
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Water-soluble luminescent platinum(II) complexes for guanine quadruplex binding†
Simon
Kroos
a,
Marian
Hebenbrock
 a,
Alexander
Hepp
a,
Marcus
Layh
a,
Joschua
Lüke
ab,
Ali R.
Tonkul
a,
Cristian A.
Strassert
abc and
Jens
Müller
*ac
a,
Alexander
Hepp
a,
Marcus
Layh
a,
Joschua
Lüke
ab,
Ali R.
Tonkul
a,
Cristian A.
Strassert
abc and
Jens
Müller
*ac
aUniversität Münster, Institut für Anorganische und Analytische Chemie, Corrensstr. 28/30, 48149 Münster, Germany. E-mail: mueller.j@uni-muenster.de
bUniversität Münster, Center for Nanotechnology (CeNTech), Heisenbergstr. 11, 48149 Münster, Germany
cUniversität Münster, Center for Soft Nanoscience (SoN) and Cells in Motion Interfaculty Centre (CiMIC), Corrensstr. 28/30, 48149 Münster, Germany
First published on 27th February 2025
Abstract
A family of 16 platinum(II) complexes was synthesized with the aim of obtaining water-soluble luminescent coordination compounds for guanine quadruplex (G4) binding. The complexes share a common tridentate N^N^C-donor ligand (based on 2-phenyl-6-(1H-1,2,3-triazol-4-yl)pyridine) bearing different substituents for solubilization, and an additional monodentate ancillary ligand (either phenylacetylide or 3-(trimethylammonium)prop-1-yne-1-ide). Single-crystal X-ray diffraction analyses confirm that the substituents do not interfere with the central planar core of the complexes required for π stacking interactions with the DNA. The interaction of the complexes with four DNA oligonucleotides that fold into various G4 topologies was evaluated using luminescence and circular dichroism spectroscopy as well as cryo-ESI mass spectrometry. The data indicate a complex correlation between type of substituent and ability of the complex to interact with G4 DNA.
Introduction
Guanine-rich DNA sequences, which are ubiquitous in the genome, can fold into tetra-stranded helical structures, so-called guanine quadruplexes (G4). G4 consist of guanine tetrads, each of which comprises four guanine residues arranged in a planar pattern, assembled via Watson–Crick and Hoogsteen hydrogen bonding. The G quadruplexes are further stabilized by electrostatic interactions with monovalent cations. K(I) coordinates between two G tetrads, whereas Na(I) mostly coordinates in the plane of a G tetrad (Chart 1).1,2 | ||
| Chart 1 Formation of a guanine tetrad via hydrogen bonds. The grey sphere represents a central cation. | ||
G4 structures form in guanine-rich nucleic acid sequences, which occur in human telomeres, in various oncogene promoter regions, and in non-human genomes as well as in artificial sequences proposed to be relevant in biological mechanisms.3–5 The sequences can adopt different topologies with parallel, antiparallel and hybrid strand orientation.6,7 Telomeres contain a repetitive overhang at the 3′ end of eukaryotic chromosomes, in humans composed of the repeating hexanucleotide sequence d(TTAGGG)n.8 This overhang plays an important role in the control of cell replication and cell division.9 In healthy cells, replication leads to incomplete duplication and the resulting shortening of the telomere sequence. At a critical telomere length, apoptosis is initiated. In 80% of cancer cells, the enzyme telomerase is overexpressed, preventing apoptosis.10,11 The formation of G quadruplexes in the telomere region can inhibit telomerase and thus represents an elegant target for research into new cancer therapeutics.12,13 Likewise, the targeting of G4 in a promoter region is known to be able to suppress the transcription of the respective gene.14 Here, G4 formation is typically suppressed under normal conditions, due to duplex formation. However, molecular crowding or negative superhelicity during transcription can enhance G4 formation,15 as can the addition of suitable small molecules. Hence, the induction and stabilization of G quadruplexes in promoter regions is also an attractive research area in cancer therapy.
Among the molecules that can target quadruplexes, a variety of heteroaromatic ligands have been reported.16–18 They interact through external π stacking, making use of their large aromatic surface, e.g., in porphyrin- or pyrene-based molecules.19,20 Metal complexes based on the salen chelator were also proposed as attractive potential G4 binders.21 They offer the additional possibility of extending the positively charged channel along the G4 helical axis.22,23 A number of metal complexes were investigated as G4 binders, including Mn(II),24,25 Ni(II),26 Cu(II),1 Pd(II),27 Ru(II),28,29 Au(I),30 Ir(III),31 and Pt(II) complexes.1,24,26,32–34 In these complexes, the ligand type goes well beyond that of a salen-based scaffold.
Recently, we showed that attaching a pendant nucleobase to the tridentate N^N^C ligand of a phosphorescent Pt(II) complex structurally related to the ligand under investigation in this project led to unique binding properties with respect to G quadruplexes.35 However, those complexes were not water-soluble and quite bulky. In the present study, luminescent Pt(II) complexes with a tridentate ligand, either mono- or difunctionalized, possibly containing a cationic pendant group, and a sterically non-demanding monodentate auxiliary ligand are investigated (Chart 2). Earlier studies on closely related complexes had shown that they are phosphorescent,36–38 which renders them interesting in the context of a time-gated imaging. Those studies also indicated that the identity of the auxiliary monodentate ligand hardly influences the phosphorescence of the Pt(II) complexes.36 Hence, it can be expected that the use of a sterically non-demanding monodentate ligand in the present study neither affects the phosphorescent properties.
 | ||
| Chart 2 Structural composition of the Pt(II) complexes. The basic structure of the ligand (black), the functionalization sites R and OR′ (blue), the auxiliary ligand L to be varied (green), and the respective counterion (red) are highlighted in a color code. | ||
The development of luminescent G4 binders is of particular interest, because they often function as luminescence “turn-on” probes upon interaction with a nucleic acid,34,39,40 allowing visualization of DNA even in cells.33,41 Alternatively (or additionally), a change in luminescence lifetime may be used to detect an interaction with G4 DNA.33,42,43 The light-switch effect occurring upon interaction of a potentially luminescent compound with a nucleic acid is based on the deactivation of the excited state by water. When interacting with a nucleic acid, the compound is shielded from the solvent and its intrinsic luminescence is restored.44
In the present study the combination of an enlarged π system and the possibility of engaging in hydrogen bonding interactions involving the pendant groups and the auxiliary ligand was expected to lead to a better target selectivity. A particular focus is given to the functionalization of the tridentate ligand, aiming at a high water-solubility of the final complex. The interaction of this family of Pt(II) complexes with four distinct G4 DNA structures was investigated by circular dichroism (CD) spectroscopy, a Förster resonance energy transfer (FRET) based melting assay, fluorescence intercalator displacement studies, and isothermal titration calorimetry (ITC).
Due to the structural complexity of the ligands and metal complexes presented herein, the IUPAC nomenclature was replaced by an independent simplified labelling. In this context, the monodentate auxiliary ligands phenylacetylide and 3-(trimethylammonium)prop-1-yne-1-ide are termed A and B, respectively. The functional groups attached to the ligand precursor (LH) are represented by superscript abbreviations, depending on the substituent(s): 3-chloropropyl (Cl), 3-bromopropyl (Br), 3-hydroxypropyl (OH), 3-morpholinopropyl (M), phosphate ester (P).
The four quadruplex-forming sequences H-telo, c-myc22, bcl2-mid and c-kit were selected for this study. They all contain three stacked guanine tetrads but adopt different topologies, they were structurally characterized, and are potential therapeutic targets. In particular, H-telo is based on the human telomeric repeat sequence d(TTAGGG). It can adopt various topologies depending on experimental conditions. Under the conditions applied in this study, it forms a hybrid structure with parallel and antiparallel strand orientation and a characteristic T![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :A:T cap.45 The other three sequences are derived from the promotor regions of the genes c-MYC, BCL2, and c-KIT. c-MYC is an oncogene that can be silenced via the formation of a G4 structure in its promotor region.46 The c-myc22 sequence investigated here is a double mutant (c-myc22-G14T/G23T) known to adopt a parallel fold.2 The bcl2-mid sequence represents a truncated and partially mutated excerpt from the promotor region of the human BCL2 gene. It adopts a hybrid structure under the experimental conditions used here.47 Finally, c-KIT is a proto-oncogene with a guanine-rich promotor region. The c-kit sequence investigated here (also referred to as c-kit87up because it is located 87 nucleotides upstream of the transcription start site) adopts a parallel fold.48 A peculiarity of this structure is the fact that it contains an isolated guanosine in one of the tetrads, whereas these are normally formed from three-guanine tracts. An overview of the structures is given in Fig. 1.
:A:T cap.45 The other three sequences are derived from the promotor regions of the genes c-MYC, BCL2, and c-KIT. c-MYC is an oncogene that can be silenced via the formation of a G4 structure in its promotor region.46 The c-myc22 sequence investigated here is a double mutant (c-myc22-G14T/G23T) known to adopt a parallel fold.2 The bcl2-mid sequence represents a truncated and partially mutated excerpt from the promotor region of the human BCL2 gene. It adopts a hybrid structure under the experimental conditions used here.47 Finally, c-KIT is a proto-oncogene with a guanine-rich promotor region. The c-kit sequence investigated here (also referred to as c-kit87up because it is located 87 nucleotides upstream of the transcription start site) adopts a parallel fold.48 A peculiarity of this structure is the fact that it contains an isolated guanosine in one of the tetrads, whereas these are normally formed from three-guanine tracts. An overview of the structures is given in Fig. 1.
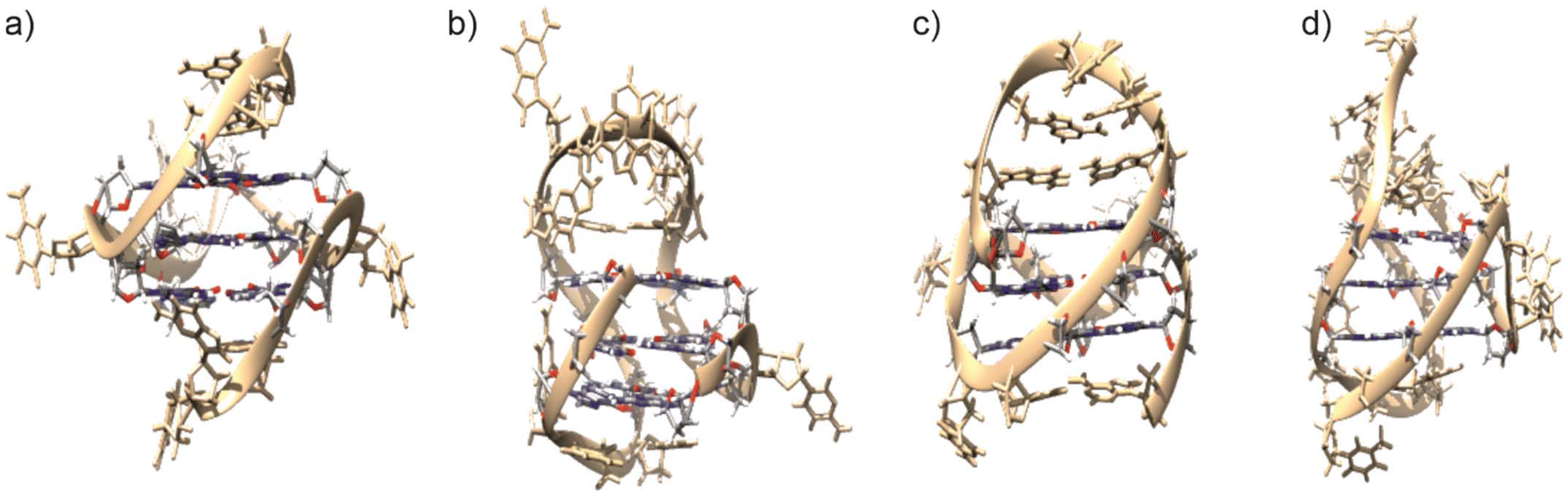 | ||
| Fig. 1 Experimental structures of the G4 sequences used in this study as previously reported for K+-containing solutions. (a) c-myc22,2 (b) bcl2-mid,47 (c) c-kit,48 (d) H-telo.45 This figure was created using UCSF Chimera49 and is reprinted from reference.50 The structures were obtained from the PDB accession codes 1XAV, 2F8U, 2O3M, and 2JPZ, respectively. | ||
Results and discussion
Synthesis of the ligand precursors
The synthesis of all ligand precursors starts from 2-ethynyl-6-phenylpyridine 1 (Scheme 1). This compound was previously reported by Hebenbrock et al.36 In three independent reactions, 1 was converted with diethyl (2-azidoethyl)phosphonate, 4-(3-azidopropyl)morpholine, and 3-azidopropan-1-ol to give compounds PLH, MLH, and OHLH, respectively. OHLH was subsequently converted to the chloroalkyl-containing ligand precursor ClLH by direct reaction of the hydroxy functionality with thionyl chloride. | ||
| Scheme 1 a) Diethyl (2-azidoethyl)phosphonate, general procedure for click reaction; (b) 4-(3-azidopropyl)morpholine, general procedure for click reaction; (c) 3-azidopropan-1-ol, general procedure for the click reaction; (d) SOCl2, CH2Cl2, 0 °C, o/n. | ||
The difunctionalized ligand precursors were synthesized starting from 4-bromophenol. Initially, 4-bromophenol was protected with 3,4-dihydro-2H-pyran in an acid-catalysed reaction.51 It was then transformed into a boronic acid by bromine lithium exchange and reaction with triisopropyl borate. Finally, a Suzuki cross-coupling reaction with 2,6-dibromopyridine followed by cleavage of the tetrahydropyranyl ether (under acidic conditions by means of concentrated hydrochloric acid) gave compound 2 in two steps (Scheme 2).52 In a Sonogashira cross-coupling reaction, compound 2 was reacted with trimethylsilyl acetylene. 4-(6-Ethynylpyridin-2-yl)phenol 3 was obtained after cleavage of the silyl protecting group under basic conditions. To access the diphosphate ester ligand precursor PLHP, intermediate 4 was formed from compound 3 and diethyl (2-azidoethyl)phosphonate in a Cu(I)-catalysed Huisgen reaction. Subsequently, the phenolic hydroxy group was functionalized to give PLHP.
