
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A mononuclear zinc(II) complex and a tetranuclear copper(II) azametallacoronate with (E)-2-((2-(quinazolin-4-yl)hydrazono)methyl)phenol: structure and biological activity†
Chrisoula
Kakoulidou
a,
Antonios G.
Hatzidimitriou
a,
Konstantina C.
Fylaktakidou
 *b and
George
Psomas
*a
*b and
George
Psomas
*a
aDepartment of General and Inorganic Chemistry, Faculty of Chemistry, Aristotle University of Thessaloniki, GR-54124 Thessaloniki, Greece. E-mail: gepsomas@chem.auth.gr
bLaboratory of Organic Chemistry, Faculty of Chemistry, Aristotle University of Thessaloniki, GR-54124 Thessaloniki, Greece. E-mail: kfylakta@chem.auth.gr
First published on 27th February 2025
Abstract
A quinazoline derivative containing a properly situated o-phenol ring, namely (E)-2-((2-(quinazolin-4-yl)hydrazono)methyl)phenol (H2L), was synthesized in order to investigate the ability of this novel ligand for metal complexation. The interaction of deprotonated HL− with Zn(II) resulted in the mononuclear complex [Zn(HL)2]·1.5CH3OH·H2O (complex 1). When doubly deprotonated ligand L−2 reacted with Cu(II), the tetranuclear complex [Cu4(L)4(DMF)4]·8H2O (complex 2) was isolated, which can be described as an azametallacoronate compound. All three compounds (H2L and complexes 1 and 2) were characterized by spectroscopic techniques and single-crystal X-ray crystallography. The biological profile of the compounds was evaluated in regard to their affinity for calf-thymus DNA, the ability to cleave supercoiled circular pBR322 plasmid DNA in the absence or presence of irradiation of various wavelengths (UVA, UVB and visible light), their binding with bovine serum albumin and their capacity to scavenge 1,1-diphenyl-picrylhydrazyl and 2,2′-azinobis-(3-ethylbenzothiazoline-6-sulfonic acid) radicals and to reduce H2O2. The compounds can bind tightly to calf-thymus DNA via intercalation, and do not induce notable cleavage of plasmid DNA. They can bind tightly and reversibly to the albumin and have shown moderate antioxidant activity.
1 Introduction
Zinc and copper are among the most abundant trace elements (second and third, respectively) in the human body.1 They both have a crucial role in physiology (copper is involved in the development of connective tissues and bones,1,2 while zinc is very important in maintaining membrane barrier structure and function3) and participate in the active center of many enzymes.1 They co-exist in the active center of Cu–Zn superoxide dismutase (SOD), an enzyme that catalyzes the dismutation of superoxide radicals into the less harmful O2 and H2O2, thus contributing to controlling oxidative damage in biological species.1–3 The application of ‘Baby Zinc’ and ZnO to treat diarrhea in Asian and African countries and skin injuries and infections, respectively, is well-known.3,4 Concerning copper, its use as a material in knobs and contact surfaces in hospitals results from its bacteriostatic activity.5 In addition, in medicine, the copper(II)-indomethacin complex, called CuAlgesal®, is used for the treatment of arthritis and a mixture of copper(II)/N,N′-donor complexes, called Casiopeinas®, is in clinical trials being tested for its anticancer activity.6,7 A series of Zn(II)8–12 and Cu(II)13–16 complexes have been reported in the literature regarding promising in vitro biological activities.Metallacrowns (MCs) constitute a class of polynuclear complexes, having a cyclic structure analogous to crown ethers with the repeating pattern [–O–N–M–] in which the methylene carbons are replaced by transition metal ions and nitrogen atoms.17–21 However, a limited class of MCs are the azametallacrowns (azaMCs) which bear the repeating unit [–N–N–M–] by replacing the O atom with a N atom.22–27 In all cases, the formation of such compounds can be designed by involving diverse (mainly) trivalent metal ions such as Fe(III), Cr(III), Mn(III), Co(III), and Er(III)23,24,26–34 or divalent metal ions such as Cu(II),22 and by choosing appropriate polydentate organic ligands such as derivatives of pyrazole, triazole, hydrazide.22–24,26,31,32 Metallacoronates (MCor) can be considered a special form of metallacrowns bearing the repeating motif [–O–C–O–M–],35–38 while just one report concerning analogous azametallacoronates (azaMCor) with a structural motif [–N–C–N–M–] was found in the literature.39
As a continuation of our previous project regarding the synthesis and characterization of derivatives (E)-4-(2-((pyridin-2-yl)methylene)hydrazinyl)quinazoline (L1, Fig. 1) and its metal complexes,40–43 we have designed, synthesized and characterized a phenol analogue of L1, namely (E)-2-((2-(quinazolin-4-yl)hydrazono)methyl)phenol (H2L, Fig. 1) as a novel ligand by replacing the pyridine ring with a phenol ring. Besides the initial structural features of L1 (nitrogen atoms providing multiple coordination sites and rigidity enabling DNA-intercalation),40–43 the insertion of the phenol group is designed to provide the possibility of single and double deprotonation of the quinazoline derivative upon reactions with metal ions. The H2L individual has been used as an intermediate for the synthesis of thiazolidinone and azetidinone heterocyclic attachments on the quinazoline scaffold taking advantage of the imine part of the hydrazone moiety, without, however, the participation of the phenol ring itself.44,45 On the contrary, the phenol part of 2-Ph-H2L derivatives have participated in reaction with Zn(OAc)2 to form complexes that were found to have enhanced fluorescent quantum yields compared to their respective ligands due to increased rigidity of the system upon the complexation.46 Finally, another 2-substituted quinazoline derivative, the 2,4-[bis-(2-hydroxy-3-methoxybenzylidene)]-dihydrazinoquinazoline was found to preferably emit fluorescence when interacted with Zn(II).47
 | ||
| Fig. 1 The syntax formula of (E)-4-(2-((pyridin-2-yl)methylene)hydrazinyl)quinazoline (L1) and (E)-2-((2-(quinazolin-4-yl)hydrazono)methyl)phenol (H2L). | ||
Therefore, the reaction of H2L, used as a novel ligand, with Zn(II) resulted in the mononuclear complex [Zn(HL)2]·1.5CH3OH·H2O (complex 1), while the reaction with Cu(II) yielded the tetranuclear complex [Cu4(L)4(DMF)4]·8H2O (complex 2) which can be described as an azametallacoronate [16-azaMCor-4] complex. The characterization of all three compounds was achieved by spectroscopic techniques (IR, UV-vis, 1H NMR) and single-crystal X-ray crystallography.
The three compounds (H2L, and novel complexes 1 and 2) were further evaluated in vitro for their: (i) interaction with calf-thymus (CT) DNA studied by UV-vis spectroscopy, viscosity measurements, and via competitive studies with ethidium bromide (EB) by fluorescence emission spectroscopy, (ii) ability to cleave supercoiled circular pBR322 plasmid DNA (pDNA) in the absence or presence of irradiation including UVA, UVB and visible light monitored by agarose gel electrophoresis, (iii) antioxidant activity (ability to scavenge the free radicals 1,1-diphenyl-picrylhydrazyl (DPPH) and 2,2′-azinobis-(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) and to reduce H2O2), and (iv) affinity (calculation of binding constant and determination of the binding site location) for bovine serum albumin (BSA) examined in vitro by fluorescence emission spectroscopy.
2 Experimental
2.1 Materials – instruments – physical measurements
All chemicals and solvents were reagent grade and were used as purchased from commercial sources; i.e. 2-aminobenzonitrile from Fluorochem; ammonium acetate, triethyl orthoformate, hydrazine hydrate, p-toluenesulfonic acid monohydrate, salicylaldehyde, CT DNA, EB, BSA, ABTS, K2S2O8, nordihydroguaiaretic acid (NDGA), butylated hydroxytoluene (BHT) from Sigma-Aldrich Co; 6-hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid (trolox) from J&K sodium warfarin, ibuprofen, DPPH from TCI; Zn(NO3)2·6H2O, CuCl2·2H2O, trisodium citrate dihydrate, NaCl, NaH2PO4 from Merck; supercoiled circular pBR322 plasmid DNA from New England Bioline; Tris base, boric acid, EDTA disodium salt dehydrate, loading buffer and H2O2 (30% w/v) from PanReac Applichem; L-ascorbic acid, Na2HPO4 and all solvents from Chemlab. All reactions were monitored on commercially available pre-coated TLC plates (layer thickness 0.25 mm) of Kieselgel 60 F254. Yields were calculated after recrystallization.CT DNA stock solution was prepared by dilution of CT DNA in buffer (containing 150 mM NaCl and 15 mM trisodium citrate at pH 7.0) followed by exhaustive stirring at 4 °C for 2 days and kept at 4 °C for no longer than a week. The stock solution of CT DNA gave a ratio of UV absorbance at 260 and 280 nm (A260/A280) of ∼1.86, indicating that the DNA was sufficiently free of protein contamination.48 The DNA concentration per nucleotide was determined by the UV absorbance at 260 nm after 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 dilution using ε = 6600 M−1 cm−1.49
:20 dilution using ε = 6600 M−1 cm−1.49
Infrared (IR) spectra (400–4000 cm−1) were recorded on a Nicolet FT-IR 6700 spectrometer with samples prepared as KBr pellets (abbreviation used: s = strong, vs = very strong, br = broad, m = medium, w = weak) (Fig. S1†). UV-visible (UV-vis) spectra were recorded as nujol mulls and in solution at concentrations in the range 50 μM–5 mM on a Hitachi U-2001 dual beam spectrophotometer. C, H and N elemental analyses were carried out on a PerkinElmer 240 B elemental analyzer. 1H NMR spectra were recorded on an Agilent 500/54 (500 MHz) spectrometer using DMSO-d6 as solvent. Chemical shifts (δ) are given in ppm and J values in Hz using solvent as an internal reference (abbreviation used: s = singlet, d = doublet, br = broad). Molar conductivity measurements were carried out in 1 mM DMSO solution of the complexes with a Crison Basic 30 conductometer. Fluorescence spectra were recorded in solution on a Hitachi F-7000 fluorescence spectrophotometer. Viscosity experiments were carried out using an ALPHA L Fungilab rotational viscometer equipped with an 18 mL LCP spindle and the measurements were performed at 100 rpm.
2.2 Synthesis of the compounds
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N); 1483 (m) ν(C–C)aromatic; 770 (m) w(N–H). UV–vis (DMSO, nm (M−1 cm−1)): λmax(ε) = 453 (1600), 402 (shoulder(sh)) (5240), 379 (8200), 361 (sh) (740), 288 (4500).
N); 1483 (m) ν(C–C)aromatic; 770 (m) w(N–H). UV–vis (DMSO, nm (M−1 cm−1)): λmax(ε) = 453 (1600), 402 (shoulder(sh)) (5240), 379 (8200), 361 (sh) (740), 288 (4500).
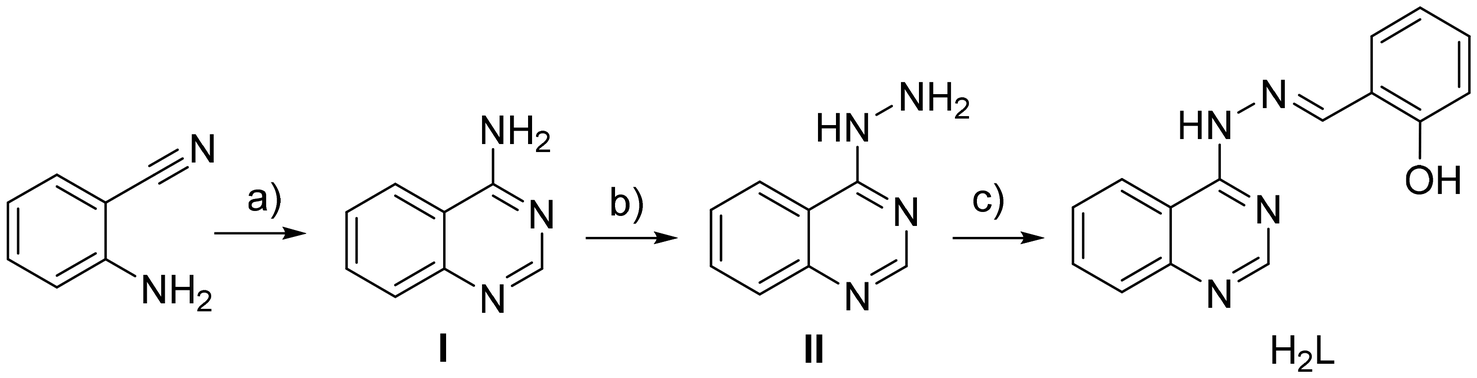 | ||
| Scheme 1 Synthesis of compound H2L. (a) NH4OAc, HC(OEt)3, EtOH reflux, 3.5 h, 78% yield; (b) NH2NH2, reflux, 24 h, 53% yield; (c) salicylaldehyde, p-TsOH, EtOH reflux, 3 h, 72% yield. | ||
N); 1473 (s) ν(C–C)aromatic; 751 (m) w(N–H); 552 (m) ν(M–N); 495 (m) ν(M–O). UV–vis (nujol, nm): λmax = 450, 425, 397; (DMSO, nm (M−1 cm−1)): λmax(ε) = 449 (1200), 423 (1500), 401 (1300), 380 (1050), 359 (1350), 341 (7500), 290 (7200). The complex is soluble in DMSO (ΛM = 9 S cm2 mol−1, 1 mM in DMSO).
N); 1471 (m) ν(C–C)aromatic; 750 (m) w(N–H); 560 (m) ν(M–N); 495 (w) ν(M–O). UV–vis: (nujol, nm): λmax = 615, 445, 415, 395; (DMSO, nm (M−1 cm−1)): λmax(ε) = 610 (160), 443 (1850), 419 (9900), 391 (5100), 360 (3700), 343 (3000), 275 (11500). The complex is soluble in DMSO (ΛM = 11 S cm2 mol−1, 1 mM in DMSO).
2.3 X-ray crystal structure determination
Suitable single-crystals of the compounds were mounted on thin glass fibers with the aid of epoxy resin. X-ray diffraction data were recorded on a Bruker Apex II CCD area-detector diffractometer, equipped with a Mo Kα (λ = 0.71073 Å) sealed tube source and a Triumph monochromator at 295 K, using the φ and ω scans technique. The program Apex2 (Bruker AXS, 2006) was used for data collection and cell refinement. The collected data were integrated with the Bruker SAINT software package,50 using a narrow-frame algorithm. Data were corrected for absorption using the numerical method SADABS,51 based on the crystal dimensions. Structures were solved using the SUPERFLIP package52 and refined with full-matrix least-squares on F2 using the Crystals program package version 14.61 build 6236.53 Anisotropic displacement parameters were applied all non-hydrogen non disordered atoms of the complexes and the non-disordered solvent atoms, while hydrogen atoms were in general found and/or positioned geometrically and refined using a riding model. All disordered atoms (water solvate molecules and coordinated DMF) were isotropically refined with fixed occupation factors. Details of crystal data and structure refinement parameters are shown in Table S1.†2.5 Study of the biological profile of the compounds
All the procedures and relevant equations used in the in vitro study of the biological activity (antioxidant activity, and interaction with CT DNA, plasmid DNA and BSA) of the compounds can be found in the ESI file (sections S1–S4†).3 Results and discussion
3.1 Synthesis and characterization
The synthesis of compound H2L was performed in three steps (Scheme 1). The synthesis of 4-aminoquinazoline (step I) and quinazolin-4-yl-hydrazine (step II) has been previously reported.40–43 Quinazolin-4-yl-hydrazine (compound II) reacted with salicylaldehyde to give in high yield (72%) (E)-2-((2-(quinazolin-4-yl)hydrazono)methyl)phenol (H2L). The data obtained for the known compounds I and II were in full accordance with the literature reported.40–43 Compound H2L was fully characterized by IR, 1H NMR, HRMS and UV-vis spectroscopies (Fig. S1–S3†) and single-crystal X-ray crystallography.The complexes were prepared via the aerobic reaction of a solution deprotonated quinazoline with the corresponding metal salts of Zn(II) and Cu(II). The reaction of Zn(II) with deprotonated quinazoline (HL−1) in a 1:2 Zn(II):HL−1 ratio resulted in complex 1, while the reaction of Cu(II) with doubly deprotonated (L−2) in a 1:1 Cu(II):L−2 ratio led to the formation of complex 2. The coordination compounds were characterized by IR and UV-vis spectroscopies, and single-crystal X-ray crystallography.
The complexes are stable in air, soluble in DMSO and insoluble in H2O. The molar conductivity values of the complexes were recorded in a 1 mM DMSO solution. Both complexes bear ΛM values in the range 9–11 S cm2 mol−1 showing their non-electrolytic nature54 which is in accordance with the formulas found from single-crystal X-ray crystallography.
The FT-IR spectra of the compounds (Fig. S1†) are complicated due to the presence of the phenol and quinazoline groups. The IR spectrum of H2L shows a weak band at 3413 cm−1 which can be assigned to the ν(O–H)phenol and the ν(N–H) group and intense bands at 1630 cm−1 and 1619 cm−1 which can be associated with the hydrazone ν(CN) group.40–43 In the IR spectra of compounds 1 and 2, these bands have shifted slightly suggesting the formation of the complexes.55 The formation of the complexes may be also verified by the appearance of medium-intensity bands in the regions 552–560 cm−1 and 494–495 cm−1 which can be assigned to ν(M–N) and ν(M–O), respectively.40–42
The UV-vis spectra of the compounds (Fig. S3†) were recorded in DMSO solution and as nujol mull. The UV-vis spectra of both complexes in solid state (nujol) showed similar patterns with those recorded in DMSO solution; therefore, their stability in solution may be proposed. In these spectra, the bands observed in the UV or near-UV region of the spectrum located at 270–290 nm and 350–453 nm may be attributed to intra-ligand transitions, typical to CN and CC bonds. In the spectra of the complexes, these bands are shifted indicating the formation of the complex. In particular, the band of H2L at 379 nm which is related to the hydrazone group suffered a bathochromic shift in the UV-vis spectra of the complexes. In addition, the bands located at 400–453 nm may be attributed to charge-transfer transitions. In the visible region of the spectrum of complex 2, the band observed at 610 nm (ε = 160 M−1 cm−1) is attributed to a d-d transition and is typical for a square pyramidal geometry for copper(II) complexes.56
3.2 Structure of the compounds
The structures of all three compounds were determined by single-crystal X-ray crystallography. | ||
| Fig. 2 The molecular structure of H2L. | ||
It should be noted that almost all atoms seem to be co-planar, since all three aromatic rings are lying in the same plane with slight deviations. Hydrogen-bonds contribute to the stabilization of the structure (Table S3†). More specifically, an intramolecular hydrogen-bond is formed between the phenolic hydrogen H11 and hydrazine N4, and an intermolecular hydrogen-bond exists between H12 of the quinazoline nitrogen N1 and quinazoline N2i of an adjacent molecule.
 | ||
| Fig. 3 The molecular structure of complex 1A and 1B. The C-bonded hydrogen atoms and solvate molecules are omitted for clarity reasons. | ||
The asymmetric unit comprises two crystallographically independent neutral mononuclear Zn(II) complexes, notated as complex 1A and 1B, as well as three methanol and one water solvate molecules. Both H2L ligands in each complex act as monoanions (HL−) after deprotonation of the phenolic hydrogen atom. All ligands are coordinated to Zn(II) in a tridentate fashion through two nitrogen atoms – coming one from the quinazoline and one from the hydrazine parts – and the phenolate oxygen, i.e. in complex 1A N1, N4 and O1 for the first ligand and N5, N8 and O2 for the second ligand respectively. This coordination mode leads to the formation of two fused chelate rings for each ligand, a five- and a six-membered one and gives rise to a six-coordinated Zn(II) cation with a N4O2 environment. In both complexes, the resulting disordered coordination octahedron has as most axial vector the Nhydrazone–Zn–Nhydrazone containing nitrogen atoms forming angles i.e. N4–Zn1–N8 = 165.41(10)° for complex 1A and N12–Zn2–N16 = 164.29(10)° for 2A.
A series of intermolecular H-bonds between adjacent molecules of the complexes and with solvate molecules result in further stabilization of the structure (Table S3†).
 | ||
| Fig. 4 The molecular structure of complex 2 (A) highlighting the metallacoronate ring (in bold) and (B) showing the coordination spheres of Cu(II) ions. The hydrogen atoms and solvate H2O molecules are omitted for clarity reasons. Symmetry codes: (i) −y + 5/4, x + 1/4, −z + 5/4; (ii) y − 1/4, −x + 5/4, −z + 5/4. | ||
Complex 2 is a neutral tetranuclear copper(II) complex consisting of four Cu(II) ions, four doubly deprotonated L2− ligands and four DMF ligands, while there are also eight solvate water molecules. In the complex, the four doubly deprotonated quinazoline ligands (L2−) are coordinated to Cu(II) ions in a tetradentate bridging manner. More specifically, the quinazoline nitrogen atom N1, the hydrazine nitrogen atom N4, and the phenolate oxygen atom O1 are coordinated to Cu1, leading to the formation of two fused chelate rings, a five- and a six-membered one. The coordination sphere of the pentacoordinate Cu(II) ion is completed by the deprotonated quinazoline nitrogen atom N2i of an adjacent ligand and the oxygen atom O2 of the DMF ligand. According to the value of the trigonality index τ5 = 0.10 (=(175.9° − 169.9°)/60°, τ5 = (φ1 − φ2)/60°, φ1 and φ2 are the largest angles in the coordination sphere;57τ5 = 0 is found for a perfect square pyramid and τ5 = 1 for a perfect trigonal bipyramid), the geometry around each Cu(II) ion can be described as slightly distorted square pyramid with O1, N1, N4 and N2i atoms forming the basal plane and O2 being at the apical of the pyramid.
The four Cu(II) ions are bridged by four L2− ligands via the quinazoline atoms N1–C1–N2 creating a 16-membered saddle-like azametallacoronate (azaMCor) ring of the type [16-azaMCor-4] with a [–N–C–N–Cu–] repeating unit. To the best of our knowledge,22–27,35–37 complex 2 is among the very few examples of an azametallacoronate compound of the type [16-azaMCor-4] reported.39 The interatomic distances between the neighboring Cu(II) ions (Cu1⋯Cu1i) and the non-neighboring Cu(II) ions (Cu1⋯Cu1ii) are 5.797 Å and 6.574 Å, respectively. Similar arrangement of the Cu(II) ions was also observed in a series of [16-MCor-4] metallacoronates of the formula [Cu4(carboxylato)4(X)4] (X = Cl−, ClO4−).35,38 The arrangement of the four Cu(II) participating in the azametallacoronate ring results in the formation of a hollow space in the [16-azaMC-4] ring where four DMF ligands are encapsulated. Each DMF ligand is coordinated to a ring Cu(II) ion via its oxygen atom O2 orientated towards the center of the [16-azaMC-4] (Fig. 4).
The extended hydrogen-bonded system involving the solvate water molecules as well as the π–π stacking interactions between the ligands from neighboring complexes offer further stabilization of the structure (Table S3†).
3.3 Interaction with DNA
Titration studies using UV-vis spectroscopy were employed in order to investigate the interaction of the compounds with CT DNA and the strength of this interaction by calculating the corresponding DNA-binding constant (Kb). Within this context, initially the UV-vis spectra of a CT DNA solution in buffer were recorded in the presence of H2L or complexes 1 and 2 at diverse ratios of [compound]/[DNA] (= r). Upon addition of increasing amounts of the compounds, a slight change of the absorbance (either hypochromism or hyperchromism) of the CT DNA band located at λmax = 258–260 nm was observed (Fig. S4†) indicating the existence of an interaction.
On the reverse titrations, the UV-vis spectra of the compounds were recorded upon addition of increasing amounts of CT DNA and a series of changes of the charge-transfer or intraligand band(s) of the compounds were observed (Fig. 5(A), and S5† and Table 1). The overall changes of these bands were rather complicated showing hypochromism and/or hyperchromism which may be accompanied by slight blue- or red-shift (Table 1). The features in the UV spectra of CT DNA confirmed the interaction between the compounds and CT DNA suggesting the formation of a new adduct between CT DNA and the compound. Because of the complicated UV spectra of the compounds bearing so many transition bands, it is not easy to suggest a discrete mode of DNA-binding and more studies such as DNA-viscosity measurements and competitive studies with EB were conducted to reach a safer conclusion regarding the interaction mode.
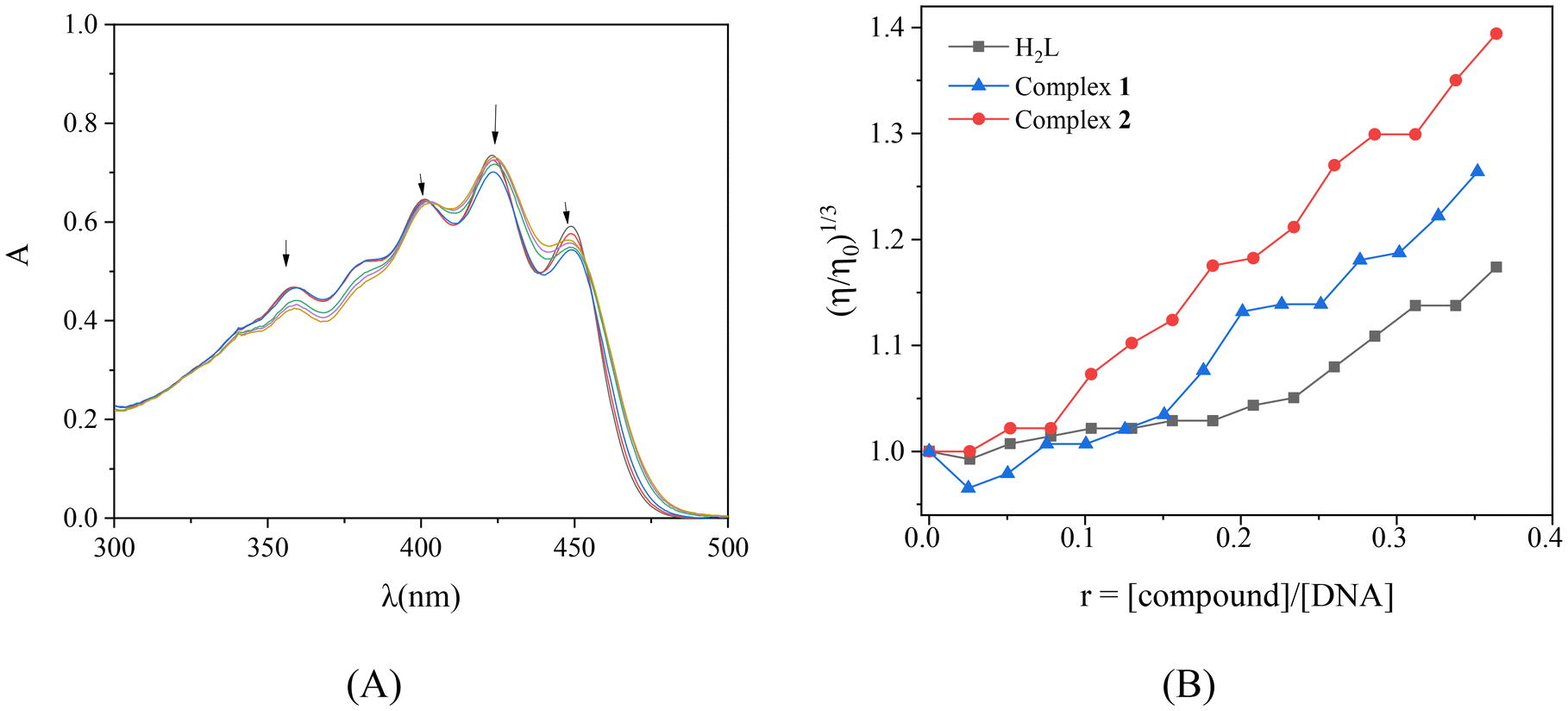 | ||
| Fig. 5 (A) UV-vis spectra of a DMSO solution of complex 1 (0.05 mM) in the presence of increasing amounts of CT DNA. The arrows show the changes upon increasing amounts of CT DNA. (B) Relative viscosity (η/η0)1/3 of CT DNA (0.1 mM) in buffer solution (150 mM NaCl and 15 mM trisodium citrate at pH 7.0) in the presence of H2L and its complexes 1 and 2 at increasing amounts (r = [complex]/[DNA]). | ||
| Compound |
λ (nm) (ΔA/A0a (%), Δλb (nm)) |
K b (M−1) |
|---|---|---|
| UV-band (λ, in nm) (percentage of the observed hyper-/hypo-chromism (ΔA/A0, in %), blue-/red-shift of the λmax (Δλ, in nm)) and DNA-binding constants (Kb, in M−1).a “+” denotes hyperchromism, “−” denotes hypochromism.b “+” denotes red-shift, “−” denotes blue–shift.c sh = shoulder.d >+50 denotes high hyperchromism.e elim = eliminated. | ||
| H2L | 361(sh)c (−12,a +1b); 379 (−12,a −1b), 401(sh) (+3, −1); 453 (>+50,d −10) |
2.04(±0.59) × 106 |
| Complex 1 | 341 (−3, +1); 359 (−9, 0); 380 (−9, −2); 401 (−2, 0); 423 (−5, +1); 449 (−8, 0) | 1.44(±0.32) × 107 |
| Complex 2 | 343 (−3, −1); 360 (−2, −1); 391 (+2, elime); 419 (−9, −2); 443 (−10, −2) | 8.38 (±0.50) × 106 |
The Wolfe–Shimer equation (eqn (S1)†)58 and the plots [DNA]/(εA − εf) versus [DNA] (Fig. S6†) were used for the calculation of the Kb values of the compounds. All three compounds presented significantly high DNA-binding constants which were higher than that of the classic intercalator EB (= 1.23 × 105 M−1).59 Both complexes exhibit high DNA-affinity than H2L (Table 1) with complex 1 showing the highest DNA-binding constant (Kb = 1.44(±0.32) × 107 M−1) among the compounds. A comparison of the Kb values derived for the compounds with reported compounds bearing a series of pyridine analogues of H2L, i.e. (E)-4-(2-(pyridin-2-ylmethylene)hydrazinyl)quinazoline and its halogenated derivatives,40–43 may reveal that the existence of the phenol ring in H2L resulted in higher affinity for CT DNA.
The viscosity of DNA-solution was examined because DNA-viscosity is sensitive to changes in the relative DNA chain-length making viscometry a useful method to clarify the DNA-interaction mode. The viscosity of a CT DNA solution (0.1 mM) was measured in the presence of incrementally increasing amounts of compounds (up to r = [compound]/[DNA] = 0.36). For all compounds, the viscosity increased significantly (Fig. 5(B)) suggesting intercalation as the most possible mode of their interaction with CT DNA; the increase of relative DNA-viscosity is related to increase of the relative DNA-length as a result from the increase of the distance between DNA-bases due to intercalation of the inserting compounds.60,61 A slight lowering of DNA-viscosity was also observed at the early steps of incremental addition of H2L and complex 1 (for r values up to 0.10), indicating an initial external interaction resulting in closer approach which was followed by intercalation.
The planar phenanthridine ring of the typical DNA-intercalator EB inserts in-between two adjacent DNA-bases resulting in an intense fluorescence emission band at 592–594 nm upon excitation of the solution at 540 nm. In order to explore the potential intercalation effect of the compounds, the fluorescence emission spectra of a solution containing EB-DNA (after 1–h pretreatment of EB (40 μM) and CT DNA (40 μM) in buffer solution) were recorded in presence of increasing amounts of solution of the compounds. The addition of the compounds in the EB-DNA solution led to a quenching of the fluorescence emission band at 594 nm (representatively shown for complex 2 in Fig. 6(A)). The quenching was significant (up to ∼69% of the initial fluorescence, Table 2 and Fig. 6(B)), and may be assigned to the displacement of EB from the EB-DNA adduct.62
 | ||
| Fig. 6 (A) Fluorescence emission spectra (λexcitation = 540 nm) for EB-DNA conjugate ([EB] = 40 μM, [DNA] = 40 μM) in buffer solution (150 mM NaCl and 15 mM trisodium citrate at pH = 7.0) in the absence and presence of increasing amounts (up to r = [complex]/[DNA] = 0.04) of complex 2. The arrow shows the changes of intensity upon increasing amounts of the complexes. (B) Plot of EB-DNA relative fluorescence emission intensity at λemission = 592 nm (I/I0, %) versus r (r = [complex]/[DNA]) in the presence of H2L and its complexes 1 and 2 (up to 37.1% of the initial EB-DNA fluorescence emission intensity H2L, 31.2% for 1, and 46.3% for 2). | ||
| Compound | ΔI/I0 (%) | K SV (M−1) | k q (M−1 s−1) |
|---|---|---|---|
| Percentage of EB-DNA fluorescence emission quenching (ΔI/I0, in %), Stern–Volmer constants (KSV, in M−1) and quenching constants (kq, in M−1 s−1). | |||
| H2L | 62.9 | 7.67(±0.15) × 105 | 3.34(±0.06) × 1013 |
| Complex 1 | 68.8 | 3.65(±0.14) × 105 | 1.59(±0.06) × 1013 |
| Complex 2 | 53.7 | 4.22(±0.09) × 105 | 1.84(±0.04) × 1013 |
The quenching ability of the compounds can be evaluated through the values of the Stern–Volmer constant (KSV) and quenching constant (kq) which were calculated with the Stern–Volmer equations (eqn (S2) and (S3)† after using the value of 23 ns as the fluorescence lifetime of EB-DNA system (τ0)),63 and the corresponding Stern–Volmer plots (Fig. S7†). The KSV values of the compounds are relatively high (Table 2) and are similar with reported compounds bearing pyridine analogues of H2L.40–43 In addition, the quenching constants are of the order 1013 M−1 s−1 (Table 2) and are three orders higher than the value of 1010 M−1 s−1, suggesting the existence of a static quenching mechanism which may confirm the formation of a new adduct between DNA and each complex.62
The potential DNA-cleaving activity of the compounds was quite negligible upon lack of irradiation (Fig. S8(A)†) or upon irradiation at 312 nm (Fig. S8(B)†). In all experiments, complex 1 proved incapable of cleaving or photocleaving pDNA. In some cases, i.e. for H2L upon irradiation at 365 nm (Fig. S8(C)†) and for complex 2 upon irradiation with UVA (Fig. S8(C)†) and visible light (Fig. S8(D)†), the pDNA-cleavage was revealed only as single-stranded (ss) nicks in the supercoiled DNA forming relaxed circular DNA (Form II), while no signal (double-stranded (ds) nick) attributed to the formation of linear DNA (Form III) was observed. For these cases, the percentages of the cleavage (shown in Fig. S8†) were calculated with eqn (S4) and (S5).† At the high concentration of 500 μM used in the experiment, these compounds present rather low-to-negligible ability to cleave DNA with complex 2 showing the highest photocleavage activity of 10% (Fig. S8(D),† lane 4).
The lack of pDNA-cleavage activity of the herein reported compounds, when compared to a series of metal complexes bearing (E)-4-(2-(pyridin-2-ylmethylene)hydrazinyl)quinazoline40–43 and its halogenated derivatives as ligands (they are the pyridine analogues of H2L), may be possibly attributed to the exchange of the pyridine ring by the phenol ring in H2L which results to formation of anionic ligands and six-membered chelate rings when coordinated to metal(II) ions.
3.4 Antioxidant activity of the compounds
Taking into consideration that compounds with antioxidant properties might play an important role in the development of effective pharmaceuticals64 as well as that anti-inflammatory activity has been reported for a series of quinazoline derivatives,65,66 the potential antioxidant activity of the compounds was assessed through monitoring their ability to scavenge DPPH and ABTS free radicals and to reduce H2O2. The results are summarized in Table 3, and the activities were compared with the activity of well-known reference compounds NDGA, BHT, trolox and L-ascorbic acid.64,67,68| Compound | DPPH% 30 min | DPPH% 60 min | ABTS% | H2O2% |
|---|---|---|---|---|
| H2L | 6.52 ± 0.12 | 7.98 ± 0.17 | 54.73 ± 1.56 | 87.98 ± 0.46 |
| Complex 1 | 9.54 ± 0.42 | 10.36 ± 0.19 | 71.63 ± 0.94 | 60.59 ± 0.47 |
| Complex 2 | Inactive | Inactive | 19.97 ± 1.32 | 67.76 ± 0.74 |
| NDGA | 87.08 ± 0.12 | 87.47 ± 0.12 | Not tested | Not tested |
| BHT | 61.30 ± 1.16 | 76.78 ± 1.12 | Not tested | Not tested |
| Trolox | Not tested | Not tested | 98.0 ± 0.48 | Not tested |
| L-Ascorbic acid | Not tested | Not tested | Not tested | 60.80 ± 0.48 |
The neutralization of DPPH radicals by potential antioxidants is often related with potential activity against cancer, aging process and/or inflammation.64 The ability of the compounds to scavenge DPPH radicals was time-independent, as shown after measuring the absorbance after 30-min and 60-min reaction. Complex 2 was proved inactive towards DPPH radicals while H2L and complex 1 showed quite low scavenging activity (up to 10.36 ± 0.19%), especially when compared with the reference compounds NDGA and BHT (Table 3).
The activity of the compounds towards the cationic ABTS radicals (the ABTS-scavenging is related to the overall antioxidant activity of compounds) is more enhanced (up to 71.63 ± 0.94% for complex 1) when compared to DPPH but still lower than the activity of the corresponding reference compound trolox (Table 3).
The scavenging or the reduction of hydrogen peroxide is related to the relief from oxidative stress and/or inhibition of reactive oxygen species.69 The interaction of the compounds with H2O2 revealed that the complexes are less active (H2O2% = 60.59–67.76%, Table 3) than free H2L (87.98 ± 0.46%). However, all compounds present equal or better activity than the reference compounds L-ascorbic acid (= 60.80 ± 0.20%).
The overall antioxidant behavior of the compounds is rather poor towards DPPH radicals, moderate towards ABTS radicals and significant towards H2O2. Such antioxidant profile is in accordance with the results reported for compounds containing the pyridine analogues of H2L.40–43
3.5 Interaction of the compounds with BSA
The fluorescence emission spectra of a BSA solution (3 mM) were recorded in the range 300–500 nm for λexcitation = 295 nm in the presence of incrementally increasing amounts of the compounds (up to r = [compound]/[BSA] = 6.2). The initial BSA emission band with λmax, emission at 343 nm showed upon addition of the compounds a significant decrease with complex 2 inducing the highest percentage (>97%) of the BSA quenching (Fig. 7). The observed quenching can be assigned to re-arrangement or modifications of the SA secondary structure which may result from the association of the compounds with BSA.62 The influence of the inner-filter effect on the measurements was calculated with eqn (S6)†71 and it was too negligible to affect the measurements.
 | ||
| Fig. 7 (A) Fluorescence emission spectra (λexcitation = 295 nm) of a buffer solution (150 mM NaCl and 15 mM trisodium citrate at pH 7.0) of BSA (3 μM) in the presence of increasing amounts of complex 2. The arrow shows the changes of intensity upon increasing amounts of the complex. (B) Plot of % relative fluorescence intensity at λem = 343 nm (I/I0, %) versus r (r = [complex]/[BSA]) for H2L and its complexes (up to 22.4% of the initial BSA fluorescence for H2L, 20.0% for 1, and 2.7% for 2). | ||
The Stern–Volmer constants (KSV), the BSA-quenching constants (kq) and the BSA-binding constants (K) of the compounds were calculated with the Stern–Volmer and Scatchard equations (eqn (S2), (S3) and (S7)†) and plots (Fig. S9 and S10†) applying the value of 10−8 s as the fluorescence lifetime of tryptophan in SAs (τ0).62 The calculated kq values of the compounds are significantly higher (in most cases by three-to-four orders) than the value of 1010 M−1 s−1 (Table 4) suggesting a static mechanism for the observed quenching,62 which confirms the interaction of the compounds with BSA.
| Compound | k q (M−1 s−1) | K (BSA) (M−1) | K (BSA,ibu) (M−1) | K (BSA,warf) (M−1) |
|---|---|---|---|---|
| BSA-quenching constants (kq, in M−1 s−1), and BSA-binding constants (K, in M−1) of the compounds in the absence (K(BSA)) or presence of a site-marker warfarin (K(BSA,warf)) or ibuprofen (K(BSA,ibu)). | ||||
| H2L | 1.84(±0.06) × 1013 | 1.30(±0.08) × 105 | 2.32(±0.03) × 105 | 1.32(±0.04) × 105 |
| Complex 1 | 2.16(±0.09) × 1013 | 1.05(±0.04) × 105 | 4.07(±0.19) × 105 | 3.16(±0.09) × 105 |
| Complex 2 | 2.05(±0.09) × 1014 | 5.60(±0.18) × 105 | 1.17(±0.06) × 106 | 1.17(±0.64) × 105 |
The values of the BSA-binding constants of the compounds are of the order 105 M−1 (Table 4) with complex 2 showing the highest K value (= 5.60(±0.18) × 105 M−1) among the compounds under study. The affinity of the compounds is within the range also reported for the pyridine analogues of H2L.40–43 However, the BSA-binding constants found for the compounds observed are lower than the value of 1015 M−1 which is considered as the limit for reversible noncovalent interactions. Within this context, the BSA-binding constants of the compounds reveal their reversible binding to the BSA in order to get transported and get released upon arrival at their potential biological targets.72
The incremental addition of the compounds in a solution containing BSA and the corresponding site-marker (warfarin or ibuprofen) resulted in a significant quenching of the initial fluorescence emission band (Fig. S11 and S12† and representatively shown for complex 2 in Fig. 8). The BSA-binding constants of the compounds were determined with the Scatchard equation (eqn (S7)†) and plot (Fig. S13 and S14†). Any decrease of the BSA-binding constant in the presence of the site-marker will show that the binding of the compound to BSA is influenced by the co-existence of this marker inhibiting the approach of the compound due to the competition for the same binding site.41,43,74 From the K values shown in Table 4, a safe conclusion concerning a preferable drug site can be given only for complex 2 which showed a significant decrease of the BSA-binding constant in the presence of warfarin indicating its selective binding to Sudlow's site 1 in subdomain IIA.
 | ||
| Fig. 8 Fluorescence emission spectra (λexcitation = 295 nm) of a buffer solution (150 mM NaCl and 15 mM trisodium citrate at pH 7.0) of BSA (3 μM) in the presence of (A) warfarin (3 μM) or (B) ibuprofen (3 μM), upon addition of increasing amounts of complex 2. The arrows show the changes of intensity upon increasing amounts of the complexes. | ||
4 Conclusions
A novel quinazoline ligand containing a phenol ring, namely (E)-2-((2-(quinazolin-4-yl)hydrazono)methyl)phenol (H2L) and its Zn(II) and Cu(II) complexes, [Zn(HL)2]·1.5CH3OH·H2O (complex 1) and [Cu4(L)4(DMF)4]·8H2O (complex 2), respectively, were isolated and characterized with spectroscopic techniques and single-crystal X-ray crystallography. In mononuclear complex 1, the quinazoline derivative is simply deprotonated acting as a tridentate ligand via the quinazoline and the hydrazone nitrogen atoms and the deprotonated phenolate oxygen atom. In tetranuclear complex 2, the doubly deprotonated quinazoline acted as a tetradentate bridging ligand via the quinazoline and the hydrazone nitrogen atoms and the deprotonated phenolic oxygen atom to a Cu(II) ion and the second quinazoline nitrogen atom to an adjacent Cu(II) ion. A cyclic 16-membered structure containing four [–N–C–N–Cu–] repeating units is formed and complex 2 is a characteristic [16-azaMCor-4] example among the few reported azametallacoronate compounds.The potential biological profile of the compounds was evaluated regarding their interaction with biomacromolecules (BSA and DNA), and their antioxidant ability. The compounds may interact with CT DNA via intercalation and the highest DNA-binding constant was found for complex 1 (Kb = 1.44(±0.32) × 107 M−1). The ability of the compounds to cleave pBR322 plasmid DNA to relaxed circular DNA was rather low-to-negligible at the high concentration of 500 μM.
The binding of compounds to BSA is tight and reversible, suggesting they can be transported by the albumins and released at potential biotargets. Furthermore, competitive studies with the typical site-markers warfarin and ibuprofen were employed to explore whether the compounds bind selectively to typical BSA-binding sites. A safe conclusion was reached only for complex 2 regarding its selective binding to Sudlow's site 1 in subdomain IIA.
Regarding the antioxidant activity of the compounds, their ability to scavenge DPPH and ABTS free radicals and to reduce H2O2 was explored. The compounds were practically inactive towards DPPH radicals; Zn(II) complex 1 was the most active compound towards ABTS radicals, albeit moderately active when compared with the reference compound trolox. Concerning the activity towards H2O2, the compounds were found more active than the reference compound L-ascorbic acid, H2L being by far the most active (H2O2% = 87.98 ± 0.46%) among the compounds.
In conclusion, the phenolic quinazoline derivative H2L and its two complexes present differentiated biological properties when compared to their pyridine quinazoline analogues which bear the skeleton of (E)-4-(2-((pyridin-2-yl)methylene)hydrazinyl)quinazoline. Apart from the structural differences and the diversity of deprotonation states, the herein reported compounds presented negligible cleavage activity of plasmid DNA and lower antioxidant potency but much higher affinity for CT DNA, a fact that may include them to potential candidates targeting DNA.
Abbreviations
| ABTS | 2,2′-Azinobis-(3-ethylbenzothiazoline-6-sulfonic acid) |
| BHT | Butylated hydroxytoluene |
| BSA | Bovine serum albumin |
| CT | Calf-thymus |
| DPPH | 1,1-Diphenyl-picrylhydrazyl |
| EB | Ethidium bromide |
| K | BSA-binding constant |
| K b | DNA-binding constant |
| k q | Quenching constant |
| K SV | Stern–Volmer constant |
| MC | Metallacrown |
| Mcor | Metallacoronate |
| NDGA | Nordihydroguaiaretic acid |
| pDNA | pBR322 plasmid DNA |
| SA | Serum albumin. |
Data availability
The data supporting this article have been included as part of the ESI.†CCDC 2401516–2401518 contain the supplementary crystallographic data for the complexes.†
Conflicts of interest
There are no conflicts to declare.References
- P. Chellan and P. J. Sadler, Philos. Trans. R. Soc., A, 2015, 373, 2014018 CrossRef PubMed.
- G. Crisponi, V. M. Nurchi, D. Fanni, C. Gerosa, S. Nemolato and G. Faa, Coord. Chem. Rev., 2010, 254, 876–889 CrossRef CAS.
- M. Jarosz, M. Olbert, G. Wyszogrodzka, K. Młyniec and T. Librowski, Inflammopharmacology, 2017, 25, 11–24 CrossRef CAS PubMed.
- C. P. Larson, U. R. Saha and H. Nazrul, PLoS Med., 2009, 6, e1000175 CrossRef PubMed.
- W. X. Tian, S. Yu, M. Ibrahim, A. W. Almonaofy, L. He, Q. Hui, Z. Bo, B. Li and G. L. Xie, J. Microbiol., 2012, 50, 586–593 CrossRef CAS PubMed.
- S. Medici, M. Peana, V. M. Nurchi, J. I. Lachowicz, G. Crisponi and M. A. Zoroddu, Coord. Chem. Rev., 2015, 284, 329–350 CrossRef CAS.
- A. Rivero-Müller, A. De Vizcaya-Ruiz, N. Plant, L. Ruiz and M. Dobrota, Chem.-Biol. Interact., 2007, 165, 189–199 CrossRef PubMed.
- J. d'Angelo, G. Morgant, N. E. Ghermani, D. Desmaële, B. Fraisse, F. Bonhomme, E. Dichi, M. Sghaier, Y. Li, Y. Journaux and J. R. J. Sorenson, Polyhedron, 2008, 27, 537–546 Search PubMed.
- Q. Zhou, T. W. Hambley, B. J. Kennedy, P. A. Lay, P. Turner, B. Warwick, J. R. Biffin and H. L. Regtop, Inorg. Chem., 2000, 39, 3742–3748 CrossRef CAS PubMed.
- H. Abu Ali, S. N. Omar, M. D. Darawsheh and H. Fares, J. Coord. Chem., 2016, 69, 1110–1122 CrossRef CAS.
- C. I. Chukwuma, S. S. Mashele, K. C. Eze, G. R. Matowane, S. M. Islam, S. L. Bonnet, A. E. M. Noreljaleel and L. M. Ramorobi, Pharmacol. Res., 2020, 155, 104744 CrossRef CAS PubMed.
- M. Porchia, M. Pellei, F. Del Bello and C. Santini, Molecules, 2020, 25, 5814 CrossRef CAS PubMed.
- B. Chudzik, I. B. Tracz, G. Czernel, M. J. Fiołka, G. Borsuk and M. Gagoś, Eur. J. Pharm. Sci., 2013, 49, 850–857 CrossRef CAS PubMed.
- J. Nagaj, R. Starosta and M. Jezowska-Bojczuk, J. Inorg. Biochem., 2015, 142, 68–74 CrossRef CAS PubMed.
- E. G. Geromichalou, D. T. Trafalis, P. Dalezis, G. Malis, G. Psomas and G. D. Geromichalos, J. Inorg. Biochem., 2022, 231, 111805 CrossRef CAS PubMed.
- O. A. Zalevskaya and Y. A. Gur'eva, Russ. J. Coord. Chem., 2021, 47, 861–880 Search PubMed.
- G. Psomas, A. J. Stemmler, C. Dendrinou-Samara, J. J. Bodwin, M. Schneider, M. Alexiou, J. W. Kampf, D. P. Kessissoglou and V. L. Pecoraro, Inorg. Chem., 2001, 40, 1562–1570 CrossRef CAS PubMed.
- A. Tarushi, C. P. Raptopoulou, V. Psycharis, C. K. Kontos, D. P. Kessissoglou, A. Scorilas, V. Tangoulis and G. Psomas, Eur. J. Inorg. Chem., 2016, 2016, 219–231 CrossRef CAS.
- A. Tarushi, A. G. Hatzidimitriou, M. Estrader, D. P. Kessissoglou, V. Tangoulis and G. Psomas, Inorg. Chem., 2017, 56, 7048–7057 CrossRef CAS PubMed.
- A. Tarushi, M. Zampakou, S. Perontsis, K. Lafazanis, A. A. Pantazaki, A. G. Hatzidimitriou, G. D. Geromichalos and G. Psomas, Inorg. Chim. Acta, 2018, 483, 579–592 CrossRef CAS.
- A. Tarushi, G. D. Geromichalos, K. Lafazanis, C. P. Raptopoulou, V. Psycharis, N. Lalioti, A. A. Pantazaki, D. P. Kessissoglou, V. Tangoulis and G. Psomas, New J. Chem., 2018, 42, 6955–6967 RSC.
- O. S. Vynohradov, V. A. Pavlenko, I. O. Fritsky, I. A. Gural'skiy and S. Shova, Russ. J. Inorg. Chem., 2020, 65, 1481–1488 CrossRef CAS.
- S. Lin, S. X. Liu, Z. Chen, B. Z. Lin and S. Gao, Inorg. Chem., 2004, 43, 2222–2224 CrossRef CAS PubMed.
- W. Yang, H. Yang, Q. Yao, S. Zeng, D. Li and J. Dou, Inorg. Chem. Commun., 2017, 82, 16–19 CrossRef CAS.
- H. Yang, Y. Q. Zhang, L. Sun, D. C. Li, S. Y. Zeng, Y. W. Li, Y. Yang and J. M. Dou, Eur. J. Inorg. Chem., 2021, 2021, 740–747 CrossRef CAS.
- H. Yang, Q. X. Yao, Y. W. Li, D. C. Li and J. M. Dou, Dalton Trans., 2014, 43, 16986–16991 RSC.
- H. Yang, F. Cao, D. Li, S. Zeng, Y. Song and J. Dou, Chem. – Eur. J., 2015, 21, 14478–14485 CrossRef CAS PubMed.
- H. Yang, D. Li, S. Zeng and J. Dou, Inorg. Chem. Commun., 2015, 58, 84–86 CrossRef CAS.
- G. J. Chen, C. Y. Gao, W. Gu, X. Liu, S. P. Yan, J. L. Tian and D. Z. Liao, Z. Anorg. Allg. Chem., 2011, 637, 374–376 CrossRef CAS.
- S. Lin, S. X. Liu, J. Q. Huanga and C. C. Lina, J. Chem. Soc., Dalton Trans., 2002, 1595–1601 RSC.
- S. Da Su, X. M. Ou-Yang, K. K. Guo, Q. M. Lin, Y. Li, K. Wang, S. Y. Luo and F. P. Liang, J. Cluster Sci., 2019, 30, 863–868 CrossRef.
- Y. Chen, J. Dou, D. Li and S. Wang, Inorg. Chem. Commun., 2010, 13, 167–170 CrossRef CAS.
- S. Lin, S. X. Liu and B. Z. Lin, Inorg. Chim. Acta, 2002, 328, 69–73 CrossRef CAS.
- A. Majumder, S. Goswami, S. R. Batten, M. S. El Fallah, J. Ribas and S. Mitra, Inorg. Chim. Acta, 2006, 359, 2375–2382 CrossRef CAS.
- G. C. Giri, S. Haldar, A. K. Ghosh, P. Chowdhury, L. Carrella, U. Ghosh and M. Bera, J. Mol. Struct., 2017, 1142, 175–184 CrossRef CAS.
- A. Beyer, M. S. von Gernler, S. Pflock, G. Türkoglu, L. Müller, A. Zahl, K. Gieb, P. Müller, T. Drewello and N. Burzlaff, Eur. J. Inorg. Chem., 2018, 2018, 765–777 CrossRef CAS.
- A. Tarushi, F. Kastanias, V. Psycharis, C. P. Raptopoulou, G. Psomas and D. P. Kessissoglou, Inorg. Chem., 2012, 51, 7460–7462 CrossRef CAS PubMed.
- E. Colacio, M. Ghazi, R. Kivekäs and J. M. Moreno, Inorg. Chem., 2000, 39, 2882–2890 CrossRef CAS PubMed.
- A. Lakma, R. N. Pradhan, S. M. Hossain, J. van Leusen, P. Kögerler and A. K. Singh, Inorg. Chim. Acta, 2019, 486, 88–94 CrossRef CAS.
- C. Kakoulidou, A. G. Hatzidimitriou, K. C. Fylaktakidou and G. Psomas, Polyhedron, 2021, 195, 114986 CrossRef CAS.
- C. Kakoulidou, C. T. Chasapis, A. G. Hatzidimitriou, K. C. Fylaktakidou and G. Psomas, Dalton Trans., 2022, 51, 16688–16705 RSC.
- C. Kakoulidou, V. R. Kosmas, A. G. Hatzidimitriou, K. C. Fylaktakidou and G. Psomas, J. Inorg. Biochem., 2021, 219, 111448 CrossRef CAS PubMed.
- C. Kakoulidou, P. S. Gritzapis, A. G. Hatzidimitriou, K. C. Fylaktakidou and G. Psomas, J. Inorg. Biochem., 2020, 211, 111194 CrossRef CAS PubMed.
- A. Jori, S. R. Dixit and G. V. Pujar, Asian – J. Chem., 2020, 32, 2617–2623 CAS.
- S. I. Kovalenko, L. M. Antypenko, A. K. Bilyi, S. V. Kholodnyak, O. V. Karpenko, O. M. Antypenko, N. S. Mykhaylova, T. I. Los and O. S. Kolomoyets, Sci. Pharm., 2012, 81, 359–392 CrossRef PubMed.
- T. V. Trashakhova, E. V. Nosova, P. A. Slepukhin, M. S. Valova, G. N. Lipunova and V. N. Charushin, Russ. Chem. Bull., 2011, 60, 2302–2307 Search PubMed.
- H. Yamada, A. Shirai, K. Kato, J. Kimura, H. Ichiba, T. Yajima and T. Fukushima, Chem. Pharm. Bull., 2010, 56, 875–878 CrossRef PubMed.
- J. Marmur, J. Mol. Biol., 1961, 3, 208–218 CrossRef CAS.
- M. E. Reichmann, S. A. Rice, C. A. Thomas and P. Doty, J. Am. Chem. Soc., 1954, 76, 3047–3053 CrossRef CAS.
- Apex2 Version 2 User Manual M86-E01078, Bruker Analytical X-ray Systems Inc., 2006, preprint Search PubMed.
- SADABS: Area–Detector Absorption Correction, Siemens Industrial Automation Inc Search PubMed.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS.
- P. W. Betteridge, J. R. Carruthers, R. I. Cooper, K. Prout and D. J. Watkin, J. Appl. Crystallogr., 2003, 36, 1487–1487 CrossRef CAS.
- W. J. Geary, Coord. Chem. Rev., 1971, 7, 81–122 CrossRef CAS.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds: Part B: Applications in Coordination, Organometallic, and Bioinorganic Chemistry, 2008, 1–408 Search PubMed.
- B. J. Hathaway, in Comprehensive Coordination Chemistry, ed. G. Wilkinson, Pergamon Press, Oxford, UK, 1987, vol. 5, pp. 533–773 Search PubMed.
- A. W. Addison, T. N. Rao, J. Reedijk, J. Van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- A. Wolfe, G. H. Shimer and T. Meehan, Biochemistry, 1987, 26, 6392–6396 CrossRef CAS PubMed.
- A. Dimitrakopoulou, C. Dendrinou-Samara, A. A. Pantazaki, M. Alexiou, E. Nordlander and D. P. Kessissoglou, J. Inorg. Biochem., 2008, 102, 618–628 CrossRef CAS PubMed.
- A. M. Pizarro and P. J. Sadler, Biochimie, 2009, 91, 1198–1211 CrossRef CAS PubMed.
- J. L. Garcia-Gimenez, M. Gonzalez-Alvarez, M. Liu-Gonzalez, B. Macías, J. Borras and G. Alzuet, J. Inorg. Biochem., 2009, 103, 923–934 CrossRef PubMed.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer, 2006 Search PubMed.
- D. P. Heller and C. L. Greenstock, Biophys. Chem., 1994, 50, 305–312 CrossRef CAS PubMed.
- C. Kontogiorgis, M. Ntella, L. Mpompou, F. Karallaki, P. Athanasios, D. Hadjipavlou-Litina and D. Lazari, J. Enzyme Inhib. Med. Chem., 2016, 31, 154–159 CrossRef CAS PubMed.
- M. M. Gineinah, M. A. El-Sherbeny, M. N. Nasr and A. R. Maarouf, Arch. Pharm., 2002, 335, 556–562 CrossRef CAS PubMed.
- A. M. Alafeefy, A. A. Kadi, O. A. Al-Deeb, K. E. H. El-Tahir and N. A. Al-Jaber, Eur. J. Med. Chem., 2010, 45, 4947–4952 CrossRef CAS PubMed.
- S. Dairi, M. A. Carbonneau, T. Galeano-Diaz, H. Remini, F. Dahmoune, O. Aoun, A. Belbahi, C. Lauret, J. P. Cristol and K. Madani, Food Chem., 2017, 237, 297–304 CrossRef CAS PubMed.
- B. M. Ali, M. Boothapandi and A. S. Sultan Nasar, Data Brief, 2020, 28, 104972 CrossRef PubMed.
- M. Wettasinghe and F. Shahidi, Food Chem., 2000, 70, 17–26 CrossRef CAS.
- R. E. Olson and D. D. Christ, Annu. Rep. Med. Chem., 1996, 31, 327–336 CAS.
- L. Stella, A. L. Capodilupo and M. Bietti, Chem. Commun., 2008, 4744–4746 RSC.
- O. H. Laitinen, V. P. Hytönen, H. R. Nordlund and M. S. Kulomaa, Cell. Mol. Life Sci., 2006, 63, 2992–3017 CrossRef CAS PubMed.
- G. Sudlow, D. J. Birkett and D. N. Wade, Mol. Pharmacol., 1976, 12, 1052–1061 CAS.
- M. Lazou, A. Tarushi, P. Gritzapis and G. Psomas, J. Inorg. Biochem., 2020, 206, 111019 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2401516–2401518. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt03518f |
| This journal is © The Royal Society of Chemistry 2025 |