2,5-disubstituted pyrroles from 1,2-amino alcohols (1) and 1.5 equiv. of secondary alcohols (2) using Pt/C containing 0.1 mol% of Pt with respect to 1,2-amino alcohols. Entries 1–10 summarize the results for the reaction of 2-amino-1-butanol (1a) with various secondary alcohols. 1-Phenylethanol (entry 1) and its derivatives with electron-donating and withdrawing groups (methyl, methoxy, chloro) at p-positions (entries 2–4) were converted to the corresponding pyrroles with good to high yields (78–92%). 1-(2-Naphthyl)ethanol (entry 5) and a cyclic aliphatic alcohol (entry 6) were also transformed to the corresponding pyrroles with moderate to high yields (64, 92%). An alcohol with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond, 6-methyl-5-hepten-2-ol (entry 7), was converted to the corresponding pyrrole with the CC group in 76% yield. The reaction of different aminoalcohols such as 2-amino-3-methyl-1-butanol (1b, entries 8–12), (R)-(−)-2-amino-3-methyl-1-butanol (1c, entries 13 and 14) and (S)-(+)-2-amino-2-phenylethanol (1d, entries 15–18) with various secondary alcohols resulted in the formation of the corresponding pyrroles with moderate to high yields (63–91%).
C double bond, 6-methyl-5-hepten-2-ol (entry 7), was converted to the corresponding pyrrole with the CC group in 76% yield. The reaction of different aminoalcohols such as 2-amino-3-methyl-1-butanol (1b, entries 8–12), (R)-(−)-2-amino-3-methyl-1-butanol (1c, entries 13 and 14) and (S)-(+)-2-amino-2-phenylethanol (1d, entries 15–18) with various secondary alcohols resulted in the formation of the corresponding pyrroles with moderate to high yields (63–91%).
|
|
||||
|---|---|---|---|---|
| Entry | 1 | 2 | Product | Yieldb (%) |
| a Conditions: a mixture of 1 mmol 1,2-amino alcohols (1), 1.5 mmol secondary alcohol (2), 1.1 mmol KOtBu and 0.1 mol% Pt/C in 1.5 mL diglyme was heated at 125 °C for 18 h under N2. b GC yields. c 24 h. | ||||
| 1 | 1a |
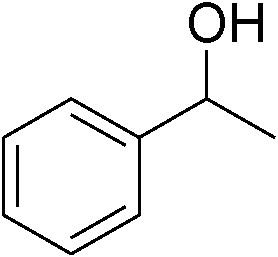
|
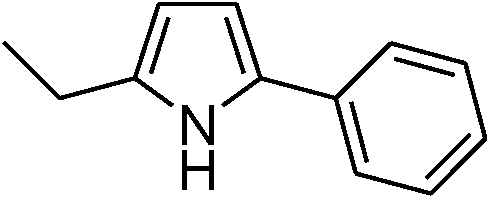
|
92 |
| 2 | 1a |
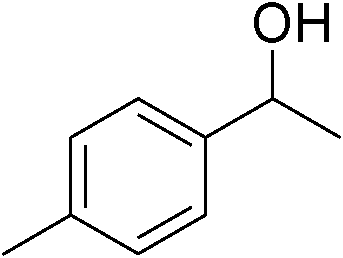
|
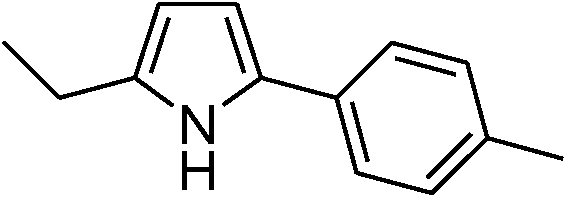
|
90 |
| 3 | 1a |
|
|
86 |
| 4 | 1a |
|
|
78 |
| 5 | 1a |
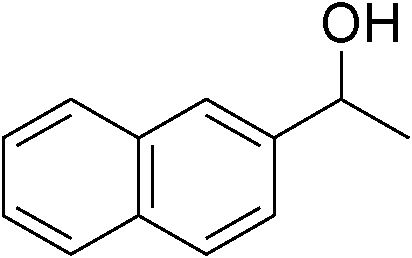
|
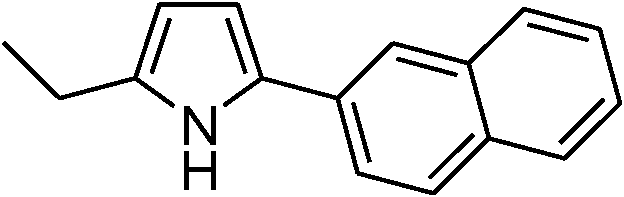
|
92 |
| 6c | 1a |
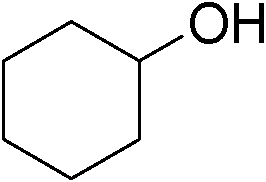
|
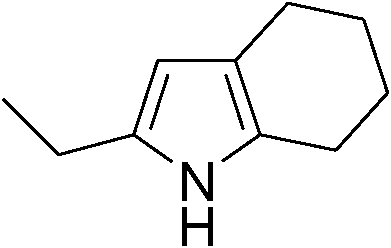
|
64 |
| 7 | 1a |
|
|
76 |
| 8 | 1b |
|
|
79 |
| 9 | 1b |
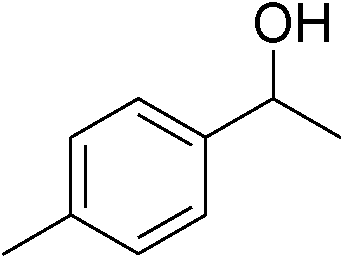
|
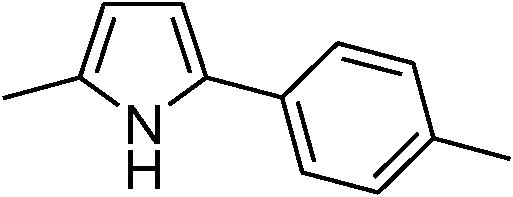
|
81 |
| 10 | 1b |
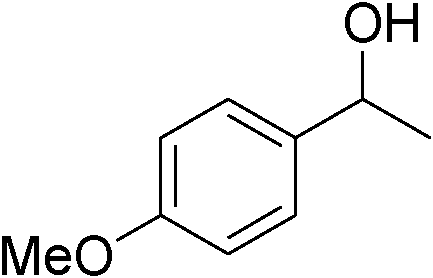
|
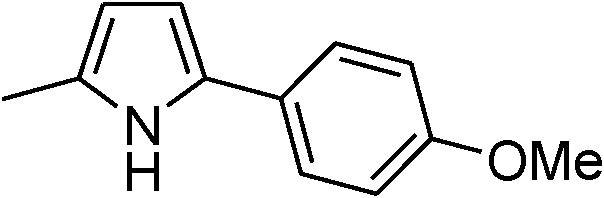
|
84 |
| 11c | 1b |
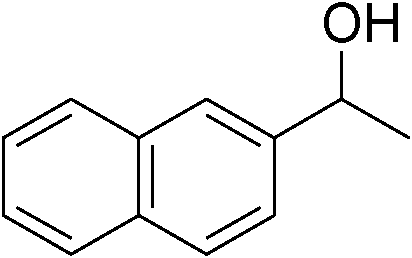
|
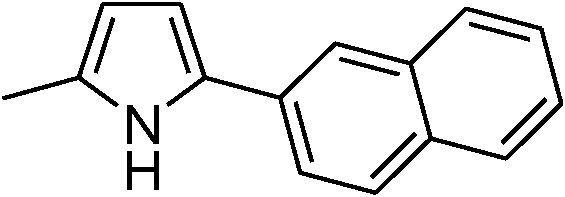
|
91 |
| 12 | 1b |
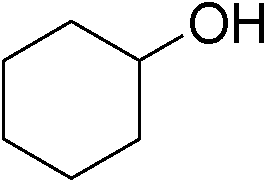
|
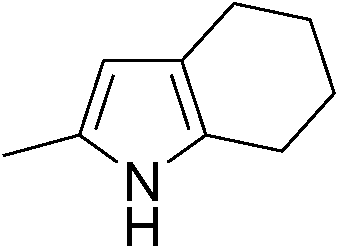
|
67 |
| 13 | 1c |
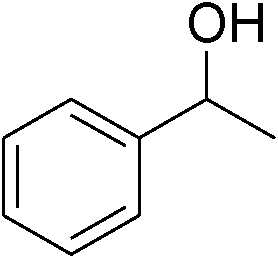
|
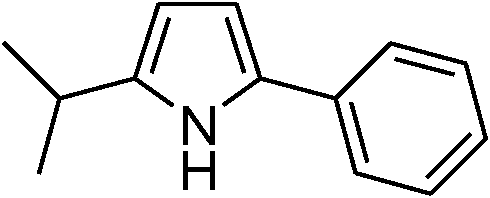
|
88 |
| 14 | 1c |
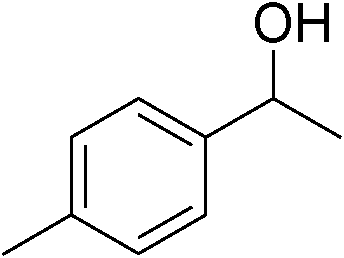
|
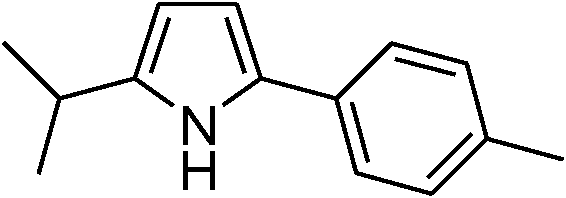
|
84 |
| 15 | 1d |
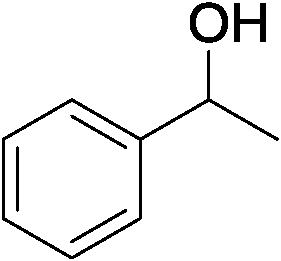
|
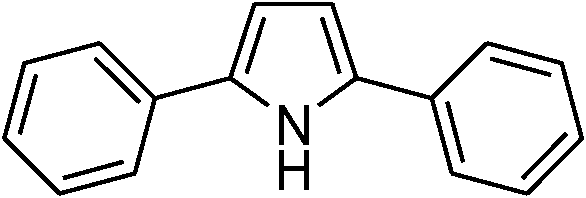
|
65 |
| 16 | 1d |
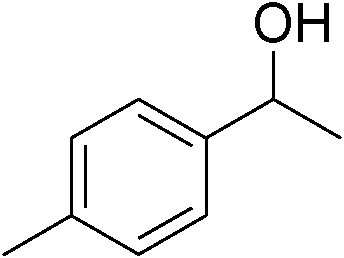
|
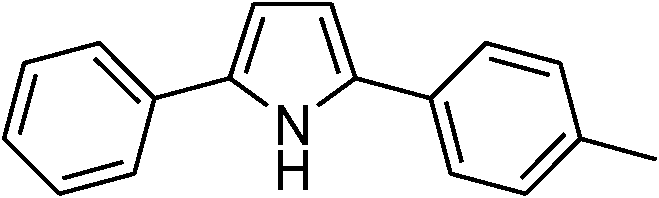
|
74 |
| 17 | 1d |
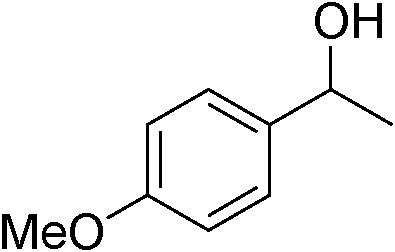
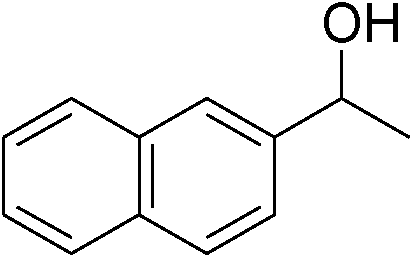
|
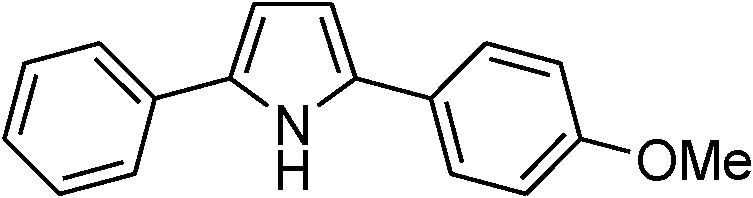
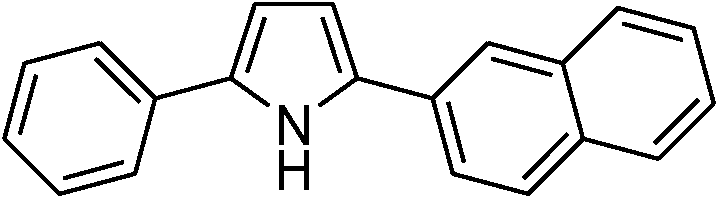
|
63 |
| 18 | 1d |
|
|
78 |
The time course of the reaction of 1a and 2a (result not shown) showed a profile characteristic of a consecutive reaction mechanism via acetophenone; acetophenone formed at the initial induction period was gradually consumed to give the product 3a. Actually, the model reaction of 1a and acetophenone in the presence of Pt/C and KOtBu under N2 for 4 h resulted in the formation of 3a in 70% yield as shown in eqn (2). As shown in eqn (3), the same reaction in the absence of Pt/C and KOtBu did not give the pyrrole 3a but gave 2-(1-phenyl-ethylideneamino)-1-butanol (4a) in 55% GC yield. Also, the reaction of 1a and 2a by Pt/C in the absence of KOtBu, shown in eqn (4), did not give the pyrrole 3a; the reaction resulted in 25% yield of acetophenone and 16% yield of 4a.
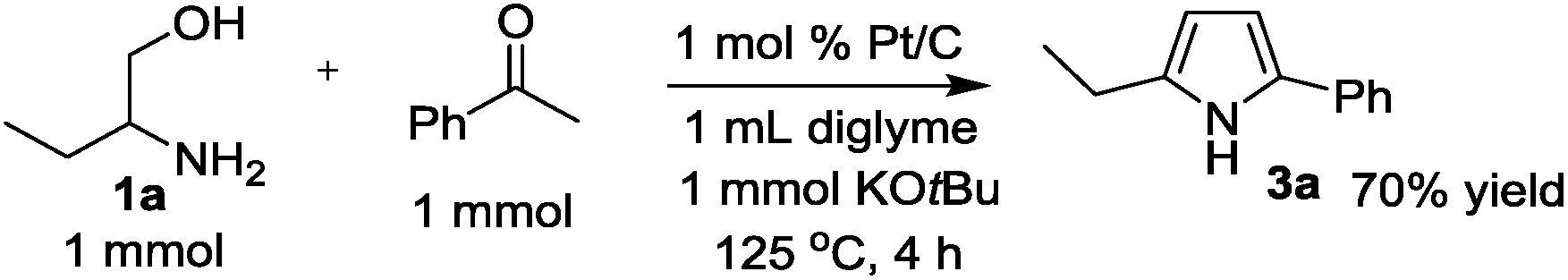 | (2) |
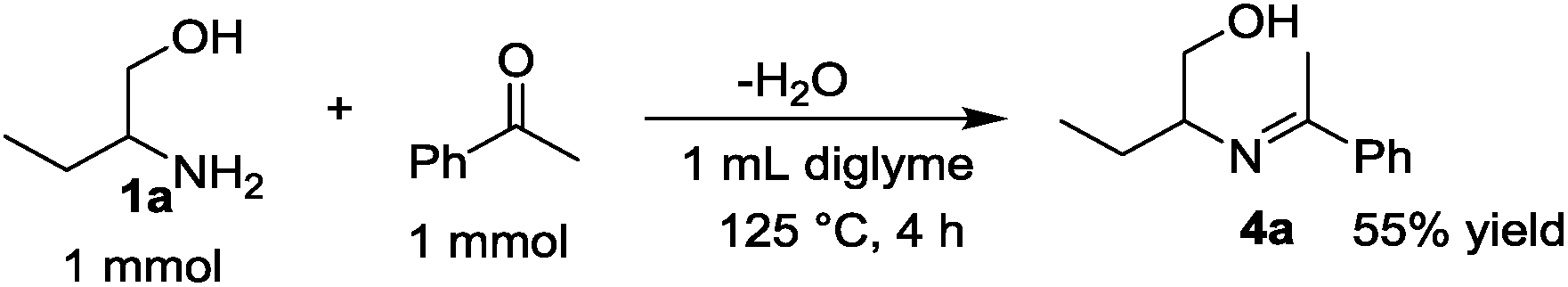 | (3) |
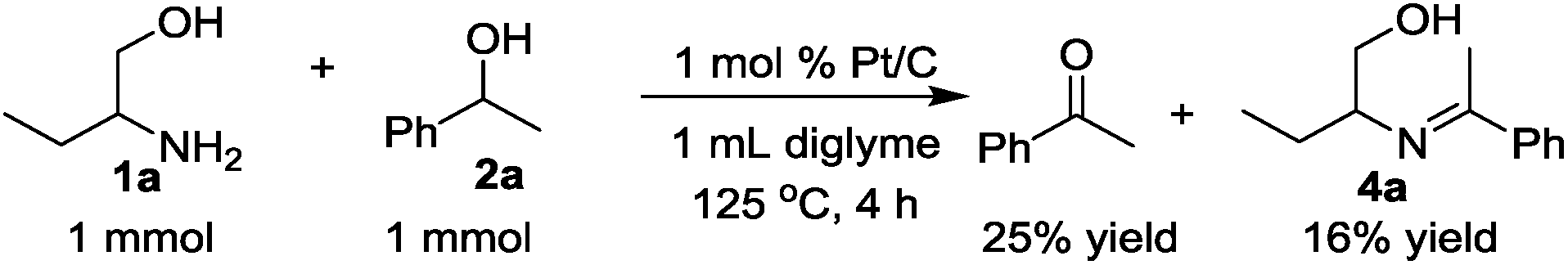 | (4) |
Taking account of the results obtained above and the previous mechanistic proposals for the acceptorless dehydrogenative synthesis of pyrroles from 1,2-amino alcohols and secondary alcohols,4,6,8 we propose a possible reaction pathway of the present catalytic reaction in Scheme 1 adopting the reaction of 1a and 2a as an example. The reaction begins with Pt-catalyzed dehydrogenation of secondary alcohol (2a) to ketone, which then undergoes condensation with 1,2-amino alcohol (1a) to afford 2-(1-phenyl-ethylideneamino)-1-butanol (4a) as an intermediate. The intermediate 4a undergoes Pt-catalyzed dehydrogenation followed by base-catalyzed condensation to give the 2,5-disubstituted pyrrole 3a. The dehydrogenation steps may also be accelerated by the basic co-catalyst, KOtBu, via activation of OH groups in 2a and 4a.
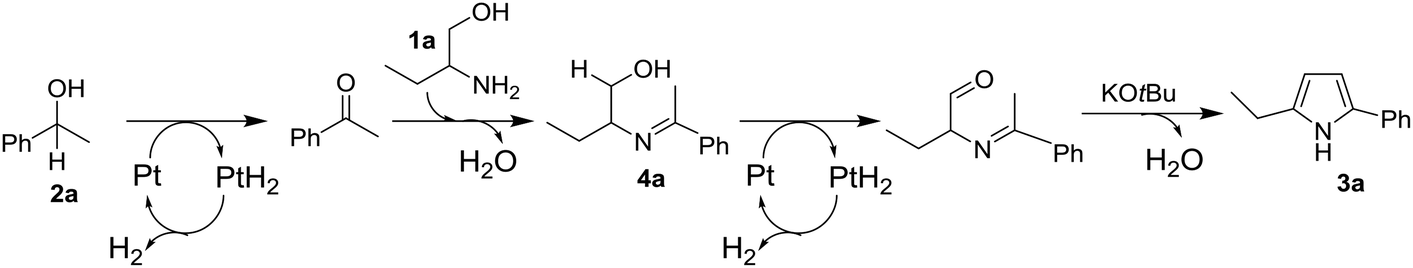 | ||
| Scheme 1 A possible reaction pathway. | ||
Conclusions
A Pt/C-based heterogeneous catalytic system has been found to be effective for direct synthesis of 2,5-disubstituted pyrroles from 1,2-amino alcohols and secondary alcohols in the presence of KOtBu as a base. The catalytic system involves acceptorless dehydrogenation of alcohols and condensation reactions. Observations made in studies of the substrate scope demonstrate that Pt/C is capable of synthesizing a wide range of pyrroles from 1,2-amino alcohols and secondary alcohols. In addition, the developed system would provide advantages such as the catalyst is prepared by a simple impregnation method and can be reused as a heterogeneous catalyst without significant loss in the catalytic activity. Furthermore, the system showed higher TON than previously-reported systems. These findings will offer a simple and sustainable synthetic method for the pyrroles.Experimental
Commercially available organic compounds (from Tokyo Chemical Industry, Sigma-Aldrich, Kishida Chemical) were used without further purification. The GC (Shimadzu GC-14B) and GCMS (Shimadzu GCMS-QP2010) analyses were carried out with an Ultra ALLOY capillary column UA+-1 (Frontier Laboratories Ltd) using nitrogen and helium as the carrier gases.The standard carbon support (296 m2 g−1, Kishida Chemical) and other carbon materials (CKB = Ketjenblack EC-600JD, Lion, 1310 m2 g−1; CVX = carbon black, Vulcan XC72, 210 m2 g−1) were commercially supplied. γ-Al2O3 was prepared by calcination of γ-AlOOH (Catapal B from Sasol) at 900 °C for 3 h. CeO2 (JRC-CEO3, 81 m2 g−1), TiO2 (JRC-TIO-4) and H+-type BEA zeolite (HBEA, SiO2/Al2O3 = 25 ± 5, JRC-Z-HB25), MgO (JRC-MGO-3), SiO2-Al2O3 (JRC-SAL-2) were supplied by the Catalysis Society of Japan. Nb2O5 was prepared by calcination of niobic acid (supplied from CBMM) at 500 °C for 3 h. ZrO2 was prepared by hydrolysis of zirconium oxynitrate 2-hydrate by an aqueous NH4OH solution, followed by filtration, washing with distilled water, drying at 100 °C for 12 h, and by calcination at 500 °C for 3 h. SiO2 (Q-10, 300 m2 g−1) was supplied from Fuji Silysia Chemical Ltd. SnO2 was prepared by calcination of H2SnO3 (Kojundo Chemical Laboratory Co., Ltd) at 500 °C for 3 h.
The precursor of Pt/C was prepared by the impregnation method; a mixture of carbon and an aqueous HNO3 solution of Pt(NH3)2(NO3)2 was evaporated at 50 °C, followed by drying at 90 °C for 12 h. Before each catalytic experiment, the Pt/C catalyst with a Pt loading of 5 wt% was prepared by pre-reduction of the precursor in a Pyrex tube under H2 flow (20 mL min−1) at 300 °C for 0.5 h. Other supported Pt catalysts with a Pt loading of 5 wt% were prepared by the same method. M/C (M = Re, Ir, Rh, Pd, Ru, Ni, Co, Cu) catalysts with a metal loading of 5 wt% were prepared by a similar manner as for Pt/C using an aqueous solution of NH4ReO4, IrCl3·nH2O, RuCl3 or metal nitrates (for Ni, Cu, Co) or an aqueous HNO3 solution of Rh(NO3)3 or Pd(NH3)2(NO3)2. A commercial Pt-loaded carbon catalyst with a Pt loading of 5 wt%, named Pt/CSA, was purchased from Sigma-Aldrich.
The typical procedure of catalytic reactions is as follows. After the pre-reduction, Pt/C (39 mg; 1 mol% Pt with respect to 1a) in the closed glass tube sealed with a septum inlet was cooled to room temperature under N2. Then, 1 mmol of 2-amino-1-butanol (1a), 1.5 mmol of 1-phenylethanol (2a) and n-dodecane (0.2 mmol) in 1.5 mL diglyme were injected into the glass tube through the septum inlet. The septum was removed, and KOtBu (1.1 mmol) and a magnetic stirrer bar were put in the tube followed by filling N2 through the septum inlet. Then, the resulting mixture was heated at 125 °C under stirring. The conversion of 1a and yields of the products (based on 1a) were determined by GC using n-dodecane as an internal standard adopting the GC sensitivity estimated using the isolated products or commercial products. After the reactions in Table 3, the substrates, solvent and byproducts were removed by column chromatography with silica gel 60 (spherical, 63–210 μm, Kanto Chemical Co. Ltd) using hexane/ethylacetate (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as the eluting solvent, and the products were identified by 1H and 13C NMR analyses as well as GC-MS equipped with the same column as GC analyses.
:1) as the eluting solvent, and the products were identified by 1H and 13C NMR analyses as well as GC-MS equipped with the same column as GC analyses.
Acknowledgements
This work was supported by JSPS KAKENHI (Grant No. 26289299), a MEXT program “Elements Strategy Initiative to Form Core Research Center” and a Grant-in-Aid for Scientific Research on Innovative Areas “Nano Informatics” (25106010) from JSPS.Notes and references
- (a) V. Estévez, M. Villacampa and J. C. Menéndez, Chem. Soc. Rev., 2014, 43, 4633 RSC; (b) V. Bhardwaj, D. Gumber, V. Abbot, S. Dhiman and P. Sharma, RSC Adv., 2015, 5, 15233 RSC.
- (a) M. C. De Jesus, Y. Fu and R. A. Weiss, Polym. Eng. Sci., 1997, 37, 1936 CrossRef CAS; (b) J. R. Reynolds, P. A. Poropatic and R. L. Toyooka, Macromolecules, 1987, 20, 958 CrossRef CAS; (c) S. Qu and H. Tian, Chem. Commun., 2012, 48, 3039 RSC; (d) A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595 CrossRef CAS PubMed; (e) H. Nishide and K. Oyaizu, Science, 2008, 319, 737 CrossRef CAS PubMed.
- (a) C. M. Park and Y. Jiang, Chem. Sci., 2014, 5, 2347 RSC; (b) Y.-H. Xu, T. He, Q.-C. Zhang and T.-P. Loh, Chem. Commun., 2014, 50, 2784 RSC; (c) M.-N. Zhao, Z.-H. Ren, Y.-Y. Wang and Z.-H. Guan, Chem. – Eur. J., 2014, 20, 1839 CrossRef CAS PubMed; (d) M.-N. Zhao, Z.-H. Ren, Y.-Y. Wang and Z.-H. Guan, Org. Lett., 2014, 16, 608 CrossRef CAS PubMed; (e) L. Ran, Z.-H. Ren, Y.-Y. Wang and Z.-H. Guan, Green Chem., 2014, 16, 112 RSC; (f) Z. Shi, M. Suri and F. Glorius, Angew. Chem., Int. Ed., 2013, 52, 4892 CrossRef CAS PubMed; (g) J. Liu, Z. Fang, Q. Zhang, Q. Liu and X. Bi, Angew. Chem., Int. Ed., 2013, 52, 6953 CrossRef CAS PubMed; (h) T. Miura, K. Hiraga, T. Biyajima, T. Nakamuro and M. Murakami, Org. Lett., 2013, 15, 3298 CrossRef CAS PubMed; (i) K. Taguchi, S. Sakaguchi and Y. Ishii, Tetrahedron Lett., 2005, 46, 4539 CrossRef CAS; (j) N. Schley, G. Dobereiner and R. H. Crabtree, Organometallics, 2011, 30, 4174 CrossRef CAS.
- S. Michlik and R. Kempe, Nat. Chem., 2013, 5, 140 CrossRef CAS PubMed.
- K. Iida, T. Miura, J. Ando and S. Saito, Org. Lett., 2013, 15, 1436 CrossRef CAS PubMed.
- D. Srimani, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2013, 52, 4012 CrossRef CAS PubMed.
- M. Zhang, X. Fang, H. Neumann and M. Beller, J. Am. Chem. Soc., 2013, 135, 11384 CrossRef CAS PubMed.
- D. Forberg, J. Obenauf, M. Friedrich, S. Hühne, W. Mader, G. Motz and R. Kempe, Catal. Sci. Technol., 2014, 4, 4188 CAS.
- C. Gunanathan and D. Milstein, Science, 2013, 341, 249 CrossRef CAS PubMed.
- (a) K. Kon, S. M. A. H. Siddiki and K. Shimizu, J. Catal., 2013, 304, 63 CrossRef CAS; (b) S. M. A. H. Siddiki, K. Kon, A. S. Touchy and K. Shimizu, Catal. Sci. Technol., 2014, 4, 1716 RSC; (c) S. K. Moromi, S. M. A. H. Siddiki, Md. A. Ali, K. Kon and K. Shimizu, Catal. Sci. Technol., 2014, 4, 3631 RSC; (d) C. Chaudhari, S. M. A. H. Siddiki, M. Tamura and K. Shimizu, RSC Adv., 2014, 4, 53374 RSC; (e) S. M. A. H. Siddiki, A. S. Touchy, K. Kon and K. Shimizu, Chem. – Eur. J., 2016, 22, 6111 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR and GC/MS analysis data. See DOI: 10.1039/c6qo00165c |
| This journal is © the Partner Organisations 2016 |