 | ||
| Scheme 2 a) 3,4-Dihydro-2H-pyran, HCl, rt, 2 h; (b) (1) nBuLi, B(OiPr)3, −78 °C, dry THF; (2) 2,6-dibromopyridine, K2CO3, PPh3, Pd(OAc)2, 1,2-dimethoxyethane, H2O, 110 °C, 18 h; (c) (1) TMS acetylene, CuI, [PdCl2(PPh3)2], dry THF, dry NEt3, 80 °C, o/n; (2) NaOH (1 M), THF, MeOH, rt, 2 h; (d) diethyl (2-azidoethyl)phosphonate, general procedure for the click reaction; (e) (diethyl phosphoric) trifluoromethanesulfonic anhydride, K2CO3, ACN, 18 h, reflux. | ||
To increase the diversity of difunctionalized ligand precursors, the phenolic hydroxy group of compound 2 was functionalized with 3-chloropropan-1-ol in the presence of K2CO3 and CuI (for halogen exchange on 3-chloropropan-1-ol to increase the leaving-group capability) to give intermediate 5 (Scheme 3).53 A Sonogashira cross-coupling reaction with trimethylsilyl acetylene and subsequent deprotection under basic conditions with aqueous NaOH solution yielded compound 6. As reported above for the monofunctionalized ligand precursor OHLH, a Cu(I)-catalysed Huisgen reaction with 3-azidopropan-1-ol resulted in the formation of the ligand precursor OHLHOH. Starting from OHLHOH, the dichlorinated species ClLHCl was synthesized by means of thionyl chloride, and the dibrominated species 7 with N-bromosuccinimide. Starting from compound 7, ligand precursor MLHM containing two pendant morpholine moieties was obtained by nucleophilic substitution with morpholine in the presence of K2CO3. Under physiological pH conditions, this compound is expected to be protonated and thereby converted into a cation.
 | ||
| Scheme 3 a) K2CO3, CuI, 3-chloropropan-1ol, 3 d, reflux; (b) (1) TMS acetylene, CuI, [PdCl2(PPh3)2], dry THF, dry NEt3, 80 °C, o/n; (2) NaOH (1 M), THF, MeOH, rt, 2 h; (c) 3-azidopropan-1-ol, CuSO4·5 H2O, Na ascorbate, tBuOH, H2O, THF, rt, o/n; (d) SOCl2, CH2Cl2, 0 °C, o/n; (e) PPh3, N-bromosuccinimide, dry DCM, 0 °C, o/n; (f) morpholine, K2CO3, ACN, reflux, o/n. | ||
Synthesis of the platinum(II) complexes
The complexation with Pt(II) was carried out by employing K2PtCl4 in boiling acetic acid under an inert atmosphere to obtain [PtCl(ML)] and [PtCl(MLM)]. For the syntheses of the other complexes, a mixture of water and 2-ethoxyethanol was used as solvent.54 The ancillary chlorido ligand was easily exchanged either via a salt elimination reaction with silver phenylacetylide or by a Sonogashira-like transfer of N,N,N-trimethyl-N-propargylammonium triflate.Molecular structures of selected platinum complexes
Molecular structures were determined for [Pt(A)(ML)] and [Pt(A)(ClL)] using single-crystal X-ray diffraction analysis (see ESI, Table S1,† for crystallographic details). As can be seen from Fig. 2a and b, the substituents do not interfere with the central planar core of the complexes required for π stacking interactions with G4 DNA (in contrast to the behaviour seen in a related earlier study).35 The phenyl moiety of the ancillary phenylacetylido ligand it oriented almost perpendicular with respect to the square-planar coordination environment of the Pt(II) ion (62.8(1)° and 79.71(5)°, respectively). Hence, it can be speculated that this moiety may serve as some kind of anchor when the complex binds non-covalently to G4 DNA. In case of the complexes with disubstituted ligands, molecular structures were determined for the intermediates [PtCl(PLP)] and [PtCl(OHLOH)] bearing a chlorido ancillary ligand (Fig. 2c and d). These complexes were not evaluated with respect to their ability to bind non-covalently to DNA because chlorido ligands are known to dissociate from Pt(II) complexes, enabling the (at this point undesired) formation of Pt–DNA bonds.55 Nevertheless, the complexes represent valid structural models for the corresponding complexes with ancillary ligands A and B. Hence, it can be concluded that again the substituents do not interfere with the central planar core of the complexes required for π stacking interactions with G4 DNA. Table 1 presents an overview of bond lengths involving the Pt(II) ion. These bond lengths are in the expected range.56 | ||
| Fig. 2 Molecular structures of (a) [Pt(A)(ML)], (b) [Pt(A)(ClL)] (a co-crystallized CH2Cl2 molecule is omitted for clarity), (c) [PtCl(PLP)], and (d) [PtCl(OHLOH)] (a co-crystalized DMF molecule (half a molecule in the asymmetric unit) is omitted for clarity) in the crystal. Displacement ellipsoids are drawn at the 50% probability level. H atoms are omitted for clarity. | ||
DFT calculations
To correlate the ability of the various complexes to engage in intermolecular interactions with their structural composition, the charge balance parameter ν (ranging from 0.000 to 0.250) was determined by means of DFT calculations.57,58 A large ν value indicates a tendency for engaging in an intermolecular interaction involving both the positive and negative regions to a similar extent. For the complexes functionalized with morpholine, the respective protonated complexes were also evaluated. Direct comparison of the ν values as a function of auxiliary ligands A and B (or of the charge density) reveals a clear trend. Auxiliary ligand A appears to strongly promote intermolecular interactions between individual complexes. With ν values between 0.227 and 0.250, an intermolecular interaction is clearly preferred compared to complexes with the auxiliary ligand B, which have ν values of merely 0.000 to 0.035. In addition to the auxiliary ligand, the charge of the complex plays a central role. The complexes with auxiliary ligand A and bearing a protonated morpholine moiety display a ν value between 0.000 and 0.015 and hence do not favour intermolecular interactions. Finally, in addition to the auxiliary ligand and the charge of the complex, the steric demand plays an important role. With increasing steric demand, the ν value decreases from 0.250 for [Pt(A)(ClLCl)] to 0.227 for [Pt(A)(PLP)] within the family of complexes with the auxiliary ligand A (Table 2). In summary, these data suggest that complexes bearing this auxiliary ligand are more likely to aggregate in solution.

|
To complement this insight, the molecular polarity index (MPI) was calculated for each complex. The MPI clearly correlates with the charge of the complex (see ESI, Table S2†). Moreover, complexes with auxiliary ligand A have a smaller MPI than the respective complexes with auxiliary ligand B, indicating the increased polarity in the latter case. In general, these data suggest that the complexes should be soluble in polar solvents. In fact, complexes bearing auxiliary ligand A turned out to be soluble in DMSO, whereas complexes with auxiliary ligand B were sufficiently water-soluble to prepare 750 μM aqueous stock solutions (except for [Pt(B)(OHLOH)]OTf, which was soluble in DMSO).
UV/vis spectroscopic and photoluminescence studies of the complexes
All complexes were characterized by UV/vis absorption and photoluminescence spectroscopies (λexc = 340 nm) to study their possible aggregation in concentrated solutions. The complexes absorb strongly in the wavelength region from 240–400 nm (see ESI, Fig. S1 and S2†). Based on the results obtained for closely related complexes, the absorbance around 270 nm can be attributed to allowed transitions into 1π–π* states, whereas the bands at 300–400 nm are attributed to the excitation into mixed 1MLCT/LC states.36,59–62As anticipated from earlier studies on closely related complexes bearing an unsubstituted tridentate ligand,35–38 the Pt(II) complexes are luminescent (see ESI, Fig. S3–S10†). They show an emission band centring around 500 nm with vibrational progression at longer wavelengths. Fig. 3 exemplarily displays concentration-dependent luminescence spectra of [Pt(A)(ML)]. At low concentrations, the main emission band at 500 nm and its vibrational progression are clearly discernible. For [Pt(A)(ML)] and a few other complexes, the formation of aggregates in aqueous solution at concentrations >15 μM can be deduced from the appearance of an additional broad red-shifted emission band at ca. 620 nm, attributed to 3MMLCT states. For some complexes, an additional emission band at ca. 400 nm is observed (vide infra). A general observation is that the identity of the auxiliary monodentate ligands hardly influences the luminescence of the complexes, thereby extending this observation to complexes with different substitution patterns on the tridentate ligand.
 | ||
| Fig. 3 Concentration-dependent emission spectra of [Pt(A)(ML)]. Conditions: metal complex (1.5–90 μM), 20 °C, λexc = 340 nm. | ||
With a potential application as probe for G4 structures in mind, the photoluminescence quantum yields of the complexes were determined. Almost all complexes bearing the auxiliary ligand A had to be dissolved in DMSO to reach the concentration necessary for performing these measurements. As a result, their quantum yields could not be determined due to quenching by the solvent. As can be seen from the overview given in Table 3, the quantum yields of the other complexes in deaerated water reach up to 36%, rendering the compounds interesting for imaging applications.
| Compound | Φ PL/air | Φ PL/Ar |
|---|---|---|
| a Conditions: Ar atmosphere, λexc = 350 nm. b Measured in DMSO. c Measured in a 10% DMSO aqueous solution. d Measured in water. e Not determined. | ||
| [Pt(A)(ClL)]b | <2 | <2 |
| [Pt(A)(ClLCl)]b | <2 | <2 |
| [Pt(A)(ML)]b | <2 | <2 |
| [Pt(A)(MLM)]b | <2 | <2 |
| [Pt(A)(OHL)]b | <2 | <2 |
| [Pt(A)(OHLOH)]c | 3 ± 2 | 14 ± 2 |
| [Pt(A)(PL)]b | <2 | <2 |
| [Pt(A)(PLP)]b | <2 | <2 |
| [Pt(B)(ClL)]OTfd | 4 ± 2 | 35 ± 4 |
| [Pt(B)(ClLCl)]OTfd | 4 ± 2 | 18 ± 2 |
| [Pt(B)(ML)]OTfd | 3 ± 2 | 25 ± 3 |
| [Pt(B)(MLM)]OTf | n.d.e | n.d.e |
| [Pt(B)(OHL)]OTfd | 4 ± 2 | 28 ± 3 |
| [Pt(B)(OHLOH)]OTfd | <2 | 20 ± 2 |
| [Pt(B)(PL)]OTfd | 6 ± 2 | 36 ± 4 |
| [Pt(B)(PLP)]OTfd | 3 ± 2 | 32 ± 3 |
In general, the experimental luminescence-spectroscopic data obtained for the monofunctionalized complexes agree well with the conclusions drawn from the calculated ν values. Unexpectedly, among the monofunctionalized complexes bearing auxiliary ligand B, the compound [Pt(B)(PL)]OTf does aggregate to a certain extent under the experimental conditions (see ESI, Fig. S10†), contrasting theoretical expectations (ν = 0.011). In all other cases, the quaternary amine with its positive charge prevents the aggregation of the metal complexes with auxiliary ligand B when compared to the corresponding complexes bearing auxiliary ligand A (see ESI, Fig. S3–S10†).
An aggregation was not observed experimentally for the difunctionalized complexes, which is in agreement with computational predictions. Among these complexes, merely [Pt(A)(ClLCl)] self-aggregates, as deduced from the pronounced absorbance at >600 nm, even though there is a decrease in intensity when increasing the concentration beyond 60 μM.
Surprisingly, a further prominent emission band at ca. 400 nm is detected for all difunctionalized Pt(II) complexes bearing auxiliary ligand A, growing in intensity with increasing concentration and partially overlapping with the band of the Raman scattering of water at 362 nm (see ESI, Fig. S3–S10†). To understand whether this emission is an intrinsic property of the metal complexes or whether it is caused by the presence of residual amounts of an unknown impurity, the Pt(II)-free ligand precursors were also evaluated with respect to their emission properties, applying the maximum concentration of 90 μM used in the complex studies. Their spectra (see ESI, Fig. S11–S14†) show that the monodentate ligand precursors do not luminesce, whereas the difunctionalized ones weakly emit at ca. 400 nm, with the emission in part being superimposed by the excitation (λexc = 340 nm). While this could imply the presence of small amounts of unreacted ligand precursor in the samples of the difunctionalized metal complexes, it does not explain that the appearance of the emission band at ca. 400 nm can in some cases also be evoked by the addition of G4 DNA (vide infra). Moreover, the complexes are pure as evidenced by NMR spectroscopy and elemental analysis. Time-dependent 1H NMR spectroscopy also confirms their stability in solution for several days (see ESI, Fig. S15–S28†), ruling out any decomposition or a possible adduct formation. Hence, the source of the unexpected emission at ca. 400 nm remains unclear for the time being.
Photoluminescence studies of the complexes in the presence of G4 DNA
The water-soluble Pt(II) complexes were developed as potential agents for the visualization of G4 DNA. Therefore, their photoluminescence spectra in the presence of the G4 DNA and dsDNA (ds26, acting as a double-stranded reference) were investigated. In most cases, the addition of an oligonucleotide to the complex leads to an increase in its luminescence at ca. 500 nm (Fig. 4; see ESI, Fig. S29–S44† for a compilation of all data). Table 4 gives an overview of the increase in luminescence intensity induced by the presence of the respective oligonucleotide. Neither the identity/number of the substituents on the tridentate nor the identity of the auxiliary monodentate ligand appears to correlate with the luminescence change. Nevertheless, the following observations can be made. The emission of [Pt(A)(ClL)] and [Pt(A)(ClLCl)] is particularly influenced by c-mcy22 and bcl2-mid, whereas that of [Pt(A)(MLM)] increases significantly in the presence of any of the investigated oligonucleotides. The luminescence of [Pt(B)(PL)]OTf is particularly strongly influenced by H-telo. Among the G4 quadruplexes investigated here, c-kit is the one with the smallest effect on the luminescence of the Pt(II) complexes and merely increasing the luminescence of [Pt(A)(MLM)] and [Pt(B)(PL)]OTf. | ||
| Fig. 4 Emission spectra of (a) [Pt(A)(ClLCl)] and (b) [Pt(A)(ClL)] upon stepwise addition of c-myc22 (0–12 equiv.). Emission spectrum of [Pt(B)(OHL)]OTf upon stepwise addition of (c) c-kit and (d) H-Telo (0–12 equiv.). Conditions: metal complex (1 μM), LiCl (90 mM), KCl (10 mM), MOPS (5 mM, pH 7.4), 20 °C, λexc = 340 nm. | ||
| a Change in luminescence determined as the ratio of the emission in the presence of 12 equiv. of oligonucleotide and the emission in the absence of oligonucleotide. b Determined at 400 nm. c Determined at 500 nm (complexes with monofunctionalized ligand) and 525 nm (complexes with difunctionalized ligand), respectively. |
|---|

|
Interestingly, the addition of DNA in most instances evokes the appearance of a new emission band at ca. 400 nm, similar to the unexpected emission seen for all difunctionalized Pt(II) complexes bearing auxiliary ligand A (vide supra). However, here the presence of this band is not limited to these Pt(II) complexes but also appears for monofunctionalized complexes and for complexes with auxiliary ligand B. While c-kit again exerts the least influence, ds26 is affecting the luminescence of almost all complexes at ca. 400 nm.
Further experiments were carried out to characterize the relevant emission bands at ca. 400 nm and at >500 nm in more detail. For this purpose, the emission spectra of selected complexes were recorded in the absence and presence of bcl2-mid. This sequence was selected out of the four quadruplexes because it shows relevant interactions with several of the complexes when taking into consideration all characterization methods applied in this study. An excited state lifetime in the ns range was measured for the luminescence at ca. 400 nm (emission from the singlet manifold), whereas the lifetime of the emission at ca. 510 nm was in the μs range (stemming from the lowest triplet state). Based on these results, it can be concluded that the emission at ca. 400 nm is due to fluorescence, whereas phosphorescence occurs at wavelengths >500 nm. It is noteworthy that no lifetimes could be measured for the ligand precursor, indicating that the ligand precursor is not responsible for the fluorescence of the difunctionalized complexes. It is also interesting to note that the phosphorescence lifetime of [Pt(A)(ML)] increases about threefold in the presence of bcl2-mid (Table 5), clearly indicating an interaction between metal complex and DNA.
| Compound | τ (Ar) (ns) @ ca. 400 nm | τ (Ar) (μs) @ ca. 510 nm |
|---|---|---|
| a Conditions: Ar atmosphere, 10 μM solution, λexc = 340 nm. b Value could not be determined due to the weak signal. c λ em = 410 nm. d λ em = 520 nm. e λ em = 510 nm. f λ em = 415 nm. Amplitude-weighted average lifetimes are indicated along with the individual components in square brackets and their relative amplitudes in parentheses. The corresponding original data, together with the fitting parameters, are given in the ESI (Fig. S45–S49).† | ||
| ClLHCl | n.d.b | n.d.b |
| [Pt(A)(ClLCl)] | 10.76 ± 0.07c [13.82 ± 0.05 (93%), 2.9 ± 0.1 (7%)] | 2.02 ± 0.01d [3.55 ± 0.04 (88%), 0.50 ± 0.01 (12%)] |
| [Pt(A)(ML)] | n.d.b | 1.37 ± 0.03e |
| bcl2-mid·[Pt(A)(ML)] | 4.71 ± 0.04f [12.37 ± 0.09 (63%), 2.29 ± 0.02 (37%)] | 3.82 ± 0.01e [6.20 ± 0.04 (74%), 1.84 ± 0.02 (26%)] |
CD spectroscopy
CD spectroscopy was used to study the conformation of the four G-DNA quadruplexes (c-myc22, bcl2-mid, c-kit, and H-telo) and the double-stranded DNA (ds26), and to investigate conformational changes in the presence of the Pt(II) complexes. In most cases, changes in the CD spectrum were found to be within experimental uncertainty (see ESI, Fig. S50–S65†). For c-myc22, H-telo, and ds26, no relevant changes are observed in the CD spectra upon addition of most of the complexes. In contrast, statistically relevant differences are found when adding some of the complexes to bcl2-mid, and to a lesser extent to c-kit. It should be noted that the absence of any change in the CD spectra does not necessarily rule out an interaction of DNA and metal complex. Likewise, CD-spectroscopic changes do not need to indicate structural changes, they may also arise from the formation of a new chromophore upon DNA/complex interaction. Table 6 presents a general overview of how the addition of the respective metal complexes influences the molar ellipticity in the CD spectra of the DNA. Clearly, bcl2-mid and c-kit are the quadruplexes whose CD spectra are most affected by the Pt(II) complexes. Two selected examples will be discussed in the following.| a Only changes with absolute values exceeding 30% are shown to exclude irrelevant changes in rather noisy regions of the spectrum. |
|---|

|
Fig. 5a shows the CD spectrum of c-myc22 in the presence of increasing amounts of [Pt(B)(ML)]OTf, i.e., the only complex significantly influencing the CD spectrum of c-myc22. The spectra display a gradual decrease of the positive Cotton effect at 266 nm in the presence of the complex, clearly indicating an interaction between complex and DNA. Similarly, [Pt(B)(PLP)]OTf affects bcl2-mid (Fig. 5b). Here, a complete flattening of the signal is observed, indicating an unfolding of the quadruplex in the presence of the complex. The minor peak appearing at ca. 358 nm is outside the wavelength region of G4-based peaks.63 It is likely an induced circular dichroism due to an interaction of the metal complex with the unfolded (yet chiral) oligonucleotide. Interestingly, this appears to be the only combination of G4 DNA and Pt(II) complex is this study where such an effect is observed (see ESI, Fig. S50–S65†).
 | ||
| Fig. 5 CD spectra of (a) c-myc22 upon stepwise addition of [Pt(B)(ML)]OTf and (b) bcl2-mid upon stepwise addition of [Pt(B)(PLP)]OTf (0–5 equiv.). Conditions: DNA (3 μM), LiCl (90 mM), KCl (10 mM), MOPS (5 mM, pH 7.4), 20 °C. | ||
Table 6 indicates that an increase in molar ellipticity upon the addition of a complex is observed almost exclusively for complexes bearing auxiliary ligand A. In contrast, interactions with complexes containing auxiliary ligand B mostly lead to decreasing molar ellipticities, indicating either less neatly stacked tetrads or the onset of an unfolding process.
FRET
Due to the changes in the photophysical properties of the Pt(II) complexes in the presence of bcl2-mid as well as its significant structural changes in the presence of selected complexes (as evident from the CD spectra, vide supra), the thermal stability of bcl2-mid was evaluated experimentally. Temperature-dependent Förster resonance energy transfer (FRET) measurements were therefore carried out using a suitably modified bcl2-mid sequence (i.e., with terminally appended carboxyfluorescein (FAM) and DABCYL moieties).64 However, the CD spectrum of FAM-bcl2-mid-DABCYL differs significantly from that of unmodified blc2-mid. Instead of adopting a hybrid topology, the labelled sequence folds into a parallel quadruplex structure, as the close resemblance of its CD spectrum with that of c-myc22 shows (Fig. S66†). Such a parallel fold is known as one possible structure of native bcl2.65 Hence, even though the results of the FRET melt assay cannot be directly correlated with the other data collected for bcl2-mid in this study, they are of potential biological relevance, as they exemplify the interaction of the Pt(II) complexes with a parallel quadruplex structure. Reference measurements in the absence of DNA confirmed that no fluorescence interference arises from the compounds themselves when excited under identical conditions (data not shown). Luminescence spectra were recorded in the absence and presence of five equiv. of the respective metal complex. In most cases, the addition of the metal complex does not influence the melting temperature Tm of FAM-bcl2-mid-DABCYL, with Tm amounting to 62 °C both in the absence and the presence of the metal complex. Amongst the complexes bearing auxiliary ligand B, only those with a phosphate ester or a hydroxy group lead to an increase in Tm. In addition, the melting temperature in the presence of the monofunctionalized complexes [Pt(B)(PL)]OTf and [Pt(B)(OHL)]OTf increases to a larger extent compared to the respective difunctionalized complexes. For example, Tm increases from an initial 62 °C by 8.8 °C to 70.8 °C (Fig. 6a) upon the addition of 5 equiv. of [Pt(B)(PL)]OTf. With [Pt(B)(OHL)]OTf, the largest increase in melting temperature of any of the monofunctionalized complexes was observed, namely by 15.1 °C to 77.1 °C. The corresponding difunctionalized complexes induce no significant melting point increase ([Pt(B)(PLP)]OTf) and a minor increase of only 1.0 °C ([Pt(B)(OHLOH)]OTf), respectively. Among the difunctionalized complexes, [Pt(A)(PLP)] and [Pt(A)(MLM)] show the largest increase in Tm, with a ΔTm of 4.1 °C and 21.8 °C, respectively. Fig. 6b shows the change in Tm on the addition of the metal complexes. A compilation of all melting curves can be found in the ESI (Fig. S67–S74).† | ||
| Fig. 6 (a) Melting curves of FAM-bcl2-mid-DABCYL in the absence (black) and presence (red) of 5 equiv. of [Pt(B)(PL)]OTf; (b) melting point increase (ΔTm) of FAM-bcl2-mid-DABCYL upon the addition of 5 equiv. of the respective metal complexes. Conditions: DNA (0.2 μM), LiCl (95 mM), KCl (5 mM), MOPS (5 mM, pH 7.4), λexc = 490 nm, λem = 520 nm. | ||
Interestingly, no obvious correlation exists between the stabilizing effect of a metal complex and its influence on the CD spectrum of G4 DNA. For example, [Pt(A)(MLM)] exerts the highest stabilizing effect on G4 DNA (Fig. 6b) but does not seem to influence their topology at all (Table 6). Similarly, [Pt(B)(PLP)]OTf strongly influences the CD spectrum of bcl2-mid (Fig. 5b), but its presence does not lead to a change of the melting temperature of this G4 DNA (see ESI, Fig. S74†). It can therefore be concluded that both, structural changes and thermal G4 DNA stabilization, should be considered independent indicators for an interaction between DNA and metal complex.
Cryo-ESI-MS
To gain further information on the interaction capacity and adduct formation ability between the Pt(II) complexes and guanine quadruplex oligonucleotide sequences, their mixtures were examined by cryo-electrospray ionization mass spectrometry (cryo-ESI-MS). This technique also enables an estimation of the stoichiometry between the bound complexes and the respective folded secondary structures. The ultra-mild ionization conditions were chosen to be able to detect weak non-covalent interactions and to improve the signal-to-noise ratio. Due to limitations elicited by the experimental conditions, i.e., the use of solutions of KCl, LiCl, and the volatile triethylammonium acetate (pH 7.0) in a mixture of water and acetonitrile (1:1, v/v), only complexes bearing the auxiliary ligand B could be analysed, as all other complexes are poorly soluble in water or acetonitrile at the concentrations required to prepare the stock solutions. Using the combination of c-myc22 and [Pt(B)(ML)]OTf as an example, a general trend of binding affinity and binding stoichiometry was established. At a ratio of 1:1 (DNA:complex), barely any signal is detectable that could be assigned to an adduct of DNA and complex. When the amount of DNA is doubled (giving a 2:1 ratio), a 1:1 adduct of DNA and complex can clearly be observed. Likewise, in the presence of excess complex (1:2 ratio), both 1:1 and 1:2 adducts are present. It can therefore be concluded that both 1:1 and 1:2 binding stoichiometries are possible. Moreover, the 1:1 adduct appears to be more stable than the 1:2 adduct (Fig. 7).66
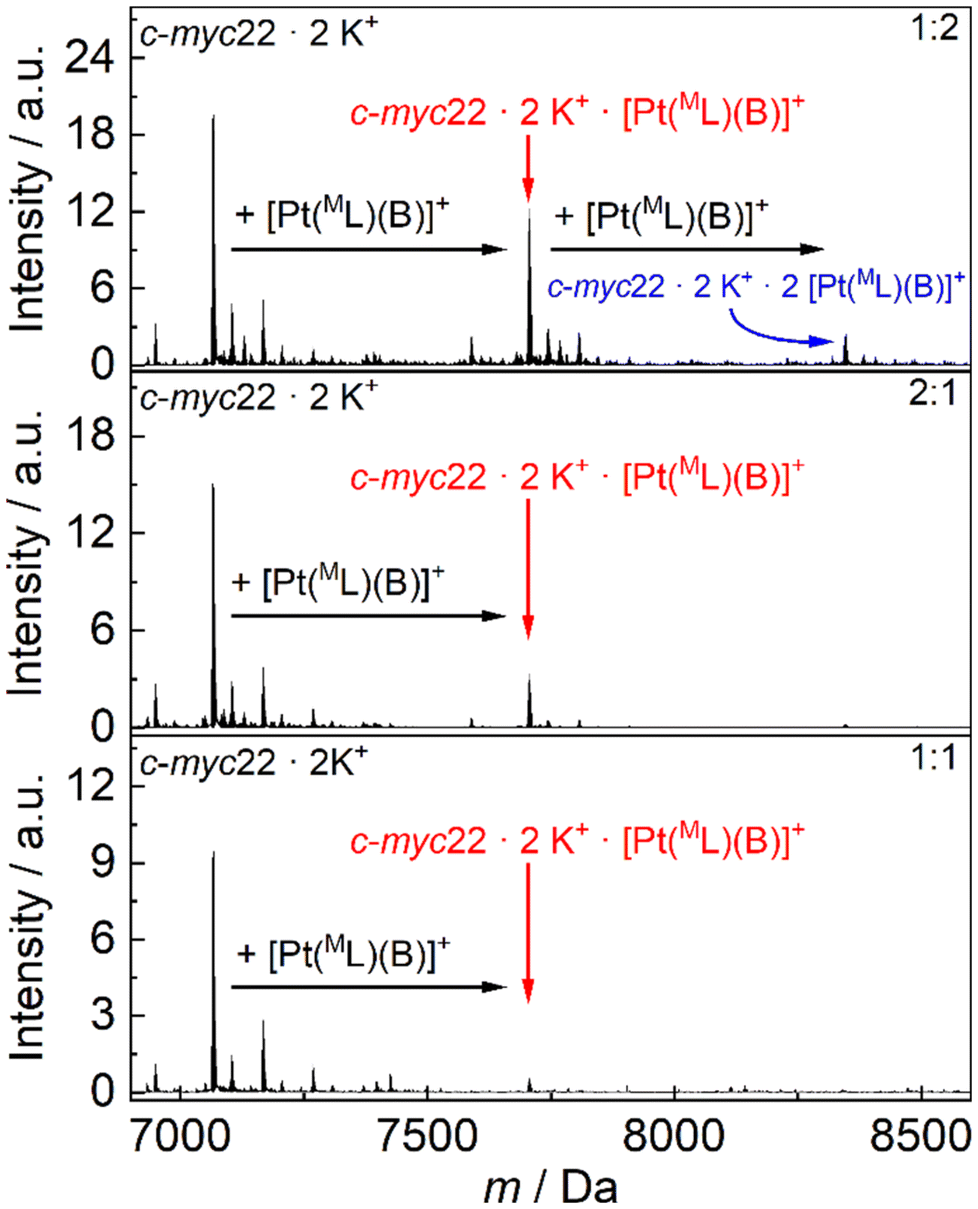 | ||
| Fig. 7 Deconvoluted cryo-ESI-MS spectra of c-myc22 and [Pt(B)(ML)]OTf in DNA:complex ratios of 1:1 (bottom), 2:1 (middle) and 1:2 (top). | ||
In all possible combinations of DNA and complex (or, as a reference measurement, of DNA and ligand precursor), a binding stoichiometry of 1:3 was never detected (data not shown), indicating a scarcity of binding sites on the DNA. Likewise, sandwich-type adducts of a 2:1 stoichiometry (DNA:complex) were not detected in any of the measurements (data not shown). In summary, the cryo-ESI-MS data confirm that an interaction of the complexes with DNA is possible. On the other hand, the fact that adducts of different stoichiometry are present simultaneously prevents a meaningful estimation of binding constants based on the data obtained in the spectroscopic titration experiments.
Unfortunately, additional experiments to quantify the affinity of the complexes and G4 DNA by means of isothermal titration calorimetry (ITC) or by thiazole orange (TO) displacement assay failed to produce meaningful data. In case of ITC, solubility issues negatively influenced the measurements. In addition, all complexes with auxiliary ligand A are only poorly soluble in water. The high concentrations required for the ITC titrations could only be achieved in DMSO. Here, the large heat of dilution of this solvent prevented any meaningful measurement (data not shown). Likewise, the heat involved with the reaction of the complexes with auxiliary ligand B (which could be provided as an aqueous solution) was too small in the applied concentration range, so that the titration data could not be fitted (data not shown). Based on the concentrations used in the experiments (150 μM bcl2-mid, 750 μM metal complex), a very rough estimation places the binding constant Ka in the range of 102 M−1 to 103 M−1, i.e., relatively low.67
In the fluorescent intercalator displacement assay, the displacement of TO from the DNA (as monitored by an increase in fluorescence at 540 nm) upon the addition of increasing amounts of the respective metal complex can be used to quantify the relative affinities of TO and metal complex to the DNA.68 Using this assay, all complexes show a rather low affinity towards G4 DNA compared to thiazole orange (see ESI, Fig. S75–S82†). In fact, most complexes do not even displace half of TO from the DNA at a complex concentration of 30 μM. A comparison with the porphyrin-derived tetracationic G4 binder TMPyP4, of which only 0.106 μM are required to achieve this effect,68 indicates the low affinity of the complexes.
Experimental and computational details
Materials and methods
Anhydrous solvents were distilled, dried, and stored following standard protocols. Silica gel column chromatography was carried out by using Silica-60, having a particle size of 40–63 μm, obtained from VWR. A particle size of 63–200 μm was used for column chromatography with aluminium oxide (MP Alumina N – Super I, neutral pH) from MP Biomedicals. Silver phenylacetylide,70 2-ethynyl-6-phenylpyridine (1),36 2-(1-(3-bromopropyl)-1H-1,2,3-triazol-4-yl)-6-phenylpyridine (2),71 4-(6-bromopyridin-2-yl)phenol (3),72MLH,71OHLH,36 [PtCl(OHL)],73 trimethylpropargylammonium hexafluorophosphate,74 and trimethylpropargylammonium triflate74 were synthesized according to literature methods. All other reagents were obtained commercially and used as supplied. All DNA solutions were heated to 85 °C for 5 min and then stored at 4 °C.
:1, v/v) consisting of DNA (30 μM), KCl (1 μM), and tetraethylammonium acetate (50 μM, pH 7.0) was used for the measurements. The data were processed according to the deconvolution algorithm “Maximum Entropy”.76
Photoluminescence lifetimes were recorded in TCSPC mode with a PicoHarp 300 with a minimum base resolution of 4 ps or in MCS mode with a TimeHarp 260. Emission and excitation spectra were corrected for the intensity of the radiation source (lamp and grating) using standard correction functions. The analysis of the lifetimes of the excited states were performed with the commercially available EasyTau 2 software from PicoQuant. The quality of the data fit was ensured by minimizing the reduced chi-square function (χ2) and by manually checking the weighted residuals and their autocorrelation. Solvents used were of spectrometric quality (Uvasol®, Merck).
Photoluminescence quantum yields were measured with a Hamamatsu Photonics absolute PL quantum yield measurement system (C9920-02) equipped with a L9799-01 CW Xe light source (150 W), a monochromator, a C7473 photonic multichannel analyser, an integrating sphere and employing U6039-05 software (Hamamatsu Photonics, Ltd, Shizuoka, Japan).
:0.4). Molecular graphics were created using the program ORTEP-3 for Windows.81,82
Synthetic procedures
:ethyl acetate, 8:2). The product was obtained as white solid. Yield: 87% (152 mg, 0.510 mmol). 1H NMR (400 MHz, CDCl3): δ (ppm) = 8.29 (s, 1H, H-15), 8.12 (d, J = 7.8 Hz, 1H, H-8), 8.10–8.03 (m, 2H, H-3), 7.84 (t, J = 7.8 Hz, 1H, H-9), 7.68 (d, J = 7.9 Hz, 1H, H-10), 7.53–7.46 (m, 2H, H-2), 7.45–7.40 (m, 1H, H-1), 4.62 (t, J = 6.6 Hz, 2H, H-16), 3.57 (t, J = 6.1 Hz, 2H, H-18), 2.49–2.40 (m, 2H, H-17). 13C{1H} NMR (101 MHz, CDCl3): δ (ppm) = 156.9 (C-5), 150.0 (C-7), 148.8 (C-11), 139.1 (C-4), 137.6 (C-9), 129.1 (C-1), 128.7 (C-2), 126.9 (C-3), 122.8 (C-15), 119.5 (C-10), 118.4 (C-8), 47.2 (C-16), 41.1 (C-18), 32.6 (C-17). 15N{1H} NMR (41 MHz, CDCl3): δ (ppm) = 363 (N-13), 348 (N-12), 298 (N-6), 246 (N-14). HRMS (m/z) [C16H15N4Cl + Na]+: calcd: 321.0877, found: 321.0881. Elem. Anal. for C16H15N4Cl (%) calcd: C 64.3, H 5.1, N 18.8, found: C 64.3, H 5.1, N 18.8.
:ethyl acetate, 8:2), and the product was obtained as a yellow solid. Yield: 64% (1.49 g, 7.65 mmol). 1H NMR (400 MHz, DMSO-d6): δ (ppm) = 9.84 (s, 1H, H-1), 7.95–7.88 (m, 2H, H-4), 7.87–7.77 (m, 2H, H-10, H-11), 7.41 (dd, J = 7.1, 1.4 Hz, 1H, H-9), 6.90–6.84 (m, 2H, H-3), 4.30 (s, 1H, H-13). 13C{1H} NMR (101 MHz, DMSO-d6): δ (ppm) = 158.9 (C-2), 156.5 (C-6), 141.2 (C-8), 137.5 (C-10), 128.6 (C-5), 128.0 (C-4), 124.8 (C-9), 119.1 (C-11), 115.5 (C-3), 83.4 (C-12), 79.6 (C-13). HRMS (m/z) [C13H9NO + H]+: calcd: 196.0757, found: 196.0757. Elem. Anal. for C13H9NO (%) calcd: C 80.0, H 4.7, N 7.2, found: C 79.9, H 4.7, N 7.0.
:CH3OH, 100:3). The product was obtained as a brown oil. Yield: 61% (500 mg, 0.905 mmol). 1H NMR (400 MHz, CD2Cl2): δ (ppm) = 8.33 (s, 1H, H-15), 8.11–8.05 (m, 2H, H-3), 8.02 (d, J = 7.7 Hz, 1H, H-8), 7.82 (t, J = 7.8 Hz, 1H, H-9), 7.65 (d, J = 7.9 Hz, 1H, H-10), 7.15–7.02 (m, 2H, H-2), 4.76–4.64 (m, 2H, H-16), 4.34 (d, 2JH,P = 10.1 Hz, 2H, H-21), 4.28–4.17 (m, 4H, H-23), 4.14–4.03 (m, 4H, H-19), 2.54–2.41 (m, 2H, H-17), 1.36 (t, J = 7.1 Hz, 6H, H-24), 1.30 (t, J = 7.1 Hz, 6H, H-20). 13C{1H} NMR (101 MHz, CD2Cl2): δ (ppm) = 160.1 (d, 3JC,P = 13.7 Hz, C-1), 156.6 (C-5), 150.4 (C-7), 149.1 (C-11), 138.0 (C-9), 133.2 (C-4), 128.6 (C-3), 123.1 (C-15), 119.1 (C-10), 118.2 (C-8), 115.0 (C-2), 63.3 (d, 3JC,P = 6.4 Hz, C-23), 62.7 (d, 1JC,P = 169.9 Hz, C-21), 62.6 (d, 3JC,P = 6.5 Hz, C-19), 45.1 (2JC,P = 1.7 Hz, C-16), 27.6 (d, 1JC,P = 141.1 Hz, C-17), 16.7 (d, 3JC,P = 5.7 Hz, C-24), 16.6 (d, 3JC,P = 6.0 Hz, C-20). 15N{1H} NMR (41 MHz, CD2Cl2): δ (ppm) = 363 (N-13), 349 (N-12), 296 (N-6), 248 (N-14). 31P{1H} NMR (162 MHz, CD2Cl2): δ (ppm) = 25.4 (P-18), 18.7 (P-22). HRMS (m/z) [C24H34N4O7P2 + H]+: calcd: 553.1975, found: 553.1979. Elem. Anal. for C24H34N4O7P2 (%) calcd: C 52.2, H 6.2, N 10.1, found: C 52.4, H 6.2, N 9.8.
:ethyl acetate, 1:1). The product was obtained as a white solid. Yield: 58% (2.86 g, 9.28 mmol). 1H NMR (400 MHz, DMSO-d6): δ (ppm) = 8.04–7.96 (m, 2H, H-3), 7.93 (d, J = 7.8 Hz, 1H, H-10), 7.77 (t, J = 7.8 Hz, 1H, H-9), 7.50 (d, J = 7.8 Hz, 1H, H-8), 7.08–7.01 (m, 2H, H-2), 4.60–4.53 (m, 1H, H-14), 4.10 (t, J = 6.4 Hz, 2H, H-11), 3.62–3.53 (m, 2H, H-13), 1.94–1.83 (m, 2H, H-12). 13C{1H} NMR (101 MHz, DMSO-d6): δ (ppm) = 160.1 (C-1), 157.2 (C-5), 141.1 (C-7), 140.3 (C-9), 129.0 (C-4), 128.0 (C-3), 125.5 (C-8), 118.5 (C-10), 114.6 (C-2), 64.7 (C-11), 57.1 (C-13), 32.0 (C-12). 15N{1H} NMR (41 MHz, DMSO-d6): δ (ppm) = 305 (N-6). HRMS (m/z) [C14H14BrNO2 + Na]+: calcd: 330.0100, found: 330.0100. Elem. Anal. for C14H14BrNO2 (%) calcd: C 54.6, H 4.6, N 4.6, found: C 54.6, H 4.7, N 4.4.
The crude product was then dissolved in a mixture of THF (20 mL) and CH3OH (20 mL) and mixed with an aqueous solution of NaOH (20 mL, 1 M). The mixture was stirred for 1 h at room temperature and the solvent removed under reduced pressure. Water was added to the residue, and the product was extracted with CH2Cl2, the combined organic layers were dried (MgSO4) and the filtrate was evaporated to dryness. The crude product was purified by column chromatography (SiO2, cyclohexane:ethyl acetate, 1:1) and the product was obtained as a yellow solid. Yield: 80% (1.32 g, 5.21 mmol). 1H NMR (400 MHz, DMSO-d6): δ (ppm) = 8.08–8.00 (m, 2H, H-3), 7.94 (d, J = 8.1 Hz, 1H, H-10), 7.87 (t, J = 7.8 Hz, 1H, H-9), 7.47 (d, J = 7.5 Hz, 1H, H-8), 7.11–7.03 (m, 2H, H-2), 4.59 (t, J = 5.1 Hz, 1H, H-16), 4.35 (s, 1H, H-12), 4.13 (t, J = 6.4 Hz, 2H, H-13), 3.61 (q, J = 6.0 Hz, 2H, H-15), 1.92 (p, J = 6.3 Hz, 2H, H-14). 13C{1H} NMR (101 MHz, DMSO-d6): δ (ppm) = 159.8 (C-1), 156.2 (C-5), 141.3 (C-7), 137.6 (C-9), 130.0 (C-4), 127.9 (C-3), 125.1 (C-8), 119.4 (C-10), 114.5 (C-2), 83.3 (C-11), 79.7 (C-12), 64.6 (C-13), 57.2 (C-15), 32.0 (C-15). 15N{1H} NMR (41 MHz, DMSO-d6): δ (ppm) = 309 (N-6). HRMS (m/z) [C16H15NO2 + H]+: calcd: 254.1176, found: 254.1176. Elem. Anal. for C16H15NO2 (%) calcd: C 75.9, H 6.0, N 5.5, found: C 75.4, H 6.1, N 5.1.
:methanol, 100:2) to give the product as a beige solid. Yield: 75% (2.20 g, 6.21 mmol). 1H NMR (500 MHz, DMSO-d6): δ (ppm) = 8.74 (s, 1H, H-15), 8.18–8.12 (m, 2H, H-3), 7.95–7.87 (m, 2H, H-8, H-9), 7.86–7.81 (m, 1H, H-10), 7.08–7.02 (m, 2H, H-2), 4.70 (t, J = 5.0 Hz, 1H, H-19), 4.58 (t, J = 5.1 Hz, 1H, H-23), 4.52 (t, J = 7.1 Hz, 2H, H-16), 4.11 (t, J = 6.4 Hz, 2H, H-20), 3.63–3.56 (m, 2H, H-20), 3.49–3.42 (m, 2H, H-18), 2.06 (p, J = 6.5 Hz, 2H, H-17), 1.90 (p, J = 6.3 Hz, 2H, H-21). 13C{1H} NMR (126 MHz, DMSO-d6): δ (ppm) = 159.7 (C-1), 155.4 (C-5), 149.6 (C-7), 147.3 (C-11), 138.0 (C-9), 130.5 (C-4), 127.9 (C-3), 123.4 (C-15), 118.1 (C-10), 117.0 (C-8), 114.4 (C-2), 64.6 (C-20), 57.4 (C-18), 57.2 (C-22), 46.8 (C-16), 32.8 (C-17), 32.0 (C-21). 15N{1H} NMR (51 MHz, DMSO-d6): δ (ppm) = 364 (N-13), 348 (N-12), 297 (N-6), 253 (N-14). HRMS (m/z) [C19H22N4O3 + H]+: calcd: 355.1765, found: 355.1765. Elem. Anal. for C19H22N4O3 (%) calcd: C 64.4, H 6.3, N 15.8, found: C 64.4, H 6.2, N 15.8.
:ethyl acetate, 8:2). The product was obtained as white solid. Yield: 95% (474 mg, 1.21 mmol). 1H NMR (500 MHz, CDCl3): δ (ppm) = 8.29 (s, 1H, H-15), 8.06 (d, J = 7.9 Hz, 1H, H-8), 8.04–7.99 (m, 2H, H-3), 7.81 (t, J = 7.9 Hz, 1H, H-9), 7.62 (d, J = 7.9 Hz, 1H, H-10), 7.04–6.98 (m, 2H, H-2), 4.63 (t, J = 6.6 Hz, 2H, H-16), 4.19 (t, J = 5.8 Hz, 2H, H-19), 3.77 (t, J = 6.3 Hz, 2H, H-21), 3.58 (t, J = 6.1 Hz, 2H, H-18), 2.46 (p, J = 6.4 Hz, 2H, H-17), 2.27 (p, J = 6.1 Hz, 2H, H-20). 13C{1H} NMR (126 MHz, CDCl3): δ (ppm) = 159.7 (C-1), 156.5 (C-5), 149.8 (C-7), 148.9 (C-11), 137.6 (C-9), 132.0 (C-4), 128.2 (C-3), 122.7 (C-15), 118.8 (C-10), 117.8 (C-8), 114.7 (C-2), 64.4 (C-19), 47.2 (C-16), 41.5 (C-21), 41.1 (C-18), 32.6 (C-17), 32.2 (C-20). 15N{1H} NMR (51 MHz, CDCl3): δ (ppm) = 363 (N-13), 348 (N-12), 246 (N-14). HRMS (m/z) [C19H20Cl2N4O + H]+: calcd: 391.1087, found: 391.1079. Elem. Anal. for C19H20Cl2N4O (%) calcd: C 58.3, H 5.2, N 14.3, found: C 58.4 H 5.4, N 14.1.
:ethyl acetate, 7:3). The product was obtained as white solid. Yield: 61% (392 mg, 0.816 mmol). 1H NMR (400 MHz, CDCl3): δ (ppm) = 8.32 (s, 1H, H-15), 8.07 (d, J = 7.8 Hz, 1H, H-8), 8.04–7.98 (m, 2H, H-3), 7.81 (d, J = 7.8 Hz, 1H, H-9), 7.61 (d, J = 7.9 Hz, 1H, H-10), 7.05–6.98 (m, 2H, H-2), 4.62 (t, J = 6.6 Hz, 2H, H-16), 4.17 (t, J = 5.8 Hz, 2H, H-19), 3.62 (t, J = 6.4 Hz, 2H, H-21), 3.41 (d, J = 6.2 Hz, 2H, H-18), 2.53 (p, J = 6.4 Hz, 2H, H-17), 2.35 (p, J = 6.1 Hz, 2H, H-20). 13C{1H} NMR (101 MHz, CDCl3): δ (ppm) = 159.7 (C-1), 156.6 (C-5), 149.8 (C-7), 148.8 (C-11), 137.7 (C-9), 131.9 (C-4), 128.3 (C-3), 122.8 (C-15), 118.9 (C-10), 117.9 (C-8), 114.7 (C-2), 65.5 (C-19), 48.4 (C-16), 32.7 (C-17), 32.4 (C-20), 30.0 (C-21), 29.4 (C-18). 15N{1H} NMR (41 MHz, CDCl3): δ (ppm) = 363 (N-13), 348 (N-12), 294 (N-6), 247 (N-14). HRMS (m/z) [C19H20Br2N4O + H]+: calcd: 479.0077, found: 479.0077. Elem. Anal. for C19H20Br2N4O (%) calcd: C 47.5, H 4.2, N 11.7, found: C 47.6, H 4.3, N 11.7.
:CH3OH, 100:5), yielding this product as a beige solid. Yield: 82% (704 mg, 1.42 mmol). 1H NMR (400 MHz, CD2Cl2): δ (ppm) = 8.29 (s, 1H, H-15), 8.06–8.02 (m, 2H, H-3), 8.01 (dd, J = 7.8, 1.0 Hz, 1H, H-8), 7.81 (t, J = 7.8 Hz, 1H, H-9), 7.63 (dd, J = 8.0, 1.0 Hz, 1H, H-10), 7.05–6.98 (m, 2H, H-2), 4.51 (t, J = 7.0 Hz, 2H, H-16), 4.10 (t, J = 6.4 Hz, 2H, H-22), 3.72–3.64 (m, 8H, H-27, H-21), 2.52 (t, J = 7.1 Hz, 2H, H-24), 2.47–2.39 (m, 8H, H-26, H-20), 2.36 (t, J = 6.7 Hz, 2H, H-18), 2.18–2.07 (m, 2H, H-17), 2.03–1.92 (m, 2H, H-23). 13C{1H} NMR (101 MHz, CD2Cl2): δ (ppm) = 160.5 (C-1), 156.8 (C-5), 150.6 (C-7), 148.9 (C-11), 137.9 (C-9), 132.0 (C-4), 128.4 (C-3), 123.0 (C-15), 118.8 (C-10), 117.8 (C-8), 114.9 (C-2), 67.4 (C-27), 67.3 (C-21), 66.7 (C-22), 55.7 (C-24), 55.4 (C-18), 54.2 (C-26), 54.0 (C-20), 48.6 (C-16), 27.4 (C-17), 26.9 (C-23). 15N{1H} NMR (41 MHz, CD2Cl2): δ (ppm) = 364 (N-13), 347 (N-12), 295 (N-6), 250 (N-14). HRMS (m/z) [C27H36N6O3 + H]+: calcd: 493.2922, found: 493.2922. Elem. Anal. for C27H36N6O3 (%) calcd: C 65.8, H 7.4, N 17.1, found: C 65.9, H 7.3, N 17.1.
:CH3OH, 100:3). The product was obtained as a yellow solid. Yield: 56% (123 mg, 0.180 mmol). 1H NMR (500 MHz, CD2Cl2): δ (ppm) = 8.25 (s, 1H, H-17), 7.93 (d, 3JH,Pt = 99 Hz, J = 7.5 Hz, 1H, H-2), 7.53–7.45 (m, 2H, H-26), 7.41 (t, J = 7.9 Hz, 1H, H-11), 7.33–7.29 (m, 2H, H-27), 7.29–7.26 (m, 1H, H-5), 7.23 (d, J = 8.2 Hz, 1H, H-12), 7.21–7.17 (m, 1H, H-28), 7.15 (t, J = 7.4 Hz, 1H, H-3), 7.09 (d, J = 7.7 Hz, 1H, H-10), 7.05 (t, J = 7.5 Hz, 1H, H-4), 4.70 (dt, 3JH,P = 13.5, J = 7.6 Hz, 2H, H-18), 4.02 (dq, 3JH,P = 8.3, J = 7.1 Hz, 4H, H-21), 2.47 (dt, 2JH,P = 18.4, J = 7.6 Hz, 2H, H-19), 1.25 (t, J = 7.1 Hz, 6H, H-22). 13C{1H} NMR (126 MHz, CD2Cl2): δ (ppm) = 163.4 (2JC,Pt = 99 Hz, C-7), 150.5 (2JC,Pt = 50 Hz, C-13), 146.7 (C-9), 146.0 (2JC,Pt = 36 Hz, C-6), 139.2 (1JC,Pt = 1124 Hz, C-1), 138.5 (C-11), 136.8 (2JC,Pt = 99 Hz, C-2), 130.9 (C-26), 130.2 (3JC,Pt = 75 Hz, C-3), 128.3 (C-25), 127.5 (C-27), 124.9 (C-17), 124.5 (C-28), 123.8 (3JC,Pt = 43 Hz, C-5), 123.1 (C-4), 116.3 (C-10), 116.2 (C-12), 104.3 (2JC,Pt = 410 Hz, C-24), 101.8 (1JC,Pt = 1541 Hz, C-23), 61.6 (d, 2JC,P = 6.5 Hz, C-21), 46.2 (d, 2JC,P = 1.7 Hz, C-18), 25.8 (d, 1JC,P = 141.2 Hz, C-19), 15.6 (d, 3JC,P = 5.8 Hz, C-22). 15N{1H} NMR (51 MHz, CD2Cl2): δ (ppm) = 359 (N-15), 298 (N-14), 248 (N-16), 245 (N-8). 31P{1H} NMR (202 MHz, CD2Cl2): δ (ppm) = 24.7 (P-20). 195Pt{1H} NMR (107 MHz, CD2Cl2): δ (ppm) = −3870. HRMS (m/z) [C27H27N4O3PPt + H]+: calcd: 682.1544, found: 682.1550. Elem. Anal. for C27H27N4O3PPt (%) calcd: C 47.6, H 4.0, N 8.2, found: C 48.1, H 4.4, N 7.7.
:CH3OH, 100:4). The product was obtained as a red solid. Yield: 41% (32 mg, 50 μmol). 1H NMR (400 MHz, CD2Cl2): δ (ppm) = 8.22 (s, 1H, H-17), 7.97 (d, J = 7.3 Hz, 1H, H-2), 7.51–7.45 (m, 2H, H-27), 7.42 (t, J = 7.9 Hz, 1H, H-11), 7.34–7.22 (m, 3H, H-5, H-28), 7.22–7.13 (m, 3H, H-3, H-12, H-29), 7.12–7.03 (m, 2H, H-4, H-10), 4.54 (t, J = 7.2 Hz, 2H, H-18), 3.59 (t, J = 4.6 Hz, 4H, H-23), 2.34–2.24 (m, 6H, H-20, H-22), 2.10 (p, J = 6.8 Hz, 2H, H-19). 13C{1H} NMR (101 MHz, CD2Cl2): δ (ppm) = 164.6 (2JC,Pt = 99 Hz, C-7), 151.3 (2JC,Pt = 40 Hz, C-13), 147.9 (C-9), 147.0 (2JC,Pt = 38 Hz, C-6), 140.3 (1JC,Pt = 1124 Hz, C-1), 139.4 (C-11), 137.9 (2JC,Pt = 100 Hz, C-2), 131.8 (C-27), 131.3 (3JC,Pt = 73 Hz, C-3), 129.4 (C-26), 128.5 (C-28), 125.6 (C-17), 125.5 (C-29), 124.8 (3JC,Pt = 45 Hz, C-5), 124.1 (C-4), 117.2 (C-12), 117.1 (C-10), 105.4 (2JC,Pt = 408 Hz, C-25), 102.8 (1JC,Pt = 1540 Hz, C-24), 67.2 (C-23), 55.3 (C-20), 53.9 (C-22), 50.9 (C-18), 30.08, 26.7 (C-19). 15N{1H} NMR (41 MHz, CD2Cl2): δ (ppm) = 359 (N-15), 297 (N-14), 250 (N-16), 245 (N-8). 195Pt{1H} NMR (86 MHz, CD2Cl2): δ (ppm) = −3869. HRMS (m/z) [C28H27N5OPt + H]+: calcd: 645.1938, found: 645.1931. Elem. Anal. for C28H27N5OPt (%) calcd: C 52.2, H 4.2, N 10.9, found: C 51.9, H 4.4, N 10.6. Single crystals for X-ray structure analysis were obtained by slow diffusion of diethyl ether into a concentrated solution of the complex in DMF.
:CH3OH, 100:2). The product was obtained as a yellow solid. Yield: 33% (38 mg, 0.66 μmol). 1H NMR (400 MHz, DMSO-d6): δ (ppm) = 9.13 (s, 1H, H-17), 8.08 (t, J = 8.0 Hz, 1H, H-11), 7.90 (d, J = 8.3 Hz, 1H, H-12), 7.87–7.80 (m, 2H, H-10, H-2), 7.62 (dd, J = 7.5, J = 1.6 Hz, 1H, H-5), 7.33–7.24 (m, 4H, H-26, H-25), 7.17–7.03 (m, 3H, H-27, H-4, H-3), 4.77 (t, J = 4.9 Hz, 1H, H-21), 4.62 (t, J = 7.1 Hz, 2H, H-18), 3.52–3.45 (m, 2H, H-20), 2.15–2.04 (m, 2H, H-19). 13C{1H} NMR (101 MHz, DMSO-d6): δ (ppm) = 163.7 (C-7), 150.6 (C-13), 147.5 (C-9), 146.6 (C-6), 140.6 (C-11), 139.7 (C-1), 137.0 (2JC,Pt = 93 Hz, C-2), 130.8 (C-25), 130.3 (C-3), 128.9 (C-24), 128.0 (C-26), 126.2 (C-17), 124.9 (C-5), 124.6 (C-27), 123.5 (C-4), 117.5 (C-12), 117.2 (C-10), 104.5 (C-22), 104.3 (C-23), 57.2 (C-20), 48.9 (C-18), 32.1 (C-19). 15N{1H} NMR (41 MHz, DMSO-d6): δ (ppm) = 361 (N-15), 296 (N-14), 252 (N-16), 245 (N-8). 195Pt{1H} NMR (86 MHz, DMSO-d6): δ (ppm) = −3894. HRMS (m/z) [C24H20N4OPt + H]+: calcd: 576.1360, found: 576.1368. Elem. Anal. for C24H20N4OPt·0.125 AgCl (%) calcd: C 48.6, H 3.4, N 9.4, found: C 48.8, H 3.7, N 9.4.
:CH3OH, 100:0.3). The product was obtained as a yellow solid. Yield: 61% (119 mg, 0.200 mmol). 1H NMR (400 MHz, CD2Cl2): δ (ppm) = 8.20 (s, 1H, H-17), 8.03–7.80 (m, 1H, H-2), 7.49–7.44 (m, 2H, H-24), 7.38–7.33 (m, 1H, H-11), 7.33–7.26 (m, 3H, H-5, H-25), 7.23–7.12 (m, 3H, H-3, H-12, H-26), 7.10–7.03 (m, 2H, H-4, H-10), 4.66 (t, J = 7.0 Hz, 2H, H-18), 3.53 (t, J = 6.1 Hz, 2H, H-20), 2.49–2.38 (m, 2H, H-19). 13C{1H} NMR (101 MHz, CD2Cl2): δ (ppm) = 164.4 (C-7), 151.6 (C-13), 147.7 (C-9), 147.0 (C-6), 140.1 (C-1), 139.3 (C-11), 137.9 (C-2), 131.8 (C-24), 131.3 (C-3), 129.2 (C-23), 128.6 (C-25), 125.8 (C-17), 125.6 (C-26), 124.8 (C-5), 124.1 (C-4), 117.4 (C-10), 117.1 (C-12), 105.4 (C-22), 102.6 (C-21), 49.9 (C-18), 41.7 (C-20), 32.3 (C-19). 15N{1H} NMR (41 MHz, CD2Cl2): δ (ppm) = 359 (N-15), 298 (N-14), 247 (N-16), 245 (N-8). 195Pt{1H} NMR (86 MHz, CD2Cl2): δ (ppm) = −3866. HRMS (m/z) [C24H19N4ClPt + H]+: calcd: 594.1021, found: 594.1014. Elem. Anal. for C24H19N4ClPt (%) calcd: C 48.5, H 3.2, N 9.4, found: C 48.9, H 3.3, N 9.3. Single crystals for X-ray structure analysis were obtained by slow diffusion of diethyl ether into a concentrated solution of the complex in DMF.
:CH3OH, 100:4). The product was obtained as a yellow solid. Yield: 49% (143 mg, 0.183 mmol). 1H NMR (400 MHz, CD2Cl2): δ (ppm) = 8.33 (s, 1H, H-17), 7.39 (t, J = 7.9 Hz, 1H, H-11), 7.17 (d, J = 2.7 Hz, 1H, H-1), 7.12 (d, J = 8.5 Hz, 1H, H-5), 7.03 (d, J = 8.3 Hz, 1H, H-12), 6.93 (d, J = 7.9 Hz, 1H, H-10), 6.65 (dd, J = 8.5, J = 2.7 Hz, 1H, H-4), 4.75 (dt, 3JH,P = 12.5, J = 7.7 Hz, 2H, H-18), 4.38 (d, 2JH,P = 9.6 Hz, 2H, H-23), 4.30–4.20 (m, 4H, H-25), 4.18–4.05 (m, 4H, H-21), 2.57 (dt, 2JH,P = 18.5, J 7.7 Hz, 1H, H-19), 1.39 (t, J = 7.1 Hz, 6H, H-22), 1.33 (t, J = 7.1 Hz, 6H, H-26). 13C{1H} NMR (101 MHz, CD2Cl2): δ (ppm) = 165.1 (C-7), 160.1 (C-3), 150.0 (C-13), 147.9 (C-9), 142.8 (C-1), 140.2 (C-6), 139.0 (C-11), 126.1 (C-5), 125.8 (C-17), 118.2 (C-2), 116.6 (C-12), 116.3 (C-10), 111.1 (C-4), 63.3, (d, 3JC,P = 6.4 Hz, C-25) 62.2 (d, 1JC,P = 141.4 Hz, C-23), 47.3 (d, 2JC,P = 1.3 Hz, C-18), 26.8 (d, 1JC,P = 141.0 Hz, C-19), 16.8 (d, 3JH,P = 5.8 Hz, C-26), 16.7 (d, 3JH,P = 6.0 Hz, C-22). 15N{1H} NMR (41 MHz, CD2Cl2): δ (ppm) = 357 (N-15), 301 (N-14), 247 (N-16), 220 (N-8). 31P{1H} NMR (162 MHz, CD2Cl2): δ (ppm) = 24.9 (P-20), 19.0 (P-24). 195Pt{1H} NMR (86 MHz, CD2Cl2): δ (ppm) = −3477. HRMS (m/z) [C24H33N4O7P2ClPt + Na]+: calcd: 804.1055, found: 804.1060. Elem. Anal. for C24H33N4O7P2ClPt (%) calcd: C 36.9, H 4.3, N 7.2, found: C 36.2, H 4.5, N 6.8. Single crystals for X-ray structure analysis were obtained by slow diffusion of diethyl ether into a concentrated solution of the complex in DMF.
:CH3OH, 100:5). The product was obtained as a yellow solid. Yield: 21% (75 mg, 88 μmol). 1H NMR (400 MHz, CD2Cl2): δ (ppm) = 8.39 (s, 1H, H-17), 7.58 (d, J = 2.7 Hz, 1H, H-2), 7.53 (t, J = 7.9 Hz, 1H, H-11), 7.50–7.45 (m, 2H, H-30), 7.32–7.25 (m, 3H, H-31, H-5), 7.23 (d, J = 8.3 Hz, 1H, H-12), 7.21–7.15 (m, 2H, H-32, H-10), 6.66 (dd, J = 8.5 Hz, J = 2.7 Hz, 1H, H-4), 4.75 (t, J = 7.5 Hz, 2H, H-18), 4.36 (d, 2JH,P = 9.7 Hz, 2H, H-23), 4.26–4.15 (m, 4H, H-25), 4.14–3.98 (m, 4H, H-21), 2.51 (dt, 2JH,P = 18.4 Hz, J = 6.8 Hz, 2H, H-19), 1.35 (t, J = 7.1 Hz, 6H, H-26), 1.27 (t, J = 7.1 Hz, 6H, H-22). 13C{1H} NMR (101 MHz, CD2Cl2): δ (ppm) = 164.2 (C-7), 160.6 (C-3), 151.9 (C-13), 147.7 (C-9), 142.7 (C-1), 140.7 (C-6), 139.6 (C-11), 131.9 (C-30), 129.2 (C-29), 128.5 (C-31), 126.3 (C-5), 125.6 (C-17), 125.5 (C-32), 122.3 (C-2), 117.0 (C-12), 116.3 (C-10), 110.7 (C-4), 105.2 (C-28), 102.6 (C-27), 63.3 (d, 3JC,P = 6.4 Hz, C-25), 62.7 (d, 3JC,P = 6.5 Hz, C-21), 62.2 (d, 1JC,P = 169.5 Hz, C-23), 47.2 (C-18), 26.9 (d, 1JC,P = 141.4 Hz, C-19), 16.7 (d, 3JC,P = 5.7 Hz, C-26), 16.6 (d, 3JC,P = 5.9 Hz, C-22). 15N{1H} NMR (41 MHz, CD2Cl2): δ (ppm) = 359 (N-15), 298 (N-14), 246 (N-16), 242 (N-8). 31P{1H} NMR (162 MHz, CD2Cl2): δ (ppm) = 24.8 (P-20), 18.9 (P-24). 195Pt{1H} NMR (86 MHz, CD2Cl2): δ (ppm) = −3863. HRMS (m/z) [C32H38N4O7P2Pt + H]+: calcd: 848.1936, found: 848.1909. Elem. Anal. for C32H38N4O7P2Pt (%) calcd: C 45.3, H 4.5, N 6.6, found: C 44.9, H 4.8, N 6.4.
:CH3OH, 100:3). The product was obtained as a yellow solid. Yield: 24% (56 mg, 86 μmol). 1H NMR (400 MHz, DMSO-d6): δ (ppm) = 9.08 (s, 1H, H-17), 8.00 (t, J = 8.0 Hz, 1H, H-11), 7.75 (d, J = 8.3 Hz, 1H, H-12), 7.71 (d, J = 7.7 Hz, 1H, H-10), 7.56 (d, J = 8.6 Hz, 1H, H-5), 7.43 (d, J = 2.7 Hz, 1H, H-2), 7.35–7.22 (m, 4H, H-30, H-29), 7.19–7.10 (m, 1H, H-31), 6.63 (dd, J = 8.5, 2.7 Hz, 1H, H-4), 4.76 (s, 1H, H-21), 4.61 (t, J = 7.1 Hz, 2H, H-18), 4.55 (s, 1H, H-25), 4.07 (t, J = 6.4 Hz, 2H, H-22), 3.60–3.52 (m, 2H, H-24), 3.48 (t, J = 5.9 Hz, 2H, H-20), 2.16–2.04 (m, 2H, H-19), 1.92–1.82 (m, 2H, H-23). 13C{1H} NMR (101 MHz, DMSO-d6): δ (ppm) = 163.6 (C-7), 160.1 (C-3), 150.7 (C-13), 147.2 (C-9), 141.8 (C-1), 140.4 (C-11), 138.9 (C-6), 130.8 (C-29), 128.9 (C-28), 128.0 (C-30), 126.5 (C-5), 125.9 (C-17), 124.7 (C-31), 122.6 (C-2), 116.8 (C-12), 115.8 (C-10), 109.3 (C-4), 104.9 (C-26), 103.9 (C-27), 64.2 (C-22), 57.2 (C-24, C-20), 48.9 (C-18), 32.1 (C-23, C-19). 15N{1H} NMR (41 MHz, DMSO-d6): δ (ppm) = 361 (N-15), 297 (N-14), 253 (N-16), 242 (N-8). 195Pt{1H} NMR (86 MHz, DMSO-d6): δ (ppm) = −3884. HRMS (m/z) [C27H26N4O3Pt + H]+: calcd: 650.1725, found: 650.1722. Elem. Anal. for C27H26N4O3Pt (%) calcd: C 49.9, H 4.0, N 8.6, found: C 49.9, H 4.4, N 8.2.
:CH3OH, 100:0.3). The product was obtained as a yellow solid. Yield: 43% (143 mg, 0.203 mmol). 1H NMR (500 MHz, CD2Cl2): δ (ppm) = 8.21 (s, 1H, H-17), 7.50 (d, J = 2.6 Hz, 1H, H-2), 7.48–7.41 (m, 2H, H-27), 7.33–7.25 (m, 3H, H-28, H-11), 7.23–7.16 (m, 2H, H-29, H-5), 7.06 (d, J = 8.1 Hz, 1H, H-10), 6.96 (d, J = 7.7 Hz, 1H, H-12), 6.60 (dd, J = 8.5, 2.7 Hz, 1H, H-4), 4.65 (t, J = 7.1 Hz, 2H, H-18), 4.20 (t, J = 5.9 Hz, 2H, H-21), 3.77 (t, J = 6.4 Hz, 2H, H-23), 3.52 (t, J = 6.1 Hz, 2H, H-20), 2.43 (p, J = 6.6 Hz, 2H, H-19), 2.25 (p, J = 6.2 Hz, 2H, H-22). 13C{1H} NMR (126 MHz, CD2Cl2): δ (ppm) = 164.1 (C-7), 160.9 (C-3), 151.6 (C-13), 147.3 (C-9), 142.3 (C-1), 139.7 (C-6), 139.1 (C-11), 131.8 (C-27), 129.2 (C-26), 128.6 (C-28), 126.4 (C-5), 125.7 (C-17), 125.6 (C-29), 123.0 (C-2), 116.5 (C-10), 116.2 (C-12), 110.3 (C-4), 105.1 (C-25), 102.8 (C-24), 64.6 (C-21), 50.0 (C-18), 42.3 (C-23), 41.8 (C-20), 32.7 (C-22), 32.3 (C-19). 15N{1H} NMR (51 MHz, CD2Cl2): δ (ppm) = 358 (N-15), 297 (N-14), 247 (N-16), 241 (N-8). 195Pt{1H} NMR (107 MHz, CD2Cl2) δ (ppm) = −3852. HRMS (m/z) [C27H24Cl2N4OPt + H]+: calcd: 686.1048, found: 686.1043. Elem. Anal. for C27H24Cl2N4OPt (%) calcd: C 47.2, H 3.5, N 8.2, found: C 47.5, H 3.9, N 8.2.
:CH3OH, 100:7). The complex was obtained as a yellow solid. Yield: 29% (63 mg, 87 μmol). 1H NMR (400 MHz, DMF-d7): δ (ppm) = 9.25 (s, 1H, H-17), 8.08–8.01 (m, 1H, H-11), 7.80–7.72 (m, 2H, H-10, H-12), 7.59 (d, J = 8.6 Hz, 1H, H-5), 7.27 (d, J = 2.6 Hz, 1H, H-2), 6.70 (dd, J = 8.5, J = 2.6 Hz, 1H, H-4), 4.74 (t, J = 6.9 Hz, 2H, H-18), 4.14 (t, J = 6.2 Hz, 2H, H-24), 3.87 (br, 4H, H-29), 3.70 (s, 4H, H-23), 2.97 (s, 2H, H-26), 2.59 (br, 10H, H-20, H-22, H-28), 2.32 (br, 2H, H-19), 2.21 (br, 2H, H-25). 13C{1H} NMR (101 MHz, DMF-d7): δ (ppm) = 166.2 (C-7), 160.7 (C-3), 150.2 (C-13), 148.8 (C-9), 142.9 (C-1), 140.5 (C-11), 140.0 (C-6), 127.1 (C-5), 126.9 (C-17), 120.1 (C-2), 117.6 (C-12), 116.8 (C-10), 110.3 (C-4), 66.5 (C-23), 65.6 (C-29), 65.4 (C-24), 55.3 (C-20, C-26), 53.7 (C-22), 53.2 (C-28), 50.8 (C-18), 26.2 (C-19), 25.4 (C-25). 15N{1H} NMR (41 MHz, DMF-d7): δ (ppm) = 301 (N-14), 251 (N-16), 220 (N-8). 195Pt{1H} NMR (107 MHz, DMF-d7): δ (ppm) = 3486. HRMS (m/z) [C27H35ClN6O3Pt + H]+: calcd: 722.2180, found: 722.2180. Elem. Anal. for C27H35ClN6O3Pt (%) calcd: C 44.9, H 4.9, N 11.6, found: C 44.8 H 4.9, N 11.4.
:CH3OH, 100:7). The product was obtained as a yellow solid. Yield: 43% (28 mg, 36 μmol). 1H NMR (400 MHz, CD2Cl2): δ (ppm) = 8.23 (s, 1H, H-17), 7.56 (d, J = 2.7 Hz, 1H, H-2), 7.52–7.43 (m, 3H, H-11, H-33), 7.33–7.26 (m, 3H, H-5, H-34), 7.24–7.14 (m, 2H, H-12, H-35), 7.09 (d, J = 7.7 Hz, 1H, H-10), 6.62 (dd, J = 8.5, J = 2.7 Hz, 1H, H-4), 4.58 (t, J = 7.2 Hz, 2H, H-18), 4.12 (t, J = 6.4 Hz, 2H, H-24), 3.65 (t, J = 4.7 Hz, 4H, H-29), 3.61 (t, J = 4.7 Hz, 4H, H-23), 2.49 (t, J = 7.2 Hz, 2H, H-26), 2.44–2.38 (m, 4H, H-28), 2.37–2.27 (m, 6H, H-20, H-22), 2.14 (p, J = 6.8 Hz, 2H, H-19), 1.95 (p, J = 6.7 Hz, 2H, H-25). 13C{1H} NMR (101 MHz, CD2Cl2): δ (ppm) = 164.7 (2JC,Pt = 97 Hz, C-7), 161.4 (3JC,Pt = 91 Hz, C-3), 151.5 (2JC,Pt = 41 Hz, C-13), 147.6 (C-9), 142.5 (1JC,Pt = 1124 Hz, C-1), 139.5 (C-11), 139.4 (C-6), 131.9 (C-33), 129.4 (C-32), 128.5 (C-33), 126.4 (3JC,Pt = 52 Hz, C-5), 125.5 (C-35), 125.2 (C-17), 123.0 (2JC,Pt = 107 Hz, C-2), 116.6 (C-12), 115.7 (C-10), 110.6 (C-4), 105.2 (2JC,Pt = 407 Hz, C-31), 102.9 (1JC,Pt = 1530 Hz, C-30), 67.3 (C-29), 67.2 (C-23), 66.4 (C-24), 55.8 (C-26), 55.2 (C-20), 54.2 (C-28), 53.9 (C 22), 50.9 (C-18), 26.9 (C-25) 26.8 (C-19). 15N{1H} NMR (41 MHz, CD2Cl2): δ (ppm) = 360 (N-15), 297 (N-14), 250 (N-16), 243 (N-8), 44 (N-27), 42 (N-21). 195Pt{1H} NMR (86 MHz, CD2Cl2): δ (ppm) = −3862. HRMS (m/z) [C35H40N6O3Pt + H]+: calcd: 788.2882, found: 788.2889. Elem. Anal. for C35H40N6O3Pt (%) calcd: C 53.4, H 5.1, N 10.7, found: C 53.1, H 5.3, N 10.5.
Conclusions
By varying the substitution pattern of a tridentate N^N^C ligand as well as the identity of a monodentate ancillary ligand, 16 new luminescent platinum(II) complexes for the selective binding of guanine quadruplexes were designed and synthesized. Based on DFT studies, the ability of the complexes to aggregate was predicted computationally and then (mostly) confirmed experimentally in aqueous medium. As expected, complexes bearing positively charged substituents were less likely to aggregate in solution. Four guanine quadruplexes that are currently being discussed as potential therapeutic agents were exemplarily chosen for the interaction studies.Cryo-ESI mass spectrometry indicates two possible binding stoichiometries of G4 DNA per complex, namely 1:1 and 1:2, with the former being more prominent. A phosphorescence turn-on is observed upon the addition of G4 DNA to most of the complexes, with the complexes bearing a phosphate ester substituent being a notable exception (except for [Pt(B)(PL)]OTf). Interestingly, the presence of c-kit hardly affects the luminescence of the Pt(II) complexes, whereas c-myc22 particularly affects that of complexes with auxiliary ligand A. The luminescence data also suggest a significant interaction of [Pt(A)(MLM)] (with its two morpholino-group containing substituents) with all oligonucleotides under investigation. This correlates well with the use of morpholine as a protonatable group in earlier studies on G4 binders.22 As the overall increase in luminescence depends on the nature of the G4 quadruplex, some selectivity towards the different G4 topologies can be anticipated.
CD spectroscopic studies do not indicate major structural changes of the G4 DNA upon addition of complexes with luminescence turn-on. However, in c-myc22 and bcl2-mid a structural change does occur (based on CD spectra) in the presence of [Pt(B)(ML)]OTf and [Pt(B)(PLP)]OTf, respectively, even though this change is neither accompanied by a stabilizing effect nor by a significant change in luminescence. In the case of bcl2-mid and [Pt(B)(PLP)]OTf, a complete unfolding of the G4 DNA is proposed.
[Pt(A)(MLM)] and [Pt(B)(OHL)]OTf are the two complexes inducing the greatest G4 DNA stabilization (based on ΔTm for FAM-bcl2-mid-DABCYL). However, there appears to be no particular selectivity of these two complexes for any of the four G4 DNA sequences under investigation.
In general, the affinity towards G4 DNA and the selectivity for targeting G4 DNA over double-stranded DNA depends on the functionalization of both the tridentate ligand and the ancillary ligand. Complexes with one or two protonatable/polar substituents appear to interact best with G4 DNA, as can be seen from the data obtained for the morpholino- and hydroxy-functionalized complexes. This points at the relevance of an electrostatic interaction with the phosphate backbone. Future studies will need to focus on increasing the binding affinity of these water-soluble complexes and on improving their binding selectivity towards G4 DNA over dsDNA.
Author contributions
SK: investigation (syntheses, spectroscopy, experiments), writing – original draft; MH: investigation (calculations, ESI-MS measurements); ML: investigation (X-ray diffraction); AH: investigation (NMR spectroscopy); JL: investigation (lifetime measurements); ART: investigation (spectroscopy, experiments); CAS: supervision; JM: supervision, conceptualization; writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for [Pt(A)(ML)], [Pt(A)(ClL)], [PtCl(PLP)], and [PtCl(OHLOH)] have been deposited at the CCDC under 2392538–2392541.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Calculations for this publication were performed on the HPC cluster PALMA II of the University of Münster, subsidised by the DFG (INST 211/667-1). We thank Bart Jan Ravoo for access to the ITC instrument and Katharina Ziegler and Bastian Stövesand for an introduction into the instrument. J. M. and C. A. S. thank the DFG for funding (SFB 858, STR 1186/7-2).References
- Quadruplex Nucleic Acids, ed. S. Neidle and S. Balasubramanian, RSC Publishing, 2006 Search PubMed.
- A. Ambrus, D. Chen, J. Dai, R. A. Jones and D. Yang, Biochemistry, 2005, 44, 2048–2058 CrossRef CAS PubMed.
- D. Rhodes and H. J. Lipps, Nucleic Acids Res., 2015, 43, 8627–8637 CrossRef CAS PubMed.
- M. L. Bochman, K. Paeschke and V. A. Zakian, Nat. Rev. Genet., 2012, 13, 770–780 Search PubMed.
- R. C. Monsen, J. O. Trent and J. B. Chaires, Acc. Chem. Res., 2022, 55, 3242–3252 Search PubMed.
- S. Burge, G. N. Parkinson, P. Hazel, A. K. Todd and S. Neidle, Nucleic Acids Res., 2006, 34, 5402–5415 CrossRef CAS PubMed.
- D. Yang and K. Okamoto, Future Med. Chem., 2010, 2, 619–646 CrossRef CAS PubMed.
- R. K. Moyzis, J. M. Buckingham, L. S. Cram, M. Dani, L. L. Deaven, M. D. Jones, J. Meyne, R. L. Ratliff and J.-R. Wu, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 6622–6626 CrossRef CAS PubMed.
- M. A. Shammas, Curr. Opin. Clin. Nutr. Metab. Care, 2011, 14, 28–34 CrossRef CAS PubMed.
- N. W. Kim, M. A. Piatyszek, K. R. Prowse, C. B. Harley, M. D. West, P. L. C. Ho, G. M. Coviello, W. E. Wright, S. L. Weinrich and J. W. Shaw, Science, 1994, 266, 2011–2015 Search PubMed.
- M. A. Blasco, Eur. J. Cell Biol., 2003, 82, 441–446 CrossRef CAS PubMed.
- S. Neidle, Nat. Rev. Chem., 2017, 1, 0041 CrossRef CAS.
- T. R. Cech, Angew. Chem., Int. Ed., 2000, 39, 34–43 Search PubMed.
- A. Siddiqui-Jain, C. L. Grand, D. J. Bearss and L. H. Hurley, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 11593–11598 Search PubMed.
- D. Sun and L. H. Hurley, J. Med. Chem., 2009, 52, 2863–2874 Search PubMed.
- S. Hu, W. Ma, J. Wang, Z. Zhou, Y. Ma, R. Zhang, K. Du, H. Zhang, M. Sun, X. Jiang, H. Tu, X. Tang, X. Yao and P. Chen, Eur. J. Pharmacol., 2022, 915, 174514 Search PubMed.
- B. R. Vummidi, J. Alzeer and N. W. Luedtke, ChemBioChem, 2013, 14, 540–558 Search PubMed.
- R. Hänsel-Hertsch, M. Di Antonio and S. Balasubramanian, Nat. Rev. Mol. Cell Biol., 2017, 18, 279–284 Search PubMed.
- U. Chilakamarthi, D. Koteshwar, S. Jinka, N. V. Krishna, K. Sridharan, N. Nagesh and L. Giribabu, Biochemistry, 2018, 57, 6514–6527 Search PubMed.
- L. D'Anna, S. Rubino, C. Pipitone, G. Serio, C. Gentile, A. Palumbo Piccionello, F. Giannici, G. Barone and A. Terenzi, Dalton Trans., 2023, 52, 2966–2975 Search PubMed.
- A. Arola-Arnal, J. Benet-Buchholz, S. Neidle and R. Vilar, Inorg. Chem., 2008, 47, 11910–11919 Search PubMed.
- S. N. Georgiades, N. H. A. Karim, K. Suntharalingam and R. Vilar, Angew. Chem., Int. Ed., 2010, 49, 4020–4034 Search PubMed.
- J. Zegers, M. Peters and B. Albada, J. Biol. Inorg. Chem., 2023, 28, 117–138 Search PubMed.
- A. E. Engelhart, J. Plavec, Ö. Persil and N. V. Hud, in Nucleic Acid-Metal Ion Interactions, ed. N. V. Hud, RSC Publishing, Cambridge, UK, 2009, pp. 118–153 Search PubMed.
- A. Maraval, S. Franco, C. Vialas, G. Pratviel, M. A. Blasco and B. Meunier, Org. Biomol. Chem., 2003, 1, 921–927 RSC.
- T. Kench, V. Rakers, D. Bouzada, J. Gomez-González, J. Robinson, M. K. Kuimova, M. Vázquez López, M. E. Vázquez and R. Vilar, Bioconjugate Chem., 2023, 34, 911–921 CrossRef CAS PubMed.
- A. Schmidt, R. Guha, A. Hepp and J. Müller, J. Inorg. Biochem., 2017, 175, 58–66 CrossRef CAS PubMed.
- V. T. Tran, J. Turek-Herman, M. Ferreira, K. N. Martin, D. Beseiso, B. R. Williams, F. Rosu, V. Gabelica, S. J. Nieter Burgmayer and L. A. Yatsunyk, J. Inorg. Biochem., 2023, 249, 112388 CrossRef CAS PubMed.
- L. Holden, K. S. Gkika, C. S. Burke, C. Long and T. E. Keyes, Inorg. Chem., 2023, 62, 2213–2227 CrossRef CAS PubMed.
- C. Kaußler, D. Wragg, C. Schmidt, G. Moreno-Alcántar, C. Jandl, J. Stephan, R. A. Fischer, S. Leoni, A. Casini and R. Bonsignore, Inorg. Chem., 2022, 61, 20405–20423 Search PubMed.
- J. Weynand, H. Bonnet, F. Loiseau, J.-L. Ravanat, J. Dejeu, E. Defrancq and B. Elias, Chem. – Eur. J., 2019, 25, 12730–12739 CrossRef CAS PubMed.
- C. J. Castor, J. Mancini, J. Fakhoury, N. Weill, R. Kieltyka, P. Englebienne, N. Avakyan, A. Mittermaier, C. Autexier, N. Moitessier and H. F. Sleiman, ChemMedChem, 2012, 7, 85–94 CrossRef PubMed.
- J. Berrones Reyes, P. S. Sherin, A. Sarkar, M. K. Kuimova and R. Vilar, Angew. Chem., Int. Ed., 2023, 62, e202310402 Search PubMed.
- M. Saini and T. E. Keyes, Dalton Trans., 2023, 52, 7787–7791 Search PubMed.
- R. Guha, D. Defayay, A. Hepp and J. Müller, ChemPlusChem, 2021, 86, 662–673 CrossRef PubMed.
- M. Hebenbrock, L. Stegemann, J. Kösters, N. L. Doltsinis, J. Müller and C. A. Strassert, Dalton Trans., 2017, 46, 3160–3169 RSC.
- M. Hebenbrock, D. González-Abradelo, A. Hepp, J. Meadowcroft, N. Lefringhausen, C. A. Strassert and J. Müller, Inorg. Chim. Acta, 2021, 516, 119988 Search PubMed.
- I. Maisuls, F. Boisten, M. Hebenbrock, J. Alfke, L. Schürmann, B. Jasper-Peter, A. Hepp, M. Esselen, J. Müller and C. A. Strassert, Inorg. Chem., 2022, 61, 9195–9204 Search PubMed.
- A. Gil-Martínez, A. Hernández, C. Galiana-Roselló, S. López-Molina, J. Ortiz, Á. Sastre-Santos, E. García-España and J. González-García, J. Biol. Inorg. Chem., 2023, 28, 495–507 Search PubMed.
- X. Xie, O. Reznichenko, L. Chaput, P. Martin, M.-P. Teulade-Fichou and A. Granzhan, Chem. – Eur. J., 2018, 24, 12638–12651 CrossRef CAS PubMed.
- K. Suntharalingam, A. Łęczkowska, M. A. Furrer, Y. Wu, M. K. Kuimova, B. Therrien, A. J. P. White and R. Vilar, Chem. – Eur. J., 2012, 18, 16277–16282 Search PubMed.
- A. Shivalingam, M. A. Izquierdo, A. Le Marois, A. Vyšniauskas, K. Suhling, M. K. Kuimova and R. Vilar, Nat. Commun., 2015, 6, 8178 CrossRef CAS PubMed.
- L.-Y. Liu, W. Liu, K.-N. Wang, B.-C. Zhu, X.-Y. Xia, L.-N. Ji and Z.-W. Mao, Angew. Chem., Int. Ed., 2020, 59, 9719–9726 Search PubMed.
- B. M. Zeglis, V. C. Pierre and J. K. Barton, Chem. Commun., 2007, 4565–4579 Search PubMed.
- J. Dai, M. Carver, C. Punchihewa, R. A. Jones and D. Yang, Nucleic Acids Res., 2007, 35, 4927–4940 Search PubMed.
- V. González and L. H. Hurley, Annu. Rev. Pharmacol. Toxicol., 2010, 50, 111–129 Search PubMed.
- J. Dai, D. Chen, R. A. Jones, L. H. Hurley and D. Yang, Nucleic Acids Res., 2006, 34, 5133–5144 Search PubMed.
- A. T. Phan, V. Kuryavyi, S. Burge, S. Neidle and D. J. Patel, J. Am. Chem. Soc., 2007, 129, 4386–4392 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- D. Escher, M. N. Hossain, H.-B. Kraatz and J. Müller, J. Biol. Inorg. Chem., 2021, 26, 659–666 CrossRef CAS PubMed.
- A. Ouedraogo and J. Lessard, Can. J. Chem., 1991, 69, 474–480 Search PubMed.
- T. L. J. Huang and D. G. Brewer, Can. J. Chem., 1981, 59, 1689–1700 CrossRef CAS.
- M. B. Smith, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, Wiley, 8th edn, 2020 Search PubMed.
- J.-Y. Cho, K. Y. Suponitsky, J. Li, T. V. Timofeeva, S. Barlow and S. R. Marder, J. Organomet. Chem., 2005, 690, 4090–4093 Search PubMed.
- B. Lippert, Cisplatin – Chemistry and Biochemistry of a Leading Anticancer Drug, Wiley-VCH and Verlag Helvetica Chimica Acta, Zürich, 1999 Search PubMed.
- A. G. Orpen, L. Brammer, F. H. Allen, O. Kennard, D. G. Watson and R. Taylor, J. Chem. Soc., Dalton Trans., 1989, S1–S83 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 Search PubMed.
- J. S. Murray, T. Brinck, P. Lane, K. Paulsen and P. Politzer, J. Mol. Struct.:THEOCHEM, 1994, 307, 55–64 CrossRef.
- J. A. G. Williams, A. Beeby, E. S. Davies, J. A. Weinstein and C. Wilson, Inorg. Chem., 2003, 42, 8609–8611 Search PubMed.
- A. Galstyan, A. R. Naziruddin, C. Cebrián, A. Iordache, C. G. Daniliuc, L. De Cola and C. A. Strassert, Eur. J. Inorg. Chem., 2015, 5822–5831 CrossRef CAS.
- N. K. Allampally, C.-G. Daniliuc, C. A. Strassert and L. De Cola, Inorg. Chem., 2015, 54, 1588–1596 CrossRef CAS PubMed.
- C. A. Strassert, C.-H. Chien, M. D. Galvez Lopez, D. Kourkoulos, D. Hertel, K. Meerholz and L. De Cola, Angew. Chem., Int. Ed., 2011, 50, 946–950 CrossRef CAS PubMed.
- R. del Villar-Guerra, J. O. Trent and J. B. Chaires, Angew. Chem., Int. Ed., 2018, 57, 7171–7175 CrossRef CAS PubMed.
- A. De Rache and J.-L. Mergny, Biochimie, 2015, 115, 194–202 Search PubMed.
- P. Agrawal, C. Lin, R. I. Mathad, M. Carver and D. Yang, J. Am. Chem. Soc., 2014, 136, 1750–1753 Search PubMed.
- H. Li, in G-Quadruplex Nucleic Acids. Methods in Molecular Biology, ed. D. Yang and C. Lin, Humana, New York, NY, 2019, vol. 2035, pp. 105–116 Search PubMed.
- J. C. Martinez, J. Murciano-Calles, E. S. Cobos, M. Iglesias-Bexiga, I. Luque and J. Ruiz-Sanz, in Applications of Calorimetry in a Wide Context – Differential Scanning Calorimetry, Isothermal Titration Calorimetry and Microcalorimetry, ed. A. A. Elkordy, InTech, 2013, pp. 73–104 Search PubMed.
- D. Monchaud, C. Allain and M.-P. Teulade-Fichou, Bioorg. Med. Chem. Lett., 2006, 16, 4842–4845 Search PubMed.
- D. A. Megger, C. Fonseca Guerra, J. Hoffmann, B. Brutschy, F. M. Bickelhaupt and J. Müller, Chem. – Eur. J., 2011, 17, 6533–6544 Search PubMed.
- B. K. Teo, Y. H. Xu, B. Y. Zhong, Y. K. He, H. Y. Chen, W. Qian, Y. J. Deng and Y. H. Zou, Inorg. Chem., 2001, 40, 6794–6801 CrossRef CAS PubMed.
- S. Kroos and M. Hebenbrock, Z. Kristallogr., 2024, 239, 225–237 CAS.
- J. E. M. Lewis, R. J. Bordoli, M. Denis, C. J. Fletcher, M. Galli, E. A. Neal, E. M. Rochette and S. M. Goldup, Chem. Sci., 2016, 7, 3154–3161 Search PubMed.
- F. Boisten, PhD thesis, Universität Münster, 2022.
- C. Yu, K. H.-Y. Chan, K. M.-C. Wong and V. W.-W. Yam, Chem. – Eur. J., 2008, 14, 4577–4584 CrossRef CAS PubMed.
- R. B. Martin, Science, 1963, 139, 1198–1203 CrossRef CAS PubMed.
- L. V. Le, T. J. Kim, Y. D. Kim and D. E. Aspnes, Entropy, 2022, 24, 1238 CrossRef PubMed.
- APEX4 (Version 2021.10.0), Bruker AXS Inc., Madison, Wisconsin, USA, 2021 Search PubMed.
- (a) SAINT (Version 8.40B, includes Xprep and SADABS), Bruker AXS Inc., Madison, Wisconsin, USA, 2001 Search PubMed; (b) G. M. Sheldrick, SADABS, University of Göttingen, Germany, 1996 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 Search PubMed.
- ORTEP-III – M. N. Burnett and C. K. Johnson, Oak Ridge Thermal Ellipsoid Plot Program for Crystal Structure Illustrations, Oak Ridge National Laboratory Report ORNL-6895, 1996.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- T. H. Dunning Jr. and P. J. Hay, in Modern Theoretical Chemistry, ed. H. F. Schaefer III, Plenum, New York, 1977, vol. 3, pp. 1–28 Search PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data, molecular polarity index, UV/Vis spectra, emission spectra, time-resolved photoluminescence decay curves with fitting parameters; CD spectra, melting curves, TO displacement assays, HPLC chromatograms, NMR spectra. CCDC 2392538–2392541. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt03067b |
| This journal is © The Royal Society of Chemistry 2025 |