
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExtension of the π-conjugated core of methylchalcogenolated polycyclic aromatic hydrocarbons: synthesis and characterization of 1,4,7,10-tetrakis(methylthio)- and tetramethoxy-coronene†
Prasanta
Pal‡
 ab,
Kirill
Bulgarevich‡
a,
Ryota
Hanaki
c,
Kohsuke
Kawabata
ac and
Kazuo
Takimiya
*abc
ab,
Kirill
Bulgarevich‡
a,
Ryota
Hanaki
c,
Kohsuke
Kawabata
ac and
Kazuo
Takimiya
*abc
aRIKEN Center for Emergent Matter Science (CEMS), 2-1 Hirosawa, Wako, Saitama 351-0198, Japan. E-mail: takimiya@riken.jp
bTohoku University Advanced Institute for Materials Research (AIMR), 2-1-1 Katahira, Aoba-ku, Sendai, Miyagi 980-8577, Japan
cDepartment of Chemistry, Graduate School of Science, Tohoku University, 6-3 Aoba, Aramaki, Aoba-ku, Sendai, Miyagi 980-8578, Japan
First published on 21st July 2025
Abstract
Controlling the crystal structures of polycyclic aromatic hydrocarbons (PAHs) by regioselective methyl chalcogenolation is an effective strategy for realizing superior molecular semiconductors showing ultrahigh mobility, as exemplified by methylthiolated pyrene and peropyrene. Following the strategy, we designed and synthesized 1,4,7,10-tetrakis(methylthio)coronene (MT-coronene) and 1,4,7,10-tetramethoxycoronene (MO-coronene) as potential candidates for high-performance molecular semiconductors. Since the coronene core is highly symmetric, the regioselective functionalization at the 1, 4, 7, and 10-positions seemed to be challenging, and thus, we tested two strategies for constructing such regio-selectively functionalized coronene derivatives: one was the direct functionalization of the parent coronene via the iridium-catalyzed borylation reaction, and the other was the stepwise construction of the coronene core with functionalized naphthalene derivatives. Interestingly, the former was suitable for the synthesis of MT-coronene, whereas the latter was suitable for MO-coronene. The crystal structures of MT- and MO-coronene were significantly different from the γ-structure of their parent and were classified into the brickwork and the sandwich herringbone structure, respectively. In accordance with the brickwork crystal structures with isotropic but small intermolecular HOMO overlaps, the MT-coronene-based single-crystal field-effect transistors (SC-FETs) showed decent transistor responses with a carrier mobility of up to 0.5 cm2 V−1 s−1. On the other hand, the SC-FETs of MO-coronene, the solid-state electronic structure of which was zero-dimensional due to the sandwich herringbone structure, were far inferior to those of MT-coronene. Based on the crystal structures and theoretical calculations on MT- and MO-coronene, we analyzed the tendency of the intermolecular interactions and intermolecular HOMO overlaps in the solid state, which explains the performances of the coronene system as molecular semiconductors. Furthermore, we compared the solid-state structures of a series of methylthiolated PAHs, pyrene, perylene, peropyrene, and coronene, to determine the differences in their performances as molecular semiconductors, which gave us new insights into the relationship between the molecular structure, packing, and electronic structure in the solid state, providing perspectives for superior molecular semiconductors.
Introduction
The control of molecular arrangement in the solid state based on molecular design has been a challenging issue in molecular solid-related fields, including pharmaceuticals and organic electronics.1,2 The optical3 and carrier transport properties4 of molecular electronic materials are affected not only by the molecular electronic structures but also by molecular arrangements in the solid state. In particular, carrier transport properties of molecular electronic materials are crucially influenced by the solid-state structure, i.e., the crystal structure of molecular materials. This is because the transport properties are the direct outcome of solid-state electronic structure, namely, the dimensionality and the extent of molecular orbital overlap.5 Thus, reliable and practical measures to control and predict the solid-state structures of given molecules have been eagerly pursued.6–10We have recently found that regio-selective methylchalcogenolation of arenes and heteroarenes is a powerful tool to control the crystal structures of molecular semiconductors.11 For example, the crystal structures of linear acenes12,13 and heteroacenes14–16 changed from the herringbone structures of the parent compounds to the pitched π-stacking structures by peri-methylchalcogenolation. On the other hand, the end-methylthiolation on acenes gave the brickwork crystal structures.17 Similar brickwork crystal structures were also realized by four-fold methylthiolation on a series of peri-condensed polycyclic aromatic hydrocarbons (PAHs), such as pyrene,18,19 perylene,20 and peropyrene (Fig. 1),21 and among them, the methylthiolated pyrene (MT-pyrene) and peropyrene (MT-peropyrene) gave single-crystal field-effect transistors (SC-FETs) showing ultrahigh mobility (∼30 cm2 V−1 s−1) and band-like transport behaviors. The characteristic feature of the brickwork crystal structures of these PAH derivatives is that they are mostly classified into the triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group with Z = 1, which means that all the molecules in the crystal structures are related only by the translation symmetry operation. This led us to simulate molecular packing structures by successively calculating bimolecular intermolecular interactions to find energetically stable mutual positions of molecular clusters in a three-dimensional manner. The resulting protocol for simulating the brickwork crystal structures, which we call “in silico crystallization”, turned out to be a useful tool for finding promising molecular semiconductors with a brickwork crystal structure21 and simulating polymorph candidates by introducing conditional branches for several typical patterns of brickwork-related crystal structures.22
space group with Z = 1, which means that all the molecules in the crystal structures are related only by the translation symmetry operation. This led us to simulate molecular packing structures by successively calculating bimolecular intermolecular interactions to find energetically stable mutual positions of molecular clusters in a three-dimensional manner. The resulting protocol for simulating the brickwork crystal structures, which we call “in silico crystallization”, turned out to be a useful tool for finding promising molecular semiconductors with a brickwork crystal structure21 and simulating polymorph candidates by introducing conditional branches for several typical patterns of brickwork-related crystal structures.22
 | ||
| Fig. 1 Molecular structures of methylchalcogenated PAHs. | ||
To further extend the chemical space for molecular semiconductors with brickwork crystal structures, we applied the in silico crystallization protocol to coronene derivatives such as 1,4,7,10-tetrakis(methylthio)coronene (MT-coronene) and 1,4,7,10-tetramethoxycoronene (MO-coronene) (Fig. 1). The simulated crystal structures and, thus, the estimated electronic structure in the solid state suggested that these methylchalcogenolated coronenes are promising molecular semiconductors (Fig. S1†). We therefore chose MT-coronene and MO-coronene as the synthetic targets to explore the potential of the coronene system as a molecular semiconductor by elucidating their crystal structures and evaluating their SC-FETs.
Results and discussion
Regio-selective functionalization of coronene
Coronene is a prototypical two-dimensionally extended PAH, and all the peripheral positions are chemically equivalent; thus, regio-selective functionalization seems to be a difficult task. Yet, there have been several references that claim regioselective bromination of the parent coronene, e.g., the successful synthesis of 1,4,7,10-tetrabromocoroene23 or 1,4,5,8,9,12-hexabromocoronene.24 All our attempts at direct bromination on coronene, according to these references, however, failed to afford such regio-selectively brominated coronene derivatives. On the other hand, the iridium-catalyzed direct borylation reaction25 on coronene was reported to give 1,4,7,10-tetrakis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)coronene (1, Scheme 1) in low yields (10–20%).26 Later, 1 was utilized in the Suzuki–Miyaura cross-coupling reaction to develop a four-perylene diimide-connected molecule as an electron acceptor for organic solar cell applications.27 In addition to the utility in the Suzuki–Miyaura cross-coupling reactions, aryl boronate ester is utilized as a precursor for other functional groups, such as halogen,28 hydroxy,29 and sulfide groups.30,31 In fact, 2,5,8,11-tetrakis(methylthio)perylene (MT-perylene, Fig. 1)20 was successfully synthesized from the corresponding pinacol boronate ester (Bpin) derivative.32 For these reasons, 1 seemed to be a promising precursor for MT-coronene and MO-coronene, and thus, we examined the conversion of the four Bpin groups of 1 into methylthio and methoxy groups (Scheme 1). By employing the copper-catalyzed deborylthiolation reaction,30,31 the Bpin groups of 1 were successfully converted into methylthio groups to afford MT-coronene, though the isolated yield after thorough purification by repetitive vacuum sublimation was around 10%. The molecular structure of MT-coronene was fully characterized by 1H NMR, high-resolution MS spectra, and single-crystal X-ray analysis (vide infra).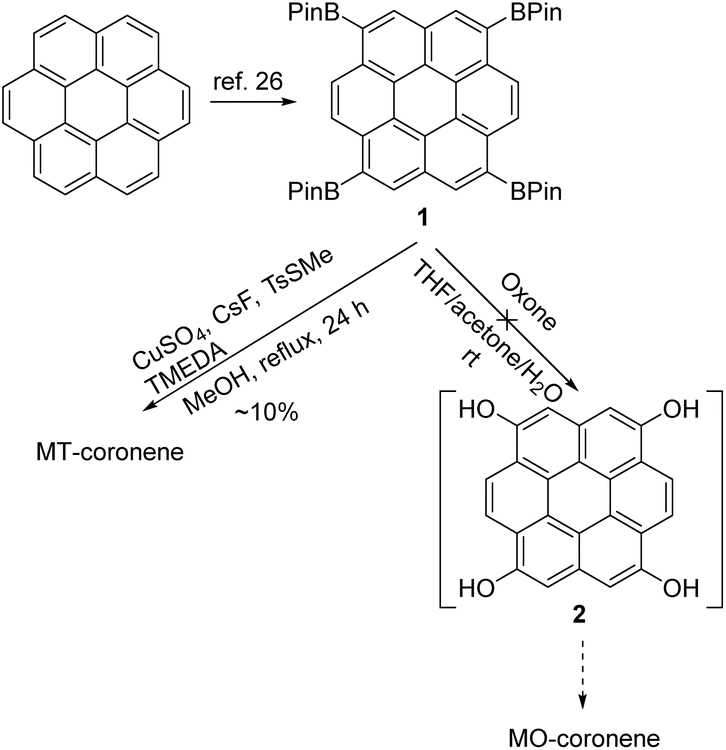 | ||
| Scheme 1 Synthesis routes of MT-coronene and MO-coronene from tetrakis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)coronene (1). | ||
In parallel, attempts have been made to convert 1 into MO-coronene via the corresponding tetrahydroxy coronene (2) followed by methylation (Scheme 1).33 However, the poor solubility of the intermediates, such as di- and tri-hydroxy compounds, likely hampered the complete conversion of all four Bpin groups into hydroxy groups, and the desired MO-coronene was not obtained (see the ESI† for details). In contrast, regio-selectively methoxylated coronene derivatives, such as 1,2-dimethoxy, 1,2,5,6-tetramethoxy, and 1,2,5,6,9,10-hexamethoxycoronene, were synthesized via the formation of the coronene core from appropriately methoxylated benzene derivatives.34 Inspired by this strategy, we designed the synthesis of MO-coronene from a dimethoxylated naphthalene derivative (Scheme 2). The key steps in the synthesis were the photochemical ring-closing reaction of a dinaphthylethene derivative to afford a benzo[ghi]perylene derivative35,36 and the construction of the final ring at its bay position via the Diels–Alder reaction followed by a decarboxylation reaction.37 Following this strategy, we carried out the synthesis from 2,7-dimethoxy-3-naphthaldehyde (3),38 which was readily converted into (E)-1,2-bis(3,6-dimethoxynaphthalen-2-yl)ethene (4) by the action of low-valent titanium.39 The photocyclization reaction of 4 gave tetramethoxybenzo[ghi]perylene (5) in a good isolated yield. The construction of the coronene core was accomplished via the Diels–Alder reaction in the presence of chloranil, followed by the decarboxylation reaction. Although the isolated yield of MO-coronene at the final step was not high, the present synthesis method is valuable since this can be an alternative method for the synthesis of regioselectively alkoxylated coronene at the 1-, 4-, 7-, and 10-positions. The molecular structures of MO-coronene and its precursor 5 were fully characterized by 1H NMR and high-resolution MS spectra (see the ESI†). Also, the single-crystal X-ray analysis explicitly confirmed their molecular and crystal structures (Fig. 2, S5, and Tables S2–S6†).
 | ||
| Scheme 2 Stepwise synthesis of MO-coronene from 2,7-dimethoxy-3-naphthaldehyde (3). | ||
 | ||
| Fig. 2 Crystal structures of the parent coronene (a), MT-coronene (b and e), and MO-coronene (c). Hirshfeld surfaces mapped with de (distance external to the surface) for coronene (d) and MO-coronene (f). White dotted circles depicted on the Hirshfeld surfaces represent the CH–π edge-to-face intermolecular contacts (d and f, left). Assignment of hydrogen atoms involved in the intermolecular contacts: the red ones are involved in the CH–π edge-to-face intermolecular contacts, whereas the blue ones are shielded by the methoxy groups (d and f, right). | ||
It is worth mentioning that a similar stepwise strategy was also tested to synthesize MT-coronene, starting from 3,6-dibromonaphthalen-2-ol (6) (Scheme S1†).40 The synthesis from 6 was performed to obtain (E)-1,2-bis(3,6-bis(methylthio)naphthalen-2-yl)ethene (11), which was then converted into the methylthio derivative of benzo[ghi]perylene (12) by a photochemical ring-closing reaction similar to the synthesis of 5. Despite our intensive efforts, the conversion of 12 to MT-coronene via the Diels–Alder reaction, followed by a decarboxylation reaction, was not successful. The reason for the failure of the conversion in the case of MT-coronene was not clear, but we speculate the substitution effects on the reactivity of benzo[ghi]perylene: in the case of MO-coronene, the high electron-donating ability of the methoxy groups enhanced the reactivity of 5 as the diene in the Diels–Alder reaction, resulting in the formation of the adduct, whereas it is likely that the methylthio groups of 12 with the inferior electron donating nature to that of the methoxy group could not facilitate the reactivity of 12 as the diene in the Diels–Alder reaction (Fig. S2†).
Crystal structures of MT-coronene and MO-coronene
Desiraju et al. systematically discussed the relationship between the molecular and crystal structures of PAHs and pointed out that reducing the number of hydrogen atoms on PAH cores relative to the carbon atoms changes the crystal structures from the herringbone (more hydrogen atoms) via sandwich herringbone to γ-structure (fewer hydrogen atoms).41 The key concept in this scheme is that the molecular factors that contribute to either the CH–π edge-to-face or the face-to-face π-stacking contacts, which are defining factors of the crystal structure of PAH molecules, originate in the molecular structure. In these relations, our previous results on the crystal structure change from the sandwich herringbone structures of pyrene,18,19 perylene,20 and peropyrene21 into brickwork structures by methylchalcogenolation can be explained as follows: the methylchalcogeno groups introduced on the PAH cores shield hydrogen atoms available for intermolecular CH–π edge-to-face contacts, rendering the resulting derivatives to interact intermolecularly through the face-to-face π-stacking contacts only, resulting in the brickwork crystal structure with no CH–π edge-to-face intermolecular contacts.The parent coronene, which crystallizes into the γ-structure (Fig. 2a), still keeps the core hydrogen atoms that can contribute to the CH–π intermolecular contacts (Fig. 2d). Upon methylchalcogenolation that shields the core hydrogen atoms, it is natural to consider that the crystal structures change into brickwork structures. In fact, the crystal structure of MT-coronene was, as expected, a typical brickwork structure with P space group (Fig. 2b), as observed for MX-pyrenes (X = O, S, Se), MT-peropyrene, and MT-perylene.11 However, the packing motif in MT-coronene is notably different from that in MX-pyrenes and MT-peropyrene: the intramolecular spacing between the two methylthio groups is large enough to accommodate one methyl moiety of the adjacent MT-coronene molecule, resulting in a characteristic zig-zag packing (Fig. 2e). This is similar to the packing patterns of MT-perylene and 2,3,6,7-tetrakis(methylthio)naphthalene (Fig. S6†),17 which have similar spacings (two benzene rings) between the methylthio groups.
In sharp contrast, the crystal structure of MO-coronene was classified as a sandwich herringbone structure with the P21/n space group (Z = 4) (Fig. 2c). This was totally unexpected, as substituting hydrogen atoms on the PAH core by methylchalcogeno groups always reduced the number of hydrogen atoms available for intermolecular CH–π edge-to-face contacts, as discussed above. However, we found that the present methoxylation on coronene practically increases the hydrogen atoms that participate in the intermolecular CH–π edge-to-face contacts. The Hirshfeld surface mapped with de (distance external to the surface) apparently demonstrated that the hydrogen atoms contributing to the CH–π edge-to-face contacts are three for MO-coronene (Fig. 2f) and two for the parent coronene (Fig. 2d). This is likely due to the size of the methoxy groups, which are not large enough to shield the peri-hydrogen atoms (2-, 3-, 8-, and 9-positions) from the π-faces of the adjacent molecules. Furthermore, one of the methyl hydrogen atoms in the methoxy groups participates in the CH–π edge-to-face contacts, making the sandwich herringbone structure favorable (Fig. 2c and f). Similar cooperative participation of the methyl hydrogen atoms, together with aromatic core hydrogen atoms, in intermolecular CH–π interactions, was already observed in the case of pitched π-stacking structures of methoxylated and methylthiolated acenes and acenedithiophenes at the end part of the molecules.12,13,15,16 As the methoxy groups have a more compact size and tend to take a co-planar structure with π-cores,42 they could be useful for controlling crystal structures by regio-selective methoxylation. Particularly, the appearance of the sandwich herringbone structure in MO-coronene implies that edge-to-face CH–π interactions can be induced by the methoxy groups, which widens the utility of methoxy groups for controlling crystal structure.
These experimental crystal structures of MT-coronene and MO-coronene (Fig. 2) did not match the simulated crystal structures by in silico crystallization (Fig. S1†). In the case of MO-coronene, the participation of the methyl hydrogen atoms in the CH–π intermolecular interaction that likely induces the formation of the sandwich herringbone structure was initially unexpected, and, thus, this is the direct cause for the difference between the simulated and experimental crystal structure. On the other hand, the failure to simulate the MT-coronene crystal structure by in silico crystallization, despite its crystallizing into a brickwork structure, is related to the formation of voids in the simulated crystal structures, which cannot be adequately accounted for. This is a limitation of the current in silico crystallization protocol, and we are now working to improve it.
Solid-state electronic structures and SC-FETs of MT- and MO-coronene
The crystal structure of MT-coronene was a two-dimensional brickwork structure with a large overlap of π-faces in one π-stacking direction, and moderate overlap in the other π-stacking direction (Fig. 2b). Based on the crystal structure, we calculated intermolecular orbital overlaps (transfer integrals, ts) in the brickwork layer: to our disappointment, the values were not high: 20.9 and 20.4 meV for the two π-stacking directions (Fig. 3a).43 These ts were much lower than that in the π-stacking of the parent coronene (170 meV).44 On the other hand, the ts in the two π-stacking directions were similar to each other, indicating high two-dimensionality in the solid-state electronic structure, despite its one-dimensional-like π-stacking structure (Fig. 2b). The carrier mobility simulated based on the hopping model (μhop) was 0.5 cm2 V−1 s−1 (Fig. 3a),45 which is similar to that of MT-perylene (1.0 cm2 V−1 s−1),20 but much lower than that of MT-pyrene (4.3 cm2 V−1 s−1).18 | ||
| Fig. 3 Evaluation of MT-coronene as a semiconducting material. (a) Calculated ts and estimated mobility based on the hopping model. (b) Transfer and (c) output characteristics of SC-FET of MT-coronene measured in air. (d) Transfer characteristics measured at various temperatures in a vacuum. (e) Temperature dependence of carrier mobility. | ||
The intrinsic carrier transport properties of MT-coronene were experimentally evaluated using SC-FETs (Fig. 3b), fabricated with thin-plate single crystals of MT-coronene grown by the physical vapor transport method.46 The SC-FETs showed typical p-channel characteristics, and the hole mobilities extracted from the saturated regime (experimental mobility, μexp) were 0.5 cm2 V−1 s−1, in good agreement with the μhop. The non-ideal carrier injection in the OFET characteristics could be attributed to the relatively large ionization energy of MT-coronene (5.3 eV) measured by photoemission spectroscopy (Fig. S3†). The μexp of MT-coronene was similar to that of MT-perylene (0.3 cm2 V−1 s−1),20 but much lower than that of MT-pyrene and MT-peropyrene (30 cm2 V−1 s−1).18,21 The good agreement between the μhop and μexp of MT-coronene implies that the carrier transport mechanism could obey the hopping model. To further confirm this, we carried out the measurement of the temperature dependence of the carrier mobility (Fig. 3d). Interestingly, the carrier mobility of MT-coronene showed almost no temperature dependence in the range of 230–300 K. Thus, the carrier transport in MT-coronene is likely on the boundary between band-like and hopping transport.
In contrast, the electronic structure of MO-coronene was significantly different from that of MT-coronene. The intermolecular HOMO overlaps in the crystal structure of MO-coronene are typical for sandwich herringbone structures; very large ts greater than 160 meV is calculated for the π-stacking dimer, whereas negligible ts are calculated for between dimers (up to 4 meV, Fig. S7†). Thus, the solid-state electronic structure of MO-coronene is described as zero-dimensional, indicating almost no possibility for efficient carrier transport.47 In fact, the SC-FETs of MO-coronene showed the mobility of ca. 10−3 cm2 V−1 s−1 (Fig. S7†), which is consistent with the expected electronic structure based on the experimental crystal structure. No other polymorphs were expected to exist for both MO- and MT-coronene based on the out-of-plane XRD measurement of the SCs (Fig. S4†).
The crystal structure of the parent coronene with the γ-structure is characterized by a one-dimensional electronic structure with a large HOMO overlap in the π-stacking columnar direction (t = 170 meV).44 Thus, it can be said that the present methoxylation on coronene changes the crystal structure and solid-state electronic structure from one-dimensional (parent coronene) to zero-dimensional (MO-coronene). This relationship is totally opposite to those of a range of methylthiolated PAHs such as pyrene, perylene, and propyrene, where the sandwich herringbone structure of parents (zero-dimensional) changes into two-dimensional brickwork crystal structures.11
Solid-state electronic structures of a series of methylthiolated PAHs
With MT-coronene, one of the largest methylthiolated PAHs, in hand, we are now able to systematically compare the solid-state packing structure and electronic structures of a series of methylthiolated peri-condensed PAHs, namely, MT-pyrene, MT-perylene, MT-peropyrene, and MT-coronene. We thought that this could also explain the reasons for the poor intermolecular orbital overlaps and, thus, the carrier mobility in MT-coronene compared to that in MT-pyrene, MS-pyrene, and MT-peropyrene. For this purpose, we inspected the π-stacking structures of the methylchalcogenolated PAH series through the quantitative comparison of the relative positions of molecules in the brickwork layer. Fig. 4 shows the way of describing the experimental crystal structures quantitatively: one molecule in the crystal structure is placed at the origin on the xy-plane of the Cartesian system so that the molecular long axis aligns with the x-axis,21,22 and the center of gravity of two π-stacking molecules are denoted as (x1, y1, z1) and (x2, y2, z2) from the origin, respectively, which are summarized in Table 1. | ||
| Fig. 4 Definition of mutual positions of molecules in the solid state. The first molecule (black in colour) is placed at the origin on the xy-plane of the Cartesian system so that the molecular long axis aligns with the x-axis, and the positions of the π-stacking molecules are defined by the coordinates of the center of gravity of the molecules. The y-axis projection (top) and the z-axis projection (bottom). | ||
| Compound | x 1 | y 1 | z 1 | x 2 | y 2 | z 2 | μ hop | μ exp | Ref. | CCDC no. |
|---|---|---|---|---|---|---|---|---|---|---|
| a Å. b cm2 V−1 s−1. | ||||||||||
| MT-coronene | 3.62 | −1.47 | 3.48 | −9.10 | 2.36 | 3.46 | 0.5 | 0.5 | This work | 2446627 |
| MT-pyrene | 5.51 | 0.12 | 3.48 | −8.73 | 0.28 | 3.70 | 4.3 | 30 | 18 | 2076078 |
| MS-pyrene | 5.55 | −0.33 | 3.52 | −8.77 | −2.05 | 3.59 | 7.2 | 7.3 | 19 | 2302656 |
| MT-perylene | 3.33 | −1.08 | 3.38 | −9.46 | 2.67 | 3.49 | 1.0 | 0.2 | 20 | 2269569 |
| MT-peropyrene | 5.72 | 0.02 | 3.43 | −12.79 | 0.40 | 3.70 | 9.5 | 30 | 21 | 2256597 |
Table 1 shows a trend of structures with high μhop (>4 cm2 V−1 s−1), all of which have small |y1| (<0.4 Å) and x1 being around 5.5 Å (e.g., MT-pyrene, MS-pyrene, and MT-peropyrene). Generally, μhop is dominated by the largest ts in the system, and the position at around (x1, y1) = (5.5, 0) Å seems to realize efficient intermolecular orbital overlap in the methylchalcogenolated PAH series. Note that the values of z1 and z2 are not critically important for μhop. Another trend is that the structures of high μexp (∼30 cm2 V−1 s−1) have small |y2| (<0.4 Å) in addition to the above trend for x1 and y1. The small y2 is likely important for realizing large ts in the second π-stacking directions, which in turn results in the high two-dimensionality of the electronic structure. The high two-dimensionality is crucial for realizing the band-like transport in the solid state, and in fact, the band-like transport was experimentally confirmed for MT-pyrene and MT-peropyrene brickwork structures. In sharp contrast, the (|x1|, |y1|) of MT-coronene and MT-pelyrene are (3.6, 1.5) and (3.3, 1.1) Å, respectively, indicating that these are out of the first trend, and the |y2|s are larger than 2 Å, also indicating falling off from the second trend. These analyses clearly indicate that the mutual molecular positions in the brickwork layer of MT-coronene are not optimal for efficient carrier transport.
To further understand the relationship between the π-stacking configurations in methylchalcogenolated PAHs and the intermolecular orbital overlaps, we created two-dimensional transfer integral maps (2D t-maps) for these molecules by systematically calculating ts of π-stacking dimers with various mutual positions: for each molecule in the series placed on the origin as above, a copy of the molecule was placed at (0.0, 0.0, 3.5) Å. The second molecule was then moved on a grid with steps of 0.4 Å in the x and y directions while computing ts between the two molecules. In other words, we calculated ts for artificial π-stacking dimers with (x1, y1, z1) = (0.4 × n, 0.4 × m, 3.5) Å, where n and m are integers. Fig. 5 shows the results of this computation for MT-coronene, MT-pyrene, MS-pyrene, MT-perylene, and MT-peropyrene. We can observe that the ts with different signs, depicted as blue and orange circles, are distributed almost parallel to the x-axis (molecular long axis) at approximately 2.2 Å intervals in the y-axis direction of all molecules, except for MT-perylene (vide infra). For MT-coronene, reflecting its two-dimensionally extended π-core structure, an additional orange stripe exists at y = ca. 9 Å, implying its potential, even in the case of a large offset along the y-axis direction, for efficient intermolecular HOMO overlap. The above intervals (2.2 Å) roughly correspond to the spacing of the nodes in the distribution of HOMO of these molecules (Fig. 5, bottom). When the π-stacking molecule shifts in the y-axis direction by such spacing, the relative positions of HOMOs in the molecules shift, resulting in the change of sign of ts. When the shift is about half of the node spacing, the intermolecular HOMO overlap becomes very poor, resulting in regions where ts are almost zero.
 | ||
| Fig. 5 Distribution of transfer integrals for π-stacking of (a) MT-coronene, (b) MT-pyrene, (c) MS-pyrene, (d) MT-perylene, (e) MT-peropyrene. Axes represent the relative displacement of two molecules in Å. The orange and blue circles correspond to different signs of ts, while the radii of the circles are proportional to the magnitude of ts. Only the plots in the first quadrant are depicted, and the rest are omitted due to symmetry. Red crosses represent (|x1|, |y1|), (|x2|, |y2|) of the experimental brickwork structures. The distributions of HOMO of the corresponding molecules are shown at the bottom. All calculations were done using Gaussian 16 at B3LYP/6-31g(d) level. | ||
The distribution of ts for MT-perylene is markedly different from that for the other molecules in the series, which reflects the difference in the distribution of HOMO (Fig. 5d). In MT-perylene, the nodes of HOMO are aligned both in the molecular long- and short-axis direction, and thus, the phases switch both in the x- and y-axis directions. This results in a characteristic checkerboard-like distribution pattern of ts in MT-perylene. This implies that the molecular mutual positions with relatively high ts with the same sign in MT-perylene are very limited, which means that the potential of perylene core as a molecular semiconductor is not high.
On the other hand, for MT-coronene, we can see that if the y1 and y2 could be restricted to almost 0, ts greater than 100 meV could be realized (Fig. 5a), like in experimental structures of MT-pyrene, MS-pyrene, and MT-peropyrene (Fig. 5b, c, and 5e). In fact, structures with small y1 and y2 were among the simulated polymorph candidates by in silico crystallization for MT-coronene (Fig. S1†). Moreover, the regions of very large ts (>100 meV) also exist in y1 and y2 of ca. 2.2 and ca. 4.5 Å. However, the experimental crystal structure did not make use of these advantages of MT-coronene. In the experimental structure, the y1 = −1.47 Å lands almost in between the orange and blue stripes, resulting in low ts of ca. −20 meV. The small x1 = 3.62 Å “pushes” the second π-stacking molecule relatively far in the x-axis direction (x2 = −9.10), where the ts start to diminish. Thus, although the y2 = 2.36 Å is close to ideal (ca. 2.2 Å), the ts of the second π-stacking molecule also becomes moderately low (−21 meV) and of very similar value to the first π-stacking one. These analyses clearly show that further molecular design is needed to land the molecules in the π-stacking configuration at the “hot spots” in the 2D t-map to fully extract the potential of the PAH cores.
To do so, the key is to focus on the intermolecular overlap of the HOMOs, not just the overlap area of the π-planes (see Fig. S8†). In this viewpoint, the isotropically extended π-core, i.e., coronene, tends to largely overlap with the PAH core parts by strong face-to-face intermolecular interaction driven by the dispersion force. On the other hand, peropyrene, a similarly extended PAH consisting of the same seven benzene rings, with a rectangular shape, affords π-stacking structures in two directions, allowing the effective intermolecular overlap of HOMOs. This means that the shape of the PAH cores is a key parameter for the packing and electronic structures in the solid state. In addition, the relative size of the π-core and substituents, methylthio groups in the present case, is likely another key parameter, as can be deduced from the comparison of MT-coronene and MT-pyrene. The molecular structure of MT-coronene can be viewed as an extended structure of MT-pyrene along the y-axis direction (Fig. 5), which implies that the intermolecular interaction in this direction can be affected, leading to offsets in the y-axis direction in MT-coronene.48
Conclusions
In summary, we have synthesized and characterized MT-coronene and MO-coronene as molecular semiconductors with largely π-extended PAH cores modified with methylchalcogeno groups. For their syntheses, we examined two different synthetic routes: a short-step four-fold methylchalcogenolation of coronene via the corresponding 1,4,7,10-borylated coronene (1) and a stepwise construction of the coronene core from methoxylated or methylthiolated naphthalene derivatives. Interestingly, these two routes were found to be complementary: the former route was suitable for the synthesis of MT-coronene, while the latter was for MO-coronene. The crystal structure of MT-coronene, as we expected, was revealed to be a brickwork structure, similar to other methylthiolated PAHs, such as MT-pyrene, MT-perylene, and MT-peropyrene. However, intermolecular HOMO overlaps calculated based on the crystal structure were unexpectedly small, only up to ca. 20 meV, even with such a large π-conjugated core as coronene. According to the small orbital overlap, the carrier mobility evaluated by SC-FET was up to 0.5 cm2 V−1 s−1, which was almost comparable to that of methylthiolated perylene, but was inferior to those of the pyrene and peropyrene counterparts. Further unexpected result was the crystal structure of MO-coronene, which was classified into a sandwich herringbone structure. In its crystal structure, the methyl hydrogen atoms in the methoxy group contribute to forming edge-to-face intermolecular interactions together with the aromatic hydrogen atoms in the coronene core, which implies that the methoxy groups are not large enough to shield the aromatic hydrogen atoms in such a largely π-extended core like coronene.Furthermore, we compared the solid-state electronic structures of a series of methylthiolated peri-condensed PAHs, pyrene, perylene, peropyrene, and coronene, to understand the differences in the performances as molecular semiconductors, which gave us new insights into the relationship between the molecular structures and the packing and thus electronic structure in the solid state, showing viewpoints for designing superior molecular semiconductors: the key is not to widen the overlap area of π-planes by extension of π-conjugation of molecules but to design the effective intermolecular overlap of the frontier orbitals, where the position and symmetry of node and antinode of the frontier molecular orbitals should be taken into account. In addition, the mutual positions of molecules in the solid state shall be determined as the energetically stable positions, in other words, the optimal positions defined by the intermolecular forces. Such positions are not always the optimal positions for efficient orbital overlap. The present comparisons of methylthiolated PAHs clearly showed that even with similar molecular structures, the areas and positions of “hot spots” are different and not easily predictable from the molecular structure itself. Thus, the key lesson we derived from the comparison of the series of methylthiolated PAHs is that we should select molecules whose energetically favorable positions in the solid state provide the optimal position for intermolecular orbital overlap. In this sense, simulating crystal structures will be a key technology in the development of molecular semiconductors.
Data availability
Experimental and characterization data, including crystallographic data and NMR and HRMS spectra. The data supporting this article have been included as part of the ESI.†Author contributions
R. H. synthesized 5, 12, and MO-coronene, and P. P. synthesized MT-coronene. Characterizations of the compounds were performed by R. H., P. P. and K. K. All the X-ray crystallographic analyses, theoretical calculations and the fabrication/evaluation of SC-FETs were performed by K. B. K. T. designed the project and target compounds, supervised the experiments, and conducted data analyses. K. T. and K. B. directed the project. The draft manuscript was written by K. T., K. K., K. B. and P. P., and all the authors finalized the manuscript through proofreading. All authors approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by JSPS KAKENHI Grant Numbers JP20H05865, JP23H00307, and JP24KK0113. We gratefully acknowledge the Center for Computational Materials Science, Institute for Materials Research, Tohoku University for the use of MASAMUNE-IMR (MAterials science Supercomputing system for Advanced MUlti-scale simulations towards NExt-generation-Institute for Materials Research) and the Supercomputer System in the Advanced Center for Computing and Communication (ACCC) of RIKEN for theoretical calculations. Elemental analyses were carried out at the Molecular Structure Characterization Unit, RIKEN Center for Sustainable Resource Science (CSRS).Notes and references
- C. C. Pantelides, C. S. Adjiman and A. V. Kazantsev, in Prediction and Calculation of Crystal Structures: Methods and Applications, ed. S. Atahan-Evrenk and A. Aspuru-Guzik, Springer International Publishing, Cham, 2014, pp. 25–58, DOI:10.1007/128_2013_497.
- Ş. Atahan-Evrenk and A. Aspuru-Guzik, in Prediction and Calculation of Crystal Structures: Methods and Applications, ed. S. Atahan-Evrenk and A. Aspuru-Guzik, Springer International Publishing, Cham, 2014, pp. 95–138, DOI:10.1007/128_2013_526.
- K. Kawabata, K. Mashimo and K. Takimiya, Core- versus End-Alkylation: Tailoring Solid-State Structures and Properties of Near-Infrared-Absorbing Organic Semiconductors Based on Naphthodithiophenediones, Chem. Mater., 2024, 36, 11920–11933 CrossRef CAS.
- K. Takimiya, S. Shinamura, I. Osaka and E. Miyazaki, Thienoacene-based organic semiconductors, Adv. Mater., 2011, 23, 4347–4370 CrossRef CAS PubMed.
- V. Coropceanu, J. Cornil, D. A. da Silva Filho, Y. Olivier, R. Silbey and J.-L. Brédas, Charge Transport in Organic Semiconductors, Chem. Rev., 2007, 107, 926–952 CrossRef CAS PubMed.
- S. L. Price, Predicting crystal structures of organic compounds, Chem. Soc. Rev., 2014, 43, 2098–2111 RSC.
- M. K. Dudek and K. Drużbicki, Along the road to crystal structure prediction (CSP) of pharmaceutical-like molecules, CrystEngComm, 2022, 24, 1665–1678 RSC.
- A. Gavezzotti, The Crystalline States of Organic Compounds, Elsevier, 2021 Search PubMed.
- T. Mori, Classification of crystal structures of thiophene-containing organic semiconductors, CrystEngComm, 2023, 25, 6266–6278 RSC.
- T. Mori, Classification of crystal structures of extended aromatic hydrocarbons and the systematic relations, CrystEngComm, 2025, 27, 889–902 RSC.
- K. Takimiya, K. Bulgarevich and K. Kawabata, Crystal-Structure Control of Molecular Semiconductors by Methylthiolation: Toward Ultrahigh Mobility, Acc. Chem. Res., 2024, 57, 884–894 CrossRef CAS PubMed.
- K. Takimiya, T. Ogaki, C. Wang and K. Kawabata, Crystal Structures of Dimethoxyanthracens: A Clue to a Rational Design of Packing Structures of π-Conjugated Molecules, Chem.–Asian J., 2020, 15, 915–919 CrossRef CAS PubMed.
- K. Kanazawa, K. Bulgarevich, K. Kawabata and K. Takimiya, Methylthiolation of Acenes: Change of Crystal Structure from Herringbone to Rubrene-like Pitched π-Stacking Structure, Cryst. Growth Des., 2023, 23, 5941–5949 CrossRef CAS.
- C. Wang, H. Nakamura, H. Sugino and K. Takimiya, Methylthionated benzo[1,2-b:4,5-b']dithiophenes: a model study to control packing structures and molecular orientation in thienoacene-based organic semiconductors, Chem. Commun., 2017, 53, 9594–9597 RSC.
- H. Takenaka, T. Ogaki, C. Wang, K. Kawabata and K. Takimiya, Selenium-Substituted β-Methylthiobenzo[1,2-b:4,5-b']dithiophenes: Synthesis, Packing Structure, and Transport Properties, Chem. Mater., 2019, 31, 6696–6705 CrossRef CAS.
- K. Takimiya, K. Bulgarevich, K. Sahara, K. Kanazawa, H. Takenaka and K. Kawabata, What Defines a Crystal Structure? Effects of Chalcogen Atoms in 3,7-Bis(methylchalcogeno)benzo[1,2-b:4,5-b']dichalcogenophene-Based Organic Semiconductors, Chin. J. Chem., 2022, 40, 2546–2558 CrossRef CAS.
- J. Shin, K. Bulgarevich and K. Takimiya, end-Methylthiolation on Acenes: Another Approach to Brickwork Crystal Structures, Cryst. Growth Des., 2025, 25, 687–693 CrossRef CAS.
- K. Takimiya, K. Bulgarevich, M. Abbas, S. Horiuchi, T. Ogaki, K. Kawabata and A. Ablat, “Manipulation” of Crystal Structure by Methylthiolation Enabling Ultrahigh Mobility in a Pyrene-Based Molecular Semiconductor, Adv. Mater., 2021, 33, 2102914 CrossRef CAS PubMed.
- K. Bulgarevich, S. Horiuchi, T. Ogaki and K. Takimiya, 1,3,6,8-Tetrakis(methylchalcogeno)pyrenes: Effects of Chalcogen Atoms on the Crystal Structure and Transport Properties, Chem. Mater., 2022, 34, 6606–6616 CrossRef CAS.
- K. Takimiya, K. Bulgarevich and S. Horiuchi, Contrasted behaviours of methylthiolated perylene and pyrene as organic semiconductors: implications of molecular electronic structure and crystal structure, J. Mater. Chem. C, 2023, 11, 10809–10815 RSC.
- K. Bulgarevich, S. Horiuchi and K. Takimiya, Crystal-Structure Simulation of Methylthiolated Peri-Condensed Polycyclic Aromatic Hydrocarbons for Identifying Promising Molecular Semiconductors: Discovery of 1,3,8,10-tetrakis(methylthio)peropyrene Showing Ultrahigh Mobility, Adv. Mater., 2023, 35, 2305548 CrossRef CAS PubMed.
- K. Bulgarevich and K. Takimiya, Crystal-structure simulation of molecular semiconductors: brickwork-related crystal structures of methylthiolated peri-condensed polycyclic aromatic hydrocarbons, Mater. Horiz., 2023, 10, 5492–5499 RSC.
- S. Kumar and Y.-T. Tao, Coronenes, Benzocoronenes and Beyond: Modern Aspects of Their Syntheses, Properties, and Applications, Chem.–Asian J., 2021, 16, 621–647 CrossRef CAS PubMed.
- H. J. Cho, S. W. Kim, S. Kim, S. Lee, J. Lee, Y. Cho, Y. Lee, T.-W. Lee, H.-J. Shin and C. Song, Suppressing π–π stacking interactions for enhanced solid-state emission of flat aromatic molecules via edge functionalization with picket-fence-type groups, J. Mater. Chem. C, 2020, 8, 17289–17296 RSC.
- I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, C−H Activation for the Construction of C–B Bonds, Chem. Rev., 2010, 110, 890–931 CrossRef CAS PubMed.
- M. N. Eliseeva and L. T. Scott, Pushing the Ir-Catalyzed C–H Polyborylation of Aromatic Compounds to Maximum Capacity by Exploiting Reversibility, J. Am. Chem. Soc., 2012, 134, 15169–15172 CrossRef CAS PubMed.
- M.-Y. Ni, S.-F. Leng, H. Liu, Y.-K. Yang, Q.-H. Li, C.-Q. Sheng, X. Lu, F. Liu and J.-H. Wan, Ternary organic solar cells with 16.88% efficiency enabled by a twisted perylene diimide derivative to enhance the open-circuit voltage, J. Mater. Chem. C, 2021, 9, 3826–3834 RSC.
- J. M. Murphy, X. Liao and J. F. Hartwig, Meta Halogenation of 1,3-Disubstituted Arenes via Iridium-Catalyzed Arene Borylation, J. Am. Chem. Soc., 2007, 129, 15434–15435 CrossRef CAS PubMed.
- F. Shi, M. R. Smith and R. E. Maleczka, Aromatic Borylation/Amidation/Oxidation: A Rapid Route to 5-Substituted 3-Amidophenols, Org. Lett., 2006, 8, 1411–1414 CrossRef CAS PubMed.
- S. Yoshida, Y. Sugimura, Y. Hazama, Y. Nishiyama, T. Yano, S. Shimizu and T. Hosoya, A mild and facile synthesis of aryl and alkenyl sulfides via copper-catalyzed deborylthiolation of organoborons with thiosulfonates, Chem. Commun., 2015, 51, 16613–16616 RSC.
- K. Kanemoto, S. Yoshida and T. Hosoya, Modified Conditions for Copper-catalyzed ipso-Thiolation of Arylboronic Acid Esters with Thiosulfonates, Chem. Lett., 2017, 47, 85–88 CrossRef.
- J. Merz, A. Steffen, J. Nitsch, J. Fink, C. B. Schürger, A. Friedrich, I. Krummenacher, H. Braunschweig, M. Moos, D. Mims, C. Lambert and T. B. Marder, Synthesis, photophysical and electronic properties of tetra-donor- or acceptor-substituted ortho-perylenes displaying four reversible oxidations or reductions, Chem. Sci., 2019, 10, 7516–7534 RSC.
- R. Yamaguchi, S. Ito, B. S. Lee, S. Hiroto, D. Kim and H. Shinokubo, Functionalization of Hexa-peri-hexabenzocoronenes: Investigation of the Substituent Effects on a Superbenzene, Chem. - Asian J., 2013, 8, 178–190 CrossRef CAS PubMed.
- R. Rieger, M. Kastler, V. Enkelmann and K. Müllen, Entry to Coronene Chemistry—Making Large Electron Donors and Acceptors, Chem.–Eur. J., 2008, 14, 6322–6325 CrossRef CAS PubMed.
- F. B. Mallory and C. W. Mallory, Organic Reactions, John Wiley and Sons, 1984 Search PubMed.
- L. Liu and T. J. Katz, Bromine auxiliaries in photosyntheses of [5]helicenes, Tetrahedron Lett., 1991, 32, 6831–6834 CrossRef CAS.
- S. Alibert-Fouet, I. Seguy, J.-F. Bobo, P. Destruel and H. Bock, Liquid-Crystalline and Electron-Deficient Coronene Oligocarboxylic Esters and Imides By Twofold Benzogenic Diels–Alder Reactions on Perylenes, Chem.–Eur. J., 2007, 13, 1746–1753 CrossRef CAS PubMed.
- A. M. Moussa, J.-R. Yang and M. E. Langmuir, Synthesis Of 3,6-Dimethoxy-2-Naphthaldehyde, Org. Prep. Proced. Int., 1993, 25, 252–255 CrossRef CAS.
- T. Mukaiyama, T. Sato and J. Hanna, Reductive Coupling of Carbonyl Compounds to Pinacols and Olefins by Using TiCl4 and Zn, Chem. Lett., 1973, 2, 1041–1044 CrossRef.
- C.-H. Chiang, D. Pangeni and E. E. Nesterov, Higher Energy Gap Control of Fluorescence in Conjugated Polymers: Turn-On Amplifying Chemosensor for Hydrogen Sulfide, Macromolecules, 2017, 50, 6961–6966 CrossRef CAS.
- G. R. Desiraju and A. Gavezzotti, Crystal structures of polynuclear aromatic hydrocarbons. Classification, rationalization and prediction from molecular structure, Acta Crystallogr., Sect. B:Struct. Sci., 1989, 45, 473–482 CrossRef.
- V. M. Bzhezovskii and E. G. Kapustin, Intramolecular Interactions in Anisole, Thioanisole, and Selenoanisole. Application of Natural Bond Orbitals Method, Russ. J. Org. Chem., 2002, 38, 564–572 CrossRef CAS.
- ADF: Powerful molecular DFT to understand chemistry: https://www.scm.com/product/adf/(accessed 2025-7-6).
- K. Takimiya, M. Nakano, H. Sugino and I. Osaka, Design and elaboration of organic molecules for high field-effect-mobility semiconductors, Synth. Met., 2016, 217, 68–78 CrossRef CAS.
- S.-H. Wen, A. Li, J. Song, W.-Q. Deng, K.-L. Han and W. A. Goddard III, First-Principles Investigation of Anistropic Hole Mobilities in Organic Semiconductors, J. Phys. Chem. B, 2009, 113, 8813–8819 CrossRef CAS PubMed.
- R. A. Laudise, C. Kloc, P. G. Simpkins and T. Siegrist, Physical vapor growth of organic semiconductors, J. Cryst. Growth, 1998, 187, 449–454 CrossRef CAS.
- C. Wang, M. Abbas, G. Wantz, K. Kawabata and K. Takimiya, “Heavy-atom effects” in the parent [1]benzochalcogenopheno[3,2-b][1]benzochalcogenophene system, J. Mater. Chem. C, 2020, 8, 15119–15127 RSC.
- K. Bulgarevich and K. Takimiya, What makes brickwork crystal structures favourable? A case study on methylthiolated arenes and heteroarenes for high-mobility molecular semiconductors, CrystEngComm, 2025, 27, 4776–4786 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2446627–2446641. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc03215f |
| ‡ P. P. and K. B. contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |