
Access to 1,2,3-triphospholide ligands by reduction of di-tert-butyldiphosphatetrahedrane†
Maria K.
Uttendorfer
 ,
Gabriele
Hierlmeier‡
,
Gábor
Balázs
and
Robert
Wolf
*
,
Gabriele
Hierlmeier‡
,
Gábor
Balázs
and
Robert
Wolf
*
Institute of Inorganic Chemistry, University of Regensburg, 93053 Regensburg, Germany. E-mail: robert.wolf@ur.de
First published on 8th May 2024
Abstract
Di-tert-butyldiphosphatetrahedrane (tBuCP)2 (A) is a reactive tetrahedral molecule which may serve as a source of new phosphaorganic molecules and ligands. However, the redox chemistry of this compound has not yet been investigated. Here, we show that the reduction of A with alkali metals (AM = Li, Na, K, Rb and Cs) affords 1,2,3-triphospholides [AM(crown ether)][1,2,3-P3C2tBu2] (1–5, [AM(crown ether)] = [Li([12]crown-4)2]+, [Na([15]crown-5)2]+, [K([18]crown-6)]+, [Rb([18]crown-6)]+, and Cs+) with 1,3-diphospholides [AM(crown ether)][1,3-P2C3tBu3] (6–10) formed as by-products. The potassium salt 3 was isolated on a preparative scale, allowing for reactivity studies. Transmetalation with iron(II) and ruthenium(II) chlorides yielded the sandwich complexes [Cp*M(η5-1,2,3-P3C2tBu2)] (11, M = Fe; 12, M = Ru, Cp* = C5Me5) featuring η5-coordinated triphospholide ligands. Treatment of 3 with [Cp2Fe][BAr4F] or [H(Et2O)2BAr4F] (BAr4F = B{C6H3(CF3)2}4) afforded the polyphosphorus compound tBu4C4P6 (13), which presumably results from the dimerisation of a 1,2,3-triphospholyl radical intermediate (1,2,3-P3C2tBu2)˙ (3˙). Tetracyclic 13 is closely structurally related to an isomer of the hydrocarbon hypostrophene (C10H10).
Introduction
Phosphatetrahedranes can be used to illustrate the diagonal relationship between carbon and phosphorus in the periodic table, and were postulated as stable compounds decades ago.1,2 Surprisingly, the first representatives for this class of tetrahedral molecules were reported only recently.3–5 We described the synthesis of di-tert-butyldiphosphatetrahedrane (tBuCP)2 (A, Fig. 1a) via the nickel-catalysed dimerisation of the phosphaalkyne tBuCP.3 In parallel, Cummins and co-workers synthesised and explored the reactivity of tri-tert-butylphosphatetrahedrane (tBu3C3P) (B) and triphosphatetrahedrane (HCP3) (C).4,5 The phosphatetrahedranes A–C are isolobal to purely carbon-based tetrahedranes such as (tBuC)4 (D), and the classical “inorganic tetrahedrane” P4 (E, white phosphorus).6,7 This has inspired the study of the reaction chemistry of A–C, which has revealed some similarities, but also differences with respect to the chemistry of white phosphorus in particular.3–5,8–14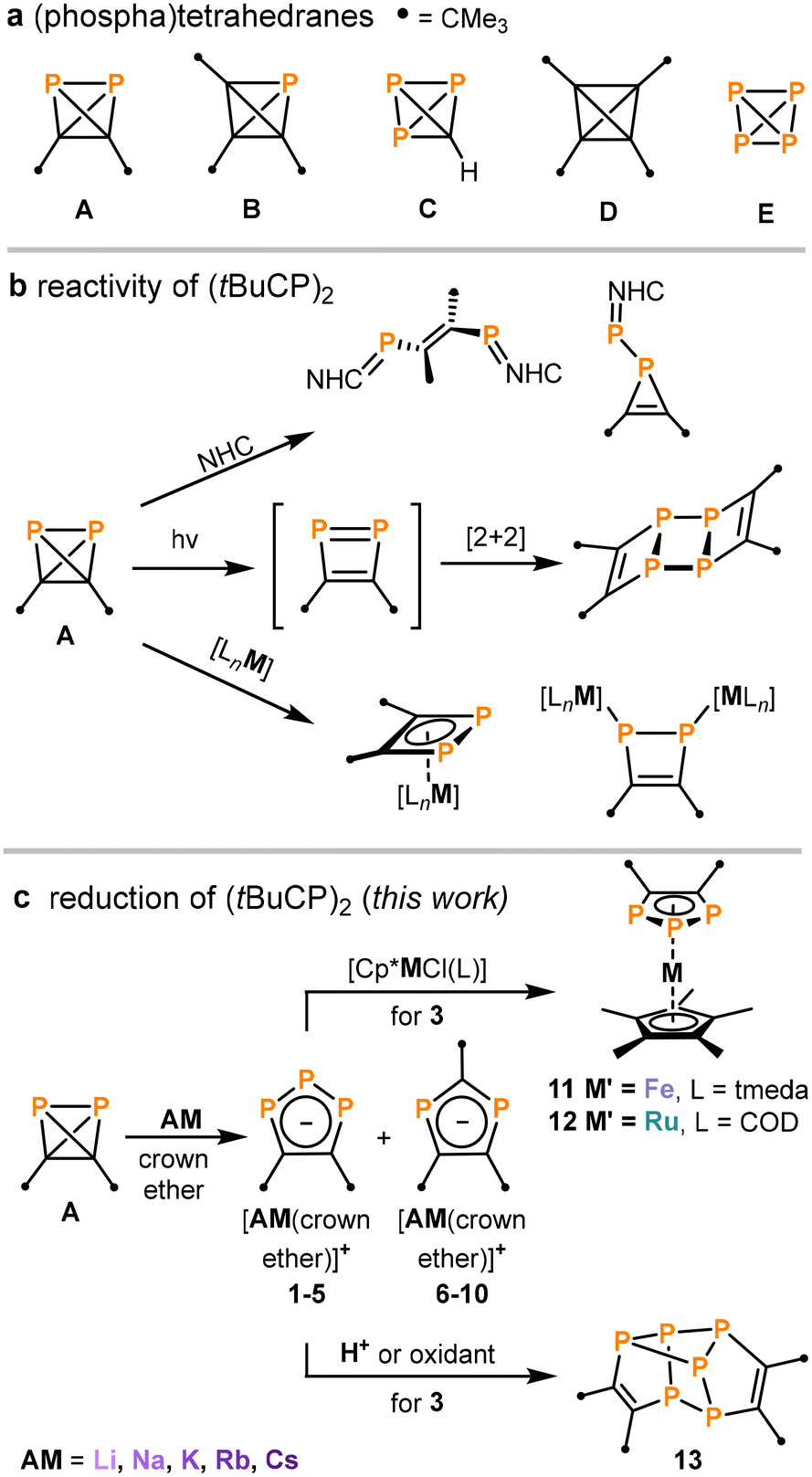 | ||
| Fig. 1 (a) Tetrahedranes comprised of phosphorus and carbon atoms;3–7 (b) reactivity of A (NHC = N-heterocyclic carbene, M = Fe, Co, Ni; L e.g. 1,5-cyclooctadiene, anthracene, cyclopentadienyl),8,11–13 (c) alkali metal reduction of A and subsequent reactivity studies. | ||
Isomerisations of the phosphatetrahedrane scaffold have been observed in reactions with main group nucleophiles. Thus, N-heterocyclic carbenes (NHCs) effect P–C or P–P bond cleavage in A, giving bis(phosphaalkenes) or phosphirenes depending on the NHC (Fig. 1b).8 An isomerisation of B to intermediary (tri-tert-butylcyclopropenyl)phosphinidene has been proposed for various reactions, e.g. with triphenylmethylenephosphorane and a base-stabilized silylene.9 The isomerisation to transient phosphacyclobutadienes is another predominant reaction path, which has been reported for the irradiation of A by UV or visible light and for the reaction of B with Lewis acids (Fig. 1b).10,11 Cyclobutadiene-type ligands are formed directly on the metal atom when A interacts with various low-valent Fe, Co and Ni complexes (Fig. 1b).12,13 Further studies have shown that phosphatetrahedranes may alternatively retain their tetrahedral structure in selected coordination compounds.3,5,14
To the best of our knowledge, there are no reports on the synthesis of organophosphorus anions by reduction of phosphatetrahedranes. In contrast, the reduction chemistry of the closely-related compounds P4 (E) and tert-butylphosphaalkyne (tBuCP, the monomer of A) is well-investigated.15–19 Reactions of P4 with sodium afford pentaphospholide, tetraphospholide and 1,2,3-triphospholide anions (among other polyphosphide species),15,16 while reduction of phosphaalkynes (RCP) with Na/Hg (and other alkali metal reductands) yields di- and 1,2,4-triphospholides.17–19 These previous results inspired us to explore similar reductions of (tBuCP)2 (A) (Fig. 1c), with the aim to access anionic ligands. Here, we report the synthesis of 1,2,3-triphospholide salts [AM(crown ether)][1,2,3-P3C2tBu2] (1–5, [AM(crown ether)] = [Li([12]crown-4)2]+, [Na([15]crown-5)2]+, [K([18]crown-6)]+, [Rb([18]crown-6)]+, and Cs+) from A and alkali metals. The utility of 3 as a suitable precursor for 1,2,3-triphospholide complexes is demonstrated by coordination to group 8 metal complexes. Furthermore, we report the synthesis and characterisation of the unusual polyphosphane (tBu4C4P6) (13), which is an oxidation product of 3 with a remarkable structure built up by two fused 1,2,3-triphospholyl units. Our results complement previous studies on the synthesis of 1,2,3-triphospholides via several other routes, including most prominently the reaction of polyphosphide anions with alkynes.16,20–29
Results and Discussion
Reduction of A with alkali metals: synthesis and characterisation of triphospholides 1–5
Initially, the redox properties of (tBuCP)2 (A) were investigated by cyclic voltammetry in toluene/THF/[nBu4N]PF6. Surprisingly, no redox process was observed between −3.0 and +1.2 V vs. Fc/Fc+ (Fc = Cp2Fe, see Fig. S47, ESI†). The absence of any redox wave suggests that strong oxidants and reductants are required to react with A, while also indicating that oxidation and reduction is likely associated with a large overpotential on the platinum working electrode. DFT calculations on A show that the HOMO lies higher and the LUMO lower in energy compared to P4.11 This suggests that, in principle, A should be more easily reduced and oxidized. P4 can be reduced at the potential E1/2 = −1.55 V (vs. bottom mercury in DMF).30 Therefore, the chemical reduction of A was pursued.Different reducing agents were evaluated in experiments performed on an NMR scale. Unfortunately, A did not react with magnesium or zinc powder. Activated Rieke zinc and Rieke magnesium gave black and brown suspensions, respectively. The reaction with Rieke zinc is very unselective, leading to an array of unidentified products according to 31P NMR spectroscopy. In contrast, the ladderane (tBuCP)4 was observed as main product in the reaction with Rieke magnesium. (tBuCP)4 is the dimerisation product of A, which can also be obtained by irradiation with UV or visible light (Fig. 1b).11,31
The use of strongly reducing alkali metals gave more promising results. Analysis of the 31P{1H} NMR spectrum of the reaction of A with lithium chunks showed full consumption of A and two new species in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (see Fig. S1, ESI†). After the addition of [12]crown-4, these species were identified as [Li([12]crown-4)2][1,2,3-P3C2tBu2] (1) and [Li([12]crown-4)2] [1,3-P2C3tBu3] (6, Scheme 1) based on the comparison of the NMR spectroscopic data with related compounds.18,20–29 The major product 1 is characterized by two doublets of doublets at 224.0 ppm and 316.2 ppm (1JAX = −463.8 Hz, 1JA′X = −465.5 Hz, 2JAA′ = 4.3 Hz), while 6 gives rise to a singlet at 196.0 ppm (see Fig. S6, ESI†). Subsequent investigations using heavier alkali metals (Na–Cs) gave similar mixtures of the tri- and diphospholide (Scheme 1).
:1 ratio (see Fig. S1, ESI†). After the addition of [12]crown-4, these species were identified as [Li([12]crown-4)2][1,2,3-P3C2tBu2] (1) and [Li([12]crown-4)2] [1,3-P2C3tBu3] (6, Scheme 1) based on the comparison of the NMR spectroscopic data with related compounds.18,20–29 The major product 1 is characterized by two doublets of doublets at 224.0 ppm and 316.2 ppm (1JAX = −463.8 Hz, 1JA′X = −465.5 Hz, 2JAA′ = 4.3 Hz), while 6 gives rise to a singlet at 196.0 ppm (see Fig. S6, ESI†). Subsequent investigations using heavier alkali metals (Na–Cs) gave similar mixtures of the tri- and diphospholide (Scheme 1).
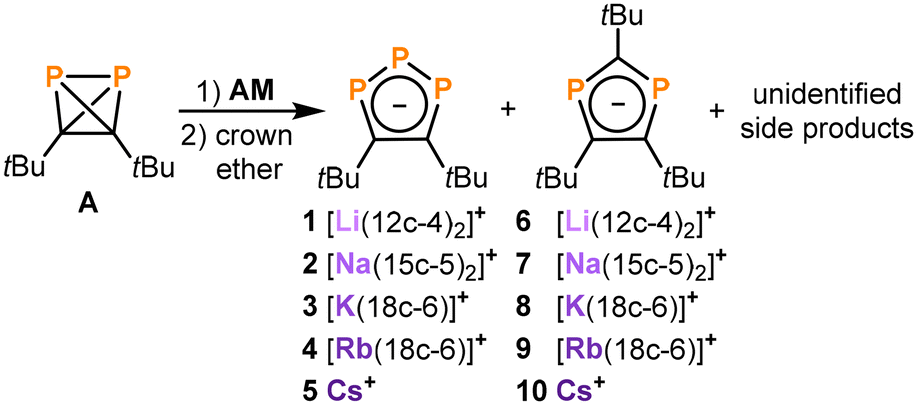 | ||
| Scheme 1 Reduction of di-tert-butyldiphosphatetrahedrane (A) yielding the triphospholides 1–5 as the major products next to the diphospholides 6–10 and other unidentified side products (AM = Li, Na, K, Rb, Cs, 12c-4 = [12]crown-4, 15c-5 = [15]crown-5, 18c-6 = [18]crown-6). Conditions: THF, −80 °C → r.t., over night. | ||
Salts 1–10 clearly result from the initial reduction of A, followed by rearrangements that involve the exchange of P and CtBu units.§ A similar process has been reported in the literature for the related reduction of tBuCP, but this yields mixtures of 1,2,4-triphospholides and 1,3-diphospholides.18 1,2,4-Triphospholides have been studied far more intensely than their 1,2,3-isomers due to the easier synthetic accessibility. The first 1,2,3-triphospholide salt was reported by Baudler and co-workers in 1990 as a side product in the reaction of P4 with sodium in diglyme.16 Since then several other 1,2,3-triphospholides have been reported, which contain aryl, alkyl, ester groups or hydrogen atoms as substituents.20–29
It is interesting to note that the ratio of di- and triphospholide in this reaction mixture does not reflect the ratio of carbon and phosphorus atoms in the starting material A. Thus, several unidentified side products were detected in the 1H NMR spectra of the reaction mixtures, among them a singlet at 1.15 ppm which likely can be attributed to the remaining tBuC fragments of this reaction (see Fig. S11, ESI†). The similar solubility of the di- and triphospholide salts hampers their clean isolation. Gratifingly, salts 1 and 3 were isolated as crystalline materials in varying yield of up to 5% and 32%, respectively, whereas attempts to cleanly isolate 2, 4 and 5 gave mixtures featuring the di- and triphospholide (see the ESI† for further details).
The molecular structures of the di- and triphospholide salts are corroborated by single-crystal X-ray diffraction (sc-XRD) studies on crystals of 1, 3, 4 and 9 obtained from n-hexane/THF or n-hexane/toluene. The molecular structure of potassium salt 3 is shown as an example in Fig. 2a. The similar structures of the lithium and rubidium triphospholides 1 and 4 and the structure of the rubidium diphospholide 9 are shown in the ESI.† The salient feature of the structures of 1, 3 and 4 are the planar five-membered P3C2 rings which are consistent with an aromatic 6 π-electron ring system with bond lengths in between typical single and double bonds (e.g.3: C–C (1.420(3) Å), C–P (1.780(2) Å), P–P (2.0740(8) Å and 2.0777(8) Å)). This is well in line with related triphospholide structures.16,20,23,25,26,29,32
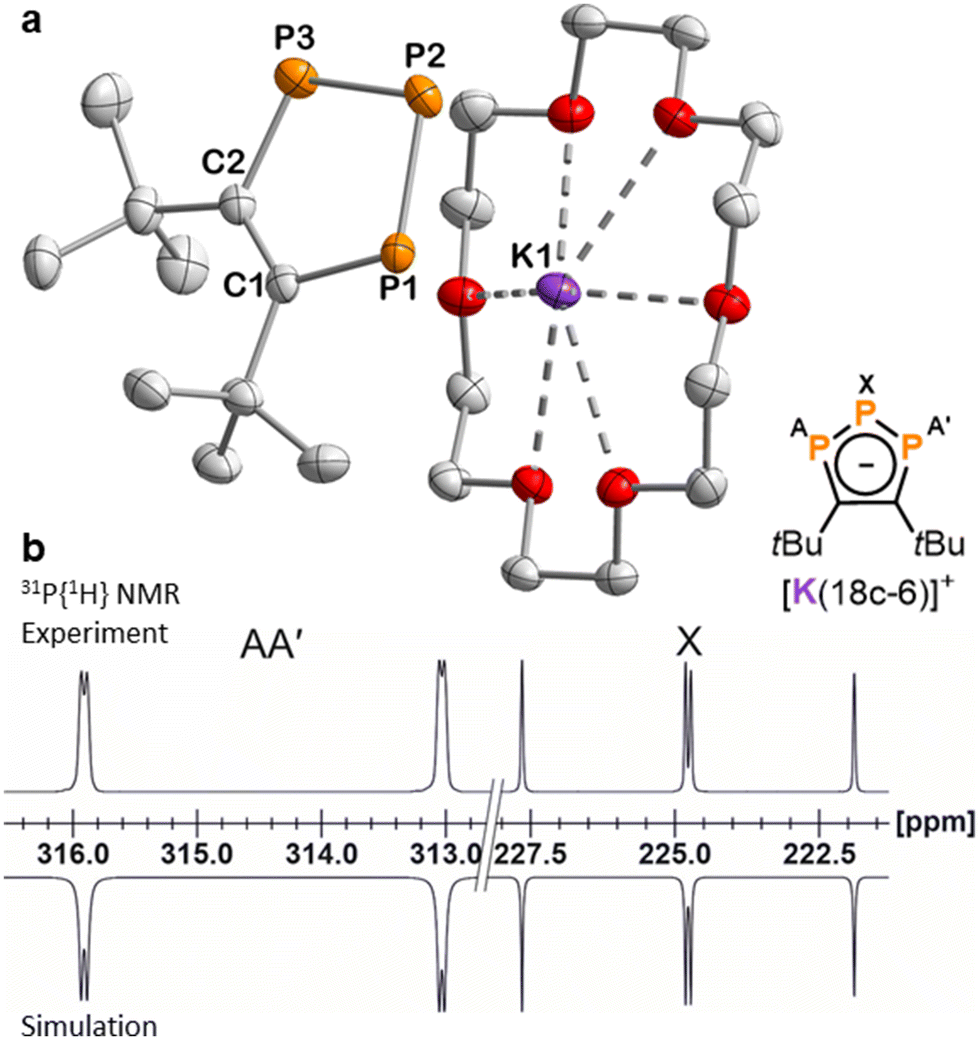 | ||
| Fig. 2 (a) Solid-state molecular structure of 3. Displacement ellipsoids are drawn at the 50% probability level; H atoms have been omitted for clarity. Selected bond lengths [Å] and angles [°]: P1–P2 2.0777(8), P2–P3 2.0740(8), P1–C1 1.780(2), P3–C2 1.780(2), C1–C2 1.420(3), P1–P2–P3 97.24(3), C1–P1–P2 102.86(8), C2–P3–P2 103.02(7), C2–C1–P1 118.52(16), C1–C2–P3 118.35(15). (b) Section of the 31P{1H} NMR spectrum of 3 with nuclei assigned to an AA′X spin system; experimental (top); simulation (bottom): δ(PX) = 224.8 ppm, δ(PAA′) = 314.4 ppm, 1JAX = −465.6 Hz, 1JA′X = −467.5 Hz, 2JAA′ = 4.3 Hz. The 31P{1H} NMR spectrum of 1 is similar (see the ESI† for further details). | ||
In contrast to other symmetrically substituted triphospholide salts,16,20,25,27,29 the 31P{1H} NMR spectra of 1–5 do not display an A2B spin system but an AA′X spin system. For example, the simulation of the spin system for 3 by an iterative fitting procedure gave the PP coupling constants: 1JAX = 465.6 Hz, 1JA′X = 467.5 Hz and 2JAA′ = 4.3 Hz (Fig. 2b). The magnetic inequivalence of the phosphorus atoms PA and PA′ is likely due to the proximity of the tBu groups which cannot rotate independently and thus differentiates the two otherwise symmetrical halves of the 1,2,3-triphospholide anion (see Fig. S44, ESI†). The 31P{1H} NMR spectrum of 3 did not change significantly at elevated temperature (25, 40 and 60 °C), which may indicate that the tBu-groups rotate in a concerted fashion (see Fig. S19, ESI†). The 1H NMR spectrum of 3 gives two partly overlapping singlets for the tBu groups at 1.75 ppm, similarly attributed to the non-equivalence of the 18 hydrogen atoms due to the hindered rotation of the tBu groups. Another singlet at 3.57 ppm arises from the hydrogen atoms of the crown ether molecule. The signal of the carbon nuclei in the five-membered P3C2 core of 3 appears as a multiplet centred at 184.4 ppm in the 13C{1H} NMR spectrum, due to coupling to the phosphorus atoms. The multiplet detected at 38.9 ppm is assigned to the magnetically inequivalent methyl groups, while quaternary carbon atoms of the tBu groups give rise to a multiplet centred at 40.5 ppm. Similar 13C NMR data were recorded for 1.† The UV-Vis absorption spectra of 1 and 3 dissolved in THF display two absorption bands in the UV region at 270 nm and 345 nm. The latter absorption tails into the visible region, causing the orange colour of 1 and 3 (see Fig. S39 and S40, ESI†).
Metal coordination of triphospholide 3: synthesis of iron and ruthenium complexes
Even though several 1,2,3-triphospholide salts were reported, the number of isolated 1,2,3-triphospholyl complexes is limited.22–25,29,33 To test the utility of the newly synthesized triphospholide salts, we investigated the synthesis of triphosphaferrocenes using 3 (which was selected among 1–5 due to the superior isolated yield).29,33 Dropwise addition of 3 to [Cp*FeCl(tmeda)] (tmeda = tetramethylethylene-1,2-diamine) afforded the complex [Cp*Fe(η5-1,2,3-P3C2tBu2)] (11, Scheme 2a), which was identified by its AA′X spin system with resonances at 116.3 ppm and at −17.8 ppm in the 31P{1H} NMR spectrum in C6D6. 11 was isolated as a purple powder in 54% yield after work-up. Similarly, the reaction of 3 with [Cp*RuCl(cod)] (cod = cycloocta-1,5-diene) led to the isolation of orange, crystalline [Cp*Ru(η5-1,2,3-P3C2tBu2)] (12) in 32% yield (Scheme 2b). Complex 12 can be identified by two multiplets (AA′X spin system) at 87.5 ppm and at −39.1 ppm in the 31P{1H} NMR spectrum.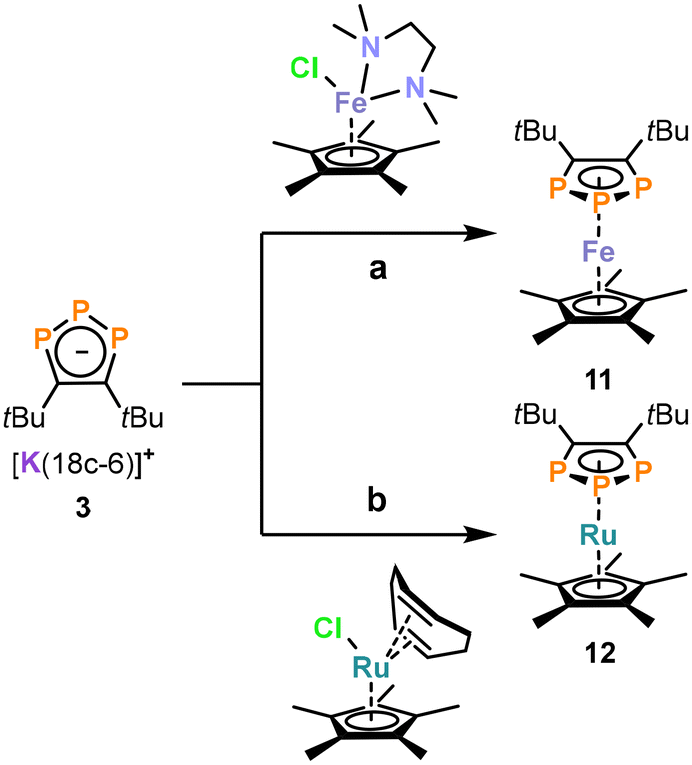 | ||
| Scheme 2 Synthesis of 11 and 12. Reagents and conditions: (a) THF, −30 °C, 1.5 h; (b) THF, −30 °C, 1.5 h. | ||
The structural and spectroscopic properties of 11 and 12 are broadly similar to related 1,2,3-triphosphaferrocenes.29,33b–d To our knowledge, only one 1,2,3-triphospholide ruthenium complex [K(2,2,2-crypt)][Ru(η5-P3C2H2){CH3C(CH2)2}2] has been reported so far, which has not been crystallographically characterised.23 In contrast to the previously reported examples,29,33b,c the (phospha-)cyclopentadienyl rings in the structures of 11 and 12 adopt a staggered conformation (Fig. 3), probably due to steric interactions between the Me groups of the Cp* ligands and the tert-butyl groups of the 1,2,3-triphospholyl ligands. It is noteworthy that the planes of the five membered ring ligands are slightly tilted with a plane-to-plane angle of 9°. In both complexes 11 and 12 the bonds in the five-membered ring of the triphospholide ligand are slightly elongated compared to the triphospholide salt 3 (e.g.3: P1–P2 = 2.0777(8) Å, C1–C2 = 1.420(3) Å, 11: P1–P2 = 2.1056(7) Å, C1–C2 = 1.437(3) Å).
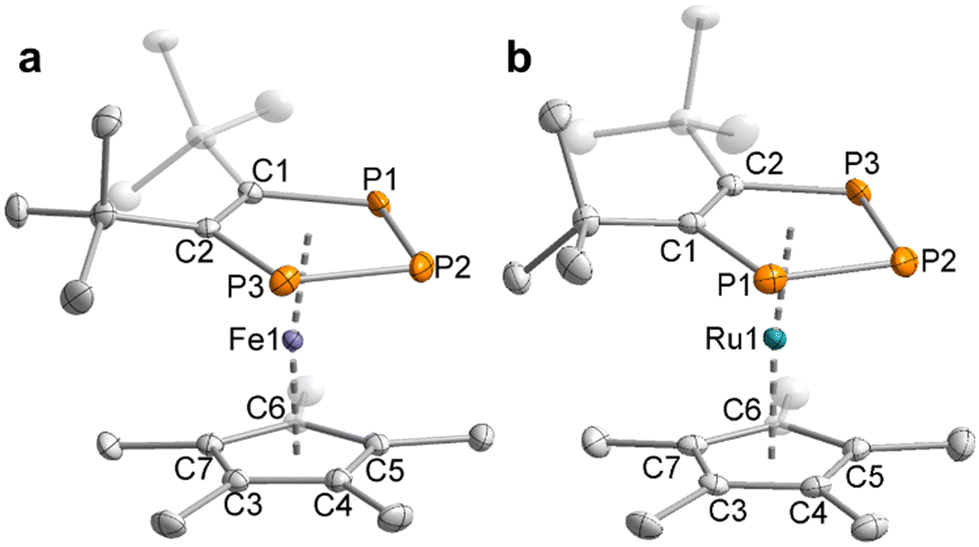 | ||
| Fig. 3 Solid-state molecular structures of (a) 11 and (b) 12. Displacement ellipsoids are drawn at the 50% probability level; H atoms have been omitted for clarity. Selected bond lengths [Å] and angles [°] for 11: P1–P2 2.1054(7), P2–P3 2.1056(7), P1–C1 1.7999(19), P3–C2 1.8104(19), C1–C2 1.437(3), Fe1–(P3C2)centroid 1.6321(6), Fe1–Cp*centroid 1.7116(9), P1–P2–P3 98.29(3), C1–P1–P2 101.85(6), C2–P3–P2 101.90(6), C1–C2–P3 118.53(13), C2–C1–P1 119.36(14), Cp*centroid–Fe1–(P3C2)centroid 172.02(4). 12: P1–P2 2.1115(8), P2–P3 2.1093(8), P1–C1 1.808(2), P3–C2 1.814(2), C1–C2 1.435(3), Ru1–(P3C2)centroid 1.7595(6), Ru1–Cp*centroid 1.8447(9), P1–P2–P3 98.25(3), C1–P1–P2 101.78(7), C2–P3–P2 101.95(7), C1–C2–P3 118.66(15), C2–C1–P1 119.33(15), Cp*centroid–Ru1–(P3C2)centroid 173.19(4). | ||
Simulation of the 31P{1H} NMR spectra revealed an AA′X spin system for 11 and 12 (Fig. 4). As reported for related complexes,23,29,33 the 31P and 13C NMR resonances are shifted to lower frequency. The 1JPP coupling constants (11: 1JAX = 397.1 Hz, 12: 1JAX = 393.4 Hz) are smaller compared to the signals of anionic 3 (1JAX = 465.6 Hz, vide supra). The UV-Vis absorption spectrum of 11 displays two absorption bands in the UV region at 240 nm and 280 nm as well as two in the visible region at 470 nm and 575 nm (see Fig. S41 and S42, ESI†).
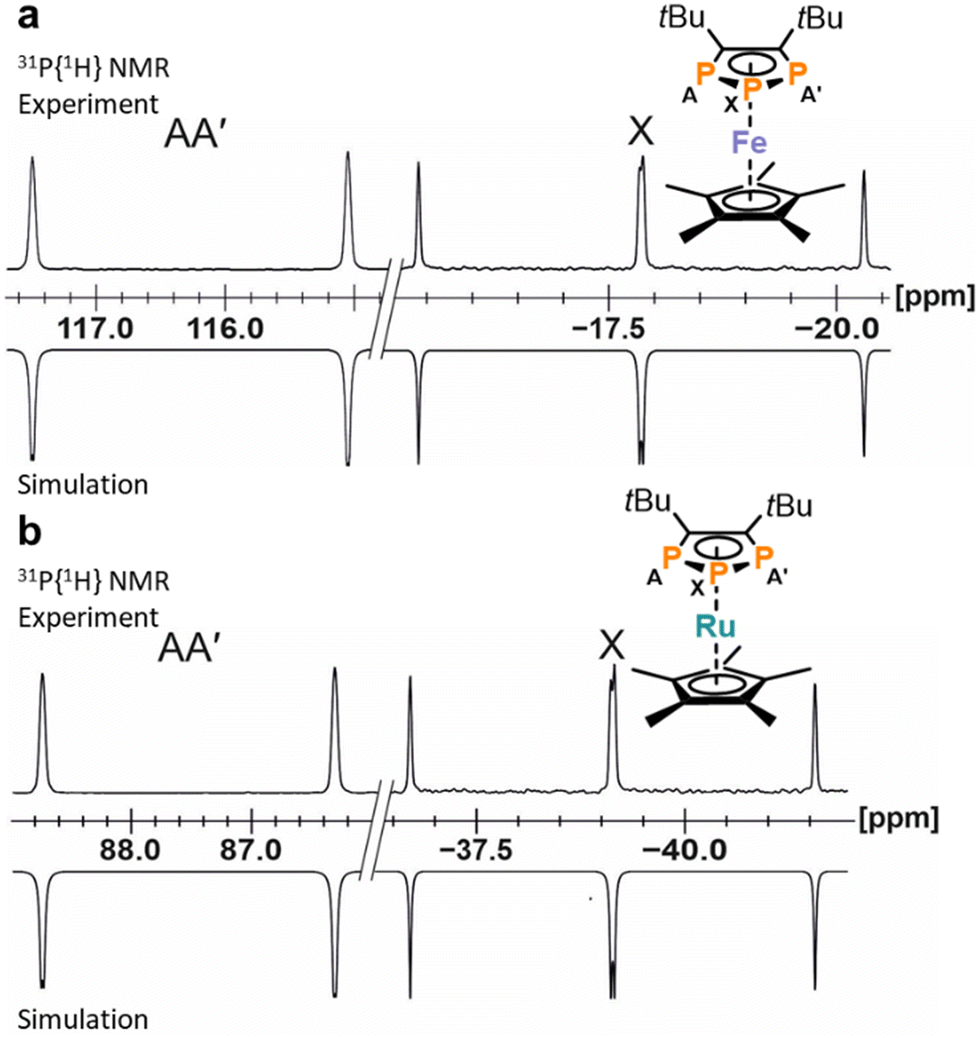 | ||
| Fig. 4 (a) Section of the 31P{1H} NMR spectrum of 11 with nuclei assigned to an AA′X spin system; experimental (top); simulation (bottom): δ(PX) = −17.8 ppm, δ(PAA′) = 116.3 ppm, 1JAX = −397.1 Hz, 1JA′X = −396.2 Hz, 2JAA = 3.4 Hz. (b) Section of the 31P{1H} NMR spectrum of 12 with nuclei assigned to an AA′X spin system; experimental (top); simulation (bottom): δ(PX) = −39.1 ppm, δ(PAA′) = 87.5 ppm, 1JAX = −393.4 Hz, 1JA′X = −393.9 Hz. | ||
Oxidation of 3 with Brookhart's acid: synthesis of hexaphosphane 13
Further reactivity studies of 3 with metal salts (e.g. [(Ph3P)2NiCl2], [Cp3Ni2]BF4, [(PPh3)CuCl]), oxidizing agents (e.g. [Cp2Fe]PF6), and organic electrophiles repeatedly revealed a set of three distinct multiplets at −63.8 ppm, 15.0 ppm and 64.2 ppm (1:1:1 integral ratio) in the 31P{1H} NMR spectra, which are assigned to a new product 13. While this pattern was accompanied by further intractable products in most cases, reactions of 3 with [Cp2Fe][BAr4F] and [H(Et2O)2BAr4F] (Brookhart's acid, BAr4F = B{C6H3(CF3)2}4) gave 13 in a selective fashion (see Fig. S31 and S37, ESI†). Isolation of crystalline 13 was possible from the reaction of 3 with [H(Et2O)2BAr4F] by extraction of the crude reaction product with n-hexane and subsequent recrystallisation from n-pentane at −30 °C (22% yield, Scheme 3).
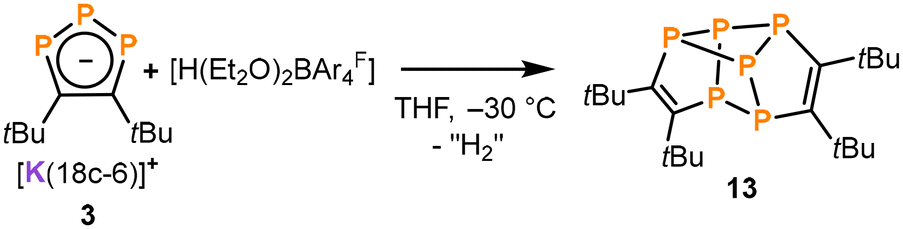 | ||
| Scheme 3 Synthesis of 13. | ||
The determination of the molecular structure of 13 by sc-XRD revealed that this complex arises from the dimerisation of two 1,2,3-triphospholyl units, resulting in a tetracyclic structure (Fig. 5). The polycyclic framework of 13 represents a phosphorus-containing isomer of the hydrocarbon hypostrophene (C10H10) reported by Pettit and co-workers.34 The P6 core features a double-envelope arrangement with covalent P–P and P–C single bonds, while the adjacent C atoms each form a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond (C1–C2 1.364(3) Å, C3–C4 1.366(3) Å).32 A similar double-envelope P6 moiety has been reported for [Cp′′2Th(μ,η3,η3-P6)ThCp′′2] (Cp′′ = η5-1,3-tBu2C5H3).35 A related P6 unit is also found in Jutzi's P6(C5Me5)2, which contains an additional P–P bond.36
C double bond (C1–C2 1.364(3) Å, C3–C4 1.366(3) Å).32 A similar double-envelope P6 moiety has been reported for [Cp′′2Th(μ,η3,η3-P6)ThCp′′2] (Cp′′ = η5-1,3-tBu2C5H3).35 A related P6 unit is also found in Jutzi's P6(C5Me5)2, which contains an additional P–P bond.36
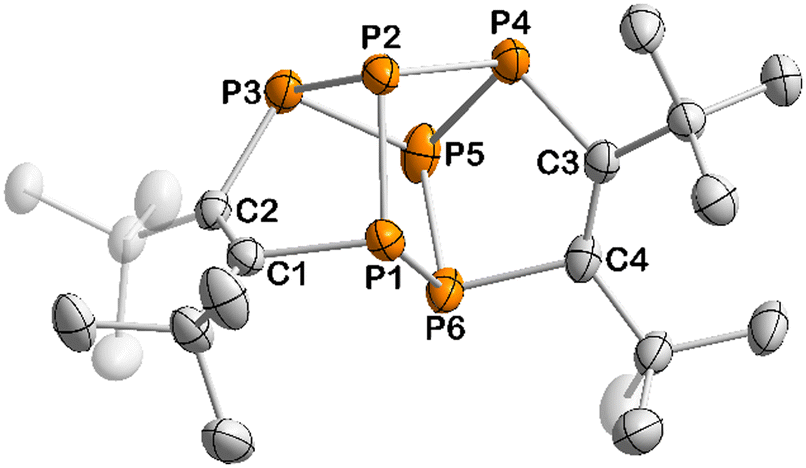 | ||
| Fig. 5 Solid-state molecular structure of 13. Displacement ellipsoids are drawn at the 50% probability level; H atoms have been omitted for clarity. Selected bond lengths [Å] and angles [°]: P1–P2 2.1709(8), P1–P6 2.2380(8), P2–P3 2.2082(8), P2–P4 2.2571(8), P3–P5 2.2540(8), P4–P5 2.2054(9), P5–P6 2.1736(9), P1–C1 1.875(2), P3–C2 1.858(2), P4–C3 1.851(2), P6–C4 1.870(2), C1–C2 1.364(3), C3–C4 1.366(3), P1–P2–P3 90.50(3), C1–P1–P2 93.58(7), C2–P3–P2 99.04(7), C1–C2–P3 115.47(15), C2–C1–P1 116.22(15), P4–P5–P6 90.47(3), C3–P4–P5 98.78(7), C4–P6–P5 93.26(7), C3–C4–P6 115.92(15), C4–C3–P4 115.81(16), P1–P2–P4 87.60(3), P3–P2–P4 83.93(3), P4–P5–P3 84.07(3), P6–P5–P3 97.62(3). | ||
The NMR spectroscopic data of 13 are fully consistent with the molecular structure, revealing two 1H NMR singlets for the chemically inequivalent tBu groups at 1.08 and 1.53 ppm and deshielded 13C nuclei in the five-membered rings (165.0 ppm and 170.5 ppm) consistent with the CC double bonds. An AA′MM′XX′ spin system is observed in the 31P{1H} NMR spectrum (Fig. 6), confirming the presence of a covalently connected P6 moiety. Note that [Cp′′2Th(μ,η3,η3-P6)ThCp′′2] gives rise to a similar AA′MM′Q2 spin system.35
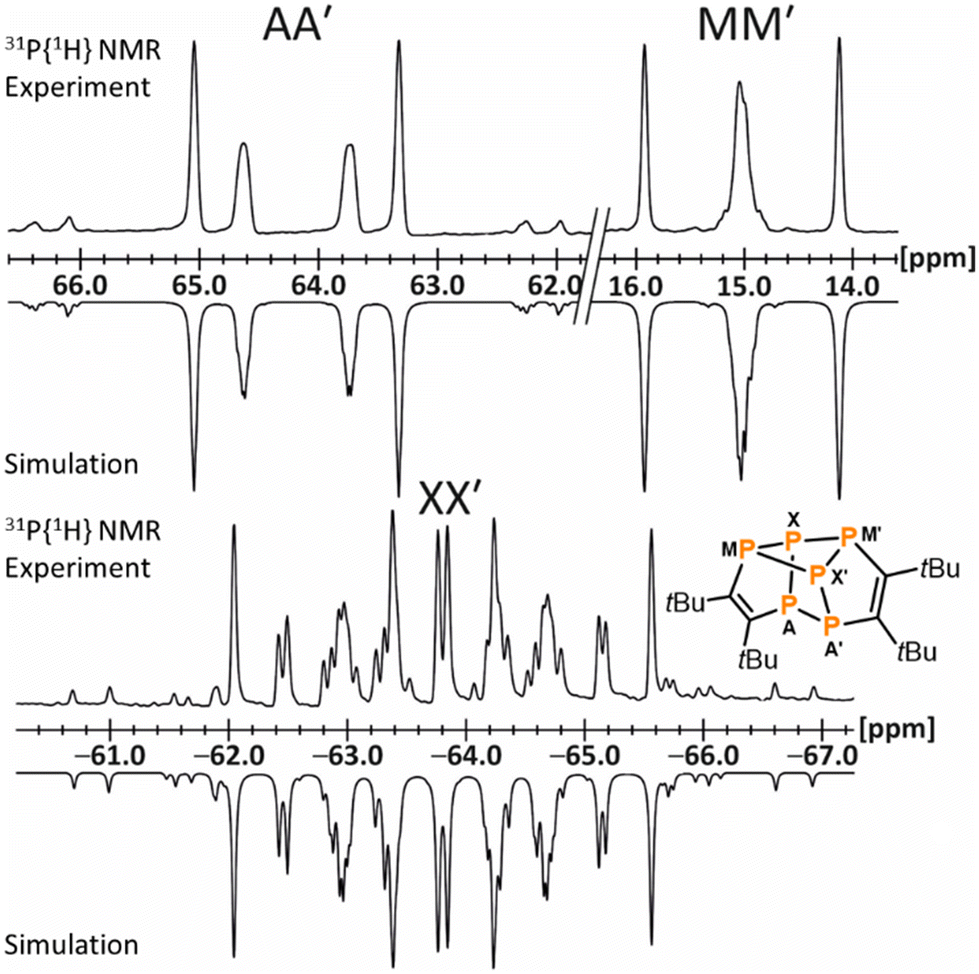 | ||
| Fig. 6 Sections of the 31P{1H} NMR spectrum of 13 with nuclei assigned to an AA′MM′XX′ spin system; experimental (top); simulation (bottom): δ(PAA′) = 64.2 ppm, δ(PMM′) = 15.0 ppm, δ(PXX′) = −63.8 ppm, 1JAA′ = −261.9 Hz, 1JAX = 1JA′X′ = −293.9 Hz, 1JM′X = 1JMX′ = −134.2 Hz, 2JAM = 2JA′M′ = −3.2 Hz, 2JA′M = 2JAM′ = 9.9 Hz, 2JMM′ = 24.9 Hz, 2JA′X = 2JAX′ = 15.3 Hz, 1JMX = 1JM′X′ = −157.7 Hz, 2JXX′ = 29.5 Hz. | ||
The possible mechanism of formation of 13 is shown in Scheme 4. We presume that [H(Et2O)2BAr4F] acts as an oxidizing agent towards 3, forming the radical (1,2,3-P3C2tBu2)˙ (3˙) and dihydrogen (H2) as a by-product.¶ Subsequently, the presumed intermediate 3˙ dimerizes under P–P bond formation, which is followed by a [2 + 2] cycloaddition to generate 13 (Scheme 4). The proposed mechanism is corroborated by DFT calculations (ωB97XD/def2-TZVPPD level of theory) on the radical 3˙, which show that the calculated spin density is mainly located on the terminal P atoms (P1/P3) of the P3 chain and to a lesser extent on the central P atom (P2).† An alternative pathway might be the protonation of 3 to form (1,2,3-P3C2tBu2H) (3-H) that would be prone to cycloaddition, followed by dihydrogen elimination (Scheme S1, ESI†).|| However, the pathway proposed in Scheme 4 is considered more likely given that 13 from 3 is also formed with aprotic oxidizing agents such as [Cp2Fe]PF6.†
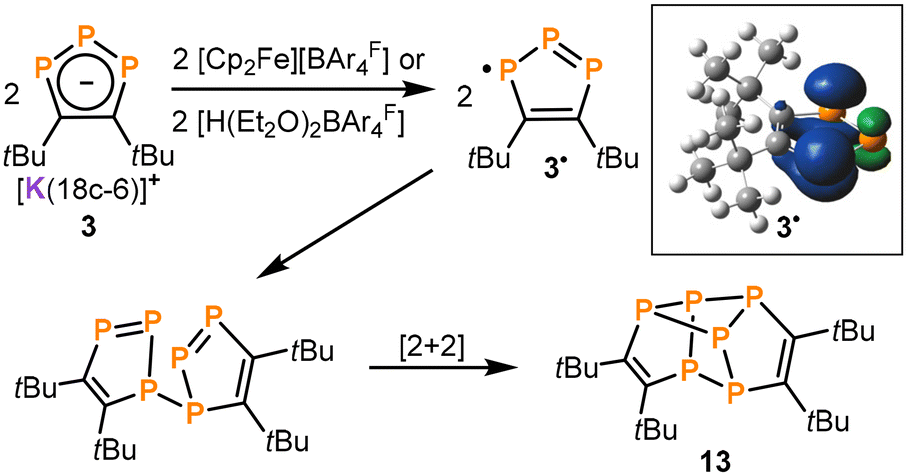 | ||
| Scheme 4 Proposed mechanism for the formation of 13 by oxidation of 3; the inset shows the calculated spin density distribution of 3˙ drawn at a surface isovalue at 0.05. | ||
Conclusions
The reduction of di-tert-butyldiphosphatetrahedrane (A) offers a new route to 1,2,3-triphospholide salts 1–5, which are obtained as mixtures with 1,3-diphospholides 6–10. This contrasts with the direct reduction of tBuCP with alkali metals which yields the related 1,2,4-triphospholides in addition to the 1,3-isomers. These findings echo the results of earlier investigations where A and tBuCP have formed isomeric reaction products.12 The synthesis of [Cp*M(η5-1,2,3-P3C2tBu2)] (11, M = Fe; 12, M = Ru) from the isolated potassium salt 3 complements earlier studies,29,33 demonstrating the potential of 3 for transition metal coordination. Furthermore, reactions of 3 with [Cp2Fe][BAr4F] and [H(Et2O)2BAr4F] resulted in oxidation and dimerisation, forming the hexaphosphane tBu4C4P6 (13). This compound has a remarkable molecular structure, which is analogous to a hypostrophene (C10H10) isomer. These results once more illustrate the potential utility of diphosphatetrahedranes as building blocks in organophosphorus chemistry. Further investigations into the reduction and oxidation of diphosphatetrahedrane A are in hand.Author contributions
MKU: investigation – experimental study and DFT calculations, writing – original draft. GH: conceptualisation, writing – review and editing. GB: investigation – DFT calculations, writing – review and editing. RW: conceptualisation, supervision, funding acquisition, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Marion Till and Lisa M. Schneider for experimental assistance and the group of Nikolaus Korber for the generous donation of rubidium and caesium. Financial support by the Deutsche Forschungsgemeinschaft (WO1496/10-1), the Fonds der Chemischen Industrie (Kekulé Fellowship for MKU), and the Studienstiftung des Deutschen Volkes (PhD Scholarship for MKU) is gratefully acknowledged. MKU is a member of the Elite Network of Bavaria Doctoral College: “IDK Chemical Catalysis with Photonic or Electric Energy Input.”References
- (a) K. B. Dillon, F. Mathey and J. F. Nixon, Phosphorus: The Carbon Copy, From Organophosphorus to Phospha-organic Chemistry, Wiley, 1998 Search PubMed; (b) F. Mathey, Angew. Chem., Int. Ed., 2003, 42, 1578–1604 CrossRef CAS PubMed.
- W. W. Schoeller and T. Busch, Angew. Chem., Int. Ed. Engl., 1993, 32, 617–619 CrossRef.
- G. Hierlmeier, P. Coburger, M. Bodensteiner and R. Wolf, Angew. Chem., Int. Ed., 2019, 58, 16918–16922 CrossRef CAS PubMed.
- M.-L. Y. Riu, R. L. Jones, W. J. Transue, P. Müller and C. C. Cummins, Sci. Adv., 2020, 6, eaaz3168 CrossRef CAS PubMed.
- M.-L. Y. Riu, M. Ye and C. C. Cummins, J. Am. Chem. Soc., 2021, 143, 16354–16357 CrossRef CAS PubMed.
- (a) G. Maier, S. Pfriem, U. Schäfer and R. Matusch, Angew. Chem., Int. Ed. Engl., 1978, 17, 520–521 CrossRef; (b) G. Maier, Angew. Chem., Int. Ed. Engl., 1988, 27, 309–332 CrossRef.
- Selected review articles: (a) M. Caporali, L. Gonsalvi, A. Rossin and M. Peruzzini, Chem. Rev., 2010, 110, 4178–4235 CrossRef CAS PubMed; (b) B. M. Cossairt, N. A. Piro and C. C. Cummins, Chem. Rev., 2010, 110, 4164–4177 CrossRef CAS PubMed; (c) M. Scheer, G. Balázs and A. Seitz, Chem. Rev., 2010, 110, 4236–4256 CrossRef CAS PubMed; (d) L. Giusti, V. R. Landaeta, M. Vanni, J. A. Kelly, R. Wolf and M. Caporali, Coord. Chem. Rev., 2021, 441, 213927 CrossRef CAS; (e) C. M. Hoidn, D. J. Scott and R. Wolf, Chem. – Eur. J., 2021, 27, 1886–1902 CrossRef CAS PubMed.
- G. Hierlmeier, M. K. Uttendorfer and R. Wolf, Chem. Commun., 2021, 57, 2356–2359 RSC.
- M.-L. Y. Riu, A. K. Eckhardt and C. C. Cummins, J. Am. Chem. Soc., 2022, 144, 7578–7582 CrossRef CAS PubMed.
- M.-L. Y. Riu, A. K. Eckhardt and C. C. Cummins, J. Am. Chem. Soc., 2021, 143, 13005–13009 CrossRef CAS PubMed.
- G. Hierlmeier, R. J. Kutta, P. Coburger, H.-G. Stammler, J. Schwabedissen, N. W. Mitzel, M. Dimitrova, R. J. F. Berger, P. Nürnberger and R. Wolf, Chem. Sci., 2024, 15, 5596–5603 RSC.
- G. Hierlmeier, P. Coburger, D. J. Scott, T. M. Maier, S. Pelties, R. Wolf, D. M. Pividori, K. Meyer, N. P. van Leest and B. de Bruin, Chem. – Eur. J., 2021, 27, 14936–14946 CrossRef CAS PubMed.
- G. Hierlmeier and R. Wolf, Angew. Chem., Int. Ed., 2021, 60, 6435–6440 CrossRef CAS PubMed.
- M. K. Uttendorfer, G. Hierlmeier and R. Wolf, Z. Anorg. Allg. Chem., 2022, 648, e202200124 CrossRef CAS.
- Reduction chemistry of P4: (a) M. Baudler, D. Düster and D. Ouzounis, Z. Anorg. Allg. Chem., 1987, 544, 87–94 CrossRef CAS; (b) M. Baudler, S. Akpapoglou, D. Ouzounis, F. Wasgestian, B. Meinigke, H. Budzikiewicz and H. Münster, Angew. Chem., Int. Ed. Engl., 1988, 27, 280–281 CrossRef; (c) V. A. Milyukov, A. V. Kataev, O. G. Sinyashin and E. Hey-Hawkins, Russ. Chem. Bull., 2006, 55, 1297–1299 CrossRef CAS.
- M. Baudler and J. Hahn, Z. Naturforsch., B: Chem. Sci., 1990, 45, 1139–1142 CrossRef CAS.
- Reviews on phosphaalkyne chemistry: (a) M. Regitz and O. J. Scherer, Multiple Bonds and Low Coordination Phosphorus Chemistry, Thieme, Stuttgart, 1990 Search PubMed; (b) J. F. Nixon, Coord. Chem. Rev., 1995, 145, 201 Search PubMed; (c) K. B. Dillon, F. Mathey and J. F. Nixon, Phosphorus: The Carbon Copy, Wiley, Chichester, 1998 Search PubMed; (d) F. Mathey, Angew. Chem., Int. Ed., 2003, 42, 1578–1604 CrossRef CAS PubMed; (e) A. Chirila, R. Wolf, J. C. Slootweg and K. Lammertsma, Coord. Chem. Rev., 2014, 270–271, 57–74 CrossRef CAS.
- Examples for the reduction of phosphaalkynes: (a) R. Bartsch, P. B. Hitchcock and J. F. Nixon, J. Chem. Soc., Chem. Commun., 1987, 1146–1148 RSC; (b) R. Bartsch and J. F. Nixon, Polyhedron, 1989, 8, 2407 CrossRef CAS; (c) R. Bartsch and J. F. Nixon, J. Organomet. Chem., 1991, 415, C15–C18 CrossRef CAS; (d) S. M. Mansell, M. Green, R. J. Kilby, M. Murray and C. A. Russell, C. R. Chim., 2010, 13, 1073–1081 CrossRef CAS.
- The Mg(I) dimer [(nacnac)Mg]2 (nacnac = HC[C(Me)N(2,6-iPr2C6H3)]2) reduces phosphaalkynes RCP to 1,3-diphosphacyclobutadienediides [(nacnac)Mg]2[1,3-P2C2R2]: D. W. N. Wilson, D. D. L. Jones, C. D. Smith, M. Mehta, C. Jones and J. M. Goicoechea, Dalton Trans., 2022, 51, 898–903 RSC.
- N. Maigrot, M. Sierra, C. Charrier and F. Mathey, Bull. Soc. Chim. Fr., 1994, 131, 397–399 CAS.
- C. P. Butts, M. Green, T. N. Hooper, R. J. Kilby, J. E. McGrady, D. A. Pantazis and C. A. Russell, Chem. Commun., 2008, 856–858 RSC.
- F. García, R. J. Less, V. Naseri, M. McPartlin, J. M. Rawson, M. Sancho Tomas and D. S. Wright, Chem. Commun., 2008, 859–861 RSC.
- R. S. P. Turbervill and J. M. Goicoechea, Chem. Commun., 2012, 48, 6100–6102 RSC.
- R. S. P. Turbervill and J. M. Goicoechea, Inorg. Chem., 2013, 52, 5527–5534 CrossRef CAS PubMed.
- R. S. P. Turbervill, A. R. Jupp, P. S. B. McCullough, D. Ergöçmen and J. M. Goicoechea, Organometallics, 2013, 32, 2234–2244 CrossRef CAS.
- A. M. Tondreau, Z. Benkő, J. R. Harmer and H. Grützmacher, Chem. Sci., 2014, 5, 1545–1554 RSC.
- I. A. Bezkishko, A. A. Zagidullin, A. V. Petrov, V. A. Miluykov, T. I. Burganov, S. A. Katsyuba and O. G. Sinyashin, J. Organomet. Chem., 2017, 844, 1–7 CrossRef CAS.
- D. Rottschäfer, S. Blomeyer, B. Neumann, H.-G. Stammler and R. S. Ghadwal, Chem. Sci., 2019, 10, 11078–11085 RSC.
- A. Petrov, L. Conrad, N. T. Coles, M. Weber, D. Andrae, A. Zagidullin, V. Miluykov and C. Müller, Chem. – Eur. J., 2022, 28, e202203056 CrossRef CAS PubMed.
- I. N. Brago and A. P. Tomilov, Sov. Electrochem., 1968, 4, 623–625 Search PubMed.
- B. Geissler, S. Barth, U. Bergsträsser, M. Slany, J. Durkin, P. B. Hitchcock, M. Hofmann, P. Binger, J. F. Nixon, P. von Ragué Schleyer and M. Regitz, Angew. Chem., Int. Ed. Engl., 1995, 34, 484–487 CrossRef CAS.
- (a) F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19 RSC; (b) D. E. C. Corbridge, Phosphorus, Chemistry, Biochemistry and Technology, CRC Press, 2013 Search PubMed.
- (a) S. Deng, C. Schwarzmaier, C. Eichhorn, O. Scherer, G. Wolmershäuser, M. Zabel and M. Scheer, Chem. Commun., 2008, 4064–4066 RSC; (b) O. J. Scherer, T. Hilt and G. Wolmershäuser, Angew. Chem., Int. Ed., 2000, 39, 1425–1427 CrossRef; (c) A. V. Petrov, A. A. Zagidullin, I. A. Bezkishko, M. N. Khrizanforov, K. V. Kholin, T. P. Gerasimova, K. A. Ivshin, R. P. Shekurov, S. A. Katsyuba, O. N. Kataeva, Y. H. Budnikova and V. A. Miluykov, Dalton Trans., 2020, 49, 17252–17262 RSC; (d) A. L. Zinnatullin, A. A. Zagidullin, L. I. Savostina, I. A. Bezkishko, A. V. Petrov, E. N. Dulov, R. R. Zairov, V. A. Miluykov and F. G. Vagizov, Organometallics, 2023, 42, 1538–1549 CrossRef CAS.
- R. Pettit, J. S. McKennis, L. Brener and J. S. Ward, J. Am. Chem. Soc., 1971, 93, 4957–4958 CrossRef CAS.
- O. J. Scherer, B. Werner, G. Heckmann and G. Wolmershäuser, Angew. Chem., Int. Ed. Engl., 1991, 30, 553–555 CrossRef.
- (a) P. Jutzi, R. Kroos, A. Müller and M. Penk, Angew. Chem., Int. Ed. Engl., 1989, 28, 600–601 CrossRef; (b) P. Jutzi, R. Kroos, A. Müller, H. Bogge and M. Penk, Chem. Ber., 1991, 124, 75–81 CrossRef CAS.
- R. B. Wexler, J. M. P. Martirez and A. M. Rappe, ACS Catal., 2017, 7, 7718–7725 CrossRef CAS.
- R. Bartsch, P. B. Hitchcock and J. F. Nixon, J. Chem. Soc., Chem. Commun., 1989, 15, 1046–1048 RSC.
- A. S. Ionkin, W. J. Marshall, B. M. Fish, A. A. Marchione, L. A. Howe, F. Davidson and C. N. McEwen, Eur. J. Inorg. Chem., 2008, 2386–2390 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2346026–2346032. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01067a |
| ‡ Present address: Institute of Inorganic Chemistry, University of Würzburg, 97074 Würzburg, Germany. |
| § Note that the ladderane (tBuCP)4 (i.e. the dimer of A) does not react with alkali metals under the conditions described in Scheme 1. |
| ¶ We could not unambiguously detect the formation of H2 by 1H NMR spectroscopy. Nevertheless, the well-documented ability of metal phosphides to catalyse hydrogen evolution reactions in acidic media supports the proposed pathway.37 |
| || Such a pathway has been proposed by Nixon and co-workers for the protonation of Li[1,2,4-P3C2tBu2] with an EtOH/MeCO2H mixture, leading to a tricyclic compound containing a P5 chain and an isolated P atom.38 Indeed, our DFT calculations for (1,2,3-P3C2tBu2)− show that P1 and P3 have a slightly higher proton affinity than C1, C2 and P2. In contrast, the carbon atoms of (1,2,4-P3C2tBu2)− have a higher proton affinity than the phosphorus atoms. The protonation of one P atom in (1,2,3-P3C2tBu2)− leads to a localized PP double bond which is prone to cycloaddition triggering the hydrogen elimination and formation of 13. Both the dimerisation of 3˙ to 13 and of protonated 3-H to 13 are exergonic with −59.1 kcal mol−1 and −23.9 kcal mol−1, respectively (see ESI†). A 1,2,4-triphospholide radical dimerisation has been reported by Ionkin and co-workers.39 |
| This journal is © The Royal Society of Chemistry 2024 |