Cu(II)-and Co(II)-containing metal–organic frameworks (MOFs) as catalysts for cyclohexene oxidation with oxygen under solvent-free conditions†
Yanghe
Fu
,
Dengrong
Sun
,
Meng
Qin
,
Renkun
Huang
and
Zhaohui
Li
*
Research Institute of Photocatalysis, Fuzhou University, State Key Laboratory Breeding Base of Photocatalysis, Fuzhou, 350002, People's Republic of China. E-mail: zhaohuili1969@yahoo.com; Fax: 86-591-83779105; Tel: 86-591-83779260
First published on 22nd February 2012
Abstract
Aerobic oxidation of cyclohexene over [Cu2(OH)(BTC)(H2O)]n·2nH2O (Cu-MOF, BTC = 1,3,5-benzenetricarboxylic acid) and [M2(DOBDC)(H2O)2]·8H2O (Co-and Ni-MOF, DOBDC = 2,5-dihydroxyterephthalic acid) in the absence of solvent under mild conditions was studied. It is observed that both Cu-MOF and Co-MOF can selectively oxidize cyclohexene to give 2-cyclohexen-1-ol and 2-cyclohexen-1-one as the main products, while Ni-MOF is totally inactive for cyclohexene oxidation. The mechanism of the catalytic oxidation of cyclohexene over Cu-MOF and Co-MOF has been proposed. These MOF-based catalysts are stable and recyclable under current reaction conditions. This study highlights the great potential of developing MOFs as highly stable, molecularly tunable, recyclable and reusable heterogeneous catalysts for alkenes oxidation.
1. Introduction
Catalytic oxidation of alkenes into value-added oxygenated derivatives is a fundamental reaction in organic chemistry that has many industrial applications. Traditional catalytic procedures involved in the oxidation of alkenes produce a great deal of environmentally undesirable wastes. The development of green processes for the catalytic oxidation of alkenes is becoming increasingly important. The uses of H2O2 or oxygen as oxidants in the catalytic oxidation reactions are considered to be green technology since no toxic by-products are produced in these reactions.As compared to H2O2, the use of oxygen as oxidant is especially attractive. Transition metal complexes containing redox active transition metals like Cu, Co, Mn and Fe, are important homogenous catalysts which can oxidize alkenes with oxygen.1–5 However, these oxidation reactions are usually performed in the liquid phase and the recovery of the catalysts from the reaction systems is tedious. To make the recovery of the catalyst easier, heterogeneous catalysts constructed by immobilizing the homogeneous complexes onto solid supports have been developed.6–8 However, the preparation procedures for these immobilized catalysts are usually troublesome and the leaching of the metal complexes to the reaction system during the catalytic reaction can not be avoided. Recently, promising heterogeneous catalysts based on the isomorphic substitutions by redox active metals in the framework positions of molecular sieves have been developed. These catalysts not only show higher stability towards leaching, the site isolation of the active metal ions in the solid matrices can also prevent their aggregation which would lead to less reactive species.9
Recently, metal–organic frameworks (MOFs) have emerged as a new class of attractive functional materials that have found great potential in applications such as gas storage/separation, molecular recognition, ion exchange, drug delivery, as well as heterogeneous catalysis.10–16 MOFs are crystalline micro-mesoporous hybrid materials constructed by poly-dentate ligands acting as linkers and (transition) metal ions or clusters acting as “nodes”. As structurally analogous to metal substituted molecular sieves, MOFs are attractive heterogeneous catalysts since they can be easily separated and recycled from the reaction systems while possessing spatially separated single catalytic active sites in their framework which is characteristic of the homogenous catalysts. Moreover, MOFs surpass metal substituted molecule sieves in that their pore sizes and thus the substrate shape and size selectivity can be systematically tailored by employing different organic linkers.17 Although the low thermal and chemical stability of MOFs as compared to their inorganic counterparts has restricted their use only in mild conditions, there already have been several reports that showed MOFs can be excellent heterogeneous catalysts for alkene oxidations.18–25 For example, Llabrés i Xamena et al. reported that Cu(II)-and Co(II)-containing MOFs, [Cu(2-pymo)2] and [Co(PhIM)2] (2-pymo = 2-hydroxypyrimidinolate; PhIM = phenylimidazolate), exhibit catalytic activity for tetralin oxidation using molecular oxygen under solvent-free condition.18 Jiang et al. reported that [Cu(bpy)(H2O)2(BF4)2(bpy)] (bpy = 4,4′-bipyridine) is a highly selective catalyst in the allylic oxidation of cyclohexene to give cyclohexene hydroperoxide with molecular oxygen under mild and solvent-free condition.19 Recently aerobic oxidations of a series of styrenes to give benzaldehyde, styrene oxide and derivatives have been observed over N-hydroxyphthalimide loaded Fe(BTC) (BTC = 1,3,5-benzenetricarboxylate) under different reaction conditions as reported by Dhakshinamoorthy et al.22 However, catalysis based on MOFs, especially their catalytic mechanism, is far less explored as compared to the large amount of MOF structures that have already been developed. The great potential of MOFs as heterogeneous catalysts is still not fully appreciated.
Herein, we report the oxidation of cyclohexene with molecular oxygen under mild and solvent-free reaction conditions over [Cu2(OH)(BTC)(H2O)]n·2nH2O (Cu-MOF, BTC = 1,3,5-benzenetricarboxylic acid) and [M2(DOBDC)(H2O)2]·8H2O (M-MOF, M = Co and Ni, DOBDC = 2,5-dihydroxyterephthalic acid). Cu-MOF is a three-dimensional porous MOF consisting of four- and five-coordinated Cu(II) ions, which contains 5 × 7 Å dimension lozenge shaped 1D open channels along crystallographic a axis (Fig. S1†).26 Iso-structural Co-MOF and Ni-MOF has a three-dimensional honeycomb-like network, which contains a channel with a diameter of ca. ∼11 Å (Fig. S2†). The metal ion (Co(II) or Ni(II)) adopts a distorted octahedral coordination mode by five oxygen atoms from the organic ligand and one oxygen atom from water.27,28 All three MOFs contain unsaturated metal centers, implying their potential applications in catalysis. The oxidation with molecular oxygen over solid catalyst in the absence of solvent is highly desirable from an environmental point of view. Our study demonstrates that Cu-MOF and Co-MOF can effectively catalyze the oxidation of cyclohexene to give 2-cyclohexen-1-ol and 2-cyclohexen-1-one, while Ni-MOF does not show any catalytic activity for cyclohexene oxidation. The mechanism of the catalytic oxidation of cyclohexene over Cu-MOF and Co-MOF has also been proposed. Our study highlights the great potential of developing MOFs as highly stable, molecularly tunable, recyclable and reusable heterogeneous catalysts for alkene oxidation.
2. Experimental
Materials
1,3,5-benzenetricarboxylic acid (H3BTC), 2,5-dihydroxyterephthalic acid (H4DOBDC) are obtained from Alfa Aesar Co. Cyclohexene (GR, Aladdin Reagent Co), copper nitrate (Cu(NO3)2·3H2O), cobaltous nitrate (Co(NO3)2·6H2O), nickel nitrate (Ni(NO3)2·6H2O), dimethylformamide (DMF) and ethanol (EtOH) were all used without further purifications.Preparations
For the preparation of Cu-MOF, H3BTC (7.65 mmol, 1.6061 g) was dissolved in 15 mL de-ionized water and the pH was adjusted to 6.7. Cu(NO3)2·3H2O (8.0 mmol, 1.9328 g) in 10 mL de-ionized water was added to the above solution and the mixture was quickly transferred into a 100 mL round bottom flask containing 32 mL of EtOH under rapid stirring. The reaction mixture was refluxed for 4 h. The obtained light blue precipitate was filtered and washed with de-ionized water and ethanol. After purification and activation, light blue Cu-MOF was obtained with 90% yield based on copper nitrate.Co-MOF and Ni-MOF were prepared according to procedures described in the literature.27 In a typical synthesis of Co-MOF, a solid mixture of H4DOBDC (0.2216 g, 1.12 mmol) and Co(NO3)2·6H2O (1.077 g, 3.70 mmol) were added to a 75 mL solution of DMF, EtOH and H2O in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 ratio. The above mixture was ultra-sonicated at room temperature to get a homogeneous solution and was transferred into a 100 mL Teflon liner and heated at 120 °C for 24 h. After reaction, the resultant suspension was filtered and the red-orange crystals obtained were washed with MeOH. MeOH was decanted and replenished four times in two days. After that, the sample was treated under vacuum at 180 °C for 5 h. The thus-treated red-purple crystalline materials were used for the catalytic reaction. A similar procedure was applied in the preparation of Ni-MOF except that cobalt nitrate was replaced with nickel nitrate.
:1:1 ratio. The above mixture was ultra-sonicated at room temperature to get a homogeneous solution and was transferred into a 100 mL Teflon liner and heated at 120 °C for 24 h. After reaction, the resultant suspension was filtered and the red-orange crystals obtained were washed with MeOH. MeOH was decanted and replenished four times in two days. After that, the sample was treated under vacuum at 180 °C for 5 h. The thus-treated red-purple crystalline materials were used for the catalytic reaction. A similar procedure was applied in the preparation of Ni-MOF except that cobalt nitrate was replaced with nickel nitrate.
Characterizations
X-ray diffraction (XRD) patterns were collected on a D8 Advance X-ray diffractometer (Bruker, Germany) with Cu-Kα radiation. The accelerating voltage and the applied current were 40 KV and 40 mA, respectively. Data were recorded at a scanning rate of 0.02° 2θ s−1 in the 2θ range of 5° to 40°. The IR experiments were carried out on a Nicolet 670 FT-IR spectrometer.Typical procedure for cyclohexene oxidation
The oxidation of cyclohexene was carried out in a glass reactor equipped with a reflux condenser and an oxygen balloon. In a typical reaction, catalyst (50 mg, ca. 0.1 mmol) and cyclohexene (5 mL, 49.3 mmol) were heated at 80 °C under atmospheric pressure. The products were analyzed on a Shimadzu gas chromatograph equipped with a flame ionization detector and a HP-5 capillary column. The amounts of all four major products were determined according to commonly used method. Cyclohexene hydroperoxide was treated with triphenylphosphine (PPh3) prior to the analyses.293. Results and discussion
XRD patterns of all three MOFs (Cu, Co and Ni) are similar to those previously reported (Fig. 1). Aerobic oxidation of cyclohexene over the three M-MOFs (Cu, Co and Ni) was studied with oxygen in absence of solvent under mild conditions. The detectable products over Co-MOF are cyclohexene oxide (A), 2-cyclohexen-1-ol (B), 2-cyclohexen-1-one (C) and cyclohexene hydroperoxide (D) (Scheme 1). Other possible products like 2,3-epoxy-cyclohexanone, phenol or cyclohexene dimer are undetectable. The conversion of cyclohexene and the yields of different products over Co-MOF are shown in Fig. 2. The conversion ratio of cyclohexene over Co-MOF increased with the reaction time and reached a conversion ratio of 32.8% after reacting for 20 h. The products distribution with the reaction time reveals that the formation of A is a minor reaction, while the allylic oxidation of cyclohexene to give B, C and D predominates over Co-MOF. However, the formation profiles for B, C and D are quite different. In the first 15 h, the amount of B, C and D all increased with the reaction time. 17.4% of cyclohexene had been converted, among which 3.8%, 5.9% and 6.6% had been converted to give B, C and D respectively after reacting for 15 h. After that (from 15 h to 20 h), the yield to B and C increased accompanied by a concomitant decrease of D, indicating the occurrence of a secondary reaction involving the transformation of D to give B and C. After reacting for 20 h, almost all D was converted to B and C. A total cyclohexene conversion ratio of 32.8% with 12.9% for B and 16.8% for C was achieved after reacting for 20 h, showing high total selectivity (90.3%) to give B and C.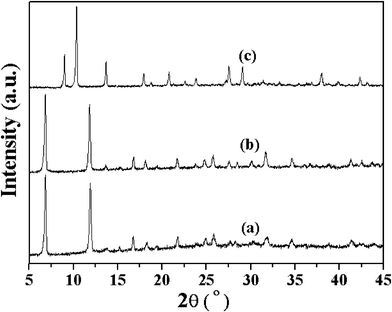 | ||
| Fig. 1 XRD patterns of M-MOF: (a) Co-MOF, (b) Ni-MOF and (c) Cu-MOF. | ||
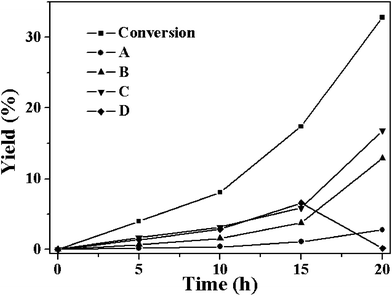 | ||
| Fig. 2 The time-dependent conversion of cyclohexene and formation of the four products A–D catalyzed by Co-MOF at 80 °C (reaction conditions: catalyst, 50 mg; cyclohexene, 5 mL; oxygen balloon; temperature, 80 °C). | ||
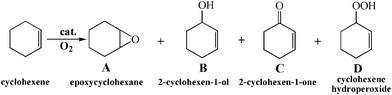 | ||
| Scheme 1 Aerobic oxidation of cyclohexene catalyzed by M-MOF at 80 °C. | ||
Although a similar selective allylic oxidation of cyclohexene was also observed when Cu-MOF was used as the catalyst (Fig. 3), there is obvious difference in the catalytic performance between Cu-MOF and Co-MOF systems. The cyclohexene conversion ratio over Cu-MOF in 20 h was only 20.8%, much lower than that over Co-MOF (32.8%). The yield to B was 8.8% and that for C was 8.0%, a lower total selectivity (80.7%) to B and C as compared to that over Co-MOF (90.3%) was achieved. In addition to this, during the reaction over Co-MOF, D reached a maximum yield (6.6%) before it is totally converted to B and C. However, over Cu-MOF system, the yield of D remained almost constant (3%) during the whole reaction process, indicating that the transformation of D to B and C over Co-MOF is much more effective than that over Cu-MOF, although it requires a longer induction time.
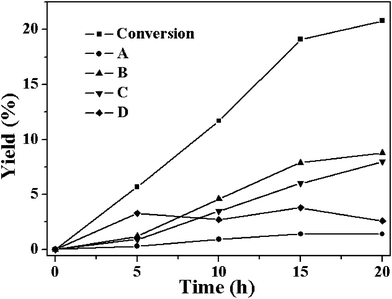 | ||
| Fig. 3 The time-dependent conversion of cyclohexene and formation of the four products A-D catalyzed by Cu-MOF at 80 °C (reaction conditions: catalyst, 50 mg; cyclohexene, 5 mL; oxygen balloon; temperature, 80 °C). | ||
It is believed that the oxidation of cyclohexene to D can occur in the absence of any catalyst although the oxidation extent is low. Therefore, the oxidation of cyclohexene in the absence of any catalyst was also carried out under otherwise similar reaction conditions and the results are shown in Fig. 4. The cyclohexene conversion in the absence of any catalysts was about 12.1%, much lower than that observed over Co-MOF and Cu-MOF. Among the converted cyclohexene, 67.1% was converted to yield D (8.1%). The much higher cyclohexene conversion ratio and the high yield of B and C observed over M-MOF (M = Cu, Co) as compared to the blank experiment indicates that these two MOFs can effectively catalyze the formation of D and the secondary transformation of D to give B and C. The formation of B and C as the main products indicates that the reaction over these two MOFs proceeds via the allylic oxidation pathway instead of the direct oxidation of the alkene double bond. Usually the allylic oxidation and epoxidation are competitive processes in the oxidation of cyclic alkenes and often occur simultaneously.30 The contribution of each reaction depends on both the substrate nature and the relative stability of the corresponding intermediates. Previous studies have revealed that the abstraction of the allylic hydrogen to give allylic oxidation products is easier as compared to its epoxidation in the catalytic oxidation of cyclohexene.
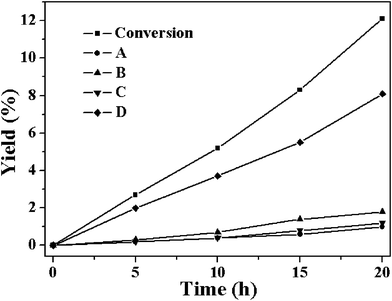 | ||
| Fig. 4 The time-dependent conversion of substrate and formation of the four major products A-D without catalyst at 80 °C (reaction conditions: catalyst, 50 mg; cyclohexene, 5 mL; oxygen balloon; temperature, 80 °C). | ||
To prove the heterogeneous character of M-MOF (M = Cu and Co), conventional filtration experiment was carried out over these two MOFs. For Co-MOF, when the solid catalyst was filtered off from the reaction mixture after reacting for 10 h, the filtrate showed a drastically decreased catalytic activity. The cyclohexene conversion ratio in the filtrate increased from 8.1% to 10.5% in 5 h and most of the cyclohexene were converted to cyclohexene hydroperoxide (Table 1). Similar result was obtained for Cu-MOF. The cyclohexene conversion ratio increased from 11.7% to 14.6% in 5 h after Cu-MOF was removed from the reaction system. The rate of the cyclohexene conversion in the filtrate was comparable to that observed in a system without any catalyst and implied that there is no leaching of metal ions in the filtrate. The absence of leaching metal ions in the filtrate was also confirmed from the ICP measurements. Both Cu-MOF and Co-MOF exhibited high stability during the aerobic oxidation of cyclohexene. The cycling results revealed that there was no obvious loss of catalytic activity of both Co-MOF and Cu-MOF for three successive runs (Fig. 5 and Fig. 6). In addition to this, the used catalysts exhibited similar XRD (Fig. S3 and Fig. S4†) and FT-IR spectra (Fig. S5 and Fig. S6†) as that of the fresh M-MOF, which indicates that the structure of M-MOFs was well preserved during the catalytic reaction. All these results indicated that M-MOF is a highly stable heterogeneous catalyst for the aerobic oxidation of cyclohexene, i.e., the catalytic transformation of cyclohexene is actually induced by solid M-MOF, instead of the metal ions dissolved in the filtrates.
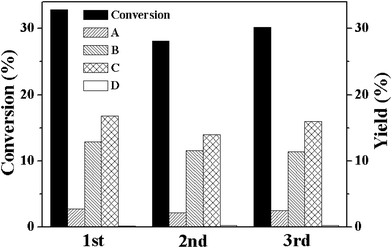 | ||
| Fig. 5 Conversion (%) and yields (%) for the oxidation of cyclohexene over Co-MOF for the fresh catalyst (1st) and for two successive reuses (2nd and 3rd). Reaction conditions: catalyst, 50 mg; no solvent; cyclohexene, 5 mL; oxygen balloon; temperature, 80 °C; time, 20 h. | ||
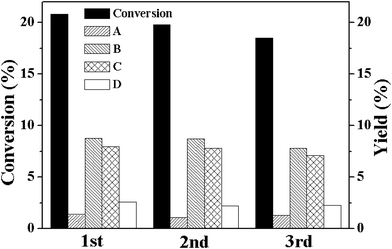 | ||
| Fig. 6 Conversion (%) and yields (%) for the oxidation of cyclohexene over Cu-MOF for the fresh catalyst (1st) and for two successive reuses (2nd and 3rd). Reaction conditions: catalyst, 50 mg; no solvent; cyclohexene, 5 mL; oxygen balloon; temperature, 80 °C; time, 20 h. | ||
| Catalyst | Time (h) | Conversion (%) | Yield (%) | |||
|---|---|---|---|---|---|---|
| A | B | C | D | |||
| a The catalyst was filtered off from the reaction mixture after reacting for 10 h and then kept reacting for 5 h). | ||||||
| Ni-MOF | 20 | 13.2 | 1.1 | 1.8 | 1.1 | 9.2 |
| Co-MOF | 10 | 8.1 | 0.4 | 1.6 | 3.2 | 2.9 |
| 15a | 10.5 | 0.5 | 2.2 | 3.5 | 4.3 | |
| Cu-MOF | 10 | 11.7 | 0.9 | 4.6 | 3.5 | 2.7 |
| 15a | 14.6 | 1.1 | 5.2 | 3.8 | 4.5 | |
A controlled experiment was also carried out over Ni-MOF, which is iso-structural to Co-MOF (Fig. 1). The cyclohexene conversion ratio over Ni-MOF was 13.2% after reacting for 20 h (Table 1), which is comparable to that observed in a system without any catalyst. The main product obtained over Ni-MOF is D. This indicates that Ni-MOF does not show obvious catalytic activity for the cyclohexene oxidation. The different catalytic activity observed over iso-structural Co-MOF and Ni-MOF suggests that Co2+ sites of Co-MOF are the catalytic active sites for the aerobic oxidation of cyclohexene and the catalytic activity is dependent on the metal ions in these MOFs.
Based on these observations, the mechanism of the aerobic oxidation of cyclohexene over M-MOFs (M = Cu or Co) can be proposed (Scheme 2) and the equations involved are listed in Scheme 3. The activation of O2 by Cu2+- or Co2+-containing complexes has previously been reported since the free paramagnetic oxygen molecules are favorable for the reactions with paramagnetic metal ions.31 As in the homogeneous Cu2+- or Co2+-containing complexes, metal centers in Cu-MOF and Co-MOF can also activate O2 to give O2 adduct of M-MOF. The O2 adduct of M-MOF (M = Cu or Co) can abstract a hydrogen from cyclohexene to form Cu- or Co-hydroperoxo complex (M(III)–OOH) while in the meantime cyclohexene is converted to cyclohexenyl radical. The cyclohexenyl radicals can be converted to cyclohexenyl peroxy radical in the presence of O2, which subsequently react with another cyclohexene to give D (Scheme 3, eqn. (1) and (2)). The fission of the M–O bond in M(III)–OOH can recover M-MOF by liberating peroxy radical (HOO·), which can react further with cyclohexenyl radical to give D (Scheme 3, Eq. (3)) or react with double bond to give A. The formation of A is a minor reaction in the catalytic oxidation of cyclohexene since the abstraction of the allylic hydrogen to give allylic oxidation products is easier.
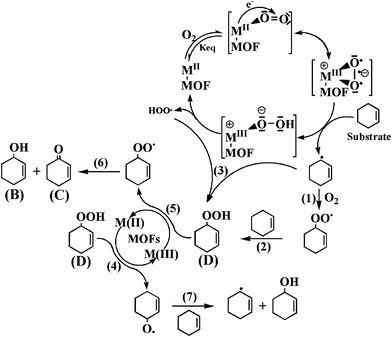 | ||
| Scheme 2 Mechanism proposed for the oxidation of cyclohexene by M-MOF. | ||
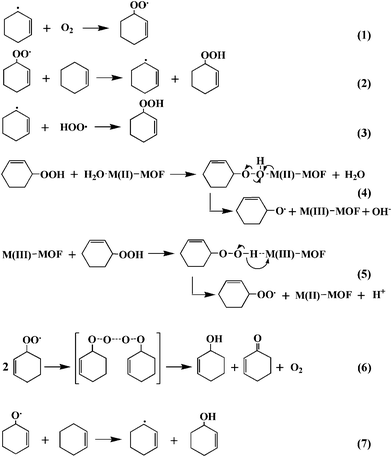 | ||
| Scheme 3 Equations involved in the proposed mechanism for the oxidation of cyclohexene by M-MOF. | ||
The subsequent step in the catalytic reaction involves the well known Haber-Weiss process for the decomposition of D to give ketone and alcohol. According to the Haber-Weiss proposal, the decomposition of the intermediate D should involve the metal catalyst cycling between two different oxidation states.2,32,33 For M-MOF (M = Cu and Co), M(II) would be involved in the formation of B via step (4) and (7), while M(III) would be involved in the formation of C via step (5) and (6). The formation of B and C in 1:1 ratio can also be obtained via step (6). That is to say, Cu(III) and Co(III) could be converted to Cu(II) and Co(II) via step (5), while the alkylperoxo complex (step (5)) could be decomposed into ketone and alcohol and regenerated Co(II) and Cu(II) for Co-MOF and Cu-MOF, respectively. In summary, the mechanism proposed for the allylic oxidation of cyclohexene over M-MOF (M = Cu and Co) includes the following steps: (a) binding of O2 to M-MOF to form O2 adduct of M-MOF; (b) O2 adduct of M-MOF (Cu or Co) abstract hydrogen from cyclohexene to form hydroperoxo complex (M(III)–OOH), while generating cyclohexenyl radicals; (c) the fission of the M–O bond in M(III)–OOH to recover M-MOF by liberating peroxy radical (HOO·); (d) reaction of cyclohexenyl radicals with peroxy radical (HOO·) to yield the intermediate 2-cyclohexene-1-hydroperoxide (D); (e) decomposition of D to alcohol and ketone (B and C) via the Haber-Weiss mechanism.
In the decomposition of D to give ketone and alcohol, the cycling of the transition metal between their different oxidation states is important. In M-MOF, the cycling of Co(II)/Co(III) and Cu(II)/Cu(III) is essential. Actually the formation of Co(III) in the Co-MOF during the catalytic reaction is confirmed from the UV/DRS spectra (Fig. 7). The UV-DRS of fresh Co-MOF shows a band at 350 nm ascribed to a Co2+–O2− charge transfer (LMCT), a broad absorption band centered at 570 nm due to the 4A2(F)→4T2(P) of tetrahedral Co2+ ions, while a band at >1200 nm for the 4A2(F)→4T2(F) transition.34,35 Used Co-MOF shows an increase of the absorption around 400–500 nm, which can be ascribed to the partial formation of Co(III) in the Co-MOF.21 The control experiment performed over the iso-structural Ni-MOF shows that almost no B and C were obtained due to the difficulty in the oxidation of Ni2+ to Ni3+.36 Besides this, since the redox potential of the transition metal ions can also be influenced by the coordinating environment, MOFs containing similar transition metal center vary a lot in their catalytic performance. For example, a pyrazolate-based cobalt-containing MOF can catalyze the oxidation of cyclohexene under aerobic condition only when N-hydroxyphthalimide (NHPI) was used as an electron-transfer mediator.21 The main product obtained is D and only less than 6% of cyclohexene was converted to C. Another Cu(II)-containing MOF [Cu(bpy)(H2O)2(BF4)2(bpy)] is also found to oxidize cyclohexene, but the yield to B and C are extremely low.19 These indicate that, like homogeneous metal complexes catalysts, the catalytic activity of the MOFs depends on the metal center as well as its coordination environment. The high intrinsic structure tunability in the MOFs, which result from the availability of different organic ligands and the versatile coordination chemistry of the transition metals, surely will make MOFs highly promising heterogeneous catalysts.
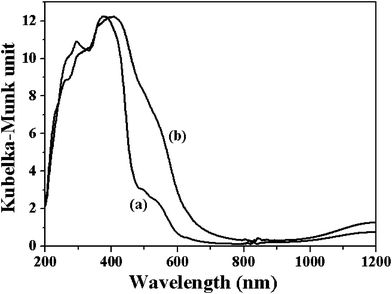 | ||
| Fig. 7 UV/VIS diffuse reflectance spectrum (DRS) for Co-MOF before (a) and after (b) cyclohexene oxidation. | ||
4. Conclusions
In summary, the as-prepared Cu-MOF and Co-MOF can selectively oxidize cyclohexene to give allylic products under mild aerobic condition. The use of molecular oxygen as a final oxidant under solvent-free conditions is significant and offers practical advantages for this process. These MOF-based catalysts undergo no metal leaching, and can be readily recovered from the catalytic reaction system. This study highlights the potential of developing MOFs as highly stable, molecularly tunable, recyclable and reusable heterogeneous catalysts for alkene oxidation. Due to the existence of almost unlimited MOF structures, which result from the availability of different organic ligands and the versatile coordination chemistry of the transition metals, we believe that MOFs-based heterogeneous catalysts with high efficiency can be developed.Acknowledgements
This work was supported by the National Natural Science Foundation of China (20977016), 973 program (2011CB612314), Program for Changjiang Scholars and Innovative Research Team in University (PCSIRT0818). The Award Program of Minjiang Scholar Professorship and the NSF of Fujian province for Distinguished Young Investigator Grant (2009J06004) to Z. Li are also acknowledged.References
- C. Wang, Y. Zhang, B. Yuan and J. Zhao, J. Mol. Catal. A: Chem., 2010, 333, 173 CrossRef CAS.
- D. E. Hamilton, R. S. Drago and A. Zombeck, J. Am. Chem. Soc., 1987, 109, 374 CrossRef CAS.
- X. Zhou and H. Ji, Chem. Eng. J., 2010, 156, 411 CrossRef CAS.
- X. Zhou, Q. Tang and H. Ji, Tetrahedron Lett., 2009, 50, 6601 CrossRef CAS.
- W. Nam, S. J. Baek, K. A. Lee, B. T. Ahn, J. G. Muller, C. J. Burrows and J. S. Valentine, Inorg. Chem., 1996, 35, 6632 CrossRef CAS.
- A. N. Zakharov and N. S. Zefirov, Russ. J. Gen. Chem., 2010, 79, 2563 CrossRef.
- H. Kameyama, F. Narumi, T. Hattori and H. Kameyama, J. Mol. Catal. A: Chem., 2006, 258, 172 CrossRef CAS.
- M. Salavati-Niasari and H. Babazadeh-Arani, J. Mol. Catal. A: Chem., 2007, 274, 58 CrossRef CAS.
- I. W. C. E. Arends and R. A. Sheldon, Appl. Catal., A, 2001, 212, 175 CrossRef CAS.
- S. T. Meek, J. A. Greathouse and M. D. Allendorf, Adv. Mater., 2011, 23, 249 CrossRef CAS.
- J. R. Li, R. J. Kuppler and H. C. Zhou, Chem. Soc. Rev., 2009, 38, 1477 RSC.
- B. Chen, S. Xiang and G. Qian, Acc. Chem. Res., 2010, 43, 1115 CrossRef CAS.
- A. U. Czaja, N. Trukhan and U. Müller, Chem. Soc. Rev., 2009, 38, 1284 RSC.
- L. Carlucci, G. Ciani, S. Maggini, D. M. Proserpio and M. Visconti, Chem.–Eur. J., 2010, 16, 12328 CrossRef CAS.
- S. Keskin and S. Kızılel, Ind. Eng. Chem. Res., 2011, 50, 1799 CrossRef CAS.
- A. Corma, H. Garcia and F. X. Llabres i Xamena, Chem. Rev., 2010, 110, 4606 CrossRef CAS.
- S.-H. Cho, B. Ma, S. T. Nguyen, J. T. Hupp and T. E. Albrecht-Schmitt, Chem. Commun., 2006, 2563 RSC.
- F. X. Llabres i Xamena, O. Casanova, R. Galiassotailleur, H. Garcia and A. Corma, J. Catal., 2008, 255, 220 CrossRef CAS.
- D. Jiang, T. Mallat, D. M. Meier, A. Urakawa and A. Baiker, J. Catal., 2010, 270, 26 CrossRef CAS.
- A. Zhang, L. Li, J. Li, Y. Zhang and S. Gao, Catal. Commun., 2011, 12, 1183 CrossRef CAS.
- M. Tonigold, Y. Lu, A. Mavrandonakis, A. Puls, R. Staudt, J. Möllmer, J. Sauer and D. Volkmer, Chem.–Eur. J., 2011, 17, 8671 CrossRef CAS.
- A. Dhakshinamoorthy, M. Alvaro and H. Garcia, ACS Catal., 2011, 1, 836 CrossRef CAS.
- K. Brown, S. Zolezzi, P. Aguirre, D. Venegas-Yazigi, V. Paredes-García, R. Baggio, M. A. Novak and E. Spodine, Dalton Trans., 2009, 1422 RSC.
- S. G. Baca, M. T. Reetz, R. Goddard, I. G. Filippova, Y. A. Simonov, M. Gdaniec and N. Gerbeleu, Polyhedron, 2006, 25, 1215 CrossRef CAS.
- S. Wang, L. Li, J. Zhang, X. Yuan and C.-Y. Su, J. Mater. Chem., 2011, 21, 7098 RSC.
- J. Chen, T. Yu, Z. Chen, H. Xiao, G. Zhou, L. Weng, B. Tu and D. Zhao, Chem. Lett., 2003, 32, 590 CrossRef CAS.
- P. D. C. Dietzel, Y. Morita, R. Blom and H. Fjellvåg, Angew. Chem., Int. Ed., 2005, 44, 6354 CrossRef CAS.
- P. D. C. Dietzel, B. Panella, M. Hirscher, R. Blom and H. Fjellvåg, Chem. Commun., 2006, 959 RSC.
- Georgiy B. Shul'pin, J. Mol. Catal. A: Chem., 2002, 39, 189 Search PubMed.
- E. F. Murphy, T. Mallat and A. Baiker, Catal. Today, 2000, 57, 115 CrossRef CAS.
- G. Henrici-Olivé and S. Olivé, Angew. Chem., Int. Ed. Engl., 1974, 13, 29 CrossRef.
- H. Weiner, A. Trovarelli and R. G. Finke, J. Mol. Catal. A: Chem., 2003, 191, 217 CrossRef CAS.
- C. Yin, Z. Yang, B. Li, F. Zhang, J. Wang and E. Ou, Catal. Lett., 2009, 131, 440 CrossRef CAS.
- Alfred Beverley Philip Lever, Inorganic Electronic Spectroscopy, Elsevier, Amsterdam, 1968, Chapter 9, p. 323 Search PubMed.
- L. Poul, N. Jouini and F. Fiévet, Chem. Mater., 2000, 12, 3123 CrossRef CAS.
- L. Menini, M. J. da Silva, M. F. F. Lelis, J. D. Fabris, R. M. Lago and E. V. Gusevskaya, Appl. Catal., A, 2004, 269, 117 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Schematic drawings of the structures, XRD and FTIR spectra of the fresh and reused M-MOF (Cu and Co). See DOI: 10.1039/c2ra01038k |
| This journal is © The Royal Society of Chemistry 2012 |