
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Green perspective drives the renaissance of anionic diene polymerization
Moritz
Rauschenbach†
 ,
Moritz
Meier-Merziger†
and
Holger
Frey†
*
,
Moritz
Meier-Merziger†
and
Holger
Frey†
*
Department of Chemistry, Johannes Gutenberg-University Mainz, Duesbergweg 10-14, 55128 Mainz, Germany. E-mail: hfrey@uni-mainz.de; Web: https://www.ak-frey.chemie.uni-mainz.de/
First published on 2nd October 2024
Abstract
Polymers based on 1,3-diene monomers play a pivotal role in many commercial elastomers and thermoplastic elastomers. This perspective summarizes the state of the art and recent developments in the living anionic polymerization of dienes, permitting to finely tune the properties of the resulting polymers. An emphasis is placed on novel biobased diene monomers (myrcene, farnesene, etc.) and polymerization solvents, which bear promise for more sustainable elastomers in the future. Furthermore, statistical copolymerization of dienes with vinyl monomers and the in situ monitoring of monomer gradients and formation of tapered di- and triblock copolymers due to disparate reactivity ratios is also reviewed. Thermoplastic elastomers based on tri- and multiblock architectures as well as recently reported diene-based polymer architectures are discussed as well. A summary of current challenges and future options for carbanionic diene polymerization concludes this short review article.
Moritz Rauschenbach was born in Bad Homburg v. d. Höhe (1996) and graduated in Chemistry (2021) from the Johannes-Gutenberg-University, Mainz in Germany. During his studies he spent 6 months in the labs of Rachel O'Reilly at the University of Birmingham working on the synthesis of antimicrobial hyperbranched polymer-complexes. In his PhD he focusses on the anionic polymerization of non-conventional 1,3-dienes. Hence, he is interested in the kinetics of those new monomers in comparison to established systems with the aim of elucidating their potential as thermoplastics or functional precursors. |
Moritz Meier-Merziger was born in Darmstadt, Germany (1995), and graduated in Chemistry (2021) from the Johannes-Gutenberg-University, Mainz in Germany. He spent 6 months abroad at the University of Toronto, Canada working on the post-modification of gelatine and hyaluronic acid for their application in hydrogels. His current research during his Ph.D. in the research group of Prof. Holger Frey focusses on the carbanionic polymerization of terpene derived dienes and bifunctional initiators as well as biobased thermoplastic elastomers. His interests comprise sustainable polymers, kinetic investigations, and polymer analytics. |
Holger Frey studied Chemistry at the University of Freiburg/Brsg. He obtained a Ph.D. from the Universiteit Twente, NL (1993) following a stay at Carnegie-Mellon University in Pittsburgh, PA (USA). From 1994 until 2001 he worked as a senior lecturer and assistant professor at the University of Freiburg. Since 2002, he has held a full professorship at the Johannes-Gutenberg-University, Mainz, Germany. His research interests are centered on anionic polymerization of epoxides, dienes and vinyl monomers as well as the catalytic polymerization of carbon dioxide. His current interests include the development of a non-crystalline and non-immunogenic alternative to PEG for medical applications, designated rPEG. Further interests are thermoplastic elastomers, kinetic investigation, branched polymer architectures and silicon-based polymer chemistry. |
1. Introduction
Polymers based on 1,3-diene monomers played a pivotal role in the early days of macromolecular science, when Staudinger introduced the term “macromolecule” in 1920, specifically referring to polyisoprene (PI).1,2 Although the first anionic diene polymerization was observed already a decade earlier by Matthews,3 who stored isoprene in the presence of sodium, it was IG Farben that later commercialized the alkali metal-initiated polybutadiene (PB) based elastomers as “Buna”. This development was prompted by a substantial increase in demand for natural rubber.4,5 From a molecular perspective, natural rubber derived from hevea brasiliensis consists of 1,4-cis (>98%) polyisoprene.6 Nowadays, synthetic rubber based on diene polymerization is produced on a megaton scale and is often combined or blended with natural rubber.7 Both synthetic polydienes and natural rubber possess a double bond per repeating unit, rendering them suitable for crosslinking via vulcanization. The resulting elastomers are used for applications as diverse as tires and medical gloves. The anionic polymerization of dienes continues to garner significant attention in today's industrial and scientific research.8 This leads to continuous new insights also for the established dienes such as butadiene (B) and isoprene (I). Synthetic rubber offers advantages over natural rubber due to its versatile and adjustable properties, since the exact chemical nature of the polymer can be influenced during the polymerization process.9 The commercial importance also arises from the option to produce homo- and copolymers with controlled molar masses and the capability to precisely regulate the microstructure by so called “modifiers” that coordinate the counterion to tailor a variety of material specifications.10 Moreover, 1,3-dienes are used in copolymers such as acrylonitrile–butadiene–styrene (ABS) and styrene–butadiene–styrene triblocks (SBS), which are frequently utilized materials. The thermoplastic elastomers SBS and the analogue SIS using isoprene instead of butadiene have been investigated in-depth and were commercialized under various trade names (i.e. Kraton™ or Styroflex™).11 S/I-copolymers have remained a key research topic of our group, elucidating how different polymerization conditions affect the statistical copolymerization and the resulting monomer gradients and mechanical properties.5,12–17 Consecutive monomer addition steps can be used to generate precisely defined, high molar mass tapered multiblock copolymers (MBCP) that are not accessible by other techniques.5,13–15 Precise knowledge regarding the incorporation statistics of two monomers within a polymer chain, expressed by their reactivity ratios, can be used to reduce the number of monomer addition steps to increase synthetic efficiency. Therefore, in situ kinetics measurements relying on online 1H NMR or near infrared (NIR) kinetics have become the methods of choice to determine the reactivity ratios of a large variety of copolymerizations in recent years.16–23 Furthermore, in the last decade a variety of new diene monomers has been introduced that extend the range of materials for elastomers and thermoplastic elastomers far beyond PB and PI.Polydienes are commonly synthesized by anionic or catalytic polymerization techniques. Catalytic polymerization strategies are employed on a large commercial scale. Recently the long-time challenge of incorporation of polar monomers was achieved by the groups of Cui and Mecking, who also introduced the catalytic polymerization of functional dienes.24,25 We emphasize that a detailed overview of the catalytic polymerization of 1,3-dienes will not be covered in this short review. Readers are referred to recently published, excellent summaries in this area.26,27 Characteristic polymer properties found and discussed for polydiene materials naturally apply, regardless of the polymerization technique used. The main advantage of the catalytic polymerization is the precise control of stereoselectivity.26 The development of a Gd-based catalyst enabled similarly high 99.9% cis-1,4-polyisoprene stereocontrol, as known from natural rubber.28
Due to the high basicity of the carbanion, the highest extent of control in anionic polymerization is achieved by the challenging break-seal method.29 This requires not only a synthetic skillset, but also glassblowing skills. The more accessible high vacuum technique, that has long been established, still enables exceptional control over the polymerization. This allows to synthesize new polydienes and to investigate the impact of polymerization conditions. Besides the established production of polydienes via batch synthesis, also continuous flow reactors for the carbanionic polymerization can be employed for the polymerization of dienes.30–33 Recently, Haddleton and coworkers reported on the anionic diene polymerization without monomer/solvent purification steps, making it more widely applicable, albeit for lower molar mass polymers, e.g. for polymer additives and surfactants.34,35 With respect to precision, anionic polymerization surpasses the catalytic approach, thanks to its distinctive living character, which enables to synthesize of a wide range of polymer architectures and also tailormade gradient and tapered chain compositions in copolymers.8
In recent years, there has been a notable shift in academic research as well as in industry towards bio-derived monomers. Efforts to tailor material properties for enhanced performance accompany this research direction, with the aim of matching or surpassing the properties of established fossil fuel-based materials. In this review we will cover 1,3-dienes beyond the classical representatives, butadiene and isoprene. A selection of the discussed mono- and disubstituted 1,3-dienes is shown in Fig. 1. While some can be found in nature, additional synthetic modification steps are required for others. The number of synthetically accessible 1,3-dienes for the anionic polymerization is steadily increasing.
 | ||
| Fig. 1 Overview of a selection of 1,3-dienes discussed in this perspective, including recently introduced diene monomers. Reactive diene moieties are marked for each monomer. | ||
2. Is the anionic polymerization of dienes genuinely a coordinative process?
Butadiene and isoprene have been the most investigated and industrially used diene monomers to date. They both can lead to different isomeric units along the chain, with respect to regio- and stereo structure (Scheme 1b). As detailed by Carlotti et al.,9 varying the microstructure is a powerful handle to adjust the materials’ properties, for instance for tire applications, where high degrees of elasticity over a broad temperature range are desired, which are usually correlated with a high 1,4-content. This involves minimizing the glass temperature (Tg) as much as possible to achieve the widest possible application temperature range. The overall regio- and stereochemistry of the anionic polymerization of 1,3-dienes has been widely discussed in the last decade with respect to their dependency on the counter ion,10,36,37 solvent,10,38 and the presence of a polar modifier.17,19 The formation of aggregates during the polymerization in nonpolar solvents and their suppression by the use of polar solvents or modifiers was found to be one of the key aspects of the propagation mechanism. The number of the aggregating chains evolves from the kinetic order, expressed by the following eqn (1):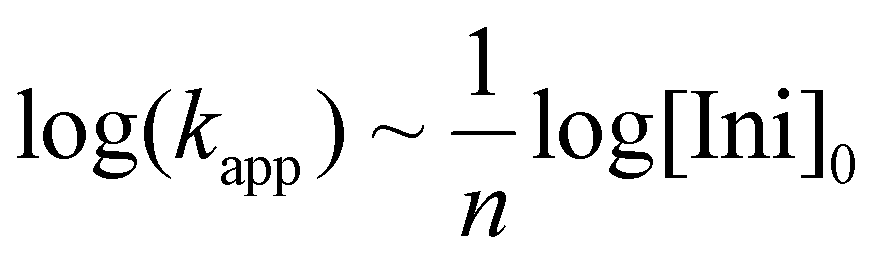 | (1) |
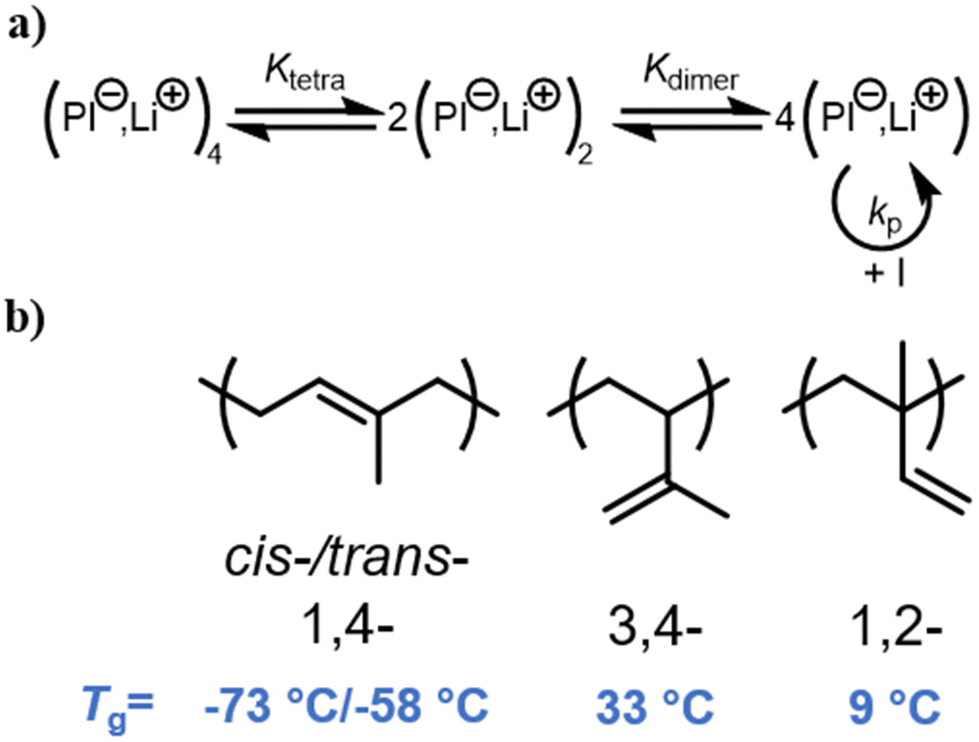 | ||
| Scheme 1 (a) Aggregation equilibrium of polyisoprenyl lithium in nonpolar solvents in analogy to Müller and Matyjaszewski; (b) possible microstructures of PI and their respective glass temperatures.38,40 | ||
k app gives the apparent rate constant of the propagation, [Ini]0 the initial concentration of the initiator and n the number of aggregating chains. Importantly, the aggregates are in an equilibration with the unimers, which are considered to be the active species capable of sustaining the propagation mechanism, see Scheme 1. M. Szwarc already reported that the relative rates of propagation for isoprene and butadiene are ordered as isoprene > butadiene, while both exhibit a number of aggregating chains of n = 4.10,39 However, it was found that polyisoprenyl lithium aggregation also depends on the concentration of active chain ends. An overall equilibration between tetramers and dimers was observed, with n approaching ∼2 with decreasing concentration.10,38 β-Myrcene and β-farnesene, although kinetically investigated under the assumption of tetramer formation, have not been explored with respect to their aggregation behavior.19,22 The observation that their propagation rate surpasses that of isoprene suggests the presence of an equilibrium solely between dimers and unimers. It should be noted that this assumption has not yet been empirically confirmed.
The coordination of the counterion to the incoming monomer and its spatial orientation mainly dictate the regioselectivity, see Scheme 2. In nonpolar media, it has been reported to undergo a six-membered transition state, where the diene is added in a cis-form, explaining overall high amount of 1,4-addition. The final cis/trans ratios have been extensively elucidated by Gebert et al. who suggested an isomerization of the cis-oriented chain end to a trans-form before the next monomer addition.38,41,42 Worsfold and Bywater further proposed that the isomerization occurs in competition with the monomer addition.10,43 Based on these fundamental studies, the final ratio of cis and trans 1,4-units can be explained.
 | ||
| Scheme 2 Proposed mechanism of the polymerization of isoprene with lithium as a counter ion in apolar or polar media, respectively; image is reproduced from Frey et al.41 with permission of the Royal Society of Chemistry, 2022. | ||
By changing the counterion from Li+ to Na+ or K+, for instance, the interionic distance increases, which reduces the 1,4-content of polydienes.10,44 Based on the industrially most popular diene representative 1,3-butadiene, Carlotti and coworkers have investigated the impact of bimetallic initiator systems with lithium and potassium or calcium, respectively. They studied the influence of the additional coordinative center on the microstructure. While the addition of potassium led to an expected increase of 1,2-units, the designed calcium-lithium initiators enabled high control towards trans-1,4-units.45,46 Reviewing the accessible microstructures of polybutadiene, Carlotti et al. also reported on the combined effect of both variation of the counterions as well as changing to polar solvents.9 The polymerization in polar solvents, e.g. THF, or the addition of polar modifiers are both associated with suppression of the coordination between the chain end, the counterion and the diene. In consequence, the resulting increased vinyl content enhances the backbone rigidity of the polydiene, which is reflected by an increase of the glass temperature (Scheme 1b).38,41 The influence of solvents on the anionic polymerization will be discussed below in greater detail.
A rather new aspect in this field is the examination of the orientation of the double bonds with respect to each other in the 1,3-diene scaffold. In this regard, DFT calculations can give mechanistic insights beyond the mere conversion in a copolymerization. In a recent publication we investigated rotationally constrained dienes.41 Two model compounds were synthesized to obtain a cisoid and a transoid monomer as shown in Fig. 2a. DFT calculations identified deviations of the torsion angle in both monomers and allow for an assumption regarding the reactivity given by the electron densities. A crucial finding from the simulation is that if a fixed transoid geometry is present, only one of the double bonds can coordinate with the lithium counterion. This crucial step should prevent the polymerization. In contrast, the cisoid model compound coordinates with both double bonds, albeit a distortion of the ring is required. Consequently, only homopolymers with the cisoid 1,2-dimethylene-cyclohexane (DMCH) were obtained and copolymerization with isoprene was achieved. DFT calculations show that the energy required for the polymerization of DMCH is significantly increased compared to isoprene due to a required distortion of the ring, see Fig. 2b. Empirically, this was observed via in situ1H NMR kinetics. In the copolymerization with isoprene, isoprene is consumed predominantly, resulting in a gradient structure along the chain. The homopolymerization of DMCH itself required unusual conditions to keep the growing chain in solution: high temperatures of 140 °C were applied in the high-boiling solvent tetralin. Noteworthy, the analysis of PDMCH confirmed the exclusive formation of 1,4-units and a Tg of 53 °C. In contrast, no polymers were obtained with the transoid monomer IMMCH. In 2015, Ishizone and coworkers described the anionic polymerization of the transoid-fixed benzofulvene in both polar and apolar media.47–49 This can be attributed to the conjugation with the aromatic ring that leads to a planar cross-conjugated π-system resulting in a higher reactivity. The removal of the aromatic ring, e.g., in methylene cyclopentane resulted in the expected reduction of the reactivity. For this monomer 70 °C and a reaction time of 4 days were required to achieve full conversion.50 Consequently, it can be concluded that transoid dienes are not suitable for the living anionic polymerization in nonpolar solvents at mild conditions.
 | ||
| Fig. 2 (a) Rotationally constrained dienes with a cisoid (DMCH) and a transoid (IMMCH) geometry; (b) reaction pathways for the four possible propagation steps of the copolymerization of isoprene and DMCH (addition of DMCH, blue; addition of isoprene, green), image is reproduced from Frey et al.41 with permission of the Royal Society of Chemistry, 2022. | ||
Moreover, already a methyl group can interfere with the coordinative process and prevent the formation of a narrow molar mass distribution. This was shown for the two isomers trans- and cis-pentadiene. They exhibit rate constants in the order cis > trans, while only the trans-isomer gives well-defined polymers (Đ ≤ 1.09).51 Surprisingly, the polymerization is gradually decelerated, if both isomers are mixed, leading to the order of the rate constants as follow: cis > trans > cis/trans.52
Taking these recent results of geometrically constrained dienes together, one can tentatively answer the initially raised question: the classic anionic polymerization shows indeed many features of a coordinative process in apolar media. Various parameters can be changed to influence the coordination, either by relying on a solvent that coordinates the counter ion or by changing the substitution pattern of the diene, which was found to influence the mechanism drastically as in the diastereomers isoprene and cis-pentadiene. These strategies are being further pursued by various groups at present.
3. Controlling the microstructure of polydienes
The resulting microstructure is directly dependent on the propagation mechanism, as introduced previously. Primarily for rubber applications, 1,4-repeating units are favored because of the known low Tg of the polymers and lower entanglement molar mass.5,44 On the other hand, for specific post-polymerization modification reactions53,54 or to suppress crystallization as for hydrogenated polybutadiene,55 an increased vinyl content is targeted. Polydienes, such as polyisoprene, were widely investigated to understand their microstructural changes with respect to solvent polarity. It is well-known that polymerization in polar solvents like THF results in significantly higher content of 3,4- (62%) and 1,2-units (24%).56 However, the monomer incorporation mode can also be tailored by using small amounts of polar modifiers in nonpolar solvents. Morton and Fetters reported that for isoprene the addition of aliquots of THF initially increases the propagation rate.57 Surprisingly, further addition leads to a subsequent decrease, as Bywater had previously demonstrated for styrene.58 Recently, we have been able to give a detailed view of the impact of THF on the statistical copolymerization of styrene and isoprene, relying on online NIR monitoring of the anionic copolymerization. When increasing the THF concentration the vinyl content of the isoprene-units increases.17 In other works based on online kinetics, polymerization of the analogous β-myrcene was investigated to compare the influence of ditetrahydrofuryl propane (DTHFP) and tetramethyl ethylene diamine (TMEDA) as polar modifiers with the established THF. As shown in Table 4, both DTHFP and TMEDA show a stronger impact on the microstructure than THF.19,59 The addition of 2 equivalents with respect to the lithium-ion concentration leads to predominant 3,4-units (>57%). Undesirably, this leads to an increase of the low glass temperature. Polydiene based elastomer chains usually exhibit a Tg below −60 °C. When gradually increasing the ratio of vinylic units, the Tg of polyisoprene can reach 0 °C.60 Although the Tg of polydienes shows this correlation with the proportion of the different microstructures, polyfarnesene (PFar), discussed later in this perspective, shows a different behavior. Due to its long side chain in every repeating unit, the Tg remains nearly unaffected by increasing the vinyl content, as illustrated in Fig. 3. | ||
| Fig. 3 Correlation of glass temperature of polydienes with the vinyl content. The values are combined from several original works.60–63 | ||
In addition to altering the system's polarity, it is well-known that increasing bulkiness of the substituent results in an almost exclusive formation of cis-1,4-units. This was demonstrated for the anionic polymerization of 2-substituted 1,3-butadiene derivatives with a methyl, ethyl, n-propyl or isopropyl group.64–66 Pendant phenyl groups, as seen in 1-phenyl butadiene (PhB), 1-phenyl isoprene (1PhI) and 4-phenyl isoprene (4PhI), have demonstrated a predominant occurrence of 1,4-addition (>80%), even in the polar solvent THF.67–69 Furthermore, Ishizone and coworkers achieved control over the incorporation by designing rather unusual allylidene monomers.70,71 The reported butadiene-derivatives were 1,1-disubstituted with increasing bulkiness of the pendant group. Various substituents, ranging from two methyl groups to a bornane ring and even the bulky adamantyl group, were attached to the dienyl-unit. Hence, the sterically hindered 1,4-addition could be further prohibited with increasing bulkiness, which is underlined by the absence of 1,2-units. The structures shown in Fig. 4a were sufficiently effective to increase 3,4-units to more than 20% in apolar media. The bulky substituents of the structures in Fig. 4b result almost exclusively in 3,4-incorporation. As a result, these polymers turned out to be very rigid materials with high glass temperatures of nearly 200 °C in case of the pendant bornane-ring. In theory, this might allow for application in high temperature resistant thermoplastic elastomers (TPEs) as a hard segment, but on the other hand this will also necessitate extremely high processing temperatures.
 | ||
| Fig. 4 1,1-Disubstituted 1,3-dienes studied with respect to their behavior in the anionic polymerization, reported by Ishizone et al.70,71 | ||
The monomer 1-vinyl cyclohexene (VCH) serves as a model compound with one double bond that is sterically constrained within a cyclic structure. The key question is whether VCH behaves as a 1,3-diene or as a styrenic vinyl monomer in the anionic polymerization in nonpolar media. In analogy to other diene structures, the homopolymerization in cyclohexane (CHx) lead to a predominant formation of 64% 1,4-units.72 The extent of 1,4-incorporation is comparable to the allylidene cyclohexane with a reported 66% 1,4-units when polymerized in benzene.71 However, the added rigidity from the ring in the backbone increased the Tg to 78 °C. The microstructure of PVCH was adjusted to enhance the vinyl content, reaching up to 78% of 3,4-PVCH upon addition of the polar modifier THF. The resulting material properties, with a Tg of up to 89 °C, are much closer to those of PS (Tg = 100 °C).73 Likewise, hydrogenation leads to a high amount of polyvinylcyclohexane (PVCH) segments, as obtained by hydrogenation of PS.
4. Bio-derived diene alternatives
The dwindling fossil resources evolved to be a major motivation in finding new substitutes for established systems such as polydiene-based (co)polymers. Wahlen and Frey presented a wide scope of terpene monomers that are amenable to the anionic polymerization.74 Predominantly, β-myrcene (Myr) and β-farnesene (Far) have gained significant attention owing to their structural similarity to the 2-substituted isoprene framework and their biobased origin from turpentine oil and sugar cane, respectively. Both monomers show similar dependencies of the polymer structures on the polymerization conditions and result in comparable polymer properties. A highly 1,4-dominated microstructure is obtained when polymerizing in apolar solvents, which results in a similarly low Tg as polyisoprene, with Tg < −60 °C (Fig. 3).60,62 This renders PFar and PMyr suitable for the application as soft phase in thermoplastic elastomers. Combinations with polystyrene or polylactide (PLA) as the vitrified plastic domains have also been reported. When polystyrene is used for the rigid phase, classical linear ABA-triblock copolymers can be synthesized. Sequential addition yields block architectures, whereas tapered structures are obtained by one-step statistical copolymerization.19,23,62 This will be discussed in greater detail in one of the following sections. By the combination with polylactide as the rigid phase even more complex architectures were realized, e.g., graft polymers and H-shaped triblock structures, see Fig. 5.75–77 | ||
| Fig. 5 Different copolymer architectures for TPEs based on block, tapered and branched triblock structures as well as graft-copolymers.22,75–78 | ||
Due to the increasing length of the side chains in PMyr and PFar that do not contribute to the polymer backbone length, the entanglement molar mass strongly increases from 7 kg mol−1 for PI, 18 kg mol−1 for PMyr to 50 kg mol−1 for PFar.79–81 This effect has considerable consequences for elastic mechanical properties that are strongly dependent on the entanglements of the polymer chains. Therefore, higher molar masses compared to PI are required for PFar to obtain similar mechanical properties.
The first report on the anionic polymerization of β-myrcene by our group focused on the behavior in statistical copolymerization with isoprene, styrene and 4-methyl styrene (4MS), respectively.23In situ1H NMR kinetics was performed, revealing β-myrcene to be a suitable substitute for isoprene with similar behavior in the copolymerization with styrene. Both systems show a steep gradient, as discussed later in this perspective. This was further investigated using the in situ NIR method.19 A series of S/Myr-copolymers were synthesized to investigate the impact of an increasing amount of THF on the nature of the gradient structure of the copolymers. It was shown that the gradient structure can be tuned by a variety of modifiers, even to complete inversion of the reactivity ratios, similar to the behavior observed earlier for isoprene.17 Furthermore, we showed that Far forms a gradient structure in the statistical copolymerization with styrene and that the polymerization in cyclohexane became significant faster when moving up the homologous series of terpenes: kp,I < kp,Myr < kp,Far.16,19,21
Although renewable feedstocks are one of the most important current research fields, their impact on nature and, above all, the possible competition with food sources should always be considered as well. β-Farnesene, relatively price competitive, as shown by its use as sustainable jet fuel besides its application in materials, is mainly derived from sugar cane.82 PFar products are already widely available in industry, e.g., PFar-diol is used for the preparation of polyurethanes.60,83 Therefore, additional feedstocks are being sought. It was already shown that heterotrophic as well as lithoautotrophic bacteria are capable of producing Far from cultivation on a variety of substrates.84
Furthermore, the additional double bonds in the side chain allow for further post-modification with respect to thiol–ene click chemistry and epoxidation, respectively.85,86 Thereby, these monomers offer the possibility of introducing functionalities along the polydiene chains, which would require suitable protecting groups if introduced prior polymerization. This will be addressed in a later section of this overview. In addition to these aforementioned examples, there are also less known representatives in the field of terpenes. For instance, our group reported on the anionic polymerization of β-ocimene (Oc).87 As Oc resembles a 1,2-disubstitued diene it naturally occurs as a mixture of the cis and trans-isomer, similar to 1,3-pentadiene. The anionic polymerization in cyclohexane resulted in an unusually high dispersity up to 2. In situ1H NMR kinetics revealed a preferred consumption of the trans-isomer over the cis-form (rtrans = 3.16 and rcis = 0.316) turning the apparent Oc homopolymerization into a copolymerization of the two stereoisomers. Surprisingly, the addition of styrene inverted the trend to an accelerated consumption of the cis-isomer. Taking advantage of these findings, both isomers were isolated by polymerization of the respective other isomer. Consequently, the NMR kinetics with styrene could be elucidated separately. The values given in Table 2 reveal the presumed consumption of styrene over the trans-isomer (rtrans = 0.628 and rS = 1.59), while unexpectedly the cis-isomer showed an almost random incorporation of both monomers (rcis = 1.01 and rS = 0.98). This phenomenon is quite uncommon in the field of carbanionic polymerization, as mostly gradient structures are obtained. An overview of a broad range of diene copolymerizations and their respective reactivity ratios is given in Table 2. For reference, the established S/I-copolymerization in cyclohexane shows a steep gradient, characterized by rI = 10 and rS = 0.015.17
1,3-Diene structures offer potential beyond low Tg and high elasticity, as their structures can be highly diverse. Ishizone et al. presented a variety of 1,1-disubstituted dienes that possess cyclic structural elements and were already discussed in more detail earlier in this perspective.70,71 Their unusually high Tgs are attributed to the rigidity of the ring structure and the resulting high content of 3,4-units. Cyclic elements can also be incorporated by 1,2-substitution as mentioned for the monomer VCH.72 With the monomer nopadiene (Nopa), 1R,5S-2-ethenyl-6,6-dimethylbicyclo[3.1.1]hept-2-ene, our group recently presented a sterically even more demanding structure, due to its bicyclic structure. Thereby, a strikingly high Tg of 160 °C for PNopa can be achieved. Nopadiene was therefore presented as a potential biobased substitute of styrene in TPE materials. Nopa can be derived from myrtenal or nopol, again originating from β-pinene. An exclusive diene structured and fully biobased TPE can be obtained using the monomers Myr and Nopa.88 Furthermore, we also developed a one-step synthesis for TPEs. By precisely controlling the reaction conditions, we achieved reactivity ratios that favor the incorporation of low Tg PFar, followed by high Tg PNopa. Since a bifunctional initiator was employed for this reaction, a telechelic polymerization was realized and thus, two-sided tapered ABA-triblock structures are accessible in a single polymerization step, enabling an extremely rapid strategy for the synthesis of biobased thermoplastic elastomers.78
The diversity of natural sourced compounds, e.g. terpenes, offers vast potential to customize material properties. However, many parameters must be considered. As many biobased monomers have expanded the portfolio of polymers accessible via carbanionic polymerization, a compilation of a variety of the resulting diene polymers in terms of their glass temperature is listed in Table 1. The data show the enormous diversity of diene structures and their potential in the field of materials science, as they can be tailored to a wide range of thermal properties. Among those listed, 4,8-dimethylnona-1,3,7-triene (DMNT) has not yet been presented in an original work yet.
| Polymer | 1,4 [%] | T g [°C] | Ref. | Polymer | 1,4 [%] | T g [°C] | Ref. |
|---|---|---|---|---|---|---|---|
| a 2-Allylidene-adamantane (Aad). b 2-Allylidene-1,1,3,3-tetramethylcyclopentane (ATMC5). | |||||||
| PB | 72 | −86 | 60 | PDMNT | 74 | −30 | — |
| PFar | 91 | −75 | 60 | POc | 15 | −26 | 87 |
| PFar | 61 | −73 | 60 | PB | 10 | −21 | 60 |
| PFar | 48 | −70 | 60 | PI | 15 | 0 | 60 |
| PMyr | 94 | −67 | 61 | PMyrDOL | 67 | 11 | 89 |
| PI | 91 | −67 | 60 | P4PhI | 94 | 48 | 69 |
| PFar | 37 | −66 | 75 | PDMCH | 100 | 53 | 41 |
| PMyr | 72 | −58 | 63 | P1PhI | 35 | 65 | 69 |
| PMyrOSi | 69 | −53 | 61 | PVCH | 22 | 89 | 72 |
| PB | 30 | −45 | 60 | PNopa | 51 | 158 | 88 |
| PMyr | 30 | −39 | 62 | PAada | 0.0 | 178 | 70 |
| PI | 44 | −32 | 60 | PATMC5b | 0.0 | 194 | 71 |
5. New solvents for the anionic polymerization of 1,3-dienes
Usually, the requirements for solvents used in the carbanionic polymerization are (i) their aprotic nature and (ii) high base-stability. Often non-polar solvents like cyclohexane or benzene are used, as is mostly the case for butadiene and isoprene systems. The reason for this is the previously discussed solvent effect on the microstructure and the associated changes in material properties. Accordingly, cyclohexane, hexane mixtures, benzene, and toluene are established solvents. Toluene is less favored due to reported transfer reactions.90 Cyclohexane is used for the industrial synthesis of thermoplastic elastomers by anionic polymerization.11 For specific demands, e.g., solubility of bifunctional initiators, functional initiators and monomers, tetrahydrofuran (THF) is primarily used to increase the polarity and to suppress aggregation. Proton abstraction at ambient temperatures restricts its use to low temperatures, usually −78 °C.91,92 However, the polymerization of dienes is thermodynamically hindered at these low temperatures. Gallei et al. took advantage of this to prepare block copolymers by using a one-pot synthesis of styrene/myrcene mixtures, applying a temperature change to polymerize the diene monomer. Only styrene polymerizes at low temperatures, whereas myrcene requires temperatures above −30 °C in THF to polymerize.93Further, the awareness of sustainability and a shift towards more biobased systems has led to a reconsideration of solvents, as they make up for a large proportion of a synthesis. Consequently, biobased, more sustainable solvents are currently under investigation, see Fig. 6.
 | ||
| Fig. 6 Established and emerging solvents for the anionic polymerization. | ||
Looking beyond butadiene and isoprene, other new and often biobased monomers may require the use of more polar solvents. As introduced above, PFar maintains a low Tg when polymerized in a polar medium, whereas the syntheses of other polydienes require an apolar solvent to achieve similar material properties. Schlaad and coworkers investigated the polymerization of isoprene and β-myrcene in a variety of “green” solvents, i.e. limonene, 2-methyl tetrahydrofuran (2-MeTHF) and cyclopentyl methyl ether (CPME).56,94 On the one hand, 2-MeTHF appeared to have almost the same impact on the microstructure as THF, affording a majority of 3,4-incoorporation (54%). On the other hand, CPME yielded a major fraction of 1,4-units of 64%. The apolar solvent limonene strikingly resulted in 90% 1,4-incorporation. In all solvents, moderate to low dispersities below 1.15 were obtained. Recently, CPME has also been employed for the copolymerization of styrene with isoprene or 1,3-pentadiene, as well as for the copolymerization of isoprene and 1,3-pentadiene. Kinetic studies were conducted to assess its suitability for use in anionic polymerization.95 Additionally, 2-MeTHF and CPME were both evaluated as additives in the anionic polymerization of 1,3-cyclohexadiene. Notably, CPME in a toluene system demonstrated promising results, with quantitative yields and successful achievement of targeted molar masses, while maintaining low dispersities of Đ < 1.12.96 In addition, Haddleton et al. investigated the use of squalane, bearing a C30 framework, in its unpurified form as a polymerization medium. Despite the high dispersity (Đ = 1.84), attributed to the avoidance of purification steps, the low polarity yielded 94% of 1,4-incorporation.35 In one of our recent works we showed that the moderately polar solvent methyl tert-butyl ether (MTBE) is ideally suited to solvate the bifunctional initiator 1,3-diisopropenyl benzene (DIB). A synergy can be found, as MTBE enables a reliable bifunctional initiation, as it prevents aggregation, while the associated increase in vinyl content does not affect the properties of PFar.75,78 MTBE, besides being cheap due to industrial scale production for petrol-additives, can also potentially be obtained from renewable feedstocks, i.e. bio-methanol and bio-isobutylene.97 It is therefore somewhat surprising that it has not received more attention in carbanionic polymerization to date. This can certainly be attributed to the prevalence of the “classical” solvents established over decades. Recently, we investigated MTBE in-depth as moderately polar solvent and additive in homo- and copolymerizations.98
6. Functional polydienes via functional monomers or post-polymerization modification
So far, all presented polydienes discussed solely consist of carbon and hydrogen. In many elastomer applications inorganic fillers are used to enhance mechanical performance (i.e. modulus and strength, etc.). A common filler material used is silica, which is poorly dispersed in a fully non-polar diene material. Silica is known to enhance the performance of tires, one of the main applications of rubber, e.g., by increasing the wet grip for higher safety standards and by reducing the rolling resistance to lower fuel consumption.99 In this context, the incompatibility of hydrophilic fillers and hydrophobic polydienes has led to an increased interest in modifying polydienes. Early works showed that functionalities have to be separated from the propagating diene unit to prevent the so-called “backside-collapse” as an undesired side-reaction (Scheme 3).44,100,101 | ||
| Scheme 3 Proposed mechanism of the backside-collapse, as described in the works of Takenaka et al.100,101 | ||
Our group reported on two approaches for functional diene-monomers. Both are based on β-myrcene as a precursor that was converted to a hydroxyl-functionalized structure. As free hydroxyl groups are incompatible with the carbanionic polymerization, suitable protecting groups must be employed. A single hydroxyl group was protected using a siloxy functionality (MyrOSi), and a dioxolane-based structure (MyrDOL) was used to incorporate two protected hydroxyl groups. Both functional monomers had a modifier effect on the polymerization, as the masked polar functionalities can interact with the counterion and thereby influence the polymerization mechanism. In this case, the diene acts both as a monomer as well as a modifier. The functionalized β-myrcene derivatives, MyrOSi and MyrDOL, showed 31% 3,4-units when polymerized in cyclohexane.61,89
Copolymers offer vast possibilities to tune, combine or design materials properties. A variety of copolymers exclusively synthesized via anionic polymerization were discussed before. Beyond this, there is a broad field of copolymers that are composed of monomers that require different polymerization techniques. This can be accomplished by preparing a macroinitiator from monomer A. Subsequently, Poly(A) serves as a macroinitiator for monomer B to form Poly(A)-b-Poly(B) block copolymers in a second reaction using a different polymerization technique. For this purpose, functional groups, e.g. hydroxyl groups, have to be incorporated at the polydiene framework. Controlled end-functionalization of carbanionic living chains is a suitable pathway. For this purpose, lithium, commonly present as a counterion in carbanionic polymerization, has the advantage of forming highly stable ion pairs with oxygen. Hence, by using an epoxide for the termination step of a carbanionic synthesis, lithium alkoxides are formed that are not capable of polymerizing any further. As a result, a single epoxide monomer reacts with the each chain end, leading to a degree of hydroxyl functionalization of up to 99%.102,103 Based on the large variety of epoxides available, this method offers vast potential for complex polymer architectures. We recently applied this method to a bifunctional system to simultaneously introduce hydroxyl groups at both chain ends. Whereas ethylene oxide is known to introduce one hydroxyl group, ethoxy ethyl glycidyl ether (EEGE) and isopropylidene glyceryl glycidyl ether (IGG) can be used to introduce two and three hydroxyl groups, respectively. Ultimately, this yields telechelic OH2-polydiene-OH2 and OH3-polydiene-OH3 macroinitiators. Via subsequent organocatalyzed chain extension relying on the ring-opening polymerization of L-lactide, elaborate architectures, e.g., H-, and super-H-shaped triblock copolymers are rapidly accessible.75,104
Another example for the subsequent formation of a second polymer is grafting from a macroinitiator bearing suitable functional groups at the chains. The high abundance of unsaturated groups present in polydienes was used to introduce functionalities in a post-polymerization pathway. This was demonstrated by the group of Li et al. using epoxidation of PMyr.77,105 Another efficient pathway is based on the use of polymers derived from functional monomers, e.g. PMyrOSi, that can be exploited as a macroinitiator after the cleavage of the protecting group. Again, by L-lactide grafting so-called bottlebrush structures are accessible.61
The coupling of polymers offers a third option for producing block copolymers, and particularly for complex polymer architectures. Coupling in the carbanionic polymerization can be achieved directly by introducing multifunctional coupling reagents, e.g. chlorosilanes, at the living polymer chains.106 In this manner, complex branched structures can be prepared, enhancing the material properties in terms of lowering viscosity in melt and solution. Coupling strategies are also employed for commercial products like Styrolux.11 Our group introduced a distillable tetra-functional chlorosilyl compound to produce well-defined multiblock star polymers.107,108 In the past, this has been pushed to the limit, with reports of up to 128-arm star polymers.109 The versatile chemistry of polydienes has recently been exploited by Hirschberg et al. in elegant work, who used epoxidized PI to couple with living polystyrenyl chains to obtain pom–pom architectures.110
7. Behavior of 1,3-dienes in the statistical copolymerization
The statistical copolymerization of styrene and butadiene in apolar solvents, leading to so-called “tapered” block copolymers was patented as early as 1958 by Phillips Petroleum Company.111,112 Further development by Shell Co. resulted in gradient copolymers for commercial application.113 The statistical copolymerization is an efficient tool to form block-like, tapered structures that still retain microphase segregation like sequential block copolymers. Therefore, the investigation of the gradient formed in a statistical anionic copolymerization is of significant interest. In recent works, we compared tapered multiblock copolymers composed of isoprene and styrene with their respective analogues obtained via sequential monomer addition. SAXS patterns revealed weaker segregation, clearly attributed to a reduced effective Flory–Huggins parameter, χeff.14 Consequently, the order–disorder transition (ODT) temperature, TODT, decreases, which is desirable for industrial applications targeting high-speed processing via melt extrusion.11 The synthesis of tapered block copolymers relies on significantly different reactivity ratios.114,115 In 2013, we reported on in situ1H NMR spectroscopy to track the individual comonomer consumption throughout the living carbanionic copolymerization. The decreasing integrals of the respective monomer signals can be used to determine the reactivity ratios of each system. By this means, one can directly monitor the copolymerization of isoprene and styrene in apolar media up to full monomer conversion, demonstrating the formation of a steep gradient. The associated reactivity ratios were determined to be rI = 11 and rS = 0.053. This can be attributed to a faster cross-propagation rate of S to I, compared to vice versa (kSI ≫ kIS).16,116 Consequently, by initiation of a monomer mixture, two blocks, PI-grad-PS, linked by a short gradient structure are obtained. This can be repeated multiple times to generate multiblock copolymers with exceptional material properties, while minimizing the number of reaction steps required.5,13–15,117 Driven by the interest for biobased monomers, we also investigated the terpenes β-myrcene and β-farnesene in the copolymerization with styrene. Interestingly, both show an even steeper gradient when combined with styrene. Table 2 summarizes 1,3-dienes that have been investigated regarding their statistical copolymerization with styrene. First, butadiene and monosubstituted 1,3-dienes form very pronounced gradients. Secondly, the homopolymerization becomes less preferred when additional substituents are introduced to the monomer scaffold. For instance, VCH with a double bond captured in the ring is consumed slower than styrene, indicating that the cross-over reaction is highly dependent on the sterics of the respective 1,3-diene. Furthermore, the aforementioned ocimene and its two isomers reveal an impact of the configuration of the double bonds. While the trans-isomer is consumed first when both isomers are polymerized together, the reactivity order inverts when both are copolymerized with styrene. This observation was supported by 1H NMR chemical titration, which revealed a surprising interaction between styrene solely with the cis-isomer.| Diene | Solvent | T [°C] | r diene | r S | Molar gradient | Ref. |
|---|---|---|---|---|---|---|
| Myr | CHx | 20 | 40 | 0.024 |
|
19 |
| β-Farnesene | CHx | 23 | 27 | 0.037 |

|
22 |
| Butadiene | CHx | 25 | 15.5 | 0.04 |

|
10 |
| Isoprene | CHx | 20 | 10.1 | 0.013 |

|
16 |
| 4PhI | CHx | 25 | 9.2 | 0.109 |

|
69 |
| MyrDOL | CHx | 25 | 5.2 | 0.19 |
|
89 |
| 1PhI | CHx | 25 | 3.38 | 0.3 |

|
69 |
| Nopadiene | CHx | 25 | 3.26 | 0.31 |

|
88 |
| cis-Ocimene | CHx | 23 | 1.015 | 0.985 |

|
87 |
| trans-Ocimene | CHx | 23 | 0.62 | 1.59 |

|
87 |
| VCH | CHx | 25 | 0.39 | 2.56 |

|
72 |
The copolymerization of two 1,3-dienes, mostly with isoprene as a comonomer, has also been investigated, see Table 3. Generally, rather weak gradients in comparison to diene/S-systems are observed. The β-myrcene derivative MyrDOL has been found to form the steepest gradients when copolymerized with either isoprene or β-myrcene, respectively. However, this monomer can hardly be compared with other systems due to its increased polarity resulting from the functional side chain, which is expected to reduce the formation of aggregates, similar to a polar modifier. Noteworthy, the copolymerization of isoprene with the DMCH and VCH, which are both rotationally constrained, results in a favored consumption of isoprene.
| Diene 1 | Diene 2 | Solvent | T [°C] | r 1 | r 2 | Molar gradient | Ref. |
|---|---|---|---|---|---|---|---|
| MyrDOL | Isoprene | CHx | 25 | 9.22 | 0.11 |

|
89 |
| β-Myrcene | CHx | 23 | 4.4 | 0.23 |

|
23 | |
| Butadiene | n-Hexane | 20 | 2.82 | 0.42 |

|
118 | |
| 1PhI | CHx | 25 | 0.865 | 1.155 |

|
69 | |
| VCH | CHx | 25 | ≪1 | ≫1 | 72 | ||
| DMCH | CHx | 25 | 0.087 | 11.51 |
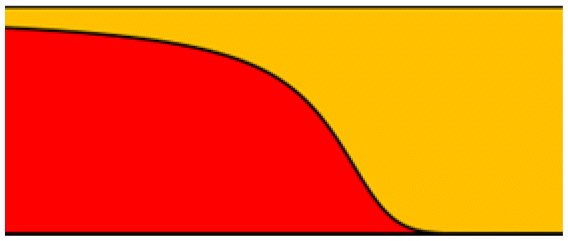
|
41 | |
| MyrDOL | β-Myrcene | CHx | 25 | 6.22 | 0.16 |
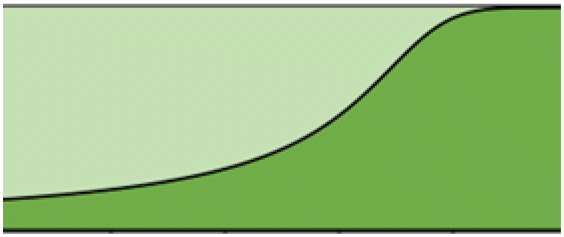
|
89 |
| Nopadiene | CHx | 25 | 0.13 | 7.49 |

|
88 | |
| Nopadiene | β-Farnesene | CHx | 25 | 0.155 | 6.50 |

|
78 |
More recently, we also implemented NIR-spectroscopy to track monomer consumption.16 The reactions can be monitored directly in the stirred polymerization solution in the reactor, which means that the product formed is directly characterized with respect to its monomer gradient in the chains. Using in situ NIR spectroscopy, we studied the influence of polar modifiers, i.e. THF and DTHFP, in the copolymerizations of I/S and Myr/S. Gradually increasing the ratio of [THF]/[Li] can be used as a tool to control the molar composition of the copolymer chains, as indicated by the profiles shown in Table 4. Both isoprene and β-myrcene are incorporated to a reduced extent, when the polarity of the system increases, resulting in the preferred consumption of styrene.17,19 In case of the copolymerization of β-myrcene and styrene, the chelating modifiers DTHFP and TMEDA were shown to have a much stronger impact compared to THF. Already 0.5 eq. of [DTHFP]/[Li] influences the copolymerization stronger than 20 eq. of THF. Only 2 eq. of either DTHFP or TMEDA already lead to an enhanced incorporation of styrene, as indicated by reactivity ratios of rS ≥ 17.5 and rMyr ≤ 0.15, respectively.19,59
| Diene | Polar modifier | eq. | T [°C] | r Diene | r S | Molar gradient | Ref. |
|---|---|---|---|---|---|---|---|
| Isoprene | THF | 0.5 | 20 | 2.856 | 0.093 |

|
17 |
| Isoprene | THF | 2 | 20 | 1.06 | 0.262 |

|
|
| Isoprene | THF | 20 | 20 | 0.374 | 0.925 |

|
|
| Isoprene | THF | 240 | 20 | 0.148 | 4.196 |

|
|
| β-Myrcene | THF | 0.5 | 20 | 12.3 | 0.082 |

|
19 |
| β-Myrcene | THF | 2 | 20 | 4.38 | 0.23 |

|
|
| β-Myrcene | THF | 20 | 20 | 0.87 | 1.15 |

|
|
| β-Myrcene | THF | 240 | 20 | 0.17 | 5.77 |

|
|
| β-Myrcene | DTHFP | 0.5 | 20 | 0.41 | 2.42 |

|
|
| β-Myrcene | DTHFP | 2 | 20 | 0.056 | 18.0 |

|
|
| β-Myrcene | TMEDA | 2 | — | 0.15 | 17.52 |
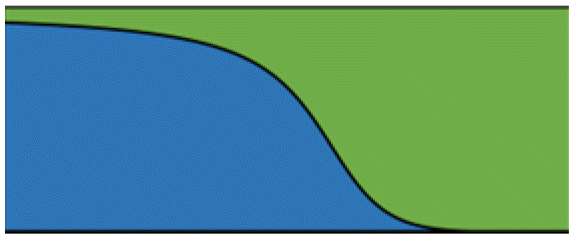
|
59 |
8. Current challenges and perspective
Anionic polymerization is still the benchmark when it comes to high precision polymer synthesis. Since the polymer chains can be obtained with high uniformity, details of structure–property correlations can be determined and consequently tailored with great accuracy. The current shift towards more sustainable feedstocks has led to an increased diversity of diene structures and transforms the deemed “old-fashioned” and fossil fuel-based status of carbanionic diene polymerization. This also motivates fundamental works regarding carbanionic polymerization to further investigate the mechanisms and eventually to develop high performance materials. As most products based on dienes are used as high-volume materials, the economy of scale is a key factor in this area.An aspect not to be neglected is the responsibility of the industrialized countries to regulate the usage of the available farmland between the production of renewable raw materials and agriculture for food production. For this reason, many different feedstocks are under investigation to obtain dienes via truly sustainable pathways. One approach relies on utilizing bacteria in fermentation processes that are capable of turning waste streams into valuable resources.84 Another approach addresses turpentine oil as a feedstock, which does not compete with food production. This shows that the future holds many challenges when fossil fuel-based isoprene and butadiene are to be replaced as standard diene building blocks. New routes are therefore mandatory to adapt the new monomers to specific material properties. Other aspects such as more sustainable solvents or more efficient processes are also driving current research.
A major challenge for the polymerization of novel dienes is the transfer of innovations from academia to industry, as this sets the starting point for economic considerations of more sustainable solutions. This involves a shift from only three primarily used monomers, i.e., styrene, butadiene, and isoprene at present towards a variety of biobased diene structures.
9. Conclusion
In this short review we have briefly described the significance of polydienes in the past and also highlighted recent developments. Capitalizing on the high precision of the anionic polymerization technique, a variety of 1,3-dienes have been successfully polymerized in a living manner. Besides matching the properties of fossil fuel-based PB or PI with biobased candidates, the synthesis of unprecedented rigid polydienes like PNopa was recently achieved. Nopadiene shows promising performance as a biobased substitute for PS. This monomer permitted the first synthesis of fully diene-based TPEs. The flexibility of polydienes can be either tuned by the addition of different polar modifiers or by changing the substitution pattern of the monomer. This has great impact on the reactivity ratios when copolymerized with styrene. While monosubstituted 1,3-dienes showed a block-like polymer composition, disubstituted 1,3-dienes reacted almost randomly with styrene in apolar media. The increase in steric demand is directly correlated to the resulting microstructure of the polydiene. Further, if the degrees of freedom are reduced to a minimum via constrained 1,3-dienes of a cisoid or transoid structure, respectively, the requirement of a cisoid-state was revealed. A large variety of copolymerizations and their respective reactivity ratios within the field of dienes in the carbanionic polymerization was presented, alongside with insights into new emerging fields including new and often biobased monomers, solvents and modifiers. We emphasize that the findings of the past years, summarized in this overview, are crucial for creating innovations in the future and are of general relevance for the field of carbanionic polymerization and polydienes.Author contributions
The manuscript was conceptualized by all authors and the original draft was written by equal contributions of Moritz Rauschenbach and Moritz Meier-Merziger. Both contributed to the visualization, data curation and formal analysis of the work. The supervision was carried out by Prof. Holger Frey as well as validation, reviewing and editing of the final work.Data availability
Since this is a review article, there are no research data available that would have to be disclosed.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We are grateful to Prof. Axel H. E. Müller for many valuable discussions. He has enriched our perspective in the whole field of carbanionic polymerization. Furthermore we much appreciate the long-term and fruitful collaboration with Prof. George Floudas of the University of Ioannina (Greece) Prof. Markus Gallei of the Saarland University (Germany) and Dr Manfred Wagner from the Max Planck Institute of Polymer Research (MPIP) in Mainz (Germany). The authors thank the company Arlanxeo for valuable collaboration in the field of diene polymerisation.References
- H. Frey and T. Johann, Polym. Chem., 2020, 11, 8–14 RSC.
- H. Staudinger and J. Fritschi, Helv. Chim. Acta, 1922, 5, 785–806 CrossRef CAS.
- F. Mollwo Perkin, J. R. Soc. Arts, 1912, 61, 85–102 Search PubMed.
- A. Holt, Angew. Chem., 1914, 27, 153 CrossRef CAS.
- M. Steube, T. Johann, R. D. Barent, A. H. E. Müller and H. Frey, Prog. Polym. Sci., 2022, 124, 101488 CrossRef CAS.
- S. Thomas, C. Han Chan, L. A. Pothen and H. J. Maria, RSC Polymer Chemistry Series Natural Rubber Materials Volume 1: Blends and IPNs, Royal Society of Chemistry, Cambridge, 2014, vol. 1 Search PubMed.
- Official Website of Malaysian Rubber Council, Rubber Industry: Industry Overview, https://www.myrubbercouncil.com/industry/world_production.php, (accessed 2 February 2024).
- K. Ntetsikas, V. Ladelta, S. Bhaumik and N. Hadjichristidis, ACS Polym. Au, 2022, 3, 158–181 CrossRef PubMed.
- A. Forens, K. Roos, C. Dire, B. Gadenne and S. Carlotti, Polymer, 2018, 153, 103–122 CrossRef CAS.
- H. Hsieh and R. Quirk, Anionic polymerization: principles and practical applications, Dekker, New York, 1996 Search PubMed.
- K. Knoll and N. Nießner, Macromol. Symp., 1998, 132, 231–243 CrossRef CAS.
- E. Galanos, C. Wahlen, H. J. Butt, H. Frey and G. Floudas, Macromol. Chem. Phys., 2022, 223, 2200033 CrossRef CAS.
- E. Grune, M. Appold, A. H. E. Müller, M. Gallei and H. Frey, ACS Macro Lett., 2018, 7, 807–810 CrossRef CAS PubMed.
- R. D. Barent, I. Perevyazko, N. Mikusheva, G. Floudas and H. Frey, Macromolecules, 2023, 56, 5792–5802 CrossRef CAS.
- M. Steube, T. Johann, E. Galanos, M. Appold, C. Rüttiger, M. Mezger, M. Gallei, A. H. E. Müller, G. Floudas and H. Frey, Macromolecules, 2018, 51, 10246–10258 CrossRef CAS.
- M. Steube, T. Johann, M. Plank, S. Tjaberings, A. H. Gröschel, M. Gallei, H. Frey and A. H. E. Müller, Macromolecules, 2019, 52, 9299–9310 CrossRef CAS.
- M. Steube, T. Johann, H. Hübner, M. Koch, T. Dinh, M. Gallei, G. Floudas, H. Frey and A. H. E. Müller, Macromolecules, 2020, 53, 5512–5527 CrossRef CAS.
- S. P. Wadgaonkar, S. Schüttner, E. Berger-Nicoletti, A. H. E. Müller and H. Frey, Macromolecules, 2022, 55, 4721–4732 CrossRef CAS.
- D. A. H. Fuchs, H. Hübner, T. Kraus, B. J. Niebuur, M. Gallei, H. Frey and A. H. E. Müller, Polym. Chem., 2021, 12, 4632–4642 RSC.
- P. Von Tiedemann, J. Blankenburg, K. Maciol, T. Johann, A. H. E. Müller and H. Frey, Macromolecules, 2019, 52, 796–806 CrossRef CAS.
- A. Natalello, M. Werre, A. Alkan and H. Frey, Macromolecules, 2013, 46, 8467–8471 CrossRef CAS.
- C. Wahlen, J. Blankenburg, P. Von Tiedemann, J. Ewald, P. Sajkiewicz, A. H. E. Müller, G. Floudas and H. Frey, Macromolecules, 2020, 53, 10397–10408 CrossRef CAS.
- E. Grune, J. Bareuther, J. Blankenburg, M. Appold, L. Shaw, A. H. E. Müller, G. Floudas, L. R. Hutchings, M. Gallei and H. Frey, Polym. Chem., 2019, 10, 1213–1220 RSC.
- C. Yao, N. Liu, S. Long, C. Wu and D. Cui, Polym. Chem., 2016, 7, 1264–1270 RSC.
- H. Leicht, I. Göttker-Schnetmann and S. Mecking, ACS Macro Lett., 2016, 5, 777–780 CrossRef CAS.
- G. Ricci, G. Pampaloni, A. Sommazzi and F. Masi, Macromolecules, 2021, 54, 5879–5914 CrossRef CAS.
- L. Friebe, O. Nuyken and W. Obrecht, Adv. Polym. Sci., 2006, 204, 1–154 CrossRef CAS.
- S. Kaita, Y. Doi, K. Kaneko, A. C. Horiuchi and Y. Wakatsuki, Macromolecules, 2004, 37, 5860–5862 CrossRef CAS.
- N. Hadjichristidis, H. Iatrou, S. Pispas and M. Pitsikalis, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 3211–3234 CrossRef CAS.
- F. Wurm, D. Wilms, J. Klos, H. Löwe and H. Frey, Macromol. Chem. Phys., 2008, 209, 1106–1114 CrossRef CAS.
- K. Iida, T. Q. Chastek, K. L. Beers, K. A. Cavicchi, J. Chun and M. J. Fasolka, Lab Chip, 2009, 9, 339–345 RSC.
- K. Pérez, S. Leveneur, F. Burel, J. Legros and D. Vuluga, React. Chem. Eng., 2023, 8, 432–441 RSC.
- C. Tonhauser, D. Wilms, F. Wurm, E. Berger-Nicoletti, M. Maskos, H. Löwe and H. Frey, Macromolecules, 2010, 43, 5582–5588 CrossRef CAS.
- J. Zhang, W. Pointer, G. Patias, L. Al-Shok, R. A. Hand, T. Smith and D. M. Haddleton, Eur. Polym. J., 2023, 183, 111755 CrossRef CAS.
- J. Zhang, C. Aydogan, G. Patias, T. Smith, L. Al-Shok, H. Liu, A. M. Eissa and D. M. Haddleton, ACS Sustainable Chem. Eng., 2022, 10, 9654–9664 CrossRef CAS PubMed.
- A. V. Tobolsky and C. E. Rogers, J. Polym. Sci., 1959, 40, 73–89 CrossRef CAS.
- A. V. Tobolsky and C. E. Rogers, J. Polym. Sci., 1959, 38, 205–207 CrossRef CAS.
- A. H. E. Müller and K. Matyjaszewski, Controlled and Living Polymerizations: From Mechanisms to Applications, Wiley-VCH Verlag, Weinheim, 2010 Search PubMed.
- M. Szwarc, Nature, 1956, 178, 1168–1169 CrossRef CAS.
- J. E. Mark, Physical properties of polymers, Springer, New York, 2007 Search PubMed.
- R. D. Barent, M. Wagner and H. Frey, Polym. Chem., 2022, 13, 5478–5485 RSC.
- W. Gebert, J. Hinz and H. Sinn, Makromol. Chem., 1971, 144, 97–115 CrossRef CAS.
- D. J. Worsfold and S. Bywater, Macromolecules, 1978, 11, 582–586 CrossRef CAS.
- N. Hadjichristidis and A. Hirao, Anionic Polymerization, Springer, Tokio, 2015 Search PubMed.
- S. Carlotti, S. Ménoret, A. Barabanova, P. Desbois and A. Deffieux, Macromol. Chem. Phys., 2004, 205, 656–663 CrossRef CAS.
- A. Forens, K. Roos, C. Dire, B. Gadenne and S. Carlotti, Chin. J. Polym. Sci., 2020, 38, 357–362 CrossRef CAS.
- Y. Kosaka, R. Goseki, S. Kawauchi and T. Ishizone, Macromol. Symp., 2015, 350, 55–66 CrossRef CAS.
- Y. Kosaka, K. Kitazawa, S. Inomata and T. Ishizone, ACS Macro Lett., 2013, 2, 164–167 CrossRef CAS.
- Y. Kosaka, S. Kawauchi, R. Goseki and T. Ishizone, Macromolecules, 2015, 48, 4421–4430 CrossRef CAS.
- S. Kobayashi, C. Lu, T. R. Hoye and M. A. Hillmyer, J. Am. Chem. Soc., 2009, 131, 7960–7961 CrossRef CAS PubMed.
- K. Liu, Q. He, L. Ren, F. Xu and W. J. Xu, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 2291–2301 CrossRef CAS.
- L. Ren, K. Liu, Q. He, E. Ou, Y. Lu and W. Xu, RSC Adv., 2016, 6, 51533–51543 RSC.
- C. Rüttiger, M. Appold, H. Didzoleit, A. Eils, C. Dietz, R. W. Stark, B. Stühn and M. Gallei, Macromolecules, 2016, 49, 3415–3426 CrossRef.
- A. H. Gabor, E. A. Lehner, G. Mao, L. A. Schneggenburger and C. K. Ober, Chem. Mater., 1994, 6, 927–934 CrossRef CAS.
- B. Rodgers, Rubber Compounding: Chemistry and Applications, CRC Press, Boca Raton, 2016 Search PubMed.
- J. Glatzel, S. Noack, D. Schanzenbach and H. Schlaad, Polym. Int., 2021, 70, 181–184 CrossRef CAS.
- M. Morton and L. J. Fetters, J. Polym. Sci., Part A: Gen. Pap., 1964, 2, 3311–3326 CrossRef.
- S. Bywater and D. J. Worsfold, Can. J. Chem., 1962, 40, 1564–1570 CrossRef CAS.
- L. Shaw and L. R. Hutchings, Polym. Chem., 2020, 11, 7020–7025 RSC.
- T. Yoo and S. K. Henning, Rubber Chem. Technol., 2017, 90, 308–324 CrossRef CAS.
- C. Wahlen, M. Rauschenbach, J. Blankenburg, E. Kersten, C. P. Ender and H. Frey, Macromolecules, 2020, 53, 9008–9017 CrossRef CAS.
- J. M. Bolton, M. A. Hillmyer and T. R. Hoye, ACS Macro Lett., 2014, 3, 717–720 CrossRef CAS PubMed.
- J. Zhang, J. Lu, K. Su, D. Wang and B. Han, J. Appl. Polym. Sci., 2019, 136, 1–10 Search PubMed.
- R. Ohno, Y. Tanaka and M. Kawakami, Polym. Chem., 1973, 4, 56–60 CAS.
- T. Suzuki, Y. Tsuji, Y. Takegami and H. J. Harwood, Macromolecules, 1979, 12, 234–239 CrossRef CAS.
- Y. X. Ding and W. P. Weber, Macromolecules, 1988, 21, 530–532 CrossRef CAS.
- T. Suzuki, Y. Tsuji and Y. Takegami, Macromolecules, 1978, 11, 639–644 CrossRef CAS.
- T. Suzuki, Y. Tsuji, Y. Watanabe and Y. Takegami, Polym. J., 1979, 11, 651–660 CrossRef CAS.
- M. Rauschenbach, L. Stein, G. M. Linden, R. Barent, K. Heinze and H. Frey, Polym. Chem., 2024, 15, 3204–3213 RSC.
- R. Goseki, S. Miyai, S. Uchida and T. Ishizone, Polym. Chem., 2021, 12, 3602–3611 RSC.
- S. Uchida, K. Togii, S. Miyai, R. Goseki and T. Ishizone, Macromolecules, 2020, 53, 10107–10116 CrossRef CAS.
- C. Hahn, M. Rauschenbach and H. Frey, Angew. Chem., Int. Ed., 2023, 62, e202302907 CrossRef CAS.
- W. M. Haynes, D. R. Lide and T. J. Bruno, CRC handbook of chemistry and physics: a ready-reference book of chemical and physical data, CRC Press, Hoboken, 95th edn, 2015 Search PubMed.
- C. Wahlen and H. Frey, Macromolecules, 2021, 54, 7323–7336 CrossRef CAS.
- M. Meier-Merziger, J. Imschweiler, F. Hartmann, B.-J. Niebuur, T. Kraus, M. Gallei and H. Frey, Angew. Chem., 2023, 62, e202310519 CrossRef CAS PubMed.
- C. Zhou, Z. Wei, X. Lei and Y. Li, RSC Adv., 2016, 6, 63508–63514 RSC.
- C. Zhou, Z. Wei, C. Jin, Y. Wang, Y. Yu, X. Leng and Y. Li, Polymer, 2018, 138, 57–64 CrossRef CAS.
- M. Meier-Merziger, N. Fotaras, I. Tzourtzouklis, C. Allouch, M. Wagner, A. H. E. Müller, G. Floudas and H. Frey, ACS Sustainable Chem. Eng., 2024, 12, 9922–9933 CrossRef CAS.
- C. Iacob, T. Yoo and J. Runt, Macromolecules, 2018, 51, 4917–4922 CrossRef CAS.
- L. J. Fetters, D. J. Lohse, D. Richter, T. A. Witten and A. Zirkel, Macromolecules, 1994, 27, 4639–4647 CrossRef CAS.
- L. J. Fetters, J. Res. Natl. Bur. Stand., Sect. A, 1964, 69, 33–37 Search PubMed.
- P. Oßwald, R. Whitside, J. Schäffer and M. Köhler, Fuel, 2017, 187, 43–50 CrossRef.
- J. Zhang, J. Chen, M. Yao, Z. Jiang and Y. Ma, J. Appl. Polym. Sci., 2019, 136, 47673 CrossRef.
- S. Milker and D. Holtmann, Microb. Cell Fact., 2021, 20, 1–7 CrossRef.
- A. Matic and H. Schlaad, Polym. Int., 2018, 67, 500–505 CrossRef CAS.
- A. Matic, A. Hess, D. Schanzenbach and H. Schlaad, Polym. Chem., 2020, 11, 1364–1368 RSC.
- S. P. Wadgaonkar, M. Wagner, L. A. Baptista, R. Cortes-Huerto, H. Frey and A. H. E. Müller, Macromolecules, 2023, 56, 664–677 CrossRef CAS.
- C. Hahn, I. Göttker-Schnetmann, I. Tzourtzouklis, M. Wagner, A. H. E. Müller, G. Floudas, S. Mecking and H. Frey, J. Am. Chem. Soc., 2023, 145, 26688–26698 CrossRef CAS PubMed.
- C. Hahn, M. Wagner, A. H. E. Müller and H. Frey, Macromolecules, 2022, 55, 4046–4055 CrossRef CAS.
- A. L. Gatzke and E. D. Vanzo, Chem. Commun., 1967, 22, 1180–1181 RSC.
- R. B. Bates, L. M. Kroposki and D. E. Potter, J. Org. Chem., 1972, 37, 560–562 CrossRef CAS.
- C. A. Ogle, F. H. Strickler and B. Gordon, Macromolecules, 1993, 26, 5803–5805 CrossRef CAS.
- J. Bareuther, M. Plank, B. Kuttich, T. Kraus, H. Frey, M. Gallei, J. Bareuther, M. Plank, B. Kuttich, T. Kraus, H. Frey and M. Gallei, Macromol. Rapid Commun., 2021, 42, 2000513 CrossRef CAS PubMed.
- A. Dev, A. Rösler and H. Schlaad, Polym. Chem., 2021, 12, 3084–3087 RSC.
- Y. Fu, S. Yang, Q. Xiong, Z. Gu, Q. Dai, H. Tan, L. Zhou, M. Geng, F. Xie, W. Yi, L. Li and K. Liu, Polym. Int., 2024, 73, 326–336 CrossRef CAS.
- I. Natori, S. Natori, J. Taehee and K. Ogino, Polymer, 2022, 250, 124821 CrossRef CAS.
- J. Wilson, S. Gering, J. Pinard, R. Lucas and B. R. Briggs, Biotechnol. Biofuels, 2018, 11, 1–11 CrossRef PubMed.
- M. Meier-Merziger, D. A. H. Fuchs, H. Frey and A. H. E. Müller, Macromolecules, 2024, 57, 8154–8161 CAS.
- C. Hayichelaeh, L. A. E. M. Reuvekamp, W. K. Dierkes, A. Blume, J. W. M. Noordermeer and K. Sahakaro, Rubber Chem. Technol., 2018, 91, 433–452 CrossRef CAS.
- K. Takenaka, Y. Akagawa, H. Takeshita, M. Miya and T. Shiomi, Polym. J., 2009, 41, 106–107 CrossRef CAS.
- K. Takenaka, D. Nakashima, M. Miya, H. Takeshita and T. Shiomi, J. Soft Matter., 2013, 9, 14–19 CrossRef.
- C. Tonhauser and H. Frey, Macromol. Rapid Commun., 2010, 31, 1938–1947 CrossRef CAS PubMed.
- P. Dreier, J. Ahn, T. Chang and H. Frey, Macromol. Rapid Commun., 2022, 43, 2200560 CrossRef CAS.
- M. Meier-Merziger, M. Fickenscher, F. Hartmann, B. Kuttich, T. Kraus, M. Gallei and H. Frey, Polym. Chem., 2023, 14, 2820–2828 RSC.
- C. Zhou, Z. Wei, Y. Wang, Y. Yu, X. Leng and Y. Li, Eur. Polym. J., 2018, 99, 477–484 CrossRef CAS.
- N. Hadjichristidis, M. Pitsikalis, S. Pispas and H. Iatrou, Chem. Rev., 2001, 101, 3747–3792 CrossRef CAS PubMed.
- P. Von Tiedemann, K. Maciol, J. Preis, P. Sajkiewicz and H. Frey, Polym. Chem., 2019, 10, 1762–1768 RSC.
- P. Von Tiedemann, J. Yan, R. D. Barent, R. J. Spontak, G. Floudas, H. Frey and R. A. Register, Macromolecules, 2020, 53, 4422–4434 CrossRef CAS.
- J. Roovers, L.-L. Zhou, P. M. Toporowski, M. van der Zwan, H. Iatrou and N. Hadjichristidis, Macromolecules, 1993, 26, 4324–4331 CrossRef CAS.
- V. Hirschberg, M. G. Schußmann, M. C. Röpert, N. Dingenouts, S. Buchheiser, H. Nirschl, J. Berson and M. Wilhelm, Macromolecules, 2024, 57, 3387–3396 CrossRef CAS.
- N. R. Legge, Rubber Chem. Technol., 1987, 60, 83–117 CrossRef.
- L. Porter, GB. Pat., 888624, 1962 Search PubMed.
- G. Holden and R. Milkovich, US 3265765, 1966 Search PubMed.
- D. J. Worsfold, J. Polym. Sci., Part A-1: Polym. Chem., 1967, 5, 2783–2789 CrossRef CAS.
- G. Kraus and K. W. Rollmann, Angew. Makromol. Chem., 1971, 16, 271–296 CrossRef.
- E. Grune, T. Johann, M. Appold, C. Wahlen, J. Blankenburg, D. Leibig, A. H. E. Müller, M. Gallei and H. Frey, Macromolecules, 2018, 51, 3527–3537 CrossRef CAS.
- W. Wang, W. Lu, A. Goodwin, H. Wang, P. Yin, N. G. Kang, K. Hong and J. W. Mays, Prog. Polym. Sci., 2019, 95, 1–31 CrossRef CAS.
- D. J. T. Hill, J. H. O'Donnell, P. W. O'Sullivan, J. E. McGrath, I. C. Wang and T. C. Ward, Polym. Bull., 1983, 9, 292–298 CrossRef CAS.
Footnote |
| † All authors have contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |