
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A rational guide to improve the activity of a hydrogen-evolving polymeric carbon nitride photocatalyst†
Kazuhiko
Maeda
 *ab,
Tomoharu
Maeda‡
a,
Chomponoot
Suppaso‡
a,
Shunta
Nishioka
a,
Yoshinobu
Kamakura
a,
Shuhei
Yasuda§
c and
Toshiyuki
Yokoi
c
*ab,
Tomoharu
Maeda‡
a,
Chomponoot
Suppaso‡
a,
Shunta
Nishioka
a,
Yoshinobu
Kamakura
a,
Shuhei
Yasuda§
c and
Toshiyuki
Yokoi
c
aDepartment of Chemistry, School of Science, Tokyo Institute of Technology, 2-12-1-NE-2 Ookayama, Meguro-ku, Tokyo 152-8550, Japan. E-mail: maedak@chem.titech.ac.jp
bLiving Systems Materialogy (LiSM) Research Group, International Research Frontiers Initiative (IRFI), Tokyo Institute of Technology, 4259 Nagatsuta-cho, Midori-ku, Yokohama, Kanagawa 226-8502, Japan
cNanospace Catalysis Unit, Institute of Innovative Research, Tokyo Institute of Technology, 4259 Nagatsuta-cho, Midori-ku, Yokohama 226-8503, Japan
First published on 21st November 2023
Abstract
Photocatalytic H2 evolution using Pt-loaded mesoporous polymeric carbon nitride (meso-PCN) was studied with respect to light intensity and the hydrogen–deuterium isotope effect, in combination with photoelectrochemical measurements. Although the photocatalytic activity was mainly limited by charge recombination in the meso-PCN, improving the oxidation reaction by photogenerated holes was critical for enhancing the H2-evolution activity. The finding of this work will be a useful guide to achieve non-sacrificial overall water splitting by PCN photocatalysts.
Introduction
Recently, semiconductor photocatalysts that produce H2 from water have been actively studied because of the increased demand for large-scale solar-to-fuel energy conversion. Among these photocatalysts, polymeric carbon nitride (PCN), an organic semiconductor, was reported in 2009 to photocatalyze the reduction and oxidation of water under visible light.1 PCN has since become an important semiconductor photocatalyst material that has spurred numerous studies, including investigations of its application to H2 evolution and CO2 reduction.2–4 Visible-light-driven overall water splitting into H2 and O2 has been achieved using urea-derived PCN as a photocatalyst with the aid of Pt and CoOx cocatalysts that promoted H2 and O2 evolution, respectively.5 Recently, single-crystalline PCN derivatives with proper cocatalyst modifications have achieved overall water splitting with an apparent quantum yield of ∼20%, although their band gap remained in the UV region (<400 nm).6,7Semiconductor photocatalysis involves several elementary steps, including the absorption of light energy, charge-carrier generation and migration and surface redox reactions with the charge carriers. Therefore, the development of a highly efficient photocatalyst requires that both its bulk and surface be appropriately designed. This situation is in fact the case for PCN, and several papers along these directions have already been published.2,3,8
The dependence of the light intensity and an isotopic assessment will provide useful information about the mechanistic bottlenecks in photocatalytic reactions. Carrying out a photocatalytic reaction with different light intensities provides information about carrier transfer and recombination in the photocatalyst.9–16 Kinetic hydrogen–deuterium (H–D) isotope effects are, in principle, sensitive to reactants if the reactants participate in surface reactions that involve bond cleavage and formation. Studies on kinetic isotope effects are therefore expected to reveal mechanistic details of surface processes and rate-determining steps.13,14,17,18 For inorganic semiconductor photocatalysts, numerous studies on kinetic aspects such as light-intensity dependence and H–D isotopic assessment have been reported.9–18 The use of hydrogen isotopes will also help to understand the physicochemical properties of hydrogen-related chemical species at the atomic level.19
By contrast, the literature contains only a few such kinetic studies using organic semiconductors such as PCN. Wang et al. investigated the light-intensity-dependent evolution of H2 from lactic acid solution over Pt-loaded urea-derived PCN under simulated sunlight with relatively high light intensity (101–103 mW cm−2 range).16 Hong et al. investigated the H2-evolution activity of RuO2-loaded PCN in a dimethylformamide (DMF)/borate buffer solution containing 1.0 M triethanolamine (TEOA) as an electron donor under 365 nm UV-LED illumination, along with its light-intensity dependence (10–60 mW cm−2 range).20 However, elucidation of the light-intensity dependence of PCN at relatively low intensities, especially under visible light, as well as elucidation of the H–D isotope effect, requires further investigation.
In most cases, the results of photocatalytic H2 evolution reactions by PCN in the presence of sacrificial donors have been discussed based on steady-state photocatalytic reaction results (i.e., by measuring the amount of evolved H2), including those reporting light intensity dependence. In addition, these reports usually start with a statement such as “charge recombination that occurs in the material is the main culprit of reducing the efficiency of PCN photocatalyst”, and end a certain level of improvement in H2 evolution activity. However, it is not always possible to explain which of reduction or oxidation reaction is the main contributor. As a photocatalytic reaction consists of a combination of oxidation and reduction reactions, product analysis based on the amount of evolved H2 is not sufficient, and individual evaluation of the reduction/oxidation reactions is necessary. In the present study, we have conducted a comprehensive examination of PCN for H2 evolution from the standpoint of light intensity dependence and H–D isotope effects, in combination with photoelectrochemical measurement to verify the oxidation behaviors.
We focused on Pt-loaded mesoporous PCN (abbreviated hereafter as Pt/meso-PCN). Pt is one of the metals most commonly used as a cocatalyst for H2 evolution with PCN-related materials3 and functions better than Ru.21 Urea-derived PCN modified with a Pt cocatalyst has been shown to become a critical building unit for visible-light-driven overall water splitting5 and for the H2-evolution system in Z-scheme water splitting.22,23 meso-PCN is a useful semiconductor photocatalyst not only for H2 evolution but also for CO2 reduction.24–27 This background information suggests that Pt/meso-PCN is suitable for investigation.
Experimental
Materials and reagents
Cyanamide (>98%; Alfa Aesar), colloidal SiO2 particles (LUDOX HS-40; Sigma-Aldrich), NH4HF2 (95%; Sigma-Aldrich), triethanolamine (TEOA; >98.0%; Kanto Chemicals), H2PtCl6·6H2O (>98.5%; FUJIFILM Wako Pure Chemical), iodine (>99.8%; Wako Pure Chemical Industries), K4[Fe(CN)6]·3H2O (>99.5%; Wako Pure Chemical Industries), KOH (>86.0%; Kanto Chemicals), acetone (>99.5%; Kanto Chemicals), NaI (>99.5%; Kanto Chemicals), ethylenediaminetetraacetic acid disodium salt dihydrate (EDTA·2Na, >99.5%; Dojindo Laboratories), methanol (>99.9%, FUJIFILM Wako Pure Chemical), methanol-d4 (≥99.8 atom% D, Sigma-Aldrich), H2 gas (>99.99999 vol%, TAIYO NIPPON SANSO), D2 gas (>99.6%; TAIYO NIPPON SANSO), D2O (99.8 atom% D, Kanto Chemicals) and Na2SO4 (99.0%, FUJIFILM Wako Pure Chemical) were used without further purification.Synthesis of meso-PCN
meso-PCN was synthesized by a hard-template approach using a previously reported method.28 Briefly, cyanamide was dissolved in water containing 40% colloidal SiO2. The cyanamide-to-SiO2 weight ratio was 1. The mixed suspension was stirred at 333 K overnight to obtain a transparent solution, which was then heated at 823 K in air for 4 h with a ramp rate of 2.3 K min−1. The resultant solid was washed with an aqueous NH4HF2 (4 M) solution for 24 h to remove the SiO2 template. Finally, the solid product was washed with H2O and ethanol three times for each.Preparation of Pt/meso-PCN
The as-prepared meso-PCN (0.3 g) was dispersed in a top-irradiation-type reaction vessel containing aqueous TEOA solution (10 vol%, 140 mL) containing dissolved H2PtCl6·6H2O (3 wt% vs. meso-PCN). The reaction vessel was connected to a closed-gas circulation system made of glass and was degassed using a vacuum pump. After a small amount of Ar gas was introduced into the reaction system, the solution was irradiated with visible light from a 300 W xenon lamp (PE300BF, Cermax) fitted with a cold mirror (CM-1) and an L42 cutoff filter. After 3 h of irradiation, the solid component was separated by filtration, washed with H2O several times and dried in air at 333 K overnight.Characterization
The prepared samples were analyzed by X-ray diffraction (XRD; Rigaku MiniFlex 600; Cu Kα), UV-vis diffuse-reflectance spectroscopy (V-770, JASCO), scanning electron microscopy (SEM; Hitachi SU9000) and energy-dispersive X-ray spectroscopy (EDS; Thermo Fisher Scientific). Nitrogen adsorption/desorption isotherms were recorded at 77 K using a gas adsorption instrument (BELSORP-mini, MicrotracBEL).H2-evolution reactions
Reactions were conducted at room temperature using 30 mL Pyrex® cuvettes containing 16 mL of solution (10 vol% TEOA) and 16 mg of Pt/meso-PCN, unless otherwise stated. Each cuvette was sealed with a rubber septum and was purged with Ar for 30 min. A MAX-400D xenon lamp (Asahi Spectra) equipped with a bandpass filter was used as the light source. After irradiation for 2 h, the gases present in the headspace of the cuvette were collected by a syringe and analyzed by gas chromatography (GL Science; model GC323; thermal conductivity detector; active carbon column) with Ar as the carrier gas. The rise of the temperature in the reactant solution after the 2 h irradiation was within 2 K.Calculation of apparent quantum yield
Apparent quantum yields (AQYs) were calculated using the following equation:where A represents the coefficient of the reaction (here, A = 2). When EDTA·2Na was used as an electron donor, we assumed a current-doubling effect (i.e., A = 1).29
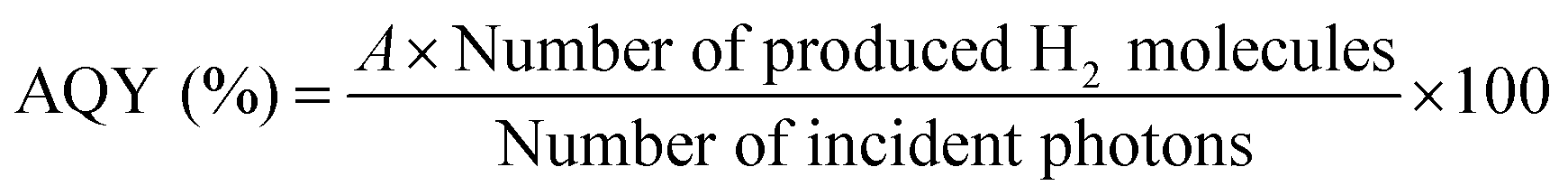
| Number of produced H2 molecules [h−1] = vH2[μmol h−1] × NA [mol−1] × 10−6 |
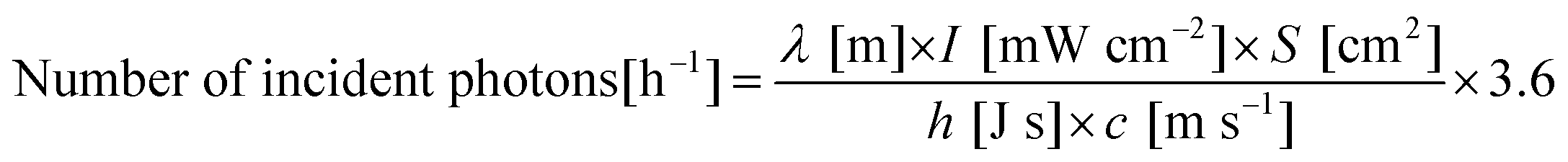
where λ, I, S, h and c are the wavelength of light, light intensity (measured by a spectrophotometer; LS-100, Eko Instruments), irradiation area, Planck's constant and the speed of light, respectively. In this work, the irradiation area was fixed to be 4.8 cm2, and the light intensity was controlled by adjusting the output power of the xenon lamp.
Calibration of H2 and D2 for gas chromatography analysis
To investigate the H–D isotope effect, calibration curves of H2 and D2 were prepared using each pure gas at room temperature. As shown in Fig. S1,† the sensitivity for D2 was slightly lower than that for H2. Schwarze et al.30 conducted photocatalytic H2-evolution experiments using Pt/meso-PCN in a D2O/TEOA mixture (85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15 v/v), which was identical to the conditions used in our work. In the study of Schwarze et al.,30 mass spectrometric analysis showed that D2 was the main product (85.4%), with minor production of HD (12.9%) and H2 (1.7%). This result indicates that, although the hydrogen originates mainly from water, H–D exchange between D2O and TEOA occurs to some extent. In the present work, we analyzed evolved hydrogen under the assumption that hydrogen produced during the reaction using D2O is D2. This approach inevitably underestimates the amount of produced hydrogen molecules to a small extent, leading to an overestimation of the H–D isotope effect.
:15 v/v), which was identical to the conditions used in our work. In the study of Schwarze et al.,30 mass spectrometric analysis showed that D2 was the main product (85.4%), with minor production of HD (12.9%) and H2 (1.7%). This result indicates that, although the hydrogen originates mainly from water, H–D exchange between D2O and TEOA occurs to some extent. In the present work, we analyzed evolved hydrogen under the assumption that hydrogen produced during the reaction using D2O is D2. This approach inevitably underestimates the amount of produced hydrogen molecules to a small extent, leading to an overestimation of the H–D isotope effect.
Photoelectrochemical measurements
Linear-sweep voltammetry (LSV) measurements were conducted in 0.1 M aqueous Na2SO4 solution containing different additives and under visible light using an HSV-110 potentiostat (Hokuto Denko). Pt/meso-PCN/FTO, Ag/AgCl and a Pt coil were used as the working, reference and counter electrodes, respectively. The Pt/meso-PCN/FTO electrode was prepared by an electrophoretic deposition method. The working electrode was irradiated using a 300 W xenon lamp (PE300BF, Cermax) equipped with a HOYA L42 cutoff filter (λ > 400 nm). The scan rate was fixed at 10 mV s−1. Before the measurement, the pH of the electrolyte solution was adjusted to a certain value by addition of NaOH and was purged with Ar gas for 30 min.Results and discussion
Structural characterization
According to the previous report, meso-PCN was synthesized via a previously reported hard-template method using 12 nm colloidal SiO2.28 Pt (3 wt%) was deposited onto the as-prepared meso-PCN via photodeposition. The details are provided in the ESI.†The X-ray diffraction (XRD) pattern of the as-synthesized meso-PCN shows reflections from the stacking of two-dimensional layers and an in-plane repeating motif (Fig. S2a†), which are characteristic of PCN. The corresponding UV-visible diffuse-reflectance spectra indicate that the absorption edge of the meso-PCN is at ∼450 nm, with a tail extending to 600 nm (Fig. S2b†), consistent with a previous report.31
As shown in Fig. S3a,† N2 adsorption/desorption isotherms of the meso-PCN show a hysteresis, which is typical of a material that possesses randomly connected spherical pores. In the corresponding pore size distribution curve obtained by the Barrett–Joyner–Halenda (BJH) method, a peak is observed at ∼12 nm (Fig. S3b†), consistent with the size of the SiO2 hard template used in the synthesis. The porous texture of the meso-PCN is evident in its scanning electron microscopy (SEM) image (Fig. S3c†).
As a result of photodeposition of Pt onto the meso-PCN, the porous texture was largely lost, accompanied by a decrease in the pore volume and size. These results indicate that most of the introduced Pt was loaded inside the pores of the meso-PCN. In the SEM observations combined with EDS analysis (Fig. S3c and S3d†), Pt deposits on the external surface of the meso-PCN are indeed difficult to observe, except for aggregated Pt particles that appear as brighter spots because of the difference in electron density between Pt and C/N.
Photocatalytic H2 evolution under different operating conditions
Using the as-prepared Pt/meso-PCN, we first conducted H2-evolution reactions in aqueous TEOA solution (10 vol%, 16 mL) under monochromatized visible light (4 mW cm−2) in a cuvette-type reactor,32 where the mass of Pt/meso-PCN was varied. TEOA is a commonly used electron donor for photocatalytic H2 evolution by PCN-related materials. The pH of the TEOA aqueous solution was approximately 11. As the equation pH = −log[H+] tells us, H+ rarely exists under these conditions and it is reasonable to assume that the reactant is H2O.As shown in Fig. 1a, the H2-evolution AQY increased when the mass concentration of Pt/meso-PCN was increased to 1.0 g L−1. A further increase of the mass concentration to 2.0 g L−1 did not change the AQY. The nearly linear increase of the AQY to 1.0 g L−1 can be explained by the increase in concentration of the Pt/meso-PCN suspension, which absorbs the incident light more effectively. At concentrations greater than 1.0 g L−1, however, an increase of the concentration is no longer effective because the additional portion does not contribute to the absorption of the incident light. Therefore, we decided to conduct the H2-evolution reaction using 16 mg of Pt/meso-PCN and 16 mL of aqueous TEOA.
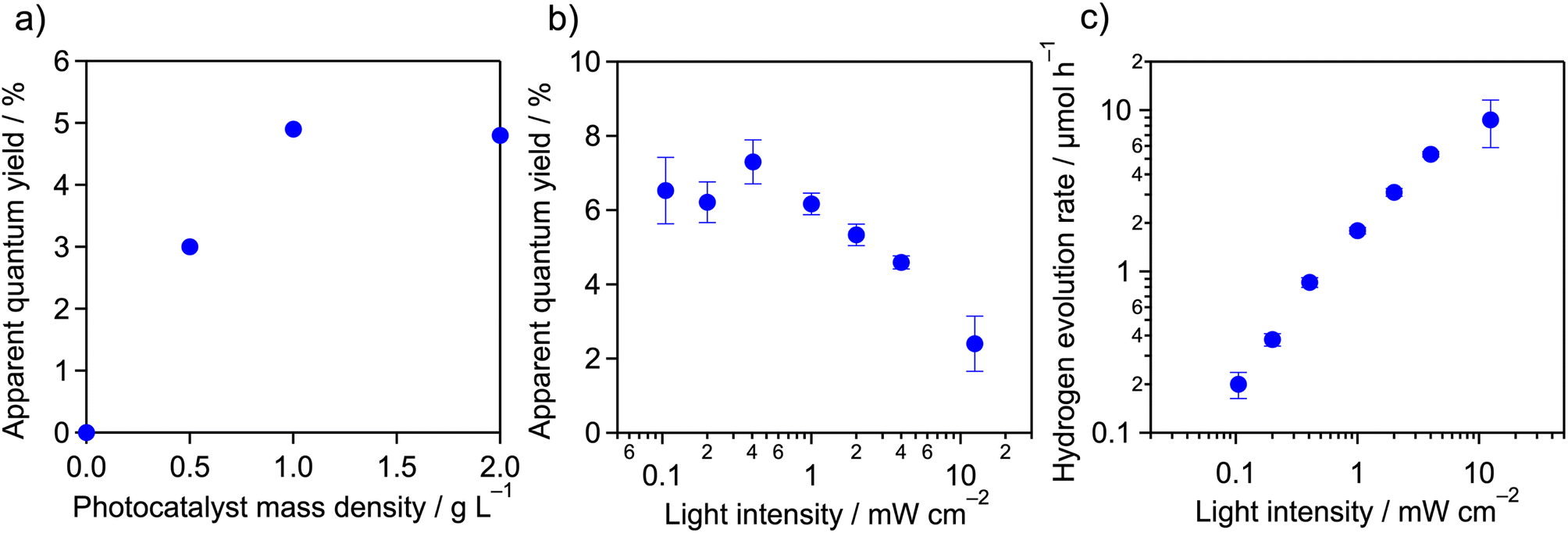 | ||
| Fig. 1 (a) Apparent quantum yields for H2 evolution under monochromatized light (∼4 mW cm−2), as measured at 400 nm as a function of the mass concentration of meso-PCN in an aqueous TEOA solution (10 vol%, 16 mL). (b) Apparent quantum yields and (c) H2-evolution rates from Pt/meso-PCN as a function of the incident-light intensity. Reaction conditions: catalyst, 16 mg; reactant solution, aqueous TEOA (10 vol%, 16 mL); wavelength of light, 400 nm. Because of a technical issue of the light source, we are unable to increase the light intensity higher than ∼10 mW cm−2. | ||
Fig. 1b shows the AQYs of H2 evolution over Pt/meso-PCN under 400 nm monochromatized light as a function of the incident-light intensity. The AQYs recorded in the light-intensity range ≤1 mW cm−2 were ∼6.5%, almost independent of the light intensity, although some experimental error was present, as reflected by the error bars in the figure. However, the AQYs decreased as the light intensity was increased beyond 1 mW cm−2.
The relationship between the rate of H2 evolution (vH2) and the incident-light intensity (I) is displayed in Fig. 1c. From the plot, two operational regimes were observed; the plot was fitted by the equation
| vH2 = aIb |
The results of the H2-evolution reaction under different light-intensity conditions indicate that the incident photons could be used for H2 evolution, with little contribution of the accumulation of photogenerated electrons and holes in Pt/meso-PCN under ≤1 mW cm−2 irradiation conditions. This feature is characteristic of a photocatalytic reaction under low-light-intensity conditions. Under ≥1 mW cm−2 irradiation conditions, this linear relationship was no longer observed because the population of photogenerated electron–hole pairs accumulated in Pt/meso-PCN became non-negligible and the electron–hole recombination affected the overall reaction rate.13 The deviation from the proportional dependence on light intensity ≥1 mW cm−2 thus corresponded to a transition from a proportional-dependence regime to square-root-dependence regime.
H–D isotope effect in H2 evolution
We next examined the H–D isotope effect in H2 evolution with respect to several electron donors that are frequently used for photocatalytic reactions involving PCN, including H2 evolution and CO2 reduction.24,33 The light intensity was set to be ∼4 mW cm−2 to avoid photoexcitation-limited situations.The H2-evolution reaction in a D2O–TEOA solution under 400 nm visible light gave AQYs of 3.9 ± 0.2%, which are lower than those obtained in H2O–TEOA solution (4.6 ± 0.2%), as listed in Table 1. The H–D isotope effect was thus calculated to be, at most, ∼1.2. The H–D isotope effect being greater than unity indicates that the overall reaction rate is influenced by surface reactions with H2O (or D2O) and its related species. The observed H–D isotope effect (∼1.2) in the case of TEOA actually indicates that the surface reaction involving species related to H2O (or D2O) affected the overall reaction rate to some extent. However, the observed value was much smaller than those reported for electrochemical H2-evolution reactions with typical H–D isotope effect values greater than 3. Hong et al. reported a H–D isotope effect of 2.7 for RuO2-loaded PCN in DMF/borate buffer solution containing 1.0 M TEOA (pH ∼13) under 365 nm UV-LED illumination with an intensity of 60 mW cm−2.20 On the basis of this result, they argued that the reduction of water to H2 is involved in the rate-determining step for the photocatalytic H2 evolution. The small H–D isotope effect observed in the present study for H2 evolution over Pt/meso-PCN indicates that the kinetically relevant steps for the reaction do not lie in bond cleavage/formation processes involving H/D atoms. It also re-confirms that, compared with Ru, Pt exhibits better performance as a H2-evolution cocatalyst.
| Entry | Electron donor | Reaction pH | AQY/% | H–D isotope effectb | |
|---|---|---|---|---|---|
| with H2O | with D2O | ||||
| a Reaction conditions: catalyst, 16 mg; reactant solution, aqueous solution containing electron donor (16 mL); wavelength of light, 400 nm; light intensity: ∼4 mW cm−2. b Defined as the ratio of the reaction rate with H2O to that of D2O. | |||||
| 1 | TEOA (10 vol%) | 10.9 | 4.6 ± 0.2 | 3.9 ± 0.2 | 1.2 ± 0.08 |
| 2 | CH3OH (10 vol%) | 7.6 | 0.48 ± 0.06 | 0.29 ± 0.01 | 1.7 ± 0.2 |
| 3 | CD3OD (10 vol%) | 7.6 | 0.35 ± 0.003 | 0.23 ± 0.04 | 1.5 ± 0.2 |
| 4 | EDTA·2Na (10 mM) | 4.5 | 1.0 ± 0.04 | 0.87 ± 0.04 | 1.1 ± 0.07 |
In the early work on Pt/PCN, the H2-evolution rates from an aqueous solution containing methanol or ethylenediaminetetraacetic acid disodium salt dihydrate (EDTA·2Na) were reported to be lower than that from an aqueous solution of TEOA.1 Similar results were obtained in the present study (Table 1). In particular, the H2-evolution AQY in aqueous methanol was an order of magnitude lower than that in aqueous TEOA. Therefore, the lower efficiency in methanol appears to arise from a difference in the kinetically relevant steps between the two reactants. Interestingly, the H–D isotope effect in the presence of methanol (∼1.7) was greater than that in the present of TEOA (∼1.2), suggesting that the oxidation of methanol on the surface of meso-PCN is less efficient than the oxidation of TEOA. When methanol-d4 (CD3OD) was used as an electron donor, a H–D isotope effect similar to that for methanol was observed but with lower AQYs compared with the nonlabelled-methanol case. This result again confirms that the oxidation process of methanol influences the overall reaction rate. Note that a certain isotope effect is expected when deuterated-TEOA is used. However, it was not possible to examine it, because we could not synthesize or buy deuterated-TEOA.
One may think that the significant difference in the H2-evolution AQY between TEOA and methanol arises from the pH difference. However, AQY in a pH ∼11 aqueous methanol (0.12%) was found to be lower than that at pH 7.6 (0.48 ± 0.06%). This is in line with the idea that the oxidation process on the meso-PCN has a strong impact on the H2 evolution activity.
We also attempted to use reversible electron donors such as FeCl2 and K4[Fe(CN)6], which can be used for Z-scheme overall water splitting.23 However, the quantity of H2 gas was not measurable under the present reaction conditions, primarily because the rates of H2 evolution from the reversible donor solution were at least an order of magnitude lower than those from the aqueous methanol solution.
Photooxidation abilities
Because meso-PCN is an n-type semiconductor,34 its photooxidation behavior can be monitored using a photoelectrochemical technique. The meso-PCN powder was deposited onto a transparent conductive glass (fluorine-doped tin oxide (FTO)) by an electrophoretic deposition method. Fig. 2 displays current–potential curves for the Pt/meso-PCN/FTO electrodes in different electrolytes. In Na2SO4 aqueous solution (pH ∼11), a clear anodic photocurrent was observed, indicating n-type semiconductivity of the material.35 Notably, photogenerated holes in PCN-related materials can be consumed by not only water oxidation but also by self-oxidation of the nitrogen component in the materials.5,21,36 When TEOA was present, the anodic photoresponse was improved, especially in the lower-potential region (Fig. 2a). This result indicates that the oxidation of TEOA is kinetically more favorable than the oxidation of water. When methanol was added as an electron donor at pH ∼7.5, the anodic photocurrent remained almost unchanged (Fig. 2b). No obvious increase in the photocurrent density was seen in a pH ∼11 aqueous methanol (Fig. S4†). Therefore, the oxidation of TEOA over meso-PCN was more efficient than that of methanol. The anodic photocurrent recorded in methanol-d4 (CD3OD)–H2O aqueous solution containing 0.1 M Na2SO4 was lower than that recorded in the methanol–H2O electrolyte, indicating an isotope effect in the photooxidation of methanol. In the case of EDTA, a photooxidation behavior similar to TEOA was observed (Fig. 2c), although the photocurrent enhancement was moderate. These trends related to photoelectrochemical oxidation qualitatively agree with those related to photocatalytic H2 evolution.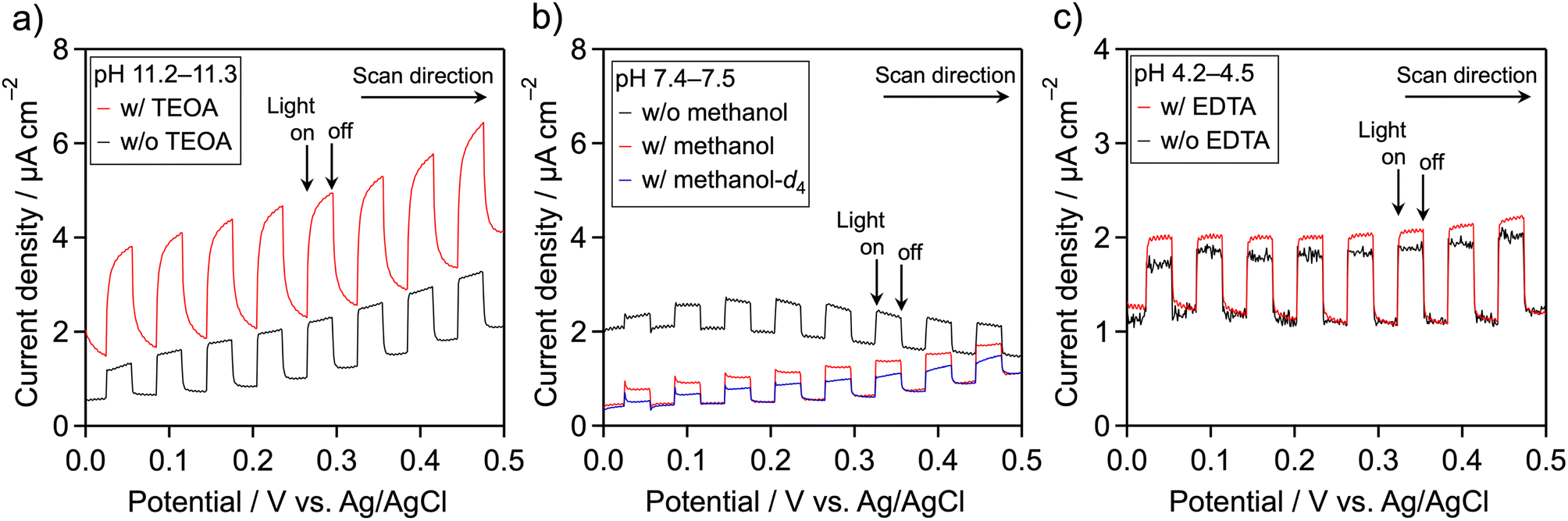 | ||
| Fig. 2 Current–potential curves for Pt/meso-PCN/FTO electrodes under an Ar atmosphere and under irradiation with chopped visible light (λ > 400 nm), as recorded in aqueous Na2SO4 (0.1 M) containing different additives: (a) TEOA, (b) methanol and (c) EDTA. Scan rate: 10 mV s−1. Note: w/and w/o in the figure indicate with and without, respectively. | ||
If high concentration of proton is the primary contributor to improving the photocatalytic H2 evolution activity of Pt/meso-PCN, the use of EDTA·2Na should give much higher activity, as compared to TEOA. However, this was not the case (see Table 1). More importantly, the photocurrent density resulting from the oxidation of EDTA·2Na was smaller than that from TEOA oxidation (Fig. 2). These facts led us to conclude that oxidation reaction by photogenerated holes is critical for enhancing the H2-evolution activity of Pt/meso-PCN.
Conclusions
In conclusion, light-intensity (I)-dependent H2-evolution experiments using Pt/meso-PCN under irradiation with 400 nm monochromatized visible light with an intensity of 0.1–4 mW cm−2 identified two operational regimes. Under light intensities lower than 1 mW cm−2, the H2-evolution AQY increased in proportion with the light intensity, with the AQY reaching a maximum of ∼8%. Under light intensities greater than or equal to 1 mW cm−2, an I∼0.6 dependence was observed. Therefore, the results show that electron–hole recombination in meso-PCN is substantial and that the adverse effect is more pronounced under high-light-intensity conditions. This result is also supported by relatively weak observed H–D isotope effects (<2) compared with those reported in electrochemical H2 evolution (typically >3).The results of the present study also provide a rational strategy toward the construction of nonsacrificial water-splitting schemes using PCN. That is, increasing the reactivity toward electron-donating species (ultimately, water) is critical. In this regard, the development of oxidation cocatalysts and/or hole-capturing materials for PCN is a reasonable approach.36–39 Actually, recent works on overall water splitting by carbon nitride-based photocatalysts rely on a water oxidation cocatalyst to induce the full potential of the photocatalyst.6,7
Author contributions
K. M. designed/supervised the project and wrote the manuscript draft. T. M. conducted photocatalytic reactions and characterization and analyzed the data with S. N. and Y. K. C. S. prepared the Pt/meso-PCN samples and conducted photocatalytic and photoelectrochemical reactions. S. Y. and T. Y. performed SEM/EDS measurements. All authors confirmed the manuscript and approved its submission.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a Grant-in-Aid for Transformative Research Areas (A) “Supra-ceramics” (JP22H05148) (JSPS). The authors thank Prof. Keigo Kamata (Tokyo Institute of Technology) for assistance with the H–D isotope effect experiments.Notes and references
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- Y. Wang, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68–89 CrossRef CAS PubMed.
- W. J. Ong, L. L. Tan, Y. H. Ng, S. T. Yong and S. P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed.
- R. Kuriki and K. Maeda, Phys. Chem. Chem. Phys., 2017, 19, 4938–4950 RSC.
- G. Zhang, Z. A. Lan, L. Lin, S. Lin and X. Wang, Chem. Sci., 2016, 7, 3062–3066 RSC.
- L. Lin, Z. Lin, J. Zhang, X. Cai, W. Lin, Z. Yu and X. Wang, Nat. Catal., 2020, 3, 649–655 CrossRef CAS.
- M. Liu, G. Zhang, X. Liang, Z. Pan, D. Zheng, S. Wang, Z. Yu, Y. Hou and X. Wang, Angew. Chem., Int. Ed., 2023, 62, e202304694 CrossRef CAS PubMed.
- J. Zhang, Y. Chen and X. Wang, Energy Environ. Sci., 2015, 8, 3092–3108 RSC.
- P. R. Harvey, R. Rudham and S. Ward, J. Chem. Soc., Faraday Trans. 1, 1983, 79, 2975–2981 RSC.
- C. Kormann, D. W. Bahnemann and M. R. Hoffmann, Environ. Sci. Technol., 1991, 25, 494–500 CrossRef CAS.
- S. Tabata, H. Ohnishi, E. Yagasaki, M. Ippommatsu and K. Domen, Catal. Lett., 1994, 28, 417–422 CrossRef CAS.
- T. Torimoto, Y. Aburakawa, Y. Kawahara, S. Ikeda and B. Ohtani, Chem. Phys. Lett., 2004, 392, 220–224 CrossRef CAS.
- T. Hisatomi, K. Maeda, K. Takanabe, J. Kubota and K. Domen, J. Phys. Chem. C, 2009, 113, 21458–21466 CrossRef CAS.
- T. Hisatomi, T. Minegishi and K. Domen, Bull. Chem. Soc. Jpn., 2012, 85, 647–655 CrossRef CAS.
- P. Ketwong, S. Yoshihara, S. Takeuchi, M. Takashima and B. Ohtani, J. Chem. Phys., 2020, 153, 124709 CrossRef CAS PubMed.
- Z. Wang, W. Qiao, M. Yuan, N. Li and J. Chen, J. Phys. Chem. Lett., 2020, 11, 2369–2373 CrossRef CAS PubMed.
- S. Nakabayashi, A. Fujishima and K. Honda, Chem. Phys. Lett., 1983, 102, 464–465 CrossRef CAS.
- R. Baba, S. Nakabayashi, A. Fujishima and K. Honda, J. Phys. Chem., 1985, 89, 1902–1905 CrossRef CAS.
- A. Vértes, S. Nagy, Z. Klencsár, R. G. Lovas and F. Rösch, Handbook of Nuclear Chemistry, SpringerNew York, 2011 Search PubMed.
- Y. Shimoyama, K. Koga, H. Tabe, Y. Yamada, Y. Kon and D. Hong, ACS Appl. Nano Mater., 2021, 4, 11700–11708 CrossRef CAS.
- K. Maeda, X. Wang, Y. Nishihara, D. Lu, M. Antonietti and K. Domen, J. Phys. Chem. C, 2009, 113, 4940–4947 CrossRef CAS.
- D. J. Martin, P. J. Reardon, S. J. Moniz and J. Tang, J. Am. Chem. Soc., 2014, 136, 12568–12571 CrossRef CAS PubMed.
- S. Nishioka, K. Shibata, Y. Miseki, K. Sayama and K. Maeda, Chin. J. Catal., 2022, 43, 2316–2320 CrossRef CAS.
- R. Kuriki, M. Yamamoto, K. Higuchi, Y. Yamamoto, M. Akatsuka, D. Lu, S. Yagi, T. Yoshida, O. Ishitani and K. Maeda, Angew. Chem., Int. Ed., 2017, 56, 4867–4871 CrossRef CAS PubMed.
- R. Kuriki, H. Matsunaga, T. Nakashima, K. Wada, A. Yamakata, O. Ishitani and K. Maeda, J. Am. Chem. Soc., 2016, 138, 5159–5170 CrossRef CAS PubMed.
- S. Roy and E. Reisner, Angew. Chem., Int. Ed., 2019, 58, 12180–12184 CrossRef CAS PubMed.
- B. Ma, G. Chen, C. Fave, L. Chen, R. Kuriki, K. Maeda, O. Ishitani, T. C. Lau, J. Bonin and M. Robert, J. Am. Chem. Soc., 2020, 142, 6188–6195 CrossRef CAS PubMed.
- F. Goettmann, A. Fischer, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2006, 45, 4467–4471 CrossRef CAS PubMed.
- K. Maeda, M. Eguchi, S.-H. A. Lee, W. J. Youngblood, H. Hata and T. E. Mallouk, J. Phys. Chem. C, 2009, 113, 7962–7969 CrossRef CAS.
- M. Schwarze, D. Stellmach, M. Schroder, K. Kailasam, R. Reske, A. Thomas and R. Schomacker, Phys. Chem. Chem. Phys., 2013, 15, 3466–3472 RSC.
- X. Wang, K. Maeda, X. Chen, K. Takanabe, K. Domen, Y. Hou, X. Fu and M. Antonietti, J. Am. Chem. Soc., 2009, 131, 1680–1681 CrossRef CAS PubMed.
- S. Nishioka, F. E. Osterloh, X. Wang, T. E. Mallouk and K. Maeda, Nat. Rev. Methods Primers, 2023, 3, 42 CrossRef CAS.
- R. Kuriki, O. Ishitani and K. Maeda, ACS Appl. Mater. Interfaces, 2016, 8, 6011–6018 CrossRef CAS PubMed.
- K. Maeda, K. Sekizawa and O. Ishitani, Chem. Commun., 2013, 49, 10127–10129 RSC.
- M. Sugiyama, K. Fujii and S. Nakamura, Solar to Chemical Energy Conversion, SpringerCham, 2016 Search PubMed.
- T. Kanazawa, K. Kato, R. Yamaguchi, T. Uchiyama, D. L. Lu, S. Nozawa, A. Yamakata, Y. Uchimoto and K. Maeda, ACS Catal., 2020, 10, 4960–4966 CrossRef CAS.
- G. Zhang, S. Zang and X. Wang, ACS Catal., 2015, 5, 941–947 CrossRef CAS.
- M. Zhang, Z. Luo, M. Zhou, G. Zhang, K. A. Alamry, L. A. Taib, A. M. Asiri and X. Wang, Appl. Catal., B, 2017, 210, 454–461 CrossRef CAS.
- G. Zhang, S. Zang, Z.-A. Lan, C. Huang, G. Li and X. Wang, J. Mater. Chem. A, 2015, 3, 17946–17950 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3se00996c |
| ‡ Equal contribution. |
| § Present address: Department of Applied Chemistry, Faculty of Engineering, University of Toyama, 3190 Gofuku, Toyama 930-8555, Japan. |
| This journal is © The Royal Society of Chemistry 2024 |