
A general photoinduced electron transfer-directed chemoselective perfluoroalkylation of N,N-dialkylhydrazones†‡
Jin
Xie
a,
Jian
Li
a,
Thomas
Wurm
a,
Vanessa
Weingand
a,
Hui-Ling
Sung
ab,
Frank
Rominger
a,
Matthias
Rudolph
a and
A. Stephen K.
Hashmi
*ac
aOrganisch-Chemisches Institut, Universität Heidelberg, Im Neuenheimer Feld 270, 69120 Heidelberg, Germany. E-mail: hashmi@hashmi.de
bDivision of Preparatory Programs for Overseas Chinese Students, National Taiwan Normal University, Taiwan
cChemistry Department, Faculty of Science, King Abdulaziz University (KAU), 21589 Jeddah, Saudi Arabia
First published on 10th May 2016
Abstract
A selective, practical and general incorporation of fluoroalkyl groups into organic frameworks is of great interest for synthesis. Herein we report the first metal-free, initiator-free, and general photochemical perfluoroalkylation of a variety of N,N-dialkylhydrazones at room temperature in the absence of any external photocatalyst. It constitutes an important advance in perfluoroalkyl radical addition to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N π bonds for the synthesis of hydrazones instead of amines. Affordable and easily available perfluoroalkyl iodides serve as effective precursors. The excellent regio-, stereo- and chemoselectivity as well as the broad substrate scope make this a very promising synthetic tool.
N π bonds for the synthesis of hydrazones instead of amines. Affordable and easily available perfluoroalkyl iodides serve as effective precursors. The excellent regio-, stereo- and chemoselectivity as well as the broad substrate scope make this a very promising synthetic tool.
Fluorine-containing organic compounds possess significant biological activities and intrinsic physical properties in pharmaceuticals,1 agrochemicals,2 material science3 and PET (positron emission tomography) imaging technology.4 Thus synthetic and medicinal chemists developed efficient methods for the selective introduction of perfluoroalkyl groups at important organic backbones by using nucleophilic or electrophilic fluoroalkyl reagents.5 Besides transition-metal-mediated cross-coupling methods,6 radical perfluoroalkylations have recently emerged as a new attractive technology to accomplish this target.7 Despite intense efforts to efficiently generate an electron-deficient perfluoroalkyl radical (
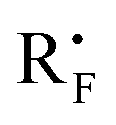 ), such conversions usually depend on the use of transition-metal catalysts,7c–h oxidants,7i–l stoichiometric radical initiators8 and/or the requirement of high reaction temperatures.9 Therefore, the development of a mild and general method to connect a perfluoroalkyl group to versatile frameworks with cheap and widely available perfluoroalkyl halides would be very appealing. Hydrazones are not only the synthetic equivalent of aldehydes, but also important organic intermediates for a series of transformations.10 Undoubtedly, the successful incorporation of a perfluoroalkyl group into hydrazones could provide excellent fluorine-containing building blocks. Very recently, the others’ group and our group developed Cu-, Ir-, Pd- and Au-catalyzed trifluoromethylation and difluoroalkylation of aromatic aldehyde hydrazones with Togni reagent and difluoromethyl bromides, respectively (Scheme 1).11,12 As our follow-up work, we herein report the first metal-free, initiator-free and general methodology for radical perfluoroalkylation of aromatic as well as non-aromatic aldehyde hydrazones with readily available and affordable perfluoroalkyl iodides in the absence of any external photocatalyst, an important progress in radical additions to CN–π-bonds for the synthesis of hydrazones instead of amines.13
), such conversions usually depend on the use of transition-metal catalysts,7c–h oxidants,7i–l stoichiometric radical initiators8 and/or the requirement of high reaction temperatures.9 Therefore, the development of a mild and general method to connect a perfluoroalkyl group to versatile frameworks with cheap and widely available perfluoroalkyl halides would be very appealing. Hydrazones are not only the synthetic equivalent of aldehydes, but also important organic intermediates for a series of transformations.10 Undoubtedly, the successful incorporation of a perfluoroalkyl group into hydrazones could provide excellent fluorine-containing building blocks. Very recently, the others’ group and our group developed Cu-, Ir-, Pd- and Au-catalyzed trifluoromethylation and difluoroalkylation of aromatic aldehyde hydrazones with Togni reagent and difluoromethyl bromides, respectively (Scheme 1).11,12 As our follow-up work, we herein report the first metal-free, initiator-free and general methodology for radical perfluoroalkylation of aromatic as well as non-aromatic aldehyde hydrazones with readily available and affordable perfluoroalkyl iodides in the absence of any external photocatalyst, an important progress in radical additions to CN–π-bonds for the synthesis of hydrazones instead of amines.13
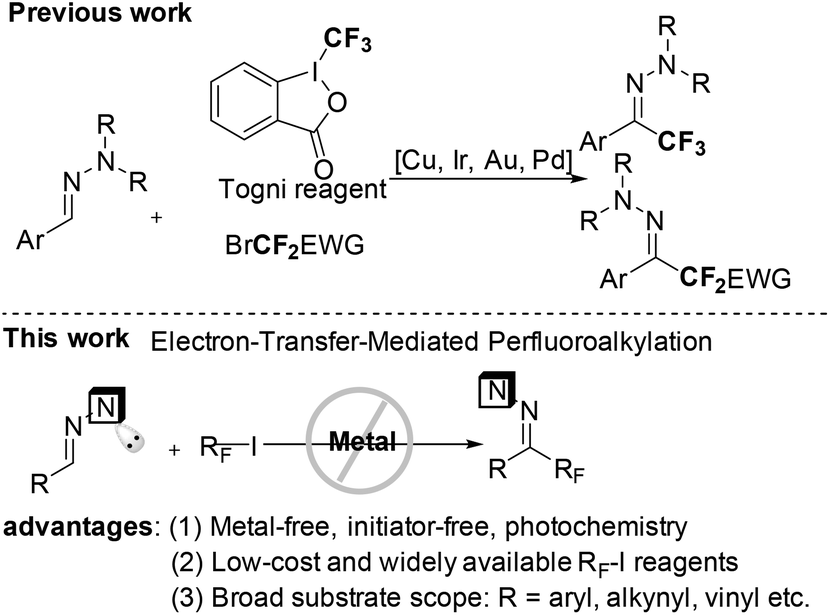 | ||
| Scheme 1 The strategies for perfluoroalkylation of hydrazones. | ||
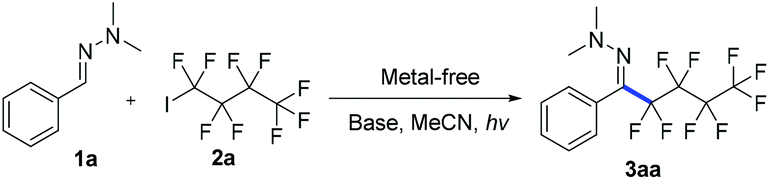
Photoredox catalysis is a powerful protocol to accomplish chemical transformations via a single electron transfer (SET) process.14 Recently, the photochemical activation of electron donor–acceptor (EDA) complexes became a new catalytic mode for clean intermolecular C–C coupling.15 In continuation of our efforts in fluoroalkylations of hydrazones,12 we hypothesized that an electron-rich hydrazone moiety could donate an electron to electron-deficient perfluoroalkyl iodides via a transiently generated EDA complex, ultimately yielding perfluoroalkylated hydrazones.
We first investigated the reaction of hydrazone 1a and nonafluoro-4-iodobutane 2a under irradiation with sunlight. The desired product 3aa was produced in 75% yield after 8 hours (Table 1, entry 1), which opens up a new route for radical addition to CN bonds not depending on metal reagents, radical initiators or toxic organotin reagents.13 The screening of different light sources showed that at 315–400 nm 3aa was formed in 94% yield (entries 1–6). Screening of different bases and solvents did not improve the reaction yield (entries 7–12, see ESI‡ for details). The control experiment demonstrated that light was essential for the reaction, and no 3aa was formed even upon heating at 60 °C for 24 hours (entry 13). Decreasing the amount of nonafluoro-4-iodobutane 2a from 2 equiv. to 1.3 equiv. reduces the yield to 70% (entry 14). Only an (E)-configuration is observed for the CN bond of 3aa (see ESI‡ for details).
|
|
||||
|---|---|---|---|---|
| Entry | Light source | Base | Time/h | Yieldb |
| a Reaction conditions: 1a (0.2 mmol), 2a (2 equiv.), base (3 equiv.), MeCN (0.6 mL). b 19F NMR yield with 1,3,5-trifluorobenzene as an internal standard; the yield in brackets is the isolated yield. c MeOH was used. d CH2Cl2 was used. e The reaction was heated to 60 °C without light. f 1.3 equiv. 2a was employed. TMG = 1,1,3,3-tetramethylguanidine. CFL = compact fluorescent light bulbs. | ||||
| 1 | Sunlight (A) | Imidazole | 8 | 75% |
| 2 | CFL (B) | Imidazole | 24 | 16% |
| 3 | λ = 400–500 nm (C) | Imidazole | 24 | 14% |
| 4 | Blue leds (D) | Imidazole | 24 | 5% |
| 5 | λ = 315–400 nm (E) | Imidazole | 12 | 94% (83%) |
| 6 | λ = 254 nm (F) | Imidazole | 12 | Trace |
| 7 | E | K2HPO4 | 12 | 87% |
| 8 | E | Cs2CO3 | 12 | 70% |
| 9 | E | 2,6-Lutidine | 12 | 88% |
| 10 | E | TMG | 12 | 54% |
| 11c | E | Imidazole | 12 | 86% |
| 12d | E | Imidazole | 12 | 90% |
| 13e | — | Imidazole | 24 | 0% |
| 14f | E | Imidazole | 12 | 70% |
| 15 | E with a bandpass filter (375 nm) | Imidazole | 24 | 38% |
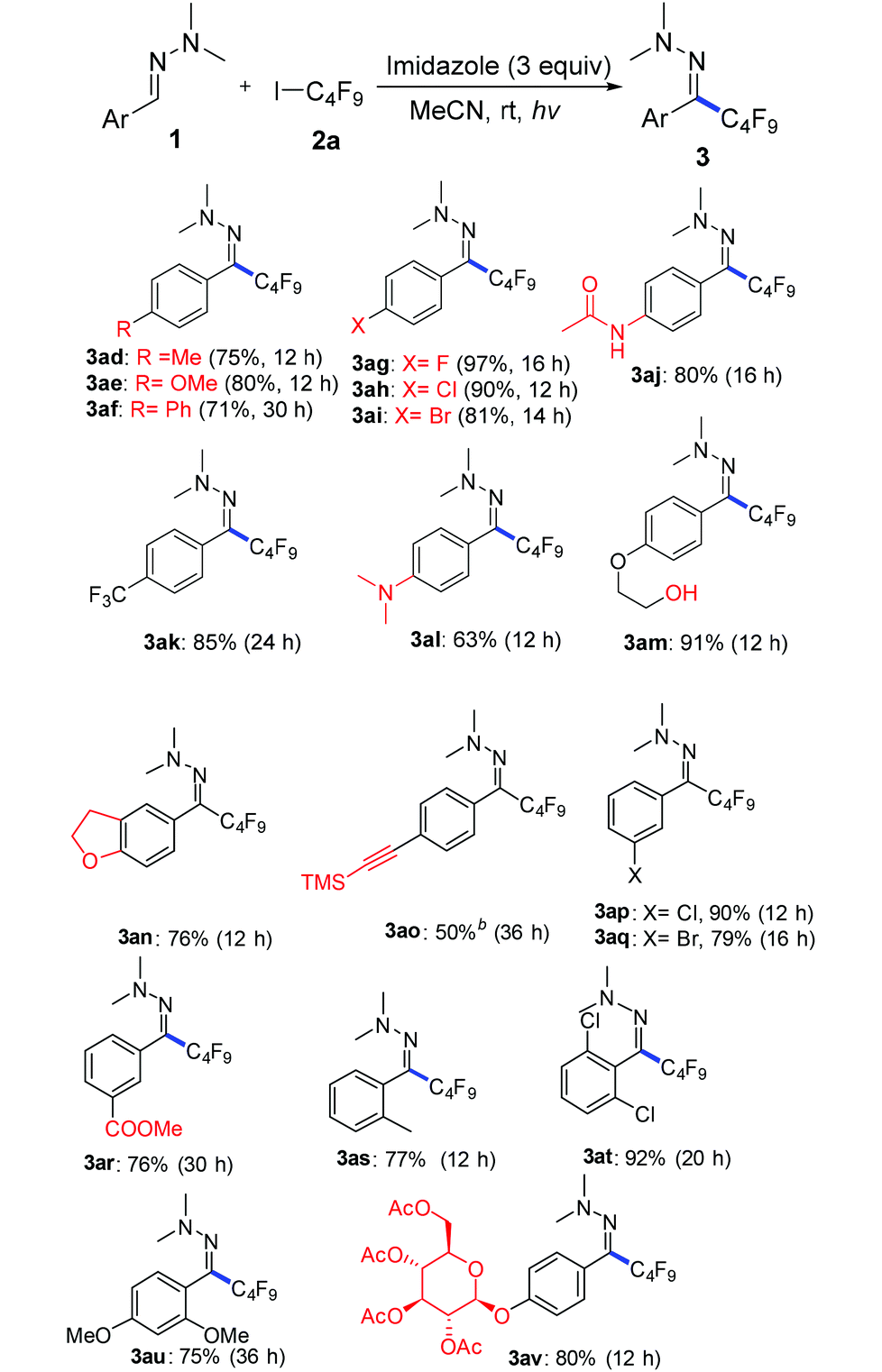
With the optimized reaction conditions (Table 1, entry 5), we first examined different electron donors (Scheme 2). N,N-Dialkyl hydrazones 1a–c were effective coupling partners, while other electron donors in 1d–f failed to show the metal-free perfluoroalkylation reaction, most probably due to their relatively weak electron donating ability. The investigation of the substrate scope of aromatic aldehyde hydrazones showed that both electron-poor and electron-rich substituents at the o-, m-, or p-position of the aromatic ring were compatible with excellent regio- and chemo-selectivity (Table 2). They can uniformly furnish the desired products 3ad–av in 50–97% yield in (E)-configuration (see Fig. 1 and ESI‡ for details). A variety of important functional-groups, such as halogens, amides, tertiary amines, ethers, esters, alcohols and alkynes were tolerated well under the mild reaction conditions. The possibility of a late-stage modification of biologically active compounds was proven by the medically important helicide derivative 3av.
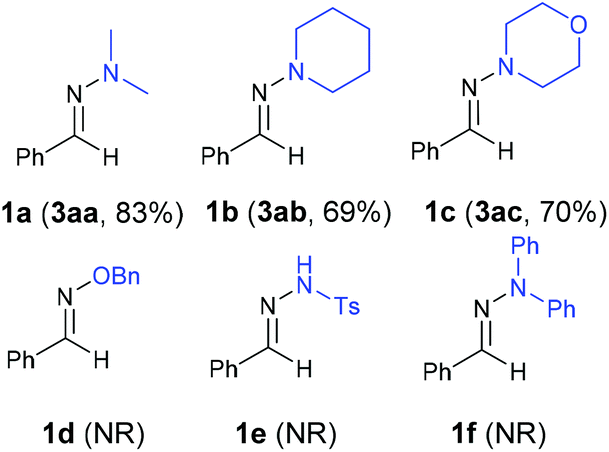 | ||
| Scheme 2 The results of various electron donors. Reaction conditions: 1 (0.2 mmol), 2a (2 equiv.), imidazole (3 equiv.), MeCN (0.6 mL), irradiation 12 hours with 315–400 nm light. Isolated yields. | ||
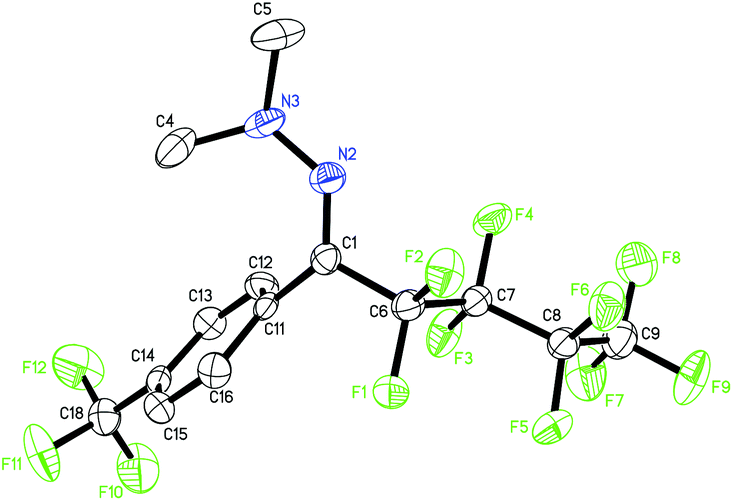 | ||
| Fig. 1 The solid-state molecular structure of 3ak.16 | ||
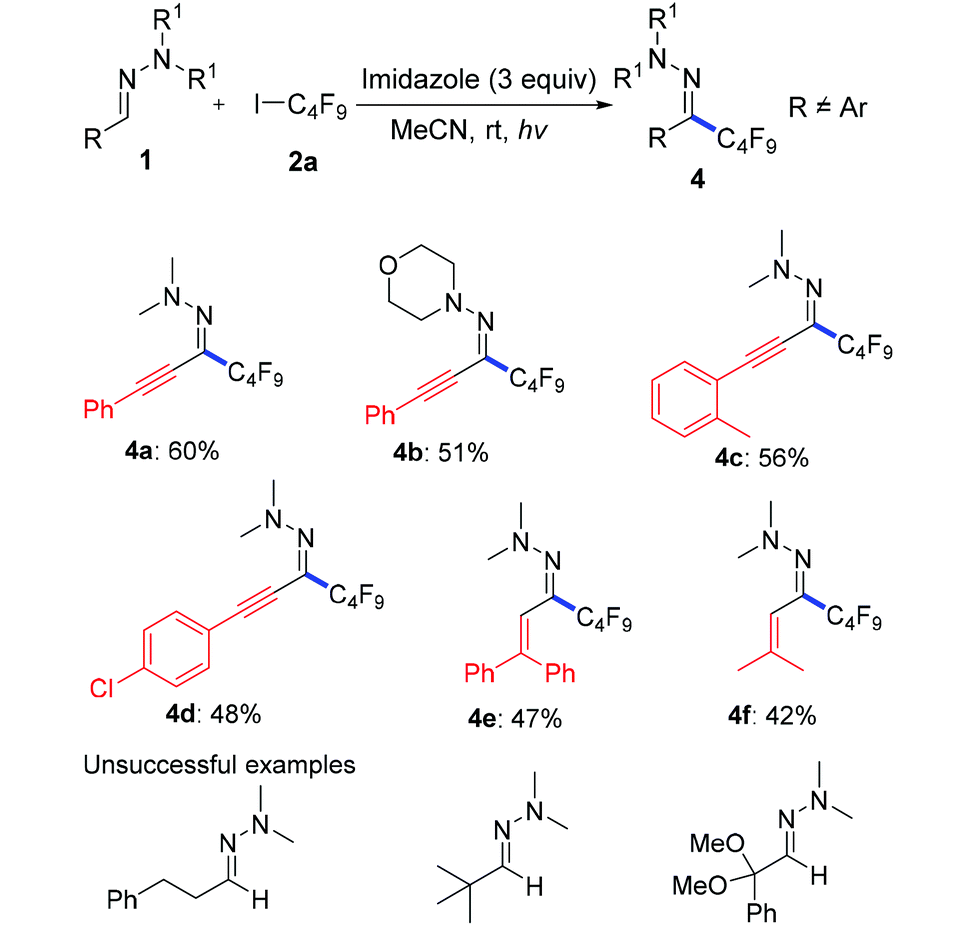
Then, we attempted to expand the substrate scope to non-aromatic aldehyde hydrazones which turned out to be unreactive in our previous work.12 As shown in Table 3, ynal and enal hydrazones indeed provided the desired products 4a–f in moderate yields. However, aliphatic aldehyde hydrazones only show a sluggish reaction.
| a Reaction conditions: 1 (0.2 mmol), 2a (3 equiv.), imidazole (3 equiv.), MeCN (0.6 mL), under 315–400 nm light for 36 hours. |
|---|
|
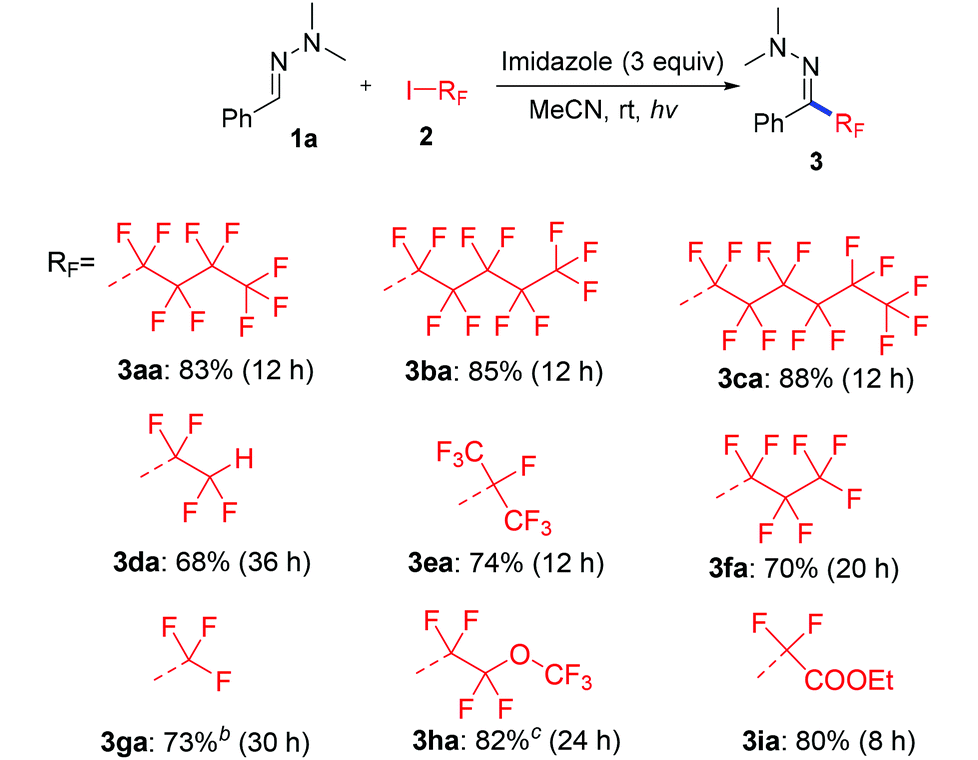
Next we investigated a variety of cheap perfluoroalkyl iodides (Table 4). With long-chain perfluoroalkyl iodides, good yields were isolated (3aa–3ca), whereas with smaller, volatile reagents only moderate yields of the perfluoroalkylated products 3da–ga were obtained. Secondary perfluoroalkyl iodides and β-ether as well as α-ester fluoroalkyl iodides readily delivered the desired products 3ea, 3ha and 3ia in 74–82% yields. Notably, normal alkyliodides such as iodomethane and iodocyclohexane as well as a perfluoroalkyl bromide (1-bromononafluorobutane) do not undergo this transformation under the optimized reaction conditions. The gram-scale application in Scheme 3 underlines the practicability. The desired coupling product 3aa was obtained in 75% yield from 20 mmol benzaldehyde. The resulting perfluoroalkylated hydrazones can smoothly be reduced or hydrolysed or undergo other organic transformations.11,12
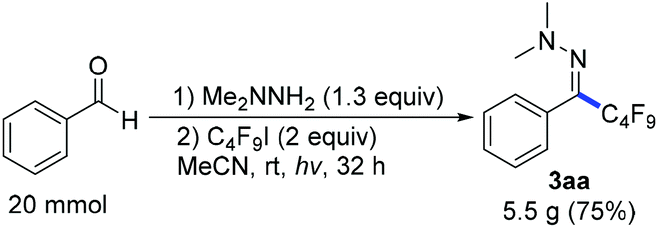 | ||
| Scheme 3 The gram-scale application. Reaction conditions: (1) benzaldehyde (20 mmol), 1,1-dimethylhydrazine (1.3 equiv.). (2) C4F9I (40 mmol), imidazole (60 mmol), MeCN (50 mL), irradiation with 315–400 nm light. | ||
Scheme 4 shows mechanistic experiments. When TEMPO was added to the model reaction, only trace amounts of 3aa were formed along with 45% of TEMPO-C4F95. It suggests that a radical pathway should be involved. Radical initiators (AIBN, tBuOOBut and Et3B/air) failed to produce the desired product 3aa upon heating to 80 °C, indicating that a radical chain propagation is less likely. The light on/off experiment (Scheme 4b) and the quantum yield (Φ = 2.3%) of the model reaction further verify this statement. The addition of another electron donor, N,N-diisopropylmethylamine (DIPMA) into the model reaction resulted in a lower yield (46% vs. 94%), and by-products 6 and 7, which were not detected in our model reaction, might arise from the H-atom abstraction of a generated aminyl radical and a perfluoroalkyl radical, respectively (see ESI‡ for discussion).17 While it is possible that hydrazone 1a and C4F9I 2a form a transient EDA complex, only slight changes were observed in UV-vis absorption spectra when mixing 1a with 2a in acetonitrile (see ESI‡ for details). The control experiments with a bandpass filter ruled out the feasibility of homolytic cleavage pathway of C4F9I.18 In the light of Melchiorre's recent work,15e direct photoexcitaion of hydrazone 1a was proposed as one possible pathway. The theory calculation of the HOMO–LUMO gap of hydrazone 1a and C4F9I 2a are 4.37 eV and 5.40 eV respectively, which indicates hydrazone 1a is more easily excited than 2a.19 As shown in Scheme 4c, one plausible mechanism starts with a SET of the excited state of 1a to 2a to form radical cation 9 and perfluoroalkyl radical 10. Meanwhile, irradiation of EDA-complex is an alternative pathway to produce 9 and 10. As these species are formed at the same time and radical addition of perfluoroalkyl radical 10 to hydrazone radical cation 9 generates intermediate 11. Then, the bulky perfluoroalkyl group forces the N,N-dimethyl group to the opposite side to give 3aa in E configuration via deprotonation by base.20
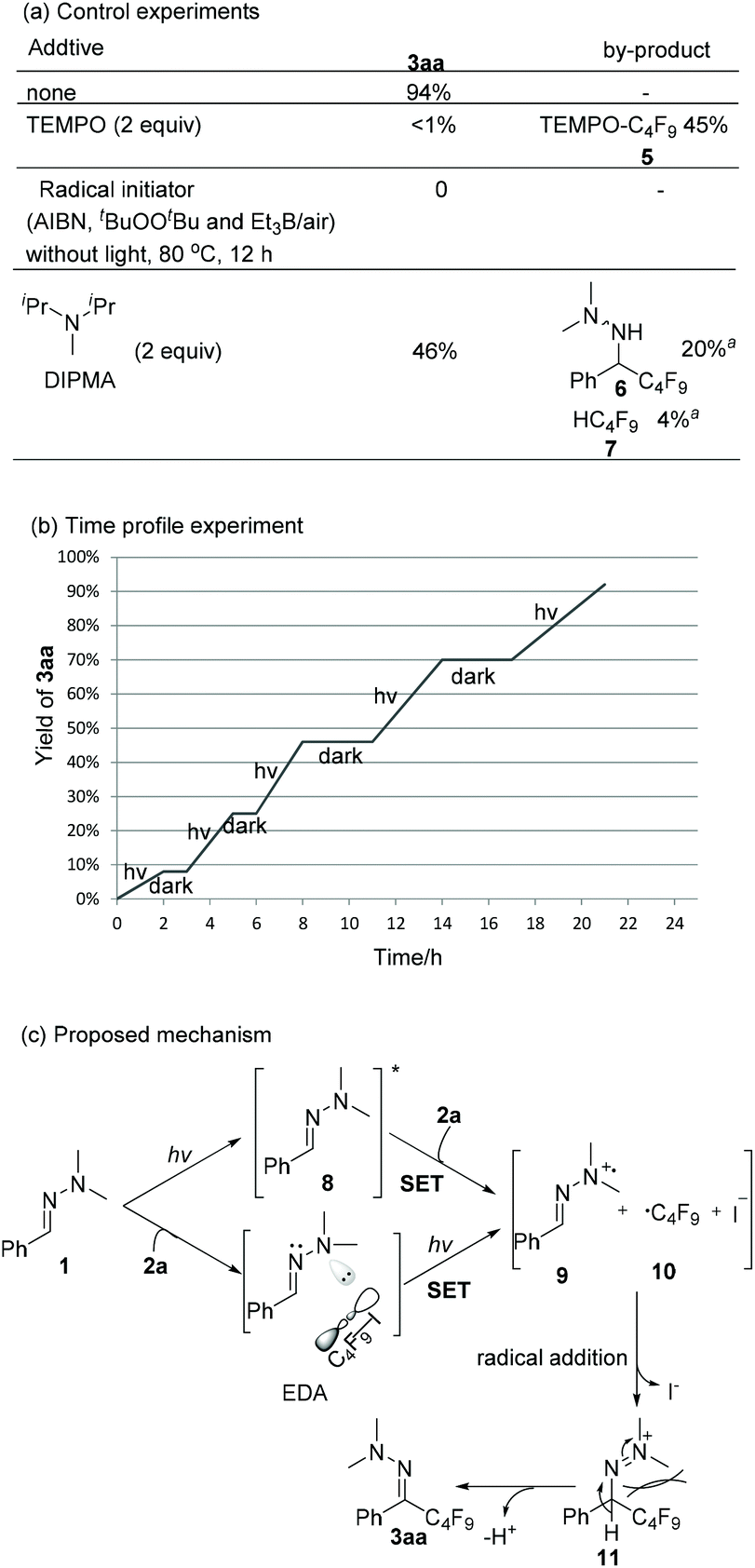 | ||
Scheme 4 Mechanistic studies of model reaction and proposed mechanism. The yields were determined by 19F NMR analysis of the reaction mixture using 1,3,5-trifluorobenzene as a reference standard. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) The yields were obtained by 1H NMR analysis of the reaction mixture. The yields were obtained by 1H NMR analysis of the reaction mixture. | ||
In summary, we have developed the first metal-free and general method for the perfluoroalkylation of aromatic and non-aromatic aldehyde hydrazones with simple and affordable perfluoroalkyl iodides by photochemistry. This opens up new options for photochemical radical C–C coupling in the absence of an external photocatalyst, a radical initiators or organotin reagents. The broad substrate scope, mild reaction conditions, excellent functional group compatibility, scale-up potential as well as the high chemoselectivity makes this protocol very practical in organic synthesis and drug discovery.
Notes and references
- (a) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC; (b) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed.
- Organofluorine Chemistry, Principles and Comercial Applications, ed. R. E. Banks, B. E. Smart, J. C. Tatlow, Springer, New York, 1994 Search PubMed.
- T. Hiyama, Organofluorine Compounds: Chemistry and Application, Springer, Berlin, 2000 Search PubMed.
- (a) S. M. Ametamey, Chem. Rev., 2008, 108, 4036–4036 CrossRef CAS; (b) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470–477 CrossRef CAS PubMed; (c) O. A. Tomashenko and V. V. Grushin, Chem. Rev., 2011, 111, 4475–4521 CrossRef CAS PubMed; (d) L. Zhu, K. Ploessl and H. F. Kung, Science, 2013, 342, 429–430 CrossRef CAS PubMed.
- Recent reviews: (a) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214–8264 CrossRef CAS PubMed; (b) E. Merino and C. Nevado, Chem. Soc. Rev., 2014, 43, 6598–6608 RSC; (c) H. Egami and M. Sodeoka, Angew. Chem., Int. Ed., 2014, 53, 8294–8308 CrossRef CAS PubMed.
- Selected examples: for Ar–X: (a) O. A. Tomashenko, E. C. Escudero-Adán, M. Martínez Belmonte and V. V. Grushin, Angew. Chem., Int. Ed., 2011, 50, 7655–7659 CrossRef CAS PubMed; (b) P. S. Fier and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 5524–5527 CrossRef CAS PubMed; (c) G. K. S. Prakash, S. K. Ganesh, J.-P. Jones, A. Kulkarni, K. Masood, J. K. Swabeck and G. A. Olah, Angew. Chem., Int. Ed., 2012, 51, 12090–12094 CrossRef CAS PubMed; (d) M. Chen and S. L. Buchwald, Angew. Chem., Int. Ed., 2013, 52, 11628–11631 CrossRef CAS PubMed; (e) M. Huiban, M. Tredwell, S. Mizuta, Z. Wan, X. Zhang, T. L. Collier, V. Gouverneur and J. Passchier, Nat. Chem., 2013, 5, 941–944 CrossRef CAS PubMed. For Ar–B(OH)2: (f) Q. Qi, Q. Shen and L. Lu, J. Am. Chem. Soc., 2012, 134, 6548–6551 CrossRef CAS PubMed; (g) N. D. Litvinas, P. S. Fier and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 536–539 CrossRef CAS PubMed; (h) Y. Ye and M. S. Sanford, J. Am. Chem. Soc., 2012, 134, 9034–9037 CrossRef CAS PubMed; (i) J. Xu, B. Xiao, C.-Q. Xie, D.-F. Luo, L. Liu and Y. Fu, Angew. Chem., Int. Ed., 2012, 51, 12551–12554 CrossRef CAS PubMed; (j) Z. Feng, Q.-Q. Min, Y.-L. Xiao, B. Zhang and X. Zhang, Angew. Chem., Int. Ed., 2014, 53, 1669–1673 CrossRef CAS PubMed; (k) Y.-B. Yu, G.-Z. He and X. Zhang, Angew. Chem., Int. Ed., 2014, 53, 10457–10461 CrossRef CAS PubMed. For C–H bonds: (l) C. Feng and T.-P. Loh, Angew. Chem., Int. Ed., 2013, 52, 12414–12417 CrossRef CAS PubMed; (m) M. Shang, S.-Z. Sun, H.-L. Wang, B. N. Laforteza, H.-X. Dai and J.-Q. Yu, Angew. Chem., Int. Ed., 2014, 53, 10439–10442 CrossRef CAS PubMed; (n) Z. Feng, Q.-Q. Min, H.-Y. Zhao, J.-W. Gu and X. Zhang, Angew. Chem., Int. Ed., 2015, 54, 1270–1274 CrossRef CAS PubMed.
- For two reviews: (a) S. Barata-Vallejo and A. Postigo, Coord. Chem. Rev., 2013, 257, 3051–3069 CrossRef CAS; (b) A. Studer, Angew. Chem., Int. Ed., 2012, 51, 8950–8958 CrossRef CAS PubMed. For selected examples: (c) D. A. Nagib, M. E. Scott and D. W. C. MacMillan, J. Am. Chem. Soc., 2009, 131, 10875–10877 CrossRef CAS PubMed; (d) D. A. Nagib and D. W. C. MacMillan, Nature, 2011, 480, 224–228 CrossRef CAS PubMed; (e) X. Wang, Y. Xu, F. Mo, G. Ji, D. Qiu, J. Feng, Y. Ye, S. Zhang, Y. Zhang and J. Wang, J. Am. Chem. Soc., 2013, 135, 10330–10333 CrossRef CAS PubMed; (f) R. Tomita, Y. Yasu, T. Koike and M. Akita, Angew. Chem., Int. Ed., 2014, 53, 7144–7148 CrossRef CAS PubMed; (g) P. Gao, Y.-W. Shen, R. Fang, X.-H. Hao, Z.-H. Qiu, F. Yang, X.-B. Yan, Q. Wang, X.-J. Gong, X.-Y. Liu and Y.-M. Liang, Angew. Chem., Int. Ed., 2014, 53, 7629–7633 CrossRef CAS PubMed; (h) N. Iqbal, J. Jung, S. Park and E. J. Cho, Angew. Chem., Int. Ed., 2014, 53, 539–542 CrossRef CAS PubMed; (i) Y. Fujiwara, J. A. Dixon, R. A. Rodriguez, R. D. Baxter, D. D. Dixon, M. R. Collins, D. G. Blackmond and P. S. Baran, J. Am. Chem. Soc., 2012, 134, 1494–1497 CrossRef CAS PubMed; (j) A. Deb, S. Manna, A. Modak, T. Patra, S. Maity and D. Maiti, Angew. Chem., Int. Ed., 2013, 52, 9747–9750 CrossRef CAS PubMed; (k) X.-Y. Jiang and F.-L. Qing, Angew. Chem., Int. Ed., 2013, 52, 14177–14180 CrossRef CAS PubMed; (l) Y.-F. Wang, G. H. Lonca and S. Chiba, Angew. Chem., Int. Ed., 2014, 53, 1067–1071 CrossRef CAS PubMed; (m) H. Egami, R. Shimizu, S. Kawamura and M. Sodeoka, Angew. Chem., Int. Ed., 2013, 52, 4000–4003 CrossRef CAS PubMed; (n) T. Xu, C. W. Cheung and X. Hu, Angew. Chem., Int. Ed., 2014, 53, 4910–4914 CrossRef CAS PubMed; (o) B. Zhang, C. M. Lichtenfeld, C. G. Daniliuc and A. Studer, Angew. Chem., Int. Ed., 2013, 52, 10792–10795 CrossRef CAS PubMed; (p) S. Barata-Vallejo, M. M. Flesia, B. Lantaño, J. E. Argüello, A. B. Peñéñory and A. Postigo, Eur. J. Org. Chem., 2013, 998–1008 CrossRef CAS.
- (a) Y. Tanabe, N. Matsuo and N. Ohno, J. Org. Chem., 1988, 53, 4582–4585 CrossRef CAS; (b) C. Lai and T. E. Mallouk, J. Chem. Soc., Chem. Commun., 1993, 1359–1361 RSC.
- For selected examples: (a) G. V. D. Tiers, J. Am. Chem. Soc., 1960, 82, 5513–5513 CrossRef CAS; (b) A. B. Cowell and C. Tamborski, J. Fluorine Chem., 1981, 17, 345–356 CrossRef CAS; (c) Y. Aihara, M. Tobisu, Y. Fukumoto and N. Chatani, J. Am. Chem. Soc., 2014, 136, 15509–15512 CrossRef CAS PubMed and ref. 7m–o.
- R. Lazny and A. Nodzewska, Chem. Rev., 2010, 110, 1386–1434 CrossRef CAS PubMed.
- (a) E. Pair, N. Monteiro, D. Bouyssi and O. Baudoin, Angew. Chem., Int. Ed., 2013, 52, 5346–5349 CrossRef CAS PubMed; (b) A. Prieto, E. Jeamet, N. Monteiro, D. Bouyssi and O. Baudoin, Org. Lett., 2014, 16, 4770–4773 CrossRef CAS PubMed; (c) P. Xu, G. Wang, Y. Zhu, W. Li, Y. Cheng, S. Li and C. Zhu, Angew. Chem., Int. Ed., 2016, 55, 2939–2943 CrossRef CAS PubMed; (d) A. Prieto, R. Melot, D. Bouyssi and N. Monteiro, Angew. Chem., Int. Ed., 2016, 55, 1885–1889 CrossRef CAS PubMed; (e) A. Prieto, R. Melot, D. Bouyssi and N. Monteiro, ACS Catal., 2016, 6, 1093–1096 CrossRef CAS.
- J. Xie, T. Zhang, F. Chen, N. Mehrkens, F. Rominger, M. Rudolph and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2016, 55, 2934–2938 CrossRef CAS PubMed.
- (a) G. K. Friestad, Tetrahedron, 2001, 57, 5461–5496 CrossRef CAS; (b) S. Torrente and R. Alonso, Org. Lett., 2001, 3, 1985–1987 CrossRef CAS PubMed; (c) D. Hager and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 16986–16989 CrossRef CAS PubMed; (d) M. Gartner, J. Ballmann, C. Damm, F. W. Heinemanna and H. Kisch, Photochem. Photobiol. Sci., 2007, 6, 159–164 RSC; (e) B. H. Deepak, G. Satish and R.-S. Liu, Adv. Synth. Catal., 2016, 358, 1348–1367 CrossRef.
- Selected reviews on photoredox catalysis: (a) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC; (b) J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828–6838 CrossRef CAS PubMed; (c) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed; (d) D. Ravelli, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2013, 42, 97–113 RSC; (e) D. P. Hari and B. König, Angew. Chem., Int. Ed., 2013, 52, 4734–4743 CrossRef CAS PubMed; (f) M. Reckenthäler and A. G. Griesbeck, Adv. Synth. Catal., 2013, 355, 2727–2744 CrossRef; (g) J. Xie, H. Jin, P. Xu and C. Zhu, Tetrahedron Lett., 2014, 55, 36–48 CrossRef CAS; (h) D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed.
- Selected examples: (a) P. V. Pham, D. A. Nagib and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2011, 50, 6119–6122 CrossRef CAS PubMed; (b) E. Arceo, I. D. Jurberg, A. Álvarez-Fernández and P. Melchiorre, Nat. Chem., 2013, 5, 750–756 CrossRef CAS PubMed; (c) M. Nappi, G. Bergonzini and P. Melchiorre, Angew. Chem., Int. Ed., 2014, 53, 4921–4925 CrossRef CAS PubMed; (d) S. R. Kandukuri, A. Bahamonde, I. Chatterjee, I. D. Jurberg, E. C. Escudero-Adán and P. Melchiorre, Angew. Chem., Int. Ed., 2015, 54, 1485–1489 CrossRef CAS PubMed; (e) M. Silvi, E. Arceo, I. D. Jurberg, C. Cassani and P. Melchiorre, J. Am. Chem. Soc., 2015, 137, 6120–6123 CrossRef CAS PubMed.
- CCDC 1053855 (3ak) contains the supplementary crystallographic data for this paper.
- It was rather difficult to isolate byproduct 6 because it was easily volatile. Its structure was determined by 1H NMR, 19F NMR, GC-MS and HRMS.
- The homolysis of C4F9I under our reaction conditions is less likely as irradiation of the model reaction with a bandpass filter (375 nm) can furnish the desired product in 38% yield (see entry 15 in Table 1).
- The HOMO and LUMO energy of hydrazone 1a and C4F9I 2a was obtained at B3LYP-D3/LanL2DZ level. For hydraznone 1a: HOMO energy, −5.21 eV and LUMO energy −0.84 eV; for C4F9I 2a: HOMO energy, −8.62 eV and LUMO energy, −3.21 eV.
- The DFT calculations suggests product 3aa in E configuration has lower energy than that in Z configuration, see ESI‡ for details.
Footnotes |
| † Dedicate to Professor Barry Trost on the occasion of his 75th birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 1053855. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00158k |
| This journal is © the Partner Organisations 2016 |