
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly efficient grafting of hetero-complementary amidinium and carboxylate hydrogen-bonding/ionic pairs onto polymer surfaces†
Ana M.
Fernandes
 a,
Manuel C.
Martos-Maldonado
a,
Javier
Araujo-Morera
a,
Claudia
Solek
a and
David
González-Rodríguez
*ab
a,
Manuel C.
Martos-Maldonado
a,
Javier
Araujo-Morera
a,
Claudia
Solek
a and
David
González-Rodríguez
*ab
aNanostructured Molecular Systems and Materials Group, Departamento de Química Orgánica, Universidad Autónoma de Madrid, Madrid 28049, Spain. E-mail: david.gonzalez.rodriguez@uam.es
bInstitute for Advanced Research in Chemical Sciences (IAdChem), Universidad Autónoma de Madrid, Madrid 28049, Spain
First published on 11th January 2024
Abstract
We describe a grafting methodology, based on thiol-fluoroarene chemistry, to efficiently incorporate complementary hydrogen-bonding carboxylate and amidinium groups into polymer backbones. The process was optimized both in solution and on the surface of processed films, with the aim to produce materials showing hetero-complementary adhesion.
Self-assembly and molecular recognition phenomena are ubiquitous in natural systems and, in some cases, adhesion is the resulting property. Noncovalent interactions are central design elements in biological adhesion, where adaptive and reversible connectivity is required.1 Examples include cell and bacterial adhesion to surfaces, and a broad range of adhesive animal secretions found in mussels, tubeworms, etc. In a bioinspired fashion, the development of synthetic adhesive materials2 using the tools and concepts of supramolecular chemistry is becoming an excellent approach to target unprecedented properties that rely on the versatile, reversible, dynamic, and selective nature of non-covalent interactions.3
Among these, hydrogen (H)-bonding interactions established between residues that present multiple H-bond donor and acceptor atoms are particularly attractive due to their high directionality, tuneable strength, and ease of incorporation in macromolecules.4 These units can be designed to exhibit a self-complementary H-bonding pattern, so that the binding unit (A) can establish multiple interactions with itself, leading to dimers (A2) or oligomers (An), which can be exploited to introduce adhesive function.4–7 However, supramolecular chemistry can also bring new, very appealing properties to polymer-based adhesives through the concept of hetero-complementary binding of dissimilar molecular fragments attached to two different surfaces. In this case, the H-bonding motifs employed for establishing noncovalent contacts in the material (A and B) are designed to present complementary patterns, so that the A:B interaction becomes stronger than A:A or B:B interactions.
The use of hetero-complementary H-bonding fragments for adhesion purposes was introduced by the Long's group with nucleobase-containing polymers.8,9 While nucleobases can self-dimerize via H-bonding, the complementary Watson–Crick binding of nucleobases is usually stronger and brings an appealing design for molecular recognition-based adhesion. More recently, Zimmerman et al. increased the strength of intermolecular interactions through synthetic heterocycles that can form strong quadruple H-bonds.10,11 However, despite the concept of producing molecular materials that function as Velcro® is very appealing, the adhesion enhancements reached so far as a result of hetero-complementary supramolecular binding are not very high. Moreover, adhesion must withstand the presence of moisture on polymer surfaces, since water is an excellent competitor for H-bonding.12,13
Along the last decade, our group has been actively exploring diverse H-bonding units, mostly nucleobase derivatives,14,15 to build well-defined nanostructures16,17 and to introduce function to self-assembled systems and materials.18,19 Among these units, we recently put our focus on the carboxylate:amidinium pair (Fig. 1)20,21 as a suited candidate to potentially provide high hetero-/homo-adhesion ratio and low moisture sensitivity to polymer materials, since it offers a balanced combination of H-bonding and ionic interactions. On one hand, hetero-complementary H-bonds are reinforced by Coulombic attraction between the carboxylate anion and the amidinium cation, while homo-complementary interactions are weakened. On the other, this charge-assisted H-bonding pair, which is reminiscent of the biological carboxylate: guanidinium “salt bridge”,22 also interacts strongly in polar solvents like water,23 which should notably reduce moisture sensitivity at interfaces.
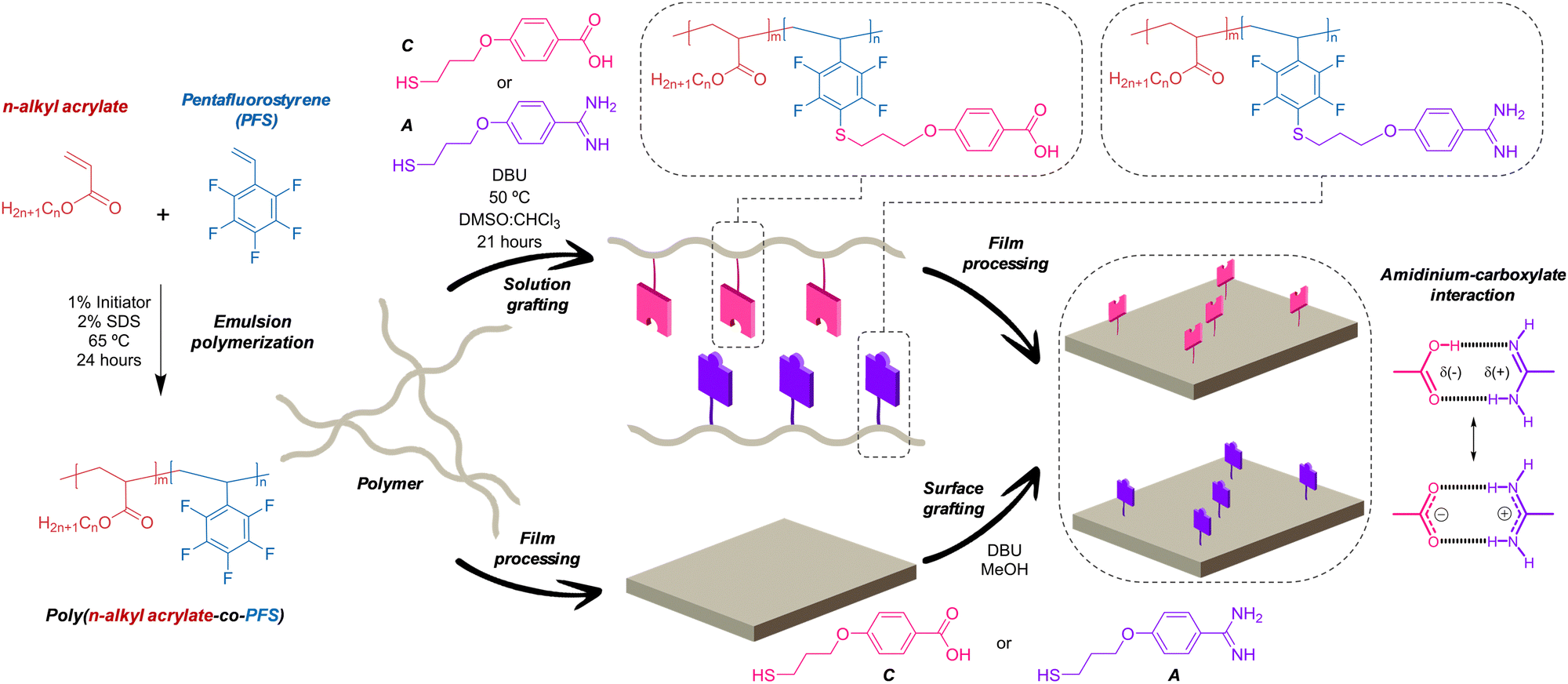 | ||
| Fig. 1 Schematic Illustration of the preparation of acrylic-PFS copolymers, grafting reaction and film processing. | ||
Here, we describe a method to efficiently incorporate these highly polar, ionizable, hetero-complementary units into polymer materials (Fig. 1). We optimized a grafting reaction between thiols containing the “sticky” carboxylate and amidinium functions, and pendant pentafluorophenyl groups present in poly(n-alkyl acrylate-co-pentafluorostyrene) copolymers. Interestingly, the procedure affords excellent grafting yields when carried out both with the dissolved polymer and directly onto polymer surfaces.
A wide range of comonomers was selected in order to obtain a good balance between the soft (acrylic moieties: methyl- (MA; n = 1), ethyl- (EA; n = 2), butyl- (BA; n = 4) or hexyl-acrylate (HA; n = 6)) and hard segments (pentafluorostyrene (PFS)), leading out to materials with good film forming ability. The final composition of the copolymers, the different conditions employed, and the most important experimental data obtained are presented in Table 1. The number-average molecular weight (Mn) and polydispersity index (PDI) ranged from 70 to 1078 kDa, and from 2 to 5.5, respectively. Lower Mn values were obtained in solution (entry 6) than in the batch conditions. The final co-monomer composition was determined by 1H-NMR (Fig. 2a and Fig. S1, ESI†), and it was found to be in good agreement with the initial monomer feeding ratio (Table 1). The increase in PFS co-monomer content also led to an enhancement of the IR band associated to C–F bond stretching (Fig. S2, ESI†). The data compiled in Table 1 also shows that the introduction of PFS and the nature of the acrylic moiety affects the glass transition temperature (Tg) of the copolymers in the expected order: Tg poly(HA-co-PFS) < Tg poly(BA-co-PFS) < Tg poly(EA-co-PFS) < Tg poly(MA-co-PFS).
| Entry | Monomer ratio | Initiatora | Final Conversion (%) | Final Compositionb (%) | M n kDa | PDI | T g °C | T g Ac °C | T g Cc °C |
|---|---|---|---|---|---|---|---|---|---|
| a 1% with respect to total monomer. b Copolymer composition was calculated by 1H NMR (see Table S1 and Fig. S1, ESI). c Data from copolymers grafted with A or C. All copolymers generated suitable thin films as long as the relative amount of PFS was kept low (< 20%). | |||||||||
| 1 | HA/PFS (90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10) :10) |
AIBN | 91 | 91:9 |
932 | 2.7 | −49 | −46 | −45 |
| 2 | BA/PFS (95:5) |
AIBN | 94 | 95:5 |
692 | 3.0 | −49 | −41 | −43 |
| 3 | BA/PFS (90:10) |
AIBN | 94 | 90:10 |
836 | 2.6 | −44 | −35 | −35 |
| 4 | BA/PFS (80:20) |
AIBN | 90 | 82:18 |
627 | 4.1 | −37 | −17 | −15 |
| 5 | BA/PFS (70:30) |
AIBN | 89 | 71:29 |
1078 | 5.5 | −33 | −8 | −10 |
| 6 | EA/PFS (90:10) |
AIBN | 82 | 91:9 |
110 | 7.4 | −13 | −6 | −13 |
| 7 | EA/PFS (80:20) |
AIBN | 79 | 81:19 |
297 | 3.3 | −10 | −3 | −1 |
| 8 | MA/PFS (95:5) |
KPS | 99 | 95:5 |
295 | 4.3 | −2 | 6 | 5 |
| 9 | MA/PFS (90:10) |
KPS | 98 | 89:11 |
556 | 4.1 | 4 | 18 | 19 |
| 10 | MA/PFS (80:20) |
KPS | 99 | 79:21 |
508 | 3.4 | 10 | 41 | 40 |
| 11 | MA/PFS (50:50) |
KPS | 98 | 52:48 |
416 | 3.7 | 35 | 51 | 52 |
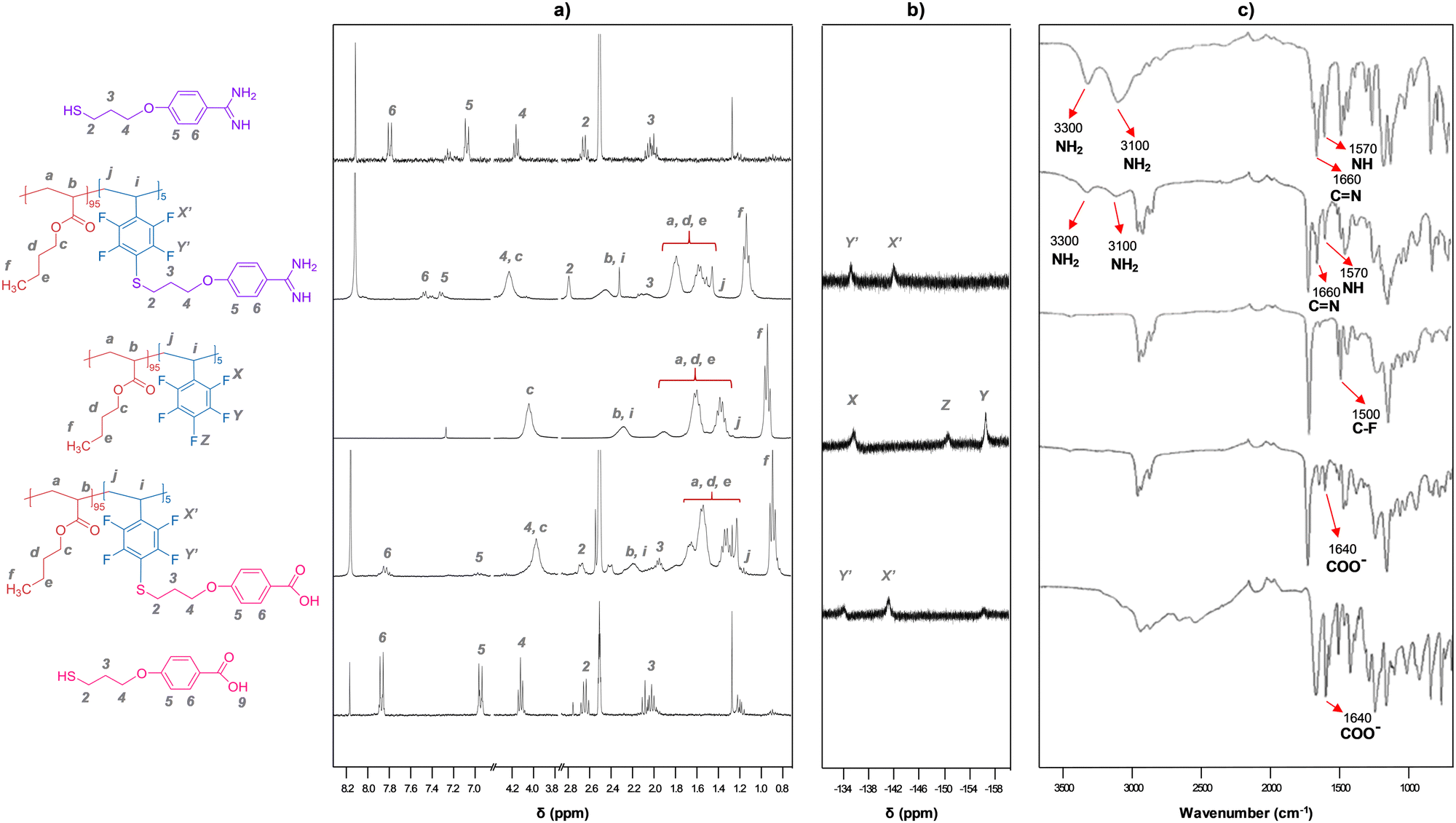 | ||
| Fig. 2 Representative (a) 1H NMR, (b) 19F NMR and (c) FT-IR spectra of the polymers grafted with A and C units, in comparison with the spectra of the precursor copolymers and the A and C thiols. NMR spectra were recorded in CDCl3:DMSO-d6 mixtures. | ||
Grafted copolymers were obtained by the addition of a small excess of a thiol that contains the amidine (A) or the carboxylic acid function (C) to the precursor copolymers in basic media (DBU), as schematically shown in Fig. 1 and detailed in the S.I. This aromatic nucleophilic substitution reaction between a thiolate and pentafluorophenyl-containing polymers, being selective for the C-F bond at the para-position,24 was optimized to proceed in a mild, high-yielding manner, as monitored using 1H NMR, 19F NMR and FT-IR spectroscopy. Fig. 2 shows representative spectra of the grafted polymers with A and C units compared to the pristine copolymers and the A and C thiols. Small chemical shifts were observed for the aromatic protons of the pendant amidinium and carboxylate groups attached to the polymers (Fig. 2a). Larger shifts were in contrast observed for the 19F nuclei of the perfluoroarene moiety (Fig. 2b and Fig. S3, ESI†). The signal corresponding to the 19F atom at the para-position (−151 ppm) disappears after complete grafting reaction, while the signals of the meta-fluorine atoms shift from −156 ppm to −134 ppm.25 The FT-IR spectra (Fig. 2c) show the expected bands associated with the functional groups of the acrylate-PFS copolymers, together with the characteristic bands of the amidinium group at ∼ 3300, 3100, 1660 and 1570 cm−1, or the carboxylate ∼ 1640 cm−1, as also observed in the A and C precursors, respectively.
Altogether, the different characterization techniques confirmed the successful incorporation of thiols A and C into the copolymer backbone. Moreover, an indirect indication that grafting occurs is the slight increase of the final Tg observed for the copolymers bearing A and C, as displayed in Table 1. Another evidence is the substantial change in solubility of the polymers, which affected their film-forming ability. While the copolymers grafted with the carboxylate function could still be solubilized under certain conditions, the amidinium-grafted copolymers were insoluble, or only partially soluble (depending on the acrylic composition), in all solvents tested and tended to form gels with polar (co)solvents. SEM images of cross sections of this material (Fig. S4, ESI†) illustrate a highly porous internal structure, which supports their performance as gels.
Due to these solubility problems and the associated loss of processability, we decided to generate the functionalized materials by grafting directly on the surface of films processed from the pristine poly(acrylate-PFS) material. This strategy has the advantage that most physicochemical properties of the bulk copolymer remain unaltered, so that processability is no longer a problem, and only the surface is modified with the hetero-complementary H-bonding/ionic units. Among the solvents tested that could dissolve the A and C thiols, but not the films, methanol was the best option. Surface grafting (Fig. 1) was therefore performed by adding the thiol and the base dissolved in methanol onto the polymer thin film, and letting stand for 2–3 days at room temperature, as explained in the S.I. (Scheme S2, ESI†). Thorough washing of the surface with abundant methanol afforded the final modified films. The copolymers containing MA were not suitable for surface grafting using this protocol.
Substantial functionalization of the thin films was evidenced in first place by some changes in their solubility. Both the film functionalized with A and C were insoluble in CHCl3, but a film treated under the same conditions in the absence of thiol (only methanol and DBU were added) was effortlessly dissolved in that solvent, just like the pristine precursor copolymers. The addition of a small amount of a polar cosolvent, like DMSO, led in contrast to complete solubilization in most cases. The 19F-NMR spectra of the re-solubilized samples showed the presence of new signals in the on-surface-grafted films, which were ascribed to the anchoring of the thiols onto the pentafluorophenyl units (Fig. S5, ESI†). The presence of multiple signals may be explained by partial and random modification of the pentafluorophenyl moieties, which leads to a complex mixture of functionalized and unmodified pentafluorophenyl moieties with different chemical environments. In addition, the 1H-NMR spectra of the functionalized films exhibited signals in the aromatic regions that were not present in the unmodified polymer, which corresponded to the aromatic moieties of the thiols (Fig. S6 and S7, ESI†).
In short, a grafting reaction between thiols and pendant pentafluorophenyl groups present in poly(n-alkyl acrylate-co-pentafluorostyrene) copolymers was optimized for both the dissolved polymers and for processed polymer surfaces, so as to introduce the hetero-complementary H-bonding carboxylate and amidinium groups. Although some preliminary ad hoc tests were made (see Fig. S8 and Video S1, ESI†), future work will be directed to characterize qualitatively and quantitatively the adhesion process between these hetero-complementarily functionalized polymers, so as to assess the impact of combining ionic and H-bonding recognition units.
Funding from MICINN (PDC2021-121487-I00) and the European Research Council (ERC-PoC-754795) is gratefully acknowledged.
Conflicts of interest
There are no conflicts to declare.References
- K. Autumn, Y. A. Liang, S. T. Hsieh, W. Zesch, W. P. Chan, T. W. Kenny, R. Fearing and R. J. Full, Nature, 2000, 405, 681 CrossRef CAS PubMed.
- K. M. Herbert, S. Schrettl, S. J. Rowan and C. Weder, Macromolecules, 2017, 50, 8845 CrossRef CAS.
- C. Heinzmann, C. Weder and L. M. de Espinosa, Chem. Soc. Rev., 2016, 45, 342 RSC.
- K. Yamauchi, J. R. Lizotte and T. E. Long, Macromolecules, 2003, 36, 1083 CrossRef CAS.
- P. Woodward, D. H. Merino, I. W. Hamley, A. T. Slark and W. Hayes, Austr. J. Chem., 2009, 62, 790 CrossRef CAS.
- G. M. L. van Gemert, J. W. Peeters, S. H. M. Söntiens, H. M. Janssen and A. W. Bosman, Macromol. Chem. Phys., 2012, 213, 234 CrossRef CAS.
- C. Heinzmann, C. Weder and L. de Espinosa, ACS Appl. Mater. Interfaces, 2014, 6, 4713 CrossRef CAS PubMed.
- K. Viswanathan, H. Ozhalici, C. L. Elkins, T. C. Ward and T. E. Long, Langmuir, 2006, 22, 1099 CrossRef CAS.
- S. Cheng, M. Zhang, N. Dixit, R. B. Moore and T. E. Long, Macromolecules, 2012, 45, 805 CrossRef CAS.
- C. A. Anderson, A. R. Jones, E. M. Briggs, E. J. Novitsky, D. W. Kuykendall, N. R. Sottos and S. C. Zimmerman, J. Am. Chem. Soc., 2013, 135, 7288 CrossRef CAS PubMed.
- Y. Zhang, C. A. Anderson and S. C. Zimmerman, Org. Lett., 2013, 15, 3506 CrossRef CAS PubMed.
- Y. Ahn, Y. Jang, N. Selvapalam, G. Yun and K. Kim, Angew. Chem., Int. Ed., 2013, 52, 3140 CrossRef CAS PubMed.
- O. Roling, L. Stricker, J. Voskuhl, S. Lamping and B. J. Ravoo, Chem. Commun., 2016, 52, 1964 RSC.
- A. del Prado, D. González-Rodríguez and Y.-L. Wu, ChemistryOpen, 2020, 9, 409 CrossRef CAS PubMed.
- J. Camacho-García, C. Montoro-García, A. M. López-Pérez, N. Bilbao, S. Romero-Pérez and D. González-Rodríguez, Org. Biomol. Chem., 2015, 13, 4506 RSC.
- F. Aparicio, M. J. Mayoral, C. Montoro-García and D. González-Rodriguez, Chem. Commun., 2019, 55, 7277 RSC.
- S. Romero-Pérez, J. Camacho-García, C. Montoro-García, A. M. López-Pérez, A. Sanz, M. J. Mayoral and D. González-Rodríguez, Org. Lett., 2015, 17, 2664 CrossRef PubMed.
- M. García-Iglesias, T. Torres and D. González-Rodríguez, Chem. Commun., 2016, 52, 9446 RSC.
- R. Chamorro, L. Juan-Fernández, B. Nieto-Ortega, M. J. Mayoral, S. Casado, L. Ruiz-González, E. M. Pérez and D. González-Rodríguez, Chem. Sci., 2018, 9, 4176 RSC.
- M. García-Iglesias, K. Peuntinger, A. Kahnt, J. Krausmann, P. Vázquez, D. González-Rodríguez, D. M. Guldi and T. Torres, J. Am. Chem. Soc., 2013, 135, 19311 CrossRef PubMed.
- I. López-Martín, J. Veiga-Herrero, F. Aparicio and D. González-Rodríguez, Chem. – Eur. J., 2023, e202302279 CrossRef PubMed.
- B. R. Crane, L. M. Siegel and E. D. Getzoff, Science, 1995, 270, 59 CrossRef CAS PubMed.
- M. Morshedi, M. Thomas, A. Tarzia, C. J. Doonan and N. G. White, Chem. Sci., 2017, 8, 3019 RSC.
- G. Delaittre and L. Barner, Polym. Chem., 2018, 9, 2679 RSC.
- C. R. Becer, K. Babiuch, D. Pilz, S. Horning, T. Heