
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCarbon spheres with catalytic silver centres as selenium hosts for stable lithium–selenium batteries†
Jahnavi M.
Sudharma‡
 ,
Sreelakshmy K.
Jayaprakash‡
,
Shruti
Suriyakumar
*,
Bhargav
Rajbongshi
and
Manikoth M.
Shaijumon
*
,
Sreelakshmy K.
Jayaprakash‡
,
Shruti
Suriyakumar
*,
Bhargav
Rajbongshi
and
Manikoth M.
Shaijumon
*
School of Physics, Indian Institute of Science Education and Research Thiruvananthapuram, Vithura, Thiruvananthapuram, Kerala 695551, India. E-mail: shaiju@iisertvm.ac.in; shruti@iisertvm.ac.in
First published on 13th December 2023
Abstract
In the quest to develop next-generation lithium batteries to meet the expansive consumer energy demands, batteries based on conversion chemistry are explored. Selenium cathodes have recently gained attention due to their high conductivity and volumetric capacity compared to the existing sulfur-based cathodes. The practical implementation of lithium–selenium batteries is often hindered by the rapid capacity fading due to the poor electronic contact and the shuttle effect caused by polyselenides. By encapsulating selenium in conducting cavities and using a suitable catalyst, we can mitigate the effect of polyselenide shuttling. Herein, we demonstrate a facile strategy to synthesise carbon sphere reactors loaded with catalytically active silver nanoparticles at the centre that act as efficient selenium hosts for Li–Se batteries. Selenium-encapsulated carbon spheres are prepared by a simple microwave synthesis method followed by carbonization and selenisation. The role of these catalytic nanoreactor centres in improving the capacity, cycling stability, and rate capability of Li–Se batteries is thoroughly investigated. The improved discharge capacity and the excellent cycling stability (249 mA h g−1 at 1C rate for 1000 cycles) are accounted for by the successful immobilization of selenides and polyselenides within the porous structure of the carbon spheres triggered by the silver catalyst.
Introduction
Exploring next-generation energy storage devices has been greatly accelerated by the increase in automotive lithium ion battery (LIB) demand and the growing global environmental concerns.1–3 The lithium-ion battery technology has made significant progress since its commercialization in 1991. However, the state-of-the-art LIBs still suffer from limited energy density to meet large-scale applications in electric transportation and smart grid realms. The increasing energy demands have led to widespread research on exploring alternatives to LIBs.4 In this regard, metal chalcogenide and metal–air batteries have been extensively studied over the past few decades.5–10 In particular, due to its high theoretical capacity (1675 mA h g−1) and high specific energy density (2600 W h kg−1), research on lithium–sulfur batteries has gained attention in the past few decades.11–13 However, the practical implementation of lithium–sulfur batteries for industrial purposes is still hindered by various intrinsic parameters like low conductivity of sulfur and polysulfide shuttling.14,15 Being an element in the same group, selenium possesses properties comparable to sulfur and thus can be considered a potential cathode material. Even though the theoretical gravimetric capacity of selenium (675 mA h g−1) is inferior to that of sulfur, it has a high theoretical volumetric capacity density of 3247 mA h g−1 on account of its higher density (4.81 g cm−1 at 25 °C)16 for lithium–selenium batteries (Li–Se).17–19 Moreover, selenium has a conductivity (1 × 10−3 S m−1) that is substantially greater than that of sulfur (5 × 10−30 S m−1), which can be attributed to higher active mass utilization and rate capability in lithium–selenium systems and other alkali metal–selenium batteries.20–22 The pioneering work in the history of lithium–selenium batteries was introduced by Khalil Amine and co-workers in the year 2012, where they prototyped the framework.23 In a typical Li–Se cell, the cathode is selenium in a matrix and the anode is lithium metal as such. The cell shows a two-step discharge process. Selenium reduction is a multistep electrochemical reaction involving the formation of Li2Sen. At the negative electrode, the oxidation of metallic lithium occurs thereby producing lithium ions (Li+) during discharge and electrodeposition during charge. During the discharge cycle, the Li+ ions in the electrolyte migrate to the cathode and reduce the selenium into lithium selenide (Li2Se). The selenide ions get re-oxidised in the charging phase. The overall reaction can be expressed as: 2Li+ + Se + 2e− ⇌ Li2Se.Though Se-based cathodes are potential alternatives to sulfur and LiCoO2, several challenges still remain to realize their practical implementation in commercial batteries.24–26 The main bottleneck associated with lithium–selenium batteries is the low utilization of selenium, which can lead to low capacity and short cycle life. Similar to the volume expansion of lithium sulfides (Li2Sn) seen in lithium–sulfur systems, lithium selenides have a high probability of expansion, thereby leading to degradation.27 Furthermore, the shuttle effect of polyselenides causes lithium corrosion, continuous depletion of active material from the cathode, leading to low Coulombic efficiency, and short battery life.28,29 Various strategies have been employed to mitigate the effect of polyselenide shuttling and low selenium utilization to overcome these issues.30 Incorporation of carbon,31,32 zeolite,33 graphene,34 CNT,35etc. in the cathode side, has been shown to enhance the performance of lithium–selenium batteries.36 There have been efforts to understand the electrochemistry of polyselenide formation in different electrolyte environments.35 Furthermore, the incorporation of catalytic additives to the selenium-based composite cathode and employing solid electrolytes have also been found effective, similar to the sulfur cathodes.37–39 Catalytic additives can enhance the rate and selectivity of the reaction and provide more stability to the product, leading to a more efficient electrochemical process. However, the choice of catalyst plays a critical role in order to achieve the optimum degree of polyselenide adsorption. Catalysts with high adsorption might hinder the release of polyselenides causing passivation and rapid capacity decay. Here, we demonstrate a facile strategy to synthesise carbon sphere reactors loaded with catalytically active silver nanoparticles at the centre that act as an efficient selenium host for Li–Se batteries. The carbon–selenium composite cathode with micropores provides short length diffusion channels and the catalytic silver centres act as a powerhouse for the polyselenide conversion kinetics paving the way for Li–Se batteries with high power and long cycle life.
Experimental
Materials
Sucrose (C12H22O11), silver nitrate (AgNO3, 99%), sodium borohydride (NaBH4,99%), polyvinyl pyrrolidone (PVP, MW-50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), and selenium (Se, 99.999%) were purchased from Sigma-Aldrich and used as received without any further purification. Deionized water was used for preparing solutions.
000), and selenium (Se, 99.999%) were purchased from Sigma-Aldrich and used as received without any further purification. Deionized water was used for preparing solutions.
Methods
000 rpm for 15 minutes. In order to improve the purity of the compound, the precipitate is then filtered using 0.2 μm filter paper and dried overnight by keeping it in an oven at 120 °C. The sample is then subjected to carbonization by closed annealing in a tube furnace at a heating rate of 5 °C min−1 up to 460 °C and dwelling for 3 h in Ar/H2 environments with a flow rate of 100 sccm.
:1 and ground well. The mixture was then selenized in a tube furnace at a heating rate of 5 °C min−1 up to 260 °C and dwelling for 2 h in an Ar/H2 environment with a flow rate of 100 sccm. After naturally cooling down to RT, the products were collected, stored, and labelled as CS/Se and Ag@CS/Se, respectively.
Material characterisation
The morphology and chemical composition of the prepared samples were analyzed and characterized by various imaging and characterization techniques. Scanning electron microscopy (SEM) and energy dispersive X-ray spectroscopy (EDS) analysis were performed using an FEI-Nova Nano SEM-450. Samples dispersed in isopropanol were drop-cast on a silicon substrate and used for SEM imaging. In order to improve the conductivity of the sample, sputtering with gold was done prior to SEM imaging. Transmission electron microscopy (TEM) was carried out by an FEI-Tecnai G2-Spirit Twin TEM 120 kV. X-ray diffraction (XRD) measurements, with a scanning step of 1° per second in the 2θ range from 10° to 80°, were carried out using a PANalytical Empyrean powder X-ray diffractometer equipped with Cu-Kα radiation. X-Ray photoelectron spectroscopy (XPS) measurements were performed on an ESCA plus XPS spectrometer (Omicron Nanotechnology Ltd, Germany) equipped with monochromatic Mg-Kα (energy 1253.6 eV). Deconvolution of the peaks was done with the assistance of CASA XPS software. The Brunauer–Emmett–Teller (BET) specific surface area and porosity characteristics were analysed using N2 adsorption/desorption isotherms measured at 77 K up to a maximum relative pressure of 1 bar with the 3-Flex surface characterization analyser (Micromeritics). Before the physisorption of nitrogen, the samples were activated at 120 °C for 12 h. Raman spectra were obtained using an HORIBA Xplora Plus Micro-Raman spectrometer system equipped with a laser source emitting 532 nm radiation. To measure the mass loading of selenium in the prepared samples, we performed thermogravimetric analysis (TGA) at a heating rate of 10 °C min−1 up to 600 °C in a N2 atmosphere.Electrochemical measurements
Electrodes were prepared by mixing a selenium–carbon sphere composite, carbon black, and polyvinylidene fluoride (PVDF) at a weight ratio of 7:2:1 in N-methyl-2-pyrrolidone (NMP) solvent. The slurry was then coated on aluminium foil that acts as a current collector and dried in a vacuum oven at 60 °C for 12 h. 14 mm electrode discs with a thickness of ∼30 μm and active mass loading of ∼1.0 mg cm−2 were cut from the coated aluminium foil and used as an electrode for the half-cell studies. The CR2032 coin cells were assembled in an argon-filled glove box (UNIlab, Germany) and used for the electrochemical measurements. The electrolyte was composed of 1 M lithium bis(trifluoro-methane-sulfonyl)imide (LiTFSI) in 1,3-dioxolane (99.8%), and 1,2-dimethoxyethane (DME, 99.5%) (1:1 volume ratio). 19 mm glass fibre Whatman filter paper was used as the separator membrane, with 80 μL electrolyte. The coin cells were galvanostatically discharged and charged in a fixed potential window of 1 to 3 V vs. Li/Li+. The cyclic voltammetry (CV), charge–discharge and electrochemical impedance spectroscopy (EIS) measurements were conducted in a BioLogic VMP3 electrochemical workstation.
Results and discussion
The silver nanoparticles were prepared separately without the carbon source, and are characterized using SEM and TEM images, as shown in Fig. S1a and b (ESI†), respectively. This is further confirmed by UV-visible spectroscopic analysis (Fig. S1c, ESI†). A sharp plasmon peak attributed to the silver nanoparticles (∼400 nm) is identified in accordance with earlier reports.40–42 It is worth mentioning that NaBH4 and PVP act as capping and stabilizing agents, respectively. The carbon spheres (CS) and catalytic silver centered carbon spheres (Ag@CS) were prepared by a simple microwave method using sucrose molecules as the carbon precursor. This method is a simple advancement of the hydrothermal carbonisation method which is commonly used to prepare carbon from biomass precursors.43–45 For the preparation of Ag@CS, the nano silver solution is added to the aqueous sucrose solution prior to the selenization process. Fig. 1a illustrates the overall synthesis procedure of the Ag@CS/Se composite. The microwave-aided single step approach for the carbon preparation and catalyst incorporation, which is reported for the first time, resulted in a great reduction in the processing time (20 min). The carbon spheres were prepared from glucose under hydrothermal conditions at 195 °C, which is higher than the normal glycosidation temperature and leads to aromatization and carbonization. The sugar precursor is firstly polymerized into polysaccharides by intermolecular dehydration and then carbonized with silver acting as the nucleation site. The synthesized CS and Ag@CS were further annealed at 460 °C in an Ar/H2 atmosphere to transform into carbon spheres completely and later subjected to selenization (selenised samples are denoted with Se). Raman spectra were obtained to confirm the structure and chemical composition (Fig. 1b and Fig. S2, ESI†).46 The composite materials have two prominent Raman peaks: one (D-band) is located at 1340.5 cm−1 (disorder caused by sp3 defect), and the other (G-band) at 1584.8 cm−1 (sp2 bond pair). In addition, the intensity ratio of the D/G band (ID/IG) increases with selenium incorporation. This could be because of the disordering as a consequence of selenium loading within the carbon matrix. The SEM images of carbon spheres are given in Fig. 1c. The narrow size distribution of the carbon spheres is attributed to the sugar-type sucrose precursor (Fig. S3, ESI†). The SEM images of Ag@CS clearly show that the silver nanoparticles are encapsulated and centrally located inside the carbon spheres and distributed uniformly, as seen in Fig. 1d. The smaller particle size of the Ag@CS sample could be attributed to the controlled growth involving Ag as the nuclei. Both CS and Ag@CS were subjected to selenization by mixing selenium in a 1:1 weight ratio and annealing at 260 °C in an argon/hydrogen atmosphere. The morphology of the spheres was retained even after selenization as shown in Fig. 1e. Traces of silver are evident from the EDX mapping of the samples as shown in the inset of Fig. 1d and e and the composition is also presented in Fig. S4 (ESI†). The X-ray diffraction patterns of the as-synthesised Ag@CS and Ag@CS/Se are shown in Fig. S5a and b (ESI†). In addition to the broad peak at 2θ = 24°, corresponding to carbon, the XRD pattern of Ag loaded CS shows small peaks reaffirming the presence of metallic silver centres. In addition to the strong peaks observed in the XRD pattern accounting for the domination of selenium in the composite cathode, a small peak at 36° in the Se loaded sample is attributed to Ag (Fig. S5b, ESI†), which is further quantified using thermogravimetric analysis (TGA). The composition of selenium in the composite cathode was found to be 26.8% and the plot is shown in Fig. 1f. It is well known that the surface area and the porosity play a significant role in the electrochemical performance of the materials. The porosity and the architecture of the pores extracted using the Brunauer–Emmett–Teller (BET) method47,48 offer significant insights into selenium encapsulation.49,50 Fig. S6 (ESI†) shows the nitrogen adsorption/desorption isotherms of CS, Ag@CS and Ag@CS/Se. The specific surface area of silver-centred carbon spheres and that of carbon spheres is found to be 492 m2 g−1 and 524 m2 g−1, respectively. The curve behaviour of CS and Ag@CS resembles a Type 1(a) isotherm. The sudden uptake of N2 at low P/P0 in CS and Ag@CS host materials is characteristic of the microporous nature as evident from the NLDFT pore distribution with micropores of size below 2 nm. Upon selenization, the surface area of the Ag@CS/Se turned out to be 7 m2 g−1. This reduction in surface area implies the micropores of the Ag@CS host being occupied by Se and can be evident from the reduced micropore distribution in the matrix.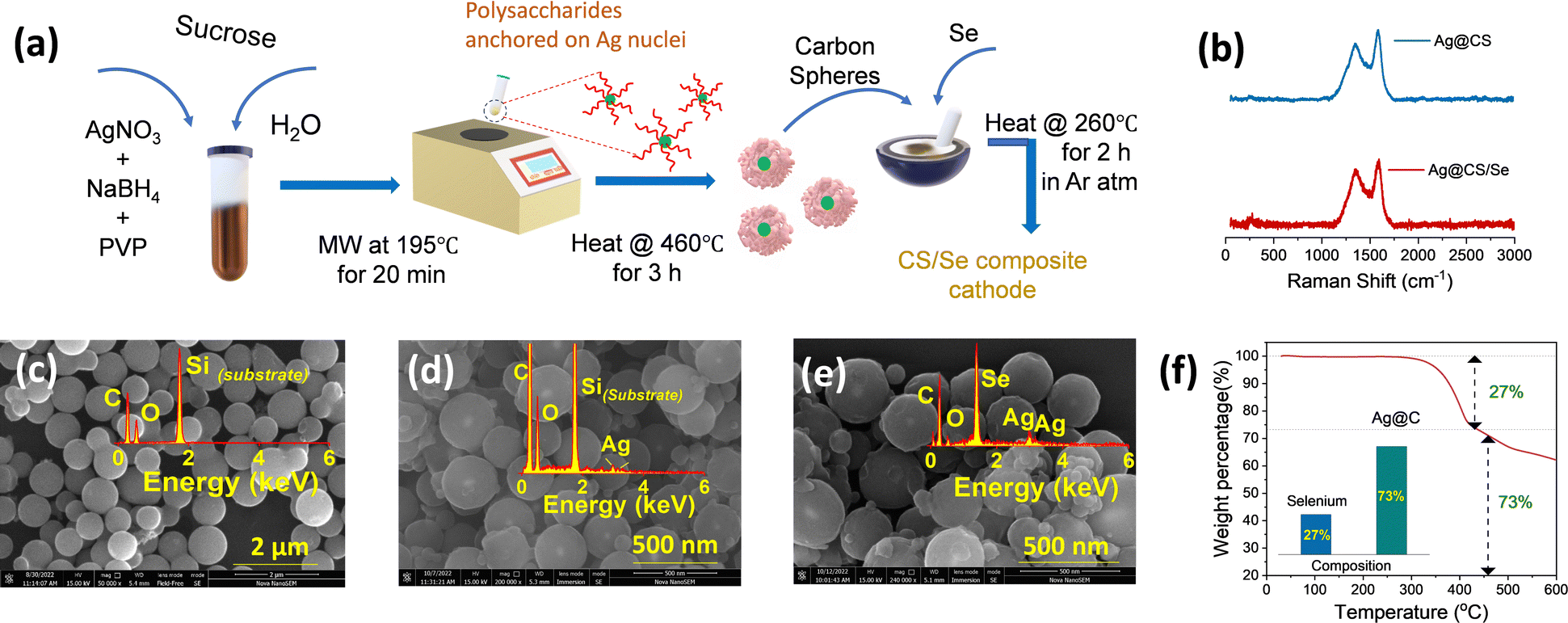 | ||
| Fig. 1 (a) Schematic representation of the microwave-assisted preparation of silver-centred CS and selenium incorporation. (b) Raman spectra of Ag@CS and Ag@CS/Se. SEM images of (c) carbon spheres, (d) Ag@CS and (e) selenium incorporated Ag/CS. The inset shows the respective EDX plots. (f) Estimation of selenium loading from TGA. | ||
To further identify the distribution of silver over the carbon spheres, TEM images were taken. It can be observed from Fig. 2a that the carbon appears as a typical spherical structure with a defined shell assembly, and has silver catalytic centres. The silver appears as a nanoparticle of ∼20 nm diameter centrally located inside the carbon spheres without causing any deformation to the structure and, the pattern was followed uniformly. The high-resolution TEM (HR-TEM) images also reveal the same structure and show that the silver is embedded inside the carbon spheres centrally (Fig. 2a). The scanning transmission electron microscopy (STEM)-energy dispersive X-ray spectroscopy (EDS) mappings for Ag@CS are recorded, which reveal the centralised uniform distribution of silver nanoparticles, as clearly seen in Fig. 2b–e. For better understanding, the TEM image of carbon spheres without silver incorporation is shown in Fig. 2f. The spheres appear highly homogenous and no evidence of a core/shell morphology is seen. The high pore size could be attributed to the channels distributed within the carbon matrix. To further study the chemical composition of the silver centres, the XPS spectra of Ag@CS and Ag@CS/Se are analysed, as depicted in Fig. 2g and h, respectively. The XPS spectra of Ag@CS show C and Ag, as well as the presence of oxygen. The C 1s spectrum shows peaks at 284.7, 285.8 and 288.1 eV corresponding to C–C, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and O–CO bonds, respectively.51 The Ag nanoparticles in Ag@CS are found to exist in the metallic state as illustrated in the Ag 3d spectra with characteristic Ag 3d5/2 and Ag 3d3/2 doublets located at 368.1 and 374.1 eV, respectively. The deconvoluted peaks in the O 1s spectra are ascribed to the O that is bonded to the C in various forms as C–O and CO, which are in agreement with the C 1s spectra. Fig. 2h depicts the XPS spectra of Ag@CS/Se. The survey spectrum shows the presence of C, O, Ag and Se, Fig. S7 (ESI†). The C 1s spectra shows a similar trend to that of Ag@CS, with peaks corresponding to C–C, CO and O–CO bonds. The Se 3d spectra exhibit sharp peaks at 55.8 and 56.7 eV that correspond to Se 3d5/2 and Se 3d3/2, respectively, signifying that Se exists in the elemental form.52 The incorporation of Se has not disrupted the electronic state of Ag from the metallic state as revealed by the Ag 3d spectra.
O and O–CO bonds, respectively.51 The Ag nanoparticles in Ag@CS are found to exist in the metallic state as illustrated in the Ag 3d spectra with characteristic Ag 3d5/2 and Ag 3d3/2 doublets located at 368.1 and 374.1 eV, respectively. The deconvoluted peaks in the O 1s spectra are ascribed to the O that is bonded to the C in various forms as C–O and CO, which are in agreement with the C 1s spectra. Fig. 2h depicts the XPS spectra of Ag@CS/Se. The survey spectrum shows the presence of C, O, Ag and Se, Fig. S7 (ESI†). The C 1s spectra shows a similar trend to that of Ag@CS, with peaks corresponding to C–C, CO and O–CO bonds. The Se 3d spectra exhibit sharp peaks at 55.8 and 56.7 eV that correspond to Se 3d5/2 and Se 3d3/2, respectively, signifying that Se exists in the elemental form.52 The incorporation of Se has not disrupted the electronic state of Ag from the metallic state as revealed by the Ag 3d spectra.
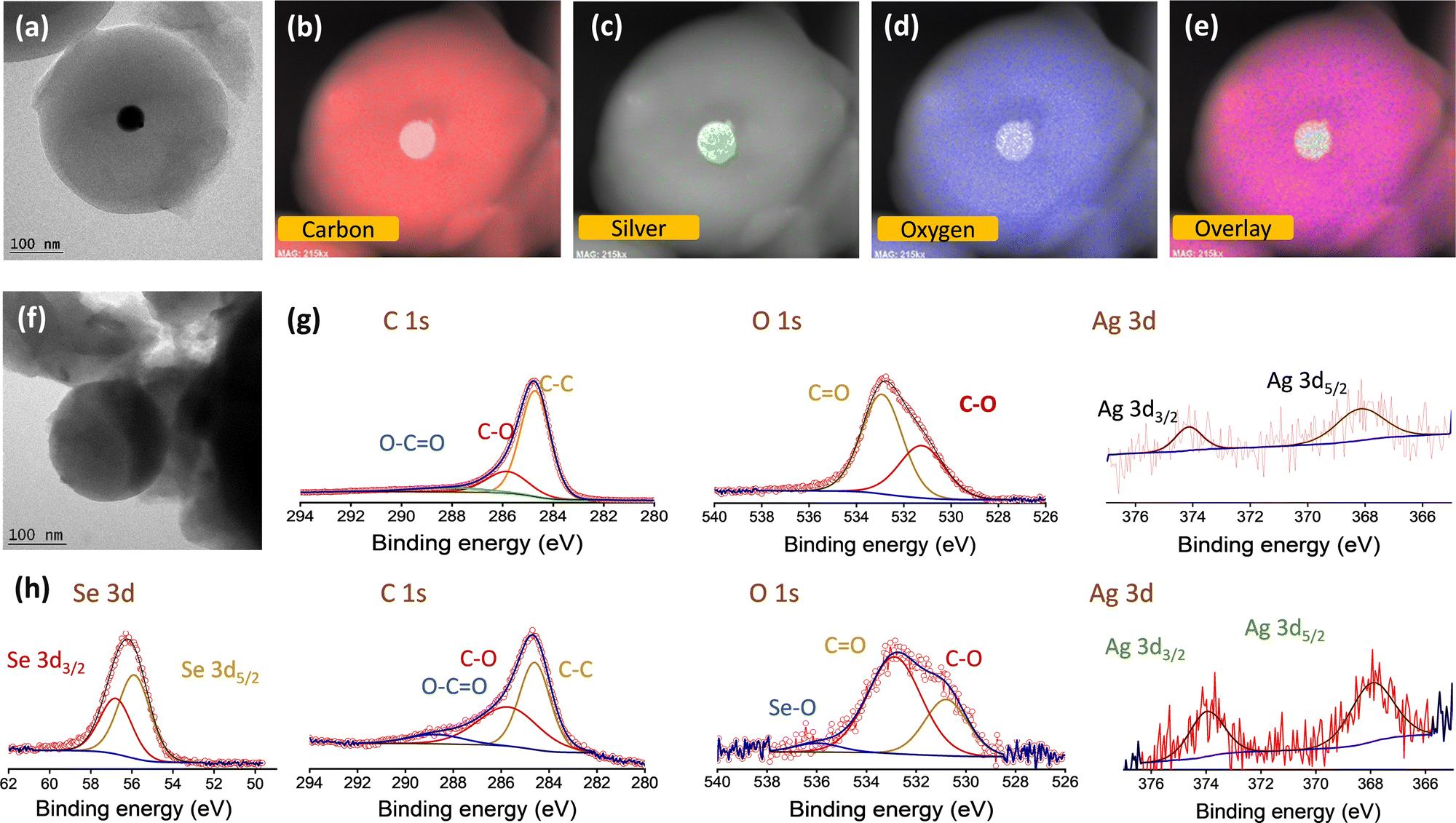 | ||
| Fig. 2 (a) TEM image and (b)–(e) elemental mapping of Ag@CS. (f) TEM image of CS. Deconvoluted XPS spectra (C 1s, O 1s, Ag 3d, Se 3d peaks) of Ag/CS (g) before and (h) after selenisation. | ||
To explore the advantages of Ag@CS/Se as an efficient composite cathode material for Li–Se batteries, the electrochemical performance of carbon spheres, silver centred carbon spheres, selenium encapsulated carbon spheres and selenium encapsulated silver centred carbon spheres were thoroughly studied vs. Li/Li+ in the half cell configuration using 2032-type coin cells. Fig. S8a–d (ESI†) shows the cyclic voltammetry curves of the carbon spheres and silver centred carbon spheres, respectively, in the potential window of 1.0–3.0 V (vs. Li+/Li) at a sweep rate of 0.5 mV s−1. Fig. S8b, e (ESI†) shows the galvanostatic discharge curves recorded at various current densities of 0.1 A g−1, 0.2 A g−1, 0.5 A g−1 and 1.0 A g−1, for CS and Ag@CS, respectively. The rate capability plots of CS and Ag@CS electrodes are given in Fig. S8c, f (ESI†), respectively. It is clear from the graphs that the introduction of an electrocatalytic additive has no significant capacity contribution. Fig. 3a and b represents the cyclic voltammetry curves of CS/Se and Ag@CS/Se recorded in the voltage range of 1–3 V vs. Li/Li+ at a scan rate of 0.2 mV s−1. Two oxidation peaks appear at the initial cycle. However, the following cycles show that the oxidation peaks shift to a lower potential position due to the formation of the solid electrolyte interphase (SEI) layer on the surface of the positive electrode and the catalytic activation of the Se electrode. The curves appeared to be overlapping after the first cycle, showing the stabilisation of the reaction mechanism. The schematic representation of the silver centres facilitating the capture of polyselenides is presented in Fig. 3c. Fig. 3d and e represent the galvanostatic discharge curves of CS/Se and Ag@CS/Se electrodes, respectively, which revealed discharge specific capacities of 437 mA h g−1 and 459 mA h g−1 at a 0.1C rate. The galvanostatic cycling profiles clearly indicate much improved electrochemical performance of the modified cathode. To understand the contribution of the host materials to the discharge capacity, half cells of CS and Ag@CS electrodes were assembled and the CV and cycling studies were carried out. Since the capacity obtained from the carbon matrix devoid of selenium is negligible (Fig. S8, ESI†), we confirm that the contribution arising from the carbon matrix with and without silver is negligible to the total capacity of selenium composite electrodes. At 1C rate, Ag@CS/Se delivered a specific capacity of 275 mA h g−1, while CS/Se showed only 137 mA h g−1.
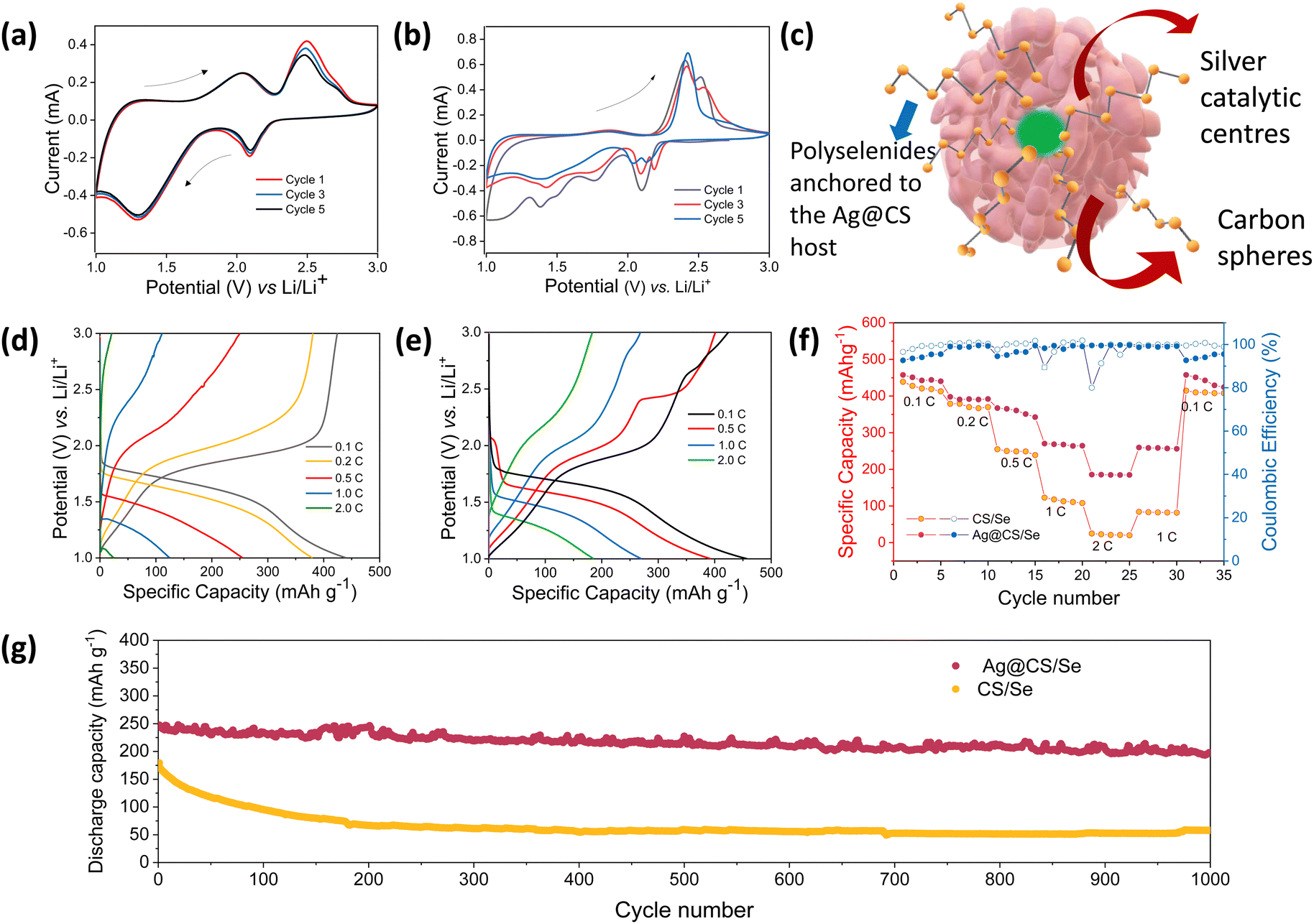 | ||
| Fig. 3 CV profiles of Li–Se cells with (a) CS/Se and (b) Ag@CS/Se cathodes at 0.2 mV s−1 sweep rate. (c) Schematic representation of the silver nanoreactors in CS. Cycling profiles of Li–Se cells with (d) CS/Se and (e) Ag@CS/Se cathodes recorded at different C-rates. Comparison of electrochemical performance in terms of (f) rate capability and (g) long cycling data at 1C rate (1000 cycles) for the CS/Se and Ag@CS/Se cathodes. | ||
The rate capability of the composite cathodes was studied (Fig. 3f) and the Ag@CS/Se electrode exhibited much improved rate performance compared to the CS/Se electrode. The catalytic silver centred selenium composite cathode further showed an excellent cycling stability at 1C rate for 1000 cycles reflecting the excellent catalytic support offered by the silver centres (Fig. 3g). Also, since cathodes with high loading53 are looked upon for better energy density, we tried to vary the selenium loading in the carbon matrix. Two different compositions with 10% and 40% selenium loading were studied and the results are presented in Fig. S9 (ESI†). The CV profile of both the electrodes showed signature peaks; however, the capacity values were strikingly different. It is found that the capacity at high rates is superior for the 10% selenium loaded cathode. On the other hand, increasing the selenium loading to 40% showed a decrement in the capacity values. This could be attributed to the accessibility of selenium to the silver centres and also the porosity of the carbon spheres, which helps in accommodating more selenium. Hence the 27% selenium loading is a trade-off and an optimum value in our case. This is further validated by fitting the Nyquist plot (Fig. S10, ESI†) recorded for the electrode, post CV measurement, which showed Rs = 34 ohms and Rct = 53 ohms.
Fig. 4a–c show the XPS spectra of C 1s, Se 3d and Se 3p for the post-cycled electrodes, respectively. The extra peaks originating in the higher binding energy side in the C 1s core spectra correspond to C–Se and C–F bonds. The peak at 290.3 eV corresponding to the C–Se bond reveals that the Se is chemically bonded to the Ag@CS host.54 In corroboration with the C 1s spectra, the Se 3d peaks show the presence of Se–C bonds as well. The Se 3d spectrum of the post-cycled Ag@CS/Se electrode has an additional feature at lower binding energy of 51.79 eV, which can be attributed to the polyselenides that are anchored on to the carbon host by virtue of the Ag catalytic centres. Post-mortem SEM and EDS mapping of the electrode before and after cycling are presented in Fig. S11 (ESI†). It is seen that the post cycled electrode is intact and has no visible cracks. There are no visible changes in the morphology of the carbon spheres either. The presence of Se in the electrode post cycling is indicative of the selenide retention in the cathode owing to the catalytic activity of silver.
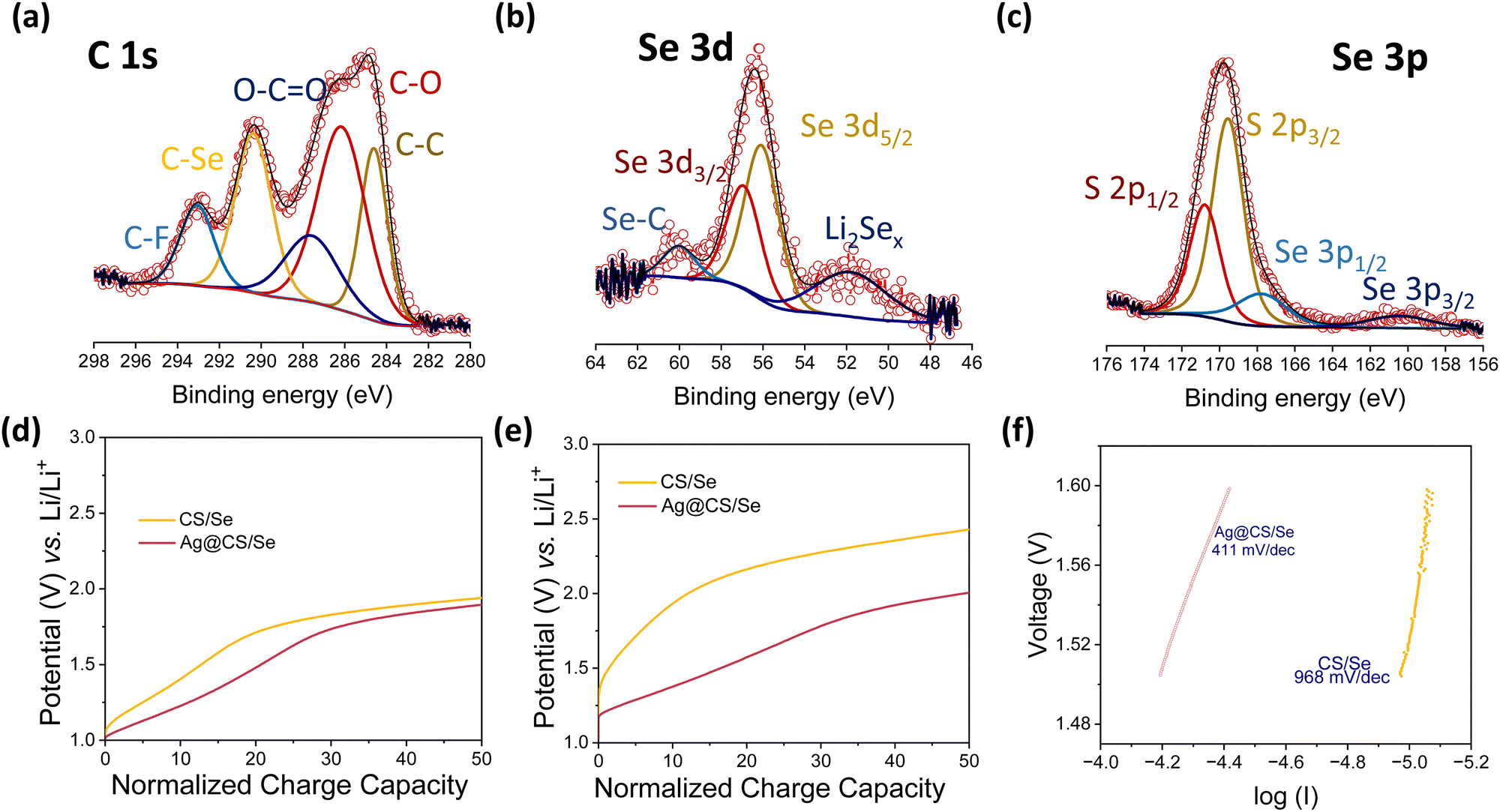 | ||
| Fig. 4 XPS spectra of the post-cycled Ag@CS/Se cathode for (a) C 1s (b) Se 3d (c) Se 3p. Zoomed in view of the normalised capacity vs. potential curve at (d) 0.1C rate and (e) 1C rate. (f) Tafel slope analysis of Li–Se cells with and without a silver centred cathode matrix. | ||
In order to get an insight into the transport kinetics of ions and electrons, the overpotentials were analysed at slow and high C-rates (0.1C and 1C), and the cycling profiles at the said rates were normalised in terms of the state-of-charge and plotted in Fig. 4d and e. It is found that the overpotentials are strikingly low for the Li–Se cells with silver centres. This is direct evidence of the catalytic performance enhancement.55–58 In addition, the Tafel slopes were deduced from the CV and are presented in Fig. 4f. Tafel measurements are commonly used to study the catalytic activity in Li–S chemistries.59,60 The Ag@CS/Se electrode showed a better Tafel value of 411 mV dec−1 again supporting our claim. To conclude, we propose that these silver centres are excellent catalysts for Li–Se batteries. A table presenting the performance of various Se-based composite cathodes is given in the ESI† (Table S1).20,33,34,49,50,61–76 The ease of silver incorporation in the porous carbon sphere matrix is another added advantage for the present work. This microwave assisted method is not just limited to the sucrose derived carbon spheres but can be extended for other carbon matrices with improved porosity and microchannels. This will not only help increase the selenium loading but also increase the performance at higher C-rates.
Conclusions
While lithium–selenium chemistries show promise as next-generation batteries, there are several challenges that need to be addressed before they can be commercialized. These challenges include optimizing the performance, stability, and cycle life of the batteries, as well as scaling up production to meet the demands of various applications. In the present work, we have attempted to explore carbon sphere reactors loaded with catalytically active silver nanoparticles at the centre as an efficient selenium host for Li–Se batteries. In the as-constructed cathode, not only is the method of preparation fast and scalable, but the performance enhancement in terms of the capacity, stability and reversibility is significant. The Li–Se cell with Ag@CS as a host shows a remarkably stable long-term cycling at 1C (249 mA h g−1) for 1000 cycles. With similar strategies, lithium–selenium batteries could potentially prove to be commercially viable and offer significant advantages over existing battery technologies. Not limited to Li–Se systems, the proposed cathode modification strategy could also be expanded to other chemistries such as Na–Se/S and Zn–Se/S.Author contributions
JMS and SKJ carried out the synthesis, electrode fabrication and were involved in the electrochemical measurements and data analysis. BR helped with the preparation of silver nanoparticles. SS designed the workflow and prepared the manuscript with inputs from SKJ and JMS. MMS was in-charge of overall direction and planning. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge IISER Thiruvananthapuram for the financial and infrastructural support. SKJ extends thanks to the Department of Science and Technology, Govt. of India, for the DST-Inspire (DST/INSPIRE/03/2018/002054) PhD fellowship. MMS and SS greatly acknowledge the financial support from Department of Science & Technology, Govt. of India [DST/TMD/IC-MAP/2K20/01].Notes and references
- P. G. Bruce, B. Scrosati and J. M. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2930–2946 CrossRef CAS PubMed
.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS
- A. Masias, J. Marcicki and W. A. Paxton, ACS Energy Lett., 2021, 6, 621–630 CrossRef CAS
- Y. Chen, Y. Kang, Y. Zhao, L. Wang, J. Liu, Y. Li, Z. Liang, X. He, X. Li, N. Tavajohi and B. Li, J. Energy Chem., 2021, 59, 83–99 CrossRef CAS
- S.-E. Cheon, K.-S. Ko, J.-H. Cho, S.-W. Kim, E.-Y. Chin and H.-T. Kim, J. Electrochem. Soc., 2003, 150, A796 CrossRef CAS
- A. Manthiram, Y. Fu, S. Chung, C. Zu and Y. Su, Chem. Rev., 2014, 114, 11751–11787 CrossRef CAS PubMed
- A. Manthiram, Y. Fu and Y.-S. Su, Acc. Chem. Res., 2013, 46, 1125–1134 CrossRef CAS
- T. Bai, D. Li, S. Xiao, F. Ji, S. Zhang, C. Wang, J. Lu, Q. Gao and L. Ci, Energy Environ. Sci., 2023, 16, 1431–1465 RSC
- A. Kondori, M. Esmaeilirad, A. M. Harzandi, R. Amine, M. T. Saray, L. Yu, T. Liu, J. Wen, N. Shan, H. H. Wang, A. T. Ngo, P. C. Redfern, C. S. Johnson, K. Amine, R. Shahbazian-Yassar, L. A. Curtiss and M. Asadi, Science, 2023, 379, 499–505 CrossRef CAS
- G. Girishkumar, B. McCloskey, A. C. Luntz, S. Swanson and W. Wilcke, J. Phys. Chem. Lett., 2010, 1, 2193–2203 CrossRef CAS
- S. Suriyakumar, K. Madasamy, M. Kathiresan, M. H. Alkordi and A. M. Stephan, J. Phys. Chem. C, 2018, 122, 27843–27849 CrossRef CAS
- S. Suriyakumar, S. Gopi, M. Kathiresan, S. Bose, E. B. Gowd, J. R. Nair, N. Angulakshmi, G. Meligrana, F. Bella, C. Gerbaldi and A. M. Stephan, Electrochim. Acta, 2018, 285, 355–364 CrossRef CAS
- S. Suriyakumar, A. M. Stephan, N. Angulakshmi, M. H. Hassan and M. H. Alkordi, J. Mater. Chem. A, 2018, 6, 14623–14632 RSC
- M. Wang, Z. Bai, T. Yang, C. Nie, X. Xu, Y. Wang, J. Yang, S. Dou and N. Wang, Adv. Energy Mater., 2022, 12, 2201585 CrossRef CAS
- G. Zhou, H. Chen and Y. Cui, Nat. Energy, 2022, 7, 312–319 CrossRef CAS
-
K. R. Nagde and S. J. Dhoble, Energy Materials: Fundamentals to Applications, 2021, pp. 335–371 Search PubMed
- A. Eftekhari, Sustainable Energy Fuels, 2017, 1, 14–29 RSC
- C. P. Yang, S. Xin, Y. X. Yin, H. Ye, J. Zhang and Y. G. Guo, Angew. Chem., Int. Ed., 2013, 52, 8363–8367 CrossRef CAS
- C. P. Yang, Y. X. Yin and Y. G. Guo, J. Phys. Chem. Lett., 2015, 6, 256–266 CrossRef CAS
- C. Luo, Y. Xu, Y. Zhu, Y. Liu, S. Zheng, Y. Liu, A. Langrock and C. Wang, ACS Nano, 2013, 7, 8003–8010 CrossRef CAS PubMed
- S. Liu, X. Zhang, S. He, Y. Tang, J. Wang, B. Wang, S. Zhao, H. Su, Y. Ren, L. Zhang, J. Huang, H. Yu and K. Amine, Nano Energy, 2019, 66, 104159 CrossRef CAS
- X. Huang, J. Sun, L. Wang, X. Tong, S. X. Dou and Z. M. Wang, Small, 2021, 17, 2004369 CrossRef CAS
- A. Abouimrane, D. Dambournet, K. W. Chapman, P. J. Chupas, W. Weng and K. Amine, J. Am. Chem. Soc., 2012, 134, 4505–4508 CrossRef CAS PubMed
- X. Gu and C. Lai, Energy Storage Mater., 2019, 23, 190–224 CrossRef
- X. Gu, T. Tang, X. Liu and Y. Hou, J. Mater. Chem. A, 2019, 7, 11566–11583 RSC
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J. M. Tarascon, Nat. Mater., 2011, 11, 19–29 CrossRef PubMed
- H. Wang, S. Chen, C. Fu, Y. Ding, G. Liu, Y. Cao and Z. Chen, ACS Mater. Lett., 2021, 3, 956–977 CrossRef CAS
- G. Di Donato, T. Ates, H. Adenusi, A. Varzi, M. A. Navarra and S. Passerini, Batteries Supercaps, 2022, 5, e202200097 CrossRef CAS
- J. Sun, Z. Du, Y. Liu, W. Ai, K. Wang, T. Wang, H. Du, L. Liu and W. Huang, Adv. Mater., 2021, 33, 2003845 CrossRef CAS
- W. Qiu, X. L. Huang, Y. Wang, C. Feng, H. Ji, H. K. Liu, S. X. Dou and Z. Wang, J. Energy Chem., 2023, 76, 528–546 CrossRef CAS
- M. H. Aboonasr Shiraz, E. Rehl, H. Kazemian and J. Liu, Nanomaterials, 2021, 11, 1976 CrossRef CAS PubMed
- Y. Du, S. Ma, J. Dai, J. Lin, X. Zhou, T. Chen and X. Gu, Batteries, 2022, 8, 123 CrossRef CAS
- H. Li, C. Li, Y. Wang, M.-H. Sun, W. Dong, Y. Li and B.-L. Su, Chem. Synth., 2022, 2, 8 CrossRef CAS
- H. C. Youn, J. H. Jeong, K. C. Roh and K. B. Kim, Sci. Rep., 2016, 6, 1–8 CrossRef
- A. Goldbach, J. Johnson, D. Meisel, L. A. Curtiss and M. L. Saboungi, J. Am. Chem. Soc., 1999, 121, 4461–4467 CrossRef CAS
- W. Pei, X. Liu, M. Khan, X. Ding, H. Zhao, Y. Wang, N. Zhang, X. Chen and J. Xu, Electrochem, 2022, 3, 285–308 CrossRef
- X. Gu, L. Deng and X. Ren, Front. Energy Res., 2021, 9, 626596 CrossRef
- X. Li, J. Liang, J. T. Kim, J. Fu, H. Duan, N. Chen, R. Li, S. Zhao, J. Wang, H. Huang and X. Sun, Adv. Mater., 2022, 34, 2200856 CrossRef CAS
- G. L. Xu, H. Sun, C. Luo, L. Estevez, M. Zhuang, H. Gao, R. Amine, H. Wang, X. Zhang, C. J. Sun, Y. Liu, Y. Ren, S. M. Heald, C. Wang, Z. Chen and K. Amine, Adv. Energy Mater., 2019, 9, 1802235 CrossRef
- Q. Zhang, N. Li, J. Goebl, Z. Lu and Y. Yin, J. Am. Chem. Soc., 2011, 133, 18931–18939 CrossRef CAS PubMed
- Q. Zhang, Y. Hu, S. Guo, J. Goebl and Y. Yin, Nano Lett., 2010, 10, 5037–5042 CrossRef CAS PubMed
- Q. Zhang, J. Ge, T. Pham, J. Goebl, Y. Hu, Z. Lu and Y. Yin, Angew. Chem., Int. Ed., 2009, 48, 3516–3519 CrossRef CAS PubMed
- M. Sevilla and A. B. Fuertes, Chem. – Eur. J., 2009, 15, 4195–4203 CrossRef CAS
- H. Tian, J. Liang, J. Liu, H. Tian, J. Liu and J. Liang, Adv. Mater., 2019, 31, 1903886 CrossRef CAS
- B. Hu, K. Wang, L. Wu, S. H. Yu, M. Antonietti and M. M. Titirici, Adv. Mater., 2010, 22, 813–828 CrossRef CAS PubMed
- P. J. Carroll and J. S. Lannin, Solid State Commun., 1981, 40, 81–84 CrossRef CAS
- Y. Zhang, R. Wang, W. Chen, K. Song, Y. Tian, J. Li and G. Shi, Int. J. Electrochem. Sci., 2023, 18, 100190 CrossRef
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS
- Z. G. Xia, J. J. Zhang, M. Q. Fan, C. J. Lv, Z. Chen and C. Li, New Carbon Mater., 2023, 38, 190–198 CrossRef CAS
- J. T. Lee, H. Kim, M. Oschatz, D. C. Lee, F. Wu, H. T. Lin, B. Zdyrko, W. Il Cho, S. Kaskel and G. Yushin, Adv. Energy Mater., 2015, 5, 1400981 CrossRef
- Y. C. Kang and J. K. Kim, ACS Nano, 2020, 14, 13203–13216 CrossRef
- Y. Cao, F. Lei, Y. Li, S. Qiu, Y. Wang, W. Zhang and Z. Zhang, J. Mater. Chem. A, 2021, 9, 16196–16207 RSC
- N. Wang, X. Zhang, Z. Ju, X. Yu, Y. Wang, Y. Du, Z. Bai, S. Dou and G. Yu, Nat. Commun., 2021, 12, 1–10 CrossRef
- J. Li, J. Song, L. Luo, H. Zhang, J. Feng, X. Zhao, X. Guo, H. Dong, S. Chen, H. Liu, G. Shao, T. D. Anthopoulos, Y. Su, F. Wang and G. Wang, Adv. Energy Mater., 2022, 12, 2200894 CrossRef CAS
- Y. Li, Z. Li, L. Yue, Y. Zhang, S. Liu, Y. Niu, S. Zhang and M. Xu, Adv. Sci., 2023, 10, 2206962 CrossRef CAS
- J. Lei, X. X. Fan, T. Liu, P. Xu, Q. Hou, K. Li, R. M. Yuan, M. Sen Zheng, Q. F. Dong and J. J. Chen, Nat. Commun., 2022, 13, 1–10 Search PubMed
- Z. Li, I. Sami, J. Yang, J. Li, R. V. Kumar and M. Chhowalla, Nat. Energy, 2023, 8, 84–93 CrossRef CAS
- Q. Fan, B. Li, Y. Si and Y. Fu, Chem. Commun., 2019, 55, 7655–7658 RSC
- K. Mahankali, N. K. Thangavel, D. Gopchenko and L. M. R. Arava, ACS Appl. Mater. Interfaces, 2020, 12, 27112–27121 CrossRef CAS
- C. X. Zhao, X. Y. Li, M. Zhao, Z. X. Chen, Y. W. Song, W. J. Chen, J. N. Liu, B. Wang, X. Q. Zhang, C. M. Chen, B. Q. Li, J. Q. Huang and Q. Zhang, J. Am. Chem. Soc., 2021, 143, 19865–19872 CrossRef CAS PubMed
- C. Luo, J. Wang, L. Suo, J. Mao, X. Fan and C. Wang, J. Mater. Chem. A, 2014, 3, 555–561 RSC
- C. Zhang, L. Liu, Y. Wei, C. Zhang, X. Li, J. Wang, L. Ling, W. Qiao and D. Long, Electrochim. Acta, 2015, 153, 140–148 CrossRef
- M. Jia, C. Mao, Y. Niu, J. Hou, S. Liu, S. Bao, J. Jiang, M. Xu and Z. Lu, RSC Adv., 2015, 5, 96146–96150 RSC
- F. Wu, J. T. Lee, Y. Xiao and G. Yushin, Nano Energy, 2016, 27, 238–246 CrossRef CAS
- J. Guo, Q. Wang, C. Qi, J. Jin, Y. Zhu and Z. Wen, Chem. Commun., 2016, 52, 5613–5616 RSC
- D. Dutta, S. Gope, D. S. Negi, R. Datta, A. K. Sood and A. J. Bhattacharyya, J. Phys. Chem. C, 2016, 120, 29011–29022 CrossRef CAS
- H. Lv, R. Chen, X. Wang, Y. Hu, Y. Wang, T. Chen, L. Ma, G. Zhu, J. Liang, Z. Tie, J. Liu and Z. Jin, ACS Appl. Mater. Interfaces, 2017, 9, 25232–25238 CrossRef CAS
- F. Sun, Y. Li, Z. Wu, Y. Liu, H. Tang, X. Li, Z. Yue and L. Zhou, RSC Adv., 2018, 8, 32808–32813 RSC
- P. Xue, Y. Zhai, N. Wang, Y. Zhang, Z. Lu, Y. Liu, Z. Bai, B. Han, G. Zou and S. Dou, Chem. Eng. J., 2020, 392, 123676 CrossRef CAS
- S. Lin, Y. Chen, Y. Wang, Z. Cai, J. Xiao, T. Muhmood and X. Hu, ACS Appl. Mater. Interfaces, 2021, 13, 9955–9964 CrossRef CAS PubMed
- A. H. A. Hoseini, M. H. A. Shiraz, L. Tao, W. Lu, M. Arjmand and J. Liu, Electrochim. Acta, 2022, 429, 140954 CrossRef CAS
- H. Yu, J. Z. Kang, L. S. Huang, J. J. Wang, X. M. Wang, X. Y. Zhao and C. F. Du, Rare Met., 2023, 42, 76–84 CrossRef CAS
- Q. Xia, J. Hu, Q. Chen and L. Zhang, J. Alloys Compd., 2022, 927, 167014 CrossRef CAS
- B. Lu, Z. R. Wang and Q. Sun, J. Electroanal. Chem., 2023, 941, 117545 CrossRef CAS
- F. Gao, X. A. Yue, X. Y. Xu, P. Xu, F. Zhang, H. Sen Fan, Z. L. Wang, Y. T. Wu, X. Liu and Y. Zhang, Rare Met., 2023, 42, 2670–2678 CrossRef CAS
- S. Pei, J. Shao, D. Wang, Z. Zhou, Z. Zhang, C. Mao, J. Liu, Z. Li and G. Li, J. Mater. Chem. A, 2023, 11, 3682–3694 RSC
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ya00487b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |