
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The CF3TMS adduct of anthraquinone as a monomer for making polymers with potential as separation membranes†
Kim Jiayi
Wu
 a,
John M.
Tobin
a,
Anli
Ji
a,
Yang
Shi
a,
Chunchun
Ye
a,
Gary S.
Nichol
a,
Alessio
Fuoco
b,
Mariagiulia
Longo
b,
Johannes C.
Jansen
b and
Neil B.
McKeown
*a
a,
John M.
Tobin
a,
Anli
Ji
a,
Yang
Shi
a,
Chunchun
Ye
a,
Gary S.
Nichol
a,
Alessio
Fuoco
b,
Mariagiulia
Longo
b,
Johannes C.
Jansen
b and
Neil B.
McKeown
*a
aEaStChem, School of Chemistry, University of Edinburgh, David Brewster Road, Edinburgh, EH9 3FJ, UK. E-mail: neil.mckeown@ed.ac.uk
bInstitute on Membrane Technology, National Research Council of Italy (CNR-ITM), via P. Bucci 17/C, Rende (CS), 87036, Italy
First published on 21st October 2024
Abstract
The readily prepared CF3TMS adduct of anthraquinone is shown to be an efficient monomer for superacid-catalysed step-growth polymerisations, as exemplified by its reaction with diphenyl ether. The resulting polymer (BTFMA-DPE) is produced rapidly, with high molecular mass, and shows promise as a gas separation membrane material.
Superacid-catalysed step-growth polymerisations have been investigated for over two decades.1–4 The mechanism of these polymerisations involves the formation of highly reactive carbocations (i.e., superelectrophiles) from a carbonyl-containing monomer, which then react with a bifunctional aromatic monomer that is activated towards aromatic electrophilic substitution. Suitable carbonyl-containing monomers include 1,1,1-trifluoroacetophenone,5 isatin and its derivatives,1,6 4-piperidone,7 1,1,1-trifluoroacetone,6 4-acetylpyridine,8,9 and acenaphthenequinone,10 and suitable aromatic monomers include biphenyl,2 terphenyl,10 4,4′-diphenoxybenzophenone1 or diphenyl ether10 (Fig. 1). Typically, the superacid used is trifluoromethylsulfonic acid (TFSA). The attractive features of superacid mediated polymerisations are their use of readily available monomers and rapid completion even at room temperature, allowing for ease of scale-up to multigram quantities or greater. Recently, such polymerisations have been used intensively to prepare membrane materials for application in a wide variety of technologies relevant to energy and environmental sustainability including gas separations,11–16 water purification,17,18 acid recovery19,20 and ion separations.21,22 For example, post-synthetic modification, such as amine quaternisation for those polymers derived from 4-piperidone23–25 or the introduction of sulfonate groups,26,27 induce ion-conducting properties that make them useful as membranes for devices such as proton exchange fuel cells,9,27–30 alkaline fuel cells,26,31–37 redox-flow batteries,38,39 zinc batteries,40 ammonia electrosynthesisers,41 and water electrolysers.42–44
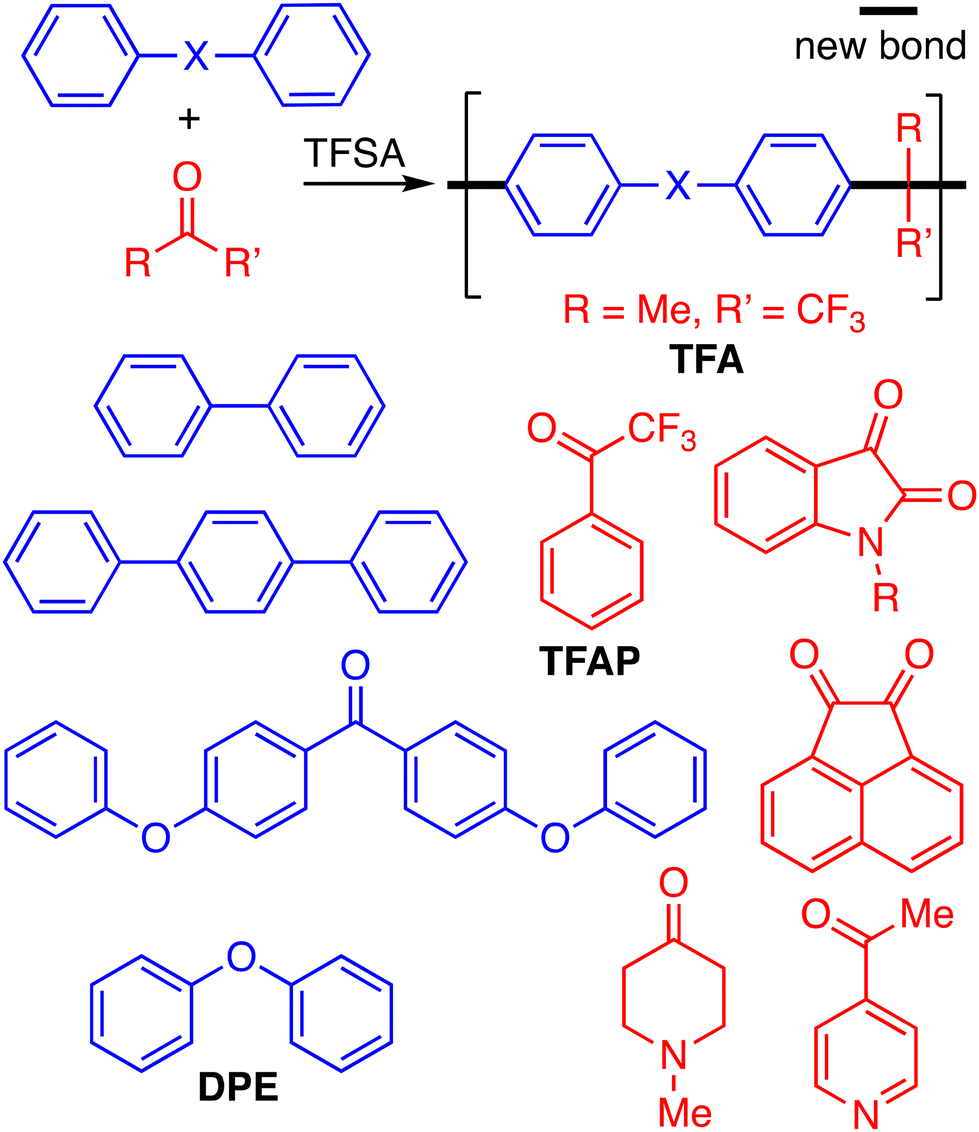 | ||
| Fig. 1 General scheme of typical superacid polymerisations with examples of suitable electrophilic monomers depicted in blue [biphenyl, terphenyl, diphenyl ether (DPE) and 4,4′-diphenoxybenzophenone] and nucleophilic monomers depicted in red [1,1,1-trifluoroacetone (TFA), 1,1,1-trifluoroacetophenone (TFAP), isatin derivatives, 4-piperidone, acenaphthenequinone and 4-acetylpyridine]. | ||
In this communication, we describe preliminary results on the use of the readily prepared CF3TMS adduct of anthraquinone (BTFMA, Fig. 2) as an electrophilic monomer for engaging in superacid mediated polymerisations as exemplified by using diphenyl ether (DPE) as co-monomer. The potential of BTFMA for making high free volume polymers for membranes is illustrated by an analysis of the gas permeability of the resulting polymer (BTFMA-DPE).
 | ||
| Fig. 2 Synthesis of model compound 1, shown with its single crystal XRD structure (CCDC 2382285†) confirming its “cis” configuration of substituents relative to the anthracene plane, and polymer BTFMA-DPE. Reagents and conditions: i. anisole, TFSA, DCM, 20 °C; ii. DPE, TFSA, DCM, 20 °C (90% yield). | ||
Previously, it was reported that two equivalents of trifluoromethyltrimethylsilane (CF3TMS, i.e., the Ruppert–Prakash reagent)45 add cleanly to anthraquinone to form 9,10-bis(trimethysiloxyl)-9,10-bis(trifluoromethyl)-9,10-dihydroanthracene (BTFMA) in high yield.46,47 On repeating this reaction, it was found that BTFMA could be purified easily, without using column chromatography, by simple recrystallisation from isopropanol to give multigram quantities of the adduct as an air-stable, white crystalline solid. Although BTFMA has been used previously as an intermediate to produce 9,10-bis(trifluoromethyl)anthracene48 and monomers for polyimide synthesis,49,50 we anticipated that it would engage directly in superacid-catalysed aromatic electrophilic substitution reactions with suitably reactive aromatic monomers. Encouragingly, the reaction between BTFMA and an excess of anisole, using TFSA as the superacid, gave a high crude yield of 9,10-bis(4′-methoxyphenyl)-9,10-bis(trifluoromethyl)-9,10-dihydroanthracene 1. It should be noted that previous uses of BTFMA as a synthetic intermediate involved the initial removal of the TMS group, via aqueous acid hydrolysis, but this proved unnecessary. Indeed, the use of the hydrolysed intermediate (i.e., 9,10-dihydroxy-9,10-bis(trifluoromethyl)-9,10-dihydroanthracene), resulted in a lower yield of model compound 1. Previous studies have also shown the benefit of using –OTMS as the leaving group for the generation of highly reactive carbocations using a superacid, which avoids the generation of water as by-product that may result in a reduction of the efficiency of TFSA.51,52 Single-crystal XRD analysis of 1 confirmed the structure of the model compound and revealed that it is composed of the “cis” isomer, in which the two CF3 substituents are placed facing the same direction relative to the anthracene plane (Fig. 2). Similar regioselectivity has been observed previously for closely related superacid mediated reactions.53 The 1H NMR of 1 obtained at ∼20 °C is deceptively simple as it shows only three peaks in the aromatic region with each of these peaks integrating to 4H relative to the 6H of the methoxy peak. Variable-temperature 1H NMR (ESI Fig. 1†) reveals that the apparently “missing” hydrogen peaks of the anisole substituents are discernible at higher and lower temperatures but are severely broadened at ambient temperature. This effect is due to the restricted rotation about the newly formed C–C bond (see discussion below).
The high yield obtained for the synthesis of model compound 1, suggested that similar TFSA mediated reactions between BTFMA and diphenyl ether (DPE) would provide a polymer, denoted as BTFMA-DPE (Fig. 2). After an aqueous work-up, fibrous colourless products are obtained that are fully soluble in chloroform. Analysis by gel permeation chromatography (GPC) of the product from polymerisation reactions carried out over a range of conditions (ESI Fig. 2 and Table 1†) showed that high molecular mass BTFMA-DPE is formed rapidly (∼0.5 h) at room temperature. For each reaction, two distinct peaks are observed by GPC: one at high molecular mass and one at low molecular mass, the latter with an apparent mass average molecular mass (Mn) of ∼2000 g mol−1, which is assumed to be due to cyclic and/or linear oligomers (ESI Fig. 2†).54 The relative height of the two peaks is dependent on the reaction conditions with deviations from the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry of monomers and lower concentration of reactants enhancing the relative height of the peak attributed to oligomers. For optimised reaction conditions, consisting of 1:1 molar stoichiometry, high concentration and a short reaction time (0.5 h), the peak attributed to higher molecular mass polymer is >5 times greater in height relative to than that attributed to the oligomers. The value of Mn calculated from the higher molecular mass peak, following calibration using polystyrene standards, is 150000 g mol−1 with a polydispersity index that is consistent with a standard step-growth mechanism (i.e., Mw/Mn = ∼2). Reprecipitation from chloroform solution using methanol reduces the amount of oligomeric material present in the polymeric product. By using these optimised reaction conditions, samples of BTFMA-DPE have been achieved rapidly and reproducibly on a multigram scale (to date up to 30 g). In contrast with previously reported superacid catalysed polymerisations, where a stoichiometric imbalance in favour of the electrophilic monomer results in higher molecular mass, a 1:1 molar equivalence of BTFMA and DPE appears optimal. Previously reported non-stoichiometric enhancement of the polymerisation was attributed to the higher reactivity of the intermediate formed after the first addition of the nucleophilic monomer.55 Therefore, for BTFMA it appears that the reactivity of the carbocation formed following the first addition of DPE is similar to that of the initially formed carbocation.
:1 stoichiometry of monomers and lower concentration of reactants enhancing the relative height of the peak attributed to oligomers. For optimised reaction conditions, consisting of 1:1 molar stoichiometry, high concentration and a short reaction time (0.5 h), the peak attributed to higher molecular mass polymer is >5 times greater in height relative to than that attributed to the oligomers. The value of Mn calculated from the higher molecular mass peak, following calibration using polystyrene standards, is 150000 g mol−1 with a polydispersity index that is consistent with a standard step-growth mechanism (i.e., Mw/Mn = ∼2). Reprecipitation from chloroform solution using methanol reduces the amount of oligomeric material present in the polymeric product. By using these optimised reaction conditions, samples of BTFMA-DPE have been achieved rapidly and reproducibly on a multigram scale (to date up to 30 g). In contrast with previously reported superacid catalysed polymerisations, where a stoichiometric imbalance in favour of the electrophilic monomer results in higher molecular mass, a 1:1 molar equivalence of BTFMA and DPE appears optimal. Previously reported non-stoichiometric enhancement of the polymerisation was attributed to the higher reactivity of the intermediate formed after the first addition of the nucleophilic monomer.55 Therefore, for BTFMA it appears that the reactivity of the carbocation formed following the first addition of DPE is similar to that of the initially formed carbocation.
Analysis by 1H- and 13C-NMR, with assignment of protons assisted by 1H-COSY, 1H/13C-HSQC, and 1H/13C-HMBC spectra (Fig. 3 and ESI Fig. 3–5†), confirmed the structure of polymer BTFMA-DPE, with the similarity of its spectra to those of model compound 1 indicating that there was selective substitution of DPE at the para-sites relative to its oxygen linker. This also confirms that there is a predominantly “cis” arrangement of the CF3 substituents relative to the dihydroanthracene plane, however, it is possible that the two weak doublet peaks observed at ∼7.55 and 7.20 ppm in DMF-d7 (Fig. 3a) and at 7.50 and 7.05 ppm in CDCl3 (Fig. 3b) may arise from a small degree of “trans” substitution of the dihydroanthracene unit (<5%).
 | ||
| Fig. 3 Variable temperature NMR of polymer BTFMA-DPE acquired in the ranges (a) 300–363 K (in DMF-d7) and (b) 243–300 K (in CDCl3). Due to hindered rotation of the benzene rings of the DE monomeric unit, the peak for the hydrogens (Ha) adjacent to the new bond only becomes distinct at higher temperatures. At lower temperatures, rotation is frozen so that each of the protons Ha1, Ha2, Hb1 and Hb2 show a distinct peak. | ||
Variable-temperature 1H-NMR spectroscopy (Fig. 3), provides clear evidence of the restricted rotation of the phenyl groups of the DPE monomeric unit within BTFMA-DPE. At room temperature, the signals associated with the 1H atoms of the DPE monomeric unit adjacent to the newly formed bond (Fig. 3, Ha) are not discernible and those adjacent to the oxygen linker (Hb) are severely broadened. At elevated temperatures (>340 K, DMF-d7, Fig. 3a), the former appears as a broad signal centred at ∼7.4 ppm and the latter as a well-resolved doublet at 7.12 ppm. In contrast, at lower temperatures (<250 K, CDCl3, Fig. 3b), four well-defined doublets are observed consistent with the frozen rotation of the phenyl groups resulting in a discrete signal for each proton. For example, the 1H atoms of the DPE monomeric unit adjacent to the newly formed bond are found at 8.05 and 6.50 ppm (Fig. 3b, Ha1 and Ha2), with the large difference in chemical shift caused by their exposure to the deshielding and shielding effects, respectively, of the ring current of the benzene rings within the dihydroanthracene unit. The coalescence temperature (Tc) of the signals for Hb1 and Hb2 (Fig. 3b) is at 275 ± 2 K, corresponding to an activation energy of 54 ± 1 kJ mol−1.56 This value of Tc for BTFMA-DPE is slightly higher than that for model compound 1 (270 ± 2 K), which may be due to the added restrictions to the motion of the phenyl rings because of their incorporation into a polymer chain.
Thermal gravimetric analysis (TGA) of BTFMA-DPE indicates no decomposition below 400 °C (ESI Fig. 6a†), with initial mass loss occurring at ∼430 °C, and no thermal transitions are discernible using differential scanning calorimetry (DSC) below this temperature (ESI Fig. 6b†). The rigidity of the components of this predominantly aromatic polymer will likely ensure a very high glass transition temperature (Tg), which will require further analysis using rapid scanning DSC techniques to determine its value.57,58 Colourless solutions of BTFMA-DPE in chloroform were used to cast robust, flexible, optically clear, self-standing films (Fig. 4) that could be used for gas permeability studies.
 | ||
| Fig. 4 A robust, optically clear, self-standing film of BTFMA-DPE fabricated from chloroform solution using the techniques of simple solvent-casting (5 cm diameter, 100 μm thick). | ||
Gas adsorption and permeability are routinely used to assess the presence of free volume in a glassy polymer. The powdered form of BTFMA-DPE showed significant N2 and CO2 adsorption (ESI Fig. 7†) with N2 uptake at 77 K allowing an apparent SABET of 450 m2 g−1 and total pore volume of 0.31 ml g−1 to be estimated. Although these values are modest compared to those of polymers of intrinsic microporosity (PIMs, e.g., the archetypal PIM-1 has an apparent SABET of ∼850 m2 g−1 and total pore volume of 0.40 ml g−1),59 they indicate that BTFMA-DPE contains a significant amount of free volume.
The potential of a new polymer for making a membrane to separate a particular gas pair (x/y) is assessed by its permeability (Px) and ideal selectivity (Px/Py). The values for He, H2, O2, CO2, CH4 and N2 permeabilities were measured for a self-standing film of BTFMA-DPE and can be compared with those from two related polymers, TFA-DPE and TFAP-DPE,11,12 prepared using DPE by a superacid catalysed polymerisation with 1,1,1-trifluoroacetone and 1,1,1-trifluoroacetophenone, respectively (Table 1). The permeability of BTFMA-DPE is much higher for all gases by a factor of 30–80. In addition, the ideal selectivities of BTFMA-DPE for the separation of the important gas pairs O2/N2 and CO2/CH4 are similar to those of TFAP-DPE, TFA-DPE, and PIM-1 but at a lower permeability for the latter, which is consistent with the lower values of N2 and CO2 adsorption noted above. Generally, polymers suffer from the well-established trade-off between gas permeability and selectivity and the performance of a new polymer is best assessed by considering the position of its data on Robeson plots relative to upper bounds, which were defined using data from the best performing PIMs.60,62 As shown for the Robeson plots of O2/N2 and CO2/CH4 (Fig. 5a and b, respectively), the gas permeability data for BTFMA-DPE lie close to the 1991 upper bounds for several gas pairs (also H2/N2 and H2/CH4),63 unlike those of TFA-DPE and TFAP-DPE. Whilst modest when compared to that of some PIMs, the permeability of BTFMA-DPE is similar to that of the best performing polyimides such as 6FDA-durene64 but with greater selectivity for key gas pairs (Table 1). Therefore, the performance of BTFMA-DPE is encouraging for a readily processed polymer that can be made easily on a large scale from readily available monomers as is the case for polymer 6FDA-durene. In addition, it is likely that replacing DPE as co-monomer, which possesses unrestricted rotational freedom about the ether linkage, with a conformationally locked monomer will result in a BTFMA based polymer with greater intrinsic microporosity. Such work and other initiatives to fulfil the potential of this new monomer for making membrane materials are ongoing.
 | ||
| Fig. 5 Robeson plots for a) CO2/CH4 and b) O2/N2 showing the data for BTFMA-DPE (red) and structurally related polymers TFA-DPE (green) and TFAP-DPE (purple). For comparison, the data points are also provided for PIM-160,61 (yellow) and the much-studied polyimide 6FDA-durene (black).64 | ||
61 and polyimide 6FDA-durene64 are included with the former being used to help define the 2008 Robeson upper bounds60 (Fig. 5)
| Polymer | Permeability Px (barrer) | Ideal selectivity Px/Py | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N2 | O2 | CO2 | CH4 | H2 | He | CO2/CH4 | CO2/N2 | O2/N2 | H2/N2 | H2/CH4 | He/N2 | |
| BTFMA-DPE | 22.9 | 97.6 | 473 | 22.3 | 463 | 327 | 21.7 | 21.7 | 4.26 | 20.2 | 20.7 | 14.3 |
|
TFA-DPE11 |
0.7 | 3.5 | 17.0 | 0.7 | 28.0 | 29.0 | 26.2 | 24.6 | 5.01 | 40.1 | 43.1 | 42.0 |
|
TFAP-DPE12 |
0.4 | 2.1 | 8.7 | 0.4 | 20 | 27 | 22.0 | 19.5 | 4.70 | 44.7 | 50.6 | 60.4 |
|
PIM-161 |
92 | 380 | 2300 | 125 | 1300 | 660 | 25.0 | 21.1 | 4.0 | 14.1 | 10.6 | 7.2 |
|
6FDA-durene64 |
39 | 135 | 678 | 38 | 585 | 355 | 20.2 | 17.4 | 3.46 | 15.0 | 15.4 | 9.1 |
To conclude, BTFMA is a readily prepared monomer that has been demonstrated to undergo efficient polymerisation mediated by a superacid. When copolymerised with DPE, a rigid polymer with good film-forming properties is rapidly produced. It is anticipated that BTFMA has excellent potential for enhancing the permeability of membrane-forming polymers that are produced using superacid polymerisations by the introduction of greater free volume.
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for model compound 1 has been deposited at the CCDC with the accession number 2382285† and can be obtained from https://www.ccdc.cam.ac.uk/structures/.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Engineering and Physical Science Research Council, UK, for funding through the programme grant SynHiSel (EP/V047078). In addition, we acknowledge funding from the UK Research and Innovation (UKRI) under the UK government's Horizon Europe funding guarantee [grant number 10091537] associated with DAM4CO2 (Double-Active Membranes for a sustainable CO2 cycle; HORIZON-EIC-2022-PATHFINDERCHALLENGES-01-number: 101115488). We also thank Dr Andrew Hall, Dr Lorna Murray, and Dr Richard York for NMR support. The authors thank Dr Dylan Wilkinson and Prof. Graeme Cooke (University of Glasgow) for their assistance with TGA and DSC characterisation of the polymer material.References
- H. M. Colquhoun, M. G. Zolotukhin, L. M. Khalilov and U. M. Dzhemilev, Macromolecules, 2001, 34, 1122–1124 CrossRef.
- M. Zolotukhin, S. Fomine, R. Salcedo and L. Khalilov, Chem. Commun., 2004, 1030–1031 RSC.
- M. G. Zolotukhin, L. Fomina, R. Salcedo, L. E. Sansores, H. M. Colquhoun and L. M. Khalilov, Macromolecules, 2004, 37, 5140–5141 CrossRef.
- L. I. Olvera, M. T. Guzmán-Gutiérrez, M. G. Zolotukhin, S. Fomine, J. Cárdenas, F. A. Ruiz-Trevino, D. Villers, T. A. Ezquerra and E. Prokhorov, Macromolecules, 2013, 46, 7245–7256 CrossRef.
- A. M. Diaz, M. G. Zolotukhin, S. Fomine, R. Salcedo, O. Manero, G. Cedillo, V. M. Velasco, M. T. Guzman, D. Fritsch and A. F. Khalizov, Macromol. Rapid Commun., 2007, 28, 183–187 CrossRef.
- M. G. Zolotukhin1, S. Fomine, L. M. Lazo, M. D. C. G. Hernández, M. T. Guzmán-Gutiérrez, A. Ruiz-Trevino, D. Fritsch, D. C. Cuellas and J. M. Fernandez-G, High Perform. Polym., 2007, 19, 638–648 CrossRef.
- A. R. Cruz, M. G. Zolotukhin, S. L. Morales, J. Cardenas, G. Cedillo, S. Fomine, M. Salmon and M. P. Carreón-Castro, Chem. Commun., 2009, 4408–4410 RSC.
- E. Cetina-Mancilla, L. I. Olvera, J. Balmaseda, M. Forster, F. A. Ruiz-Treviño, J. Cárdenas, E. Vivaldo-Lima and M. G. Zolotukhin, Polym. Chem., 2020, 11, 6194–6205 RSC.
- Y. Jin, T. Wang, X. Che, J. Dong, Q. Li and J. Yang, J. Power Sources, 2022, 526, 231131 CrossRef.
- M. G. Zolotukhin, S. Fomine, L. M. Lazo, R. Salcedo, L. E. Sansores, G. G. Cedillo, H. M. Colquhoun, J. M. Fernandez-G and A. F. Khalizov, Macromolecules, 2005, 38, 6005–6014 CrossRef.
- M. T. Guzmán-Gutiérrez, M. G. Zolotukhin, D. Fritsch, F. A. Ruiz-Treviño, G. Cedillo, E. Fregoso-Israel, C. Ortiz-Estrada, J. Chavez and C. Kudla, J. Membr. Sci., 2008, 323, 379–385 CrossRef.
- M. T. Guzmán-Gutiérrez, M. H. Rios-Dominguez, F. A. Ruiz-Treviño, M. G. Zolotukhin, J. Balmaseda, D. Fritsch and E. Prokhorov, J. Membr. Sci., 2011, 385–386, 277–284 CrossRef.
- E. C. Mancilla, H. Hernández-Martínez, M. G. Zolotukhin, F. A. Ruiz-Treviño, M. O. González-Díaz, J. Cardenas and U. Scherf, Ind. Eng. Chem. Res., 2019, 58, 15280–15287 CrossRef.
- H. Hernández-Martínez, E. Coutino-Gonzalez, F. Espejel-Ayala, F. A. Ruiz-Treviño, G. Guerrero-Heredia, A. L. García-Riego and L. I. Olvera, ACS Omega, 2021, 6, 4921–4931 CrossRef.
- I. Hossain, A. Husna, I. Jeong and T.-H. Kim, ACS Appl. Polym. Mater., 2022, 4, 3779–3790 CrossRef.
- L. Matesanz-Niño, N. Esteban, M. T. Webb, A. Martínez-Gómez, F. Suárez-García, A. González-Ortega, J. A. Miguel, L. Palacio, M. Galizia, C. Álvarez and Á. E. Lozano, Polymer, 2023, 267, 125647 CrossRef.
- Y. Dou, X. Dong, Y. Ma, P. Ge, C. Li, A. Zhu, Q. Liu and Q. Zhang, J. Membr. Sci., 2022, 659, 120779 CrossRef.
- S. Fang, H. Tang, M. Wang, Z. Xu and N. Li, J. Membr. Sci., 2023, 676, 121620 CrossRef.
- X. Du, Z. Wang, H. Zhang, Y. Yuan, H. Wang and Z. Zhang, J. Membr. Sci., 2021, 619, 118805 CrossRef.
- L. Xu, H. Wang, L. Min, W. Xu and W. Zhang, Sep. Purif. Technol., 2023, 312, 123396 CrossRef.
- W.-H. Lee, Y. S. Kim and C. Bae, ACS Macro Lett., 2015, 4, 814–818 CrossRef.
- E. J. Park, P. Jannasch, K. Miyatake, C. Bae, K. Noonan, C. Fujimoto, S. Holdcroft, J. R. Varcoe, D. Henkensmeier, M. D. Guiver and Y. S. Kim, Chem. Soc. Rev., 2024, 53, 5704–5780 RSC.
- J. S. Olsson, T. H. Pham and P. Jannasch, Adv. Funct. Mater., 2018, 28, 1702758 CrossRef.
- J. Wang, Y. Zhao, B. P. Setzler, S. Rojas-Carbonell, C. Ben Yehuda, A. Amel, M. Page, L. Wang, K. Hu, L. Shi, S. Gottesfeld, B. Xu and Y. Yan, Nat. Energy, 2019, 4, 392–398 CrossRef.
- D. Pan, T. H. Pham and P. Jannasch, ACS Appl. Energy Mater., 2021, 4, 11652–11665 CrossRef.
- T. Ryu, H. Jang, F. Ahmed, N. S. Lopa, H. Yang, S. Yoon, I. Choi and W. Kim, Int. J. Hydrogen Energy, 2018, 43, 5398–5404 CrossRef.
- W. Li, R. Zhang, X. Zhao, Z. Yue, H. Qian and H. Yang, J. Mater. Chem. A, 2023, 11, 4547–4558 RSC.
- Y. Jin, T. Wang, X. Che, J. Dong, R. Liu and J. Yang, J. Membr. Sci., 2022, 641, 119884 CrossRef.
- W. Yu, Z. Ge, K. Zhang, X. Liang, X. Ge, H. Wang, M. Li, X. Shen, Y. Xu, L. Wu and T. Xu, Ind. Eng. Chem. Res., 2022, 61, 4329–4338 CrossRef.
- J. Li, C. Yang, X. Zhang, Z. Xia, S. Wang, S. Yu and G. Sun, J. Mater. Chem. A, 2023, 11, 18409–18418 RSC.
- H. Bai, H. Peng, Y. Xiang, J. Zhang, H. Wang, S. Lu and L. Zhuang, J. Power Sources, 2019, 443, 227219 CrossRef.
- S. Zhang, X. Zhu and C. Jin, J. Mater. Chem. A, 2019, 7, 6883–6893 RSC.
- N. Chen, C. Hu, H. H. Wang, S. P. Kim, H. M. Kim, W. H. Lee, J. Y. Bae, J. H. Park and Y. M. Lee, Angew. Chem., Int. Ed., 2021, 60, 7710–7718 CrossRef CAS PubMed.
- N. Chen, S. Y. Paek, J. Y. Lee, J. H. Park, S. Y. Lee and Y. M. Lee, Energy Environ. Sci., 2021, 14, 6338–6348 RSC.
- Y. Zhao, Y. Yang, S. Wang, T. Wang, C. Liu, S. Cheng, H. Wei and Y. Ding, J. Membr. Sci., 2023, 688, 122132 CrossRef CAS.
- L. W. Lai, H. Peng, Y. N. Ding, X. B. Yue, Q. G. Zhang, A. M. Zhu and Q. L. Liu, J. Membr. Sci., 2024, 703, 122837 CrossRef CAS.
- J. Xue, J. C. Douglin, K. Yassin, T. Huang, H. Jiang, J. Zhang, Y. Yin, D. R. Dekel and M. D. Guiver, Joule, 2024, 8, 1457–1477 CrossRef CAS.
- X. Yan, H. Zhang, Z. Hu, L. Li, L. Hu, Z. A. Li, L. Gao, Y. Dai, X. Jian and G. He, ACS Appl. Mater. Interfaces, 2019, 11, 44315–44324 CrossRef CAS.
- W. Tang, T. Mu, X. Che, J. Dong and J. Yang, ACS Sustainable Chem. Eng., 2021, 9, 14297–14306 CrossRef CAS.
- Y. Wang, H. Peng, M. Hu, L. Zhuang, J. Lu and L. Xiao, J. Power Sources, 2021, 486, 229376 CrossRef CAS.
- Z. Xu, Y. Liao, M. Pang, L. Wan, Q. Xu, Y. Zhen and B. Wang, Energy Environ. Sci., 2023, 16, 3815–3824 RSC.
- E. J. Park, C. B. Capuano, K. E. Ayers and C. Bae, J. Power Sources, 2018, 375, 367–372 CrossRef CAS.
- X. Cheng, C. Li, X. Zou, B. Sun, L. Wang, W. Wang and X. Meng, Chem. Eng. J., 2024, 483, 149328 CrossRef CAS.
- X. Su, S. Nan, Y. Gu, W. Wei and R. He, Chem. Eng. J., 2024, 482, 149056 CrossRef CAS.
- G. K. S. Prakash and A. K. Yudin, Chem. Rev., 1997, 97, 757–786 CrossRef CAS.
- B. C. Ahvazi and D. S. Argyropoulos, J. Agric. Food Chem., 1996, 44, 2167–2175 CrossRef CAS.
- R. P. Singh and J. N. M. Shreeve, J. Fluorine Chem., 2012, 133, 20–26 CrossRef CAS.
- S. Yamada, K. Kinoshita, S. Iwama, T. Yamazaki, T. Kubota and T. Yajima, RSC Adv., 2013, 3, 6803–6806 RSC.
- F. Li, J. Liu, X. Liu, Y. Wang, X. Gao, X. Meng and G. Tu, Polymers, 2018, 10, 546 CrossRef.
- Y. Wang, Y. Li, F. Li, J. Shen, J. Zhao and G. Tu, Polym. Test., 2023, 129, 108269 CrossRef CAS.
- O. V. Khoroshilova, I. A. Boyarskaya and A. V. Vasilyev, J. Org. Chem., 2022, 87, 15845–15862 CrossRef CAS PubMed.
- O. V. Khoroshilova and A. V. Vasilyev, J. Org. Chem., 2020, 85, 5872–5883 CrossRef CAS PubMed.
- M. J. O'Connor, K. N. Boblak, M. J. Topinka, P. J. Kindelin, J. M. Briski, C. Zheng and D. A. Klumpp, J. Am. Chem. Soc., 2010, 132, 3266–3267 CrossRef.
- A. Cruz-Rosado, J. E. Romero-Hernández, M. Ríos-López, S. López-Morales, G. Cedillo, L. M. Ríos-Ruiz, E. Cetina-Mancilla, J. Palacios-Alquisira, M. G. Zolotukhin and E. Vivaldo-Lima, Polymer, 2022, 243, 124616 CrossRef CAS.
- M. T. Guzmán-Gutiérrez, D. R. Nieto, S. Fomine, S. L. Morales, M. G. Zolotukhin, M. C. G. Hernandez, H. Kricheldorf and E. S. Wilks, Macromolecules, 2011, 44, 194–202 CrossRef.
- M. T. Huggins, T. Kesharwani, J. Buttrick and C. Nicholson, J. Chem. Educ., 2020, 97, 1425–1429 CrossRef CAS.
- H. J. Yin, Y. Z. Chua, B. Yang, C. Schick, W. J. Harrison, P. M. Budd, M. Bohning and A. Schonhals, J. Phys. Chem. Lett., 2018, 9, 2003–2008 CrossRef CAS PubMed.
- H. Yin, B. Yang, Y. Z. Chua, P. Szymoniak, M. Carta, R. Malpass-Evans, N. B. McKeown, W. J. Harrison, P. M. Budd, C. Schick, M. Böhning and A. Schönhals, ACS Macro Lett., 2019, 8, 1022–1028 CrossRef CAS PubMed.
- P. M. Budd, B. S. Ghanem, S. Makhseed, N. B. McKeown, K. J. Msayib and C. E. Tattershall, Chem. Commun., 2004, 230–231 RSC.
- L. M. Robeson, J. Membr. Sci., 2008, 320, 390–400 CrossRef CAS.
- P. M. Budd, K. J. Msayib, C. E. Tattershall, B. S. Ghanem, K. J. Reynolds, N. B. McKeown and D. Fritsch, J. Membr. Sci., 2005, 251, 263–269 CrossRef CAS.
- B. Comesaña-Gándara, J. Chen, C. G. Bezzu, M. Carta, I. Rose, M.-C. Ferrari, E. Esposito, A. Fuoco, J. C. Jansen and N. B. McKeown, Energy Environ. Sci., 2019, 12, 2733–2740 RSC.
- L. M. Robeson, J. Membr. Sci., 1991, 62, 165–185 CrossRef CAS.
- W.-H. Lin and T.-S. Chung, J. Membr. Sci., 2001, 186, 183–193 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2382285. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4py01002g |
| This journal is © The Royal Society of Chemistry 2024 |