
Synthesis of luminescent dinaphthopentacene isomers and their application in OLEDs
Hannah V.
Anderson
a,
Felix
Brust
b,
Maksym
Fizer
a,
Jonas
Spengler
 b,
Yvonne
Wagenhäuser
b,
Matthias
Stolte
b,
Ana
de Bettencourt-Dias
a,
Sergey A.
Varganov
*a,
Frank
Würthner
*b and
Wesley A.
Chalifoux
*c
b,
Yvonne
Wagenhäuser
b,
Matthias
Stolte
b,
Ana
de Bettencourt-Dias
a,
Sergey A.
Varganov
*a,
Frank
Würthner
*b and
Wesley A.
Chalifoux
*c
aDepartment of Chemistry, University of Nevada, Reno, 1664 N. Virginia Street, Reno, Nevada 89557, USA. E-mail: svarganov@unr.edu
bInstitut für Organische Chemie and Center for Nanosystems Chemistry (CNC), Universität Würzburg, Am Hubland, 97074 Würzburg, Germany. E-mail: frank.wuerthner@uni-wuerzburg.de
cDepartment of Chemistry, University of Alberta, 11227 Saskatchewan Drive, Edmonton, Alberta T6G 2G2, Canada. E-mail: wchalifoux@ualberta.ca
First published on 3rd September 2025
Abstract
Here we report the straightforward synthesis of a novel nanographene, syn-dinaphthopentacene. This new compound was prepared along with its isomer anti-dinaphthopentacene to compare how small structural differences may affect properties such as crystal packing, photostability, and device performance. syn-Dinaphthopentacene has a twisted, chiral structure, and assembles in enantiomerically pure π-stacked columns in the solid state, while the anti-isomer is rigid and planar and forms a slip-stacked packing structure. anti-Dinaphthopentacene quickly undergoes light-induced [4+2] cycloaddition with singlet oxygen when exposed to ambient light and air, but the twisted syn-dinaphthopentacene exhibits remarkable stability under the same conditions. The two isomers have notably different UV-Vis absorbance and emission profiles, and upon probing the efficacy of these compounds as OLED emitters, the anti-isomer was found to be the better fluorophore, achieving luminance values exceeding 2000 cd m−2 at peak efficiencies of almost 1%, as compared to the syn-counterpart which only reached 250 cd m−2.
Introduction
Since the first Nobel-prize winning isolation of monolayer graphene in 2010, there has been a surge of interest in graphitic materials due to their intriguing physical properties such as conductivity, flexibility, and thermostability.1–3 Because of this interest in graphitic materials, the development of methods for preparing graphene and graphene-based materials has become an important area of synthetic chemistry.4–8 Smaller and more precisely-shaped segments of graphene, referred to as nanographenes, have become a hot topic of research due to their tunable semiconducting and optical properties, and their potential application in devices such as organic field-effect transistors (OFETs), organic light-emitting diodes (OLEDs), and organic solar cells.9,10 The performance of organic systems in these types of devices has been observed to be closely tied to specific physical properties such as edge functionalization and packing arrangements,11,12 which are characteristics that can be carefully controlled through rational synthetic design. Many synthetic techniques have been developed to prepare nanographenes with unique shapes, sizes, and functionalization,13–17 but continuous development of techniques and strategies will allow for new classes, and improved performance, to become synthetically accessible.The Chalifoux research group has previously reported a method for rapidly expanding polycyclic aromatic hydrocarbon (PAH) systems, based on the early works of Swager and coworkers.18 This method is a two-step process, involving Suzuki cross-coupling, followed by multi-fold alkyne benzannulation onto neighboring aromatic (Ar) groups, and can be used to prepare nanographenes of varying sizes, shapes, and functionalization (Fig. 1a).19,20 A variety of novel pyrene-based structures with unique optical properties have been synthesized and published using this method, such as peropyrenes and teropyrenes,21,22 configurationally stable chiral peropyrenes and teropyrenes,23–25 and graphene nanoribbons (GNRs) (Fig. 1b).26
 | ||
| Fig. 1 (a) General Suzuki coupling and alkyne benzannulation method developed by the Chalifoux group, and (b) published structures that have been prepared through this method. | ||
More recently, alkyne benzannulation methodology was applied to prepare an isomer of dinaphthopentacene,27 a small nanographene subclass with a pentacene core.28,29 Pentacene is a member of the acene family, a class of nanographenes that are of particular interest due to their impressive semiconducting capabilities and potential applicability in OFET devices, but are somewhat limited in application by issues such as poor (photo)stability and solubility.30–35 The dinaphthopentacene was synthesized by coupling BPin-functionalized aryldiyne precursors to a commercially available anthracene-2,6-diyl bis(trifluoromethanesulfonate), followed by InCl3/AgNTf2-catalyzed four-fold alkyne cyclization to produce an “anti” substituted dinaphthopentacene isomer (Fig. 2a). This specific structure displayed comparable photophysical characteristics, but notably improved photostability, as compared to pentacene.
 | ||
| Fig. 2 (a) Synthesis of anti-dinaphthopentacene,27 and (b) preparation of the new syn-dinaphthopentacene (this work). | ||
In recent years, C–H activation has emerged as an extremely powerful tool in the synthesis and preparation of PAH structures. C–H activation allows for direct functionalization of PAH structures, which can dramatically improve solubility and stabilization, and provide further opportunities for synthetic design.36 Using established methods for iridium-catalyzed C–H borylation of anthracene, both the 2,6- and 2,7-di(boronate) isomers are readily accessible,37 enabling for the direct synthesis of a new structural isomer of dinaphthopentacene (Fig. 2b). Having two isomers of the same PAH system (referred to as “anti” and “syn”) allows for a controlled and targeted exploration of the relationship between structure and physical properties such as photostability, optical properties, and device performance.
Here, we present the straightforward preparation of syn-dinaphthopentacene. By using iridium-catalyzed C–H activation techniques to prepare the aromatic cores, this new isomer was easily accessible by applying known synthetic methods to a 2,6-di(boronate) anthracene starting material. The anti-isomer was also prepared and analyzed in tandem, to allow for direct comparison of the two isomers. The photostability of each of the two structures were compared through 1H NMR spectroscopy, and the crystal structures and packing were explored through X-ray crystallography. The observed photophysical peculiarities and isomer stability were interpreted by the analysis of density functional theory (DFT) calculations. Photophysical analysis of the two isomers suggested applications as singlet emitters, so the efficacy of these molecules in OLED devices was also explored.
Synthesis
Isomers 4(syn) and 4(anti) were prepared separately through the same facile two-step synthetic method. Starting compounds 1a, 1b, and 2 were prepared according to published methods,27,37 and underwent a divergent two-fold Suzuki–Miyaura coupling to produce intermediates 3a and 3b in moderate yields of 30–51%. These intermediates then underwent InCl3/AgNTf2-mediated four-fold alkyne benzannulation to give 4(syn) and 4(anti) in good yields of 72–82% (Scheme 1). Both isomers displayed good solubility in organic solvents (dichloromethane (DCM), toluene, hexanes, chloroform), although 4(syn) was observed to be the more readily soluble of the two, with 4(anti) requiring physical agitation to dissolve completely. The two isomers displayed minor color differences, both as a solid and in solution, with 4(syn) appearing more orange, and the 4(anti) more red.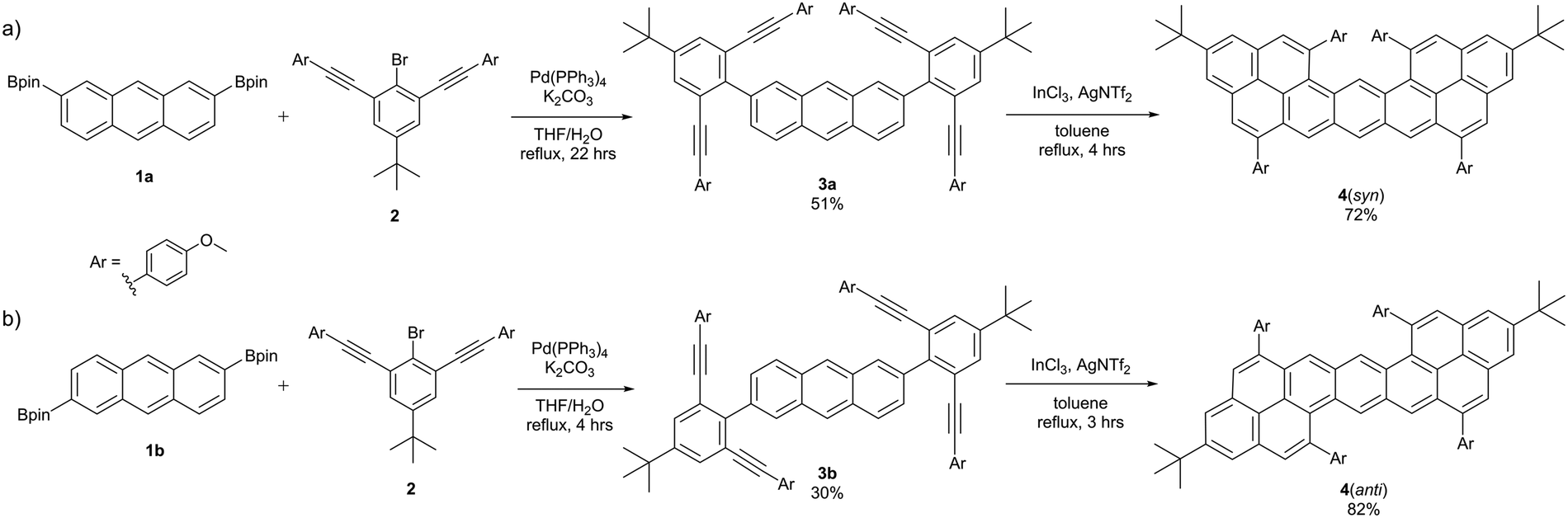 | ||
| Scheme 1 Synthesis of compounds (a) 4(syn) and (b) 4(anti). | ||
The 1H NMR and 13C NMR spectra of 4(syn) (Fig. S13 and S14, SI) display strong evidence of restricted rotation of the aryl groups within the gulf region due to steric congestion. Variable-temperature 1H NMR allowed for improved resolution of the aromatic signals, but full resolution was not achieved even at 85 °C (Fig. S15 and S16, SI). Following established methods, we used a coalescence temperature of approximately 40 °C for the aryl rotation of 4(syn) to calculate the rotational energy barrier (ΔG‡) as 13.9 kcal mol−1 (Fig. 3a).25 The Gibbs energies of activation for the proposed enantiomerization pathway were calculated at 298 K for gas phase (15.1 kcal mol−1) and for solutions in toluene (14.6 kcal mol−1), DCM (14.1 kcal mol−1), acetonitrile (13.9 kcal mol−1), and DMSO (13.9 kcal mol−1) (Fig. 3b). This low barrier of enantiomerization explains why our attempts to separate the two enantiomers at room temperature using recycling chiral HPLC were unsuccessful.
 | ||
| Fig. 3 (a) Steric crowding of aryl groups in the gulf region (red) of 4(syn), with a 13.9 kcal mol−1 rotational barrier. (b) Activation barriers (in kcal mol−1) for the isomerization of 4(syn) in the gas phase [a], toluene [b], DCM [c], acetonitrile or DMSO [d] calculated for the model compounds with tert-butyl groups replaced by hydrogen atoms at the CPCM-ωB97X-D3/def2-TZVP//“gas phase”-ωB97X-D3/def2-SVP level of theory. All values of ΔG‡ in [kcal mol−1]. | ||
Photostability
When exposed to ambient air and light, acenes and acene-like systems commonly undergo [4+2] cycloaddition with singlet oxygen to form endoperoxide intermediates.38,39 In the case of 4(anti), this reactivity was also observed, consistent with its previously reported role as a singlet oxygen sensitizer.27 Time-dependent 1H NMR studies confirmed complete conversion of 4(anti) to an endoperoxide adduct after approximately 2 hours of exposure to ambient light and air (Fig. S8, SI). In contrast, 4(syn) exhibited remarkable resistance to this transformation under identical conditions. Even after 48 hours of continuous exposure to air and light, no discernible changes were observed in the 1H NMR spectrum of 4(syn) (Fig. S7, SI). This difference in reactivity may be attributed to the interlocked structure created by the two phenyl groups of 4(syn) leading to steric hindrance in the gulf region, which could inhibit the orbital alignment required for a concerted [4+2] cycloaddition with singlet oxygen. It is also possible that 4(syn) is a less efficient singlet oxygen sensitizer than 4(anti), owing to a weaker S0 → S1 transition (vide infra).Crystallography
Single crystals suitable for X-ray analysis were grown for the two compounds 4(syn) and 4(anti) by slow diffusion of n-hexane to a chloroform solution and subsequent slow crystallization under inert conditions (Fig. 4, CCDC numbers 2467542 and 2467541, Tables S1 and S2, SI, respectively). While 4(syn) displays an intramolecular twist of about 30° of the π-core due to the strong congestion of two sterically demanding aryl substituents, 4(anti) shows an almost planar π-surface that is shielded by the four aryl substituents. The congestion of two aryl substituents in 4(syn) results in a chiral structure, forming M- and P-enantiomers. These enantiomers were found to narcissistically separate into enantiomerically pure π-stacked columns, with close π–π distances as low as 3.7 Å (Fig. S1, SI). In stark contrast, the four large substituents in the rigid 4(anti) successfully prevent π–π stacking and induce a slip-stacked molecular arrangement with distances between the π-cores of over 7 Å. | ||
| Fig. 4 Molecular structures viewed from the top (top) and along the long axis (bottom) of (a) 4(syn) (P-enantiomer) and (b) 4(anti) found in the single crystal structures. | ||
Photophysical properties
UV-Vis absorption spectroscopy of the pentacene analogues 4(syn) and 4(anti) produced two unique absorbance profiles in solution (Fig. 5 and Fig. S2, SI). Both molecules, 4(syn) and 4(anti), show vibrationally resolved excitation spectra with maxima (λabs) of their A00 transitions at 538 and 539 nm in toluene, respectively. The S0–S2 transition of 4(syn) exhibits greater oscillator strength than its S0–S1 transition, as indicated by the significantly higher absorption observed below 450 nm (see Fig. S2a, SI). In accordance with the twisted C2 geometry of 4(syn) (vide supra), its A00 is less intense than A01 and the vibrational modes less resolved (A00/A01 = 0.76) than for the planar and rigid C2h symmetric chromophore 4(anti) (1.53). In contrast, A00 is most intense for both emission spectra with maxima (λem) at 589 nm (ΔṽStokes) for 4(syn) and 552 nm for 4(anti), resulting in Stokes shifts of 420 cm−1 and 370 cm−1, respectively. Time-dependent density functional theory (TD-DFT) calculations support these results (see SI, Computational details, Tables S3, S4 and Fig. S5, S6). | ||
| Fig. 5 Normalized excitation (solid line) as well as PL spectra (solid line, shaded areas) of 4(syn) and 4(anti) in toluene at RT under inert conditions. λex are indicated by arrows. | ||
Photoluminescence (PL) quantum yields (ΦPL) could be determined in DCM solution by absolute measurements with an integration sphere setup to be 46% for 4(syn) and 64% for 4(anti) (Table 1 and Fig. S3, SI). While 4(syn) exhibits a biexponential decay (τPL) in DCM with a short- and long-lived component of 9.6 and 39.3 ns respectively, 4(anti) on the other hand shows only a monoexponential as well as faster decay of 6.7 ns (Table 1 and Fig. S4, SI). Accordingly, the PL properties of the planar and rigid molecule 4(anti) render it the superior fluorophore with respect to 4(syn) for OLED applications. It should be noted that compound 4(anti) containing a 4-methoxyphenyl substituent shows a slight hypsochromic shift (9 nm) relative to the previously reported anti-dinaphthopentacene that contains a 4-hexyloxyphenyl substituent.27 The emission lifetime, quantum yield, and absorption wavelength are nearly unchanged between these two derivatives.
| λ abs [nm] | A 00/A01,exca [1] | λ em [nm] | A 00/A01,ema [1] | ΔṽStokesa [cm−1] | Φ PL [%] | τ PL(Rel.)bd [ns (%)] | |
|---|---|---|---|---|---|---|---|
| a Measured in toluene. b Measured in freeze-pumped DCM in inert gas cuvette. c Absolute quantum yield measured with integration sphere setup, error of ±2% due to material instability. d Fluorescence excited with EPL picosecond pulsed laser diode (λex = 505.8 nm). | |||||||
| 4 (syn) | 538 | 0.76 | 589 | 2.02 | 420 | 46 | 9.6 (52), 39.3 (48) |
| 4(anti) | 539 | 1.53 | 552 | 2.49 | 370 | 64 | 6.7 (100) |
OLED fabrication
The devices were fabricated by vacuum deposition using the following multilayer structure: ITO/MoO3/TAPC/TCTA/1 wt% 4(syn)/4(anti) in mCBP/PPF/TPBi/Liq/Al. For further details including the corresponding layer thicknesses, see Fig. 6a as well as the SI. The low doping percentage of 1 wt% was deliberately selected to suppress aggregation, thereby ensuring that both chromophores are exclusively surrounded by matrix material and emit as individual monomers. For both derivatives the electroluminescence (EL) spectra observed in the OLEDs are compared to the PL spectra in solution (Fig. 6b and c). Both emitters exhibit a slight decrease in their A00/A01 ratio (4(syn): 1.33, 4(anti): 1.83) as well as minor bathochromic shifts of their EL spectra by 350 cm−1 with respect to their PL in solution due to polarization effect in the solid matric mCBP. The small additional EL signal at 500 nm presumably originates from minor decomposition during thermal sublimation. | ||
| Fig. 6 (a) Device architecture used for all OLED devices with the electronic energy levels of employed materials. HOMO and LUMO levels determined using DFT calculations (B3LYP/6-31G*). Device electroluminescence (EL) (dark color) and solution PL spectra (bright color) of (b) 4(syn) and (c) 4(anti). Displayed in the insets are photographs of operating devices. The asterisks (*) mark EL from decomposition product originating from thermal sublimation. Average (d) JVL- and (e) CE/L- and EQE/L-curves of 4(syn) and 4(anti). The shaded areas represent the standard deviation of these values over the seven devices measured on one sample substrate. | ||
Despite the structural similarities between the emitting molecules, 4(anti) was found to be the superior material in terms of device performance. Compound 4(anti) exhibits higher device efficiencies and up to tenfold greater luminance compared to 4(syn), while also operating at lower driving voltages (Fig. 6d, e and Table 2). Where 4(anti) reaches peak external quantum efficiency (EQE) values of almost 1%, 4(syn) barely surpasses 0.3% with a 2 V higher turn-on voltage. These EQE values are in the same range as a previously reported acene derivatives.40,41 At 15 V, devices based on 4(anti) achieve luminance values exceeding 2000 cd m−2, whereas those based on 4(syn) reach only 250 cd m−2. The device characteristics support the photophysical studies, confirming that 4(anti) is the superior of the two acene analogues in terms of emission properties.
| 1 wt% of: | T Sub [°C] | V TO [V] | λ EL,max [nm] | EQEmax [%] | μ L,max [cd A−1] | L 15V [cd m−2] |
|---|---|---|---|---|---|---|
| a Temperature required to produce the 1 wt% mixed layer via vacuum deposition at pressures below 5 × 10−6 mbar. b Evaluated at L = 1 cd m−2; number of investigated devices: 4(syn): 24, 4(anti): 22. | ||||||
| 4(syn) | 315 | 7.8 ± 0.5 | 550 | 0.33 ± 0.06 | 1.0 ± 0.2 | 250 ± 80 |
| 4(anti) | 300 | 5.8 ± 0.5 | 560 | 0.8 ± 0.2 | 2.5 ± 0.5 | 2400 ± 600 |
Conclusion
Here we have presented two structural isomers of dinaphthopentacene synthesized by alkyne benzannulation. Crystal structures confirm that the anti and syn isomers have different packing patterns in the solid-state, with 4(anti) taking on a slip-stacked arrangement, and 4(syn) forming enantiopure π-stacked columns with very close π–π spacing. The novel isomer, syn-dinaphthopentacene 4(syn), proved to be significantly more resistant to light-induced endoperoxide formation than its anti-counterpart, likely due to the significant steric encumbrance in the central region of the structure where the endoperoxide would form. Both isomers exhibit similar absorption and fluorescence properties with fluorescence quantum yields of 46% for 4(syn) and 64% for 4(anti) in solution. Additionally, they were also sufficiently stable for vacuum sublimation and could be applied in OLEDs, where 4(anti) showed the significantly better device performance over its 4(syn) counterpart.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the SI. See DOI: https://doi.org/10.1039/d5tc02535d.CCDC 2467541 (4(anti)) and 2467542 (4(syn)) contain the supplementary crystallographic data for this paper.42a,b
Acknowledgements
The authors thank the Alexander von Humboldt Foundation for generously providing funding to Prof. Wesley Chalifoux through the Humboldt Research Fellowship program. This material is based upon work supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences Established Program to Stimulate Competitive Research under Award Number DE-SC0022178. We acknowledge DESY (Hamburg, Germany), a member of the Helmholtz Association HGF, for providing the experimental facilities at PETRA III (proposal I-20231007). We thank Dr Guillaume Pompidor for assistance in using the P11 beamline.References
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef PubMed.
- K. S. Novoselov, V. I. Fal′ko, L. Colombo, P. R. Gellert, M. G. Schwab and K. Kim, Nature, 2012, 490, 192–200 CrossRef PubMed.
- A. K. Geim, Science, 2009, 324, 1530–1534 CrossRef PubMed.
- X.-Y. Wang, A. Narita and K. Müllen, Nat. Rev. Chem., 2017, 2, 0100 CrossRef.
- L. Zhi and K. Müllen, J. Mater. Chem., 2008, 18, 1472–1484 RSC.
- Z. Liu, S. Fu, X. Liu, A. Narita, P. Samorì, M. Bonn and H. I. Wang, Adv. Sci., 2022, 9, 2106055 CrossRef.
- A. Mateo-Alonso, Chem. Soc. Rev., 2014, 43, 6311–6324 RSC.
- R. K. Dubey, M. Marongiu, S. Fu, G. Wen, M. Bonn, H. I. Wang, M. Melle-Franco and A. Mateo-Alonso, Chem, 2023, 9, 2983–2996 Search PubMed.
- J. Wu, W. Pisula and K. Müllen, Chem. Rev., 2007, 107, 718–747 CrossRef PubMed.
- J. Mei, Y. Diao, A. L. Appleton, L. Fang and Z. Bao, J. Am. Chem. Soc., 2013, 135, 6724–6746 CrossRef PubMed.
- M. Acik and Y. J. Chabal, Jpn. J. App. Phys., 2011, 50, 070101 CrossRef.
- J. Guo, C. Shi, Y. Zhen and W. Hu, Acc. Mater. Res., 2024, 5, 907–919 CrossRef.
- H. Shinokubo, Proc. Jpn. Acad. Ser. B Phys. Biol. Sci., 2014, 90, 1–11 CrossRef.
- T. Maekawa, H. Ueno, Y. Segawa, M. M. Haley and K. Itami, Chem. Sci., 2016, 7, 650 RSC.
- R. S. Jassas, E. U. Mughal, A. Sadiq, R. I. Alsantali, M. M. Al-Rooqi, N. Naeem, Z. Moussa and S. A. Ahmed, RSC Adv., 2021, 11, 32158–32202 RSC.
- X. Y. Wang, X. Yao and K. Müllen, Sci. China: Chem., 2019, 62, 1099–1144 CrossRef.
- A. Narita, X. Y. Wang, X. Feng and K. Müllen, Chem. Soc. Rev., 2015, 44, 6616–6643 RSC.
- M. B. Goldfinger, K. B. Crawford and T. M. Swager, J. Am. Chem. Soc., 1997, 119, 4578–4593 CrossRef.
- W. Yang, R. R. Kazemi, N. Karunathilake, V. J. Catalano, M. A. Alpuche-Aviles and W. A. Chalifoux, Org. Chem. Front., 2018, 5, 2288–2295 RSC.
- R. Bam, W. Yang, G. Longhi, S. Abbate, A. Lucotti, M. Tommasini, R. Franzini, C. Villani, V. J. Catalano, M. M. Olmstead and W. A. Chalifoux, Org. Lett., 2019, 21, 8652–8656 CrossRef PubMed.
- W. Yang, J. H. S. K. Monteiro, A. de Bettencourt-Dias and W. A. Chalifoux, Can. J. Chem., 2016, 95, 341–345 CrossRef.
- W. Yang, J. H. S. K. Monteiro, A. de Bettencourt-Dias, V. J. Catalano and W. A. Chalifoux, Angew. Chem., Int. Ed., 2016, 55, 10427–10430 CrossRef PubMed.
- R. Bam, W. Yang, G. Longhi, S. Abbate, A. Lucotti, M. Tommasini, R. Franzini, C. Villani, V. J. Catalano, M. M. Olmstead and W. A. Chalifoux, Angew. Chem., Int. Ed., 2024, 63, e202404849 CrossRef PubMed.
- W. Yang, G. Longhi, S. Abbate, A. Lucotti, M. Tommasini, C. Villani, V. J. Catalano, A. O. Lykhin, S. A. Varganov and W. A. Chalifoux, J. Am. Chem. Soc., 2017, 139, 13102–13109 CrossRef PubMed.
- S. P. George, J. Spengler, R. J. Malone, J. Krzoska, F. Würthner and W. A. Chalifoux, J. Org. Chem., 2024, 89, 5159–5163 CrossRef PubMed.
- W. Yang, A. Lucotti, M. Tommasini and W. A. Chalifoux, J. Am. Chem. Soc., 2016, 138, 9137–9144 CrossRef PubMed.
- W. Yang, J. H. S. K. Monteiro, A. de Bettencourt-Dias, V. J. Catalano and W. A. Chalifoux, Chem. – Eur. J., 2019, 25, 1441–1445 CrossRef.
- Y. Ren, J. Liu, A. Zheng, X. Dong and P. Wang, Adv. Sci., 2017, 4, 1700099 CrossRef.
- X.-J. Li, M. Li, H.-Y. Lu and C.-F. Chen, Org. Biomol. Chem., 2015, 13, 7628–7632 RSC.
- J. E. Anthony, J. S. Brooks, D. L. Eaton and S. R. Parkin, J. Am. Chem. Soc., 2001, 123, 9482–9483 CrossRef.
- K. J. Thorley and J. E. Anthony, Isr. J. Chem., 2014, 54, 642–649 CrossRef.
- M. Müller, L. Ahrens, V. Brosius, J. Freudenberg and U. H. F. Bunz, J. Mater. Chem. C, 2019, 7, 14011–14034 RSC.
- H. Meng, M. Bendikov, G. Mitchell, R. Helgeson, F. Wudl, Z. Bao, T. Siegrist, C. Kloc and C. H. Chen, Adv. Mater., 2003, 15, 1090–1093 CrossRef.
- M. Watanabe, K.-Y. Chen, Y. J. Chang and T. J. Chow, Acc. Chem. Res., 2013, 46, 1606–1615 CrossRef PubMed.
- J. E. Anthony, Chem. Rev., 2006, 106, 5028–5048 CrossRef PubMed.
- B. P. Mathew and M. R. Kuram, Inorg. Chim. Acta, 2019, 490, 112–129 CrossRef.
- R. Ozawa, K. Yoza and K. Kobayashi, Chem. Lett., 2011, 40, 941–943 CrossRef.
- R. Mondal, B. K. Shah and D. C. Neckers, J. Am. Chem. Soc., 2006, 128, 9612–9613 CrossRef PubMed.
- R. Mondal, R. M. Adhikari, B. K. Shah and D. C. Neckers, Org. Lett., 2007, 9, 2505–2508 CrossRef PubMed.
- J. Li, Y. Zhao, J. Lu, G. Li, J. Zhang, Y. Zhao, X. Sun and Q. Zhang, J. Org. Chem., 2015, 80, 109–113 CrossRef.
- R. Sato, K. Chen, T. Yasuda, T. Kanbara and J. Kuwabara, Org. Chem. Front., 2023, 10, 2955–2962 RSC.
- (a) H. V. Anderson, F. Brust, M. Fizer, J. Spengler, Y. Wagenhäuser, M. Stolte, A. de Bettencourt-Dias, S. A. Varganov, F. Würthner and W. A. Chalifoux, CCDC 2467541: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2ntp39; (b) H. V. Anderson, F. Brust, M. Fizer, J. Spengler, Y. Wagenhäuser, M. Stolte, A. de Bettencourt-Dias, S. A. Varganov, F. Würthner and W. A. Chalifoux, CCDC 2467542: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2ntp4b.
| This journal is © The Royal Society of Chemistry 2025 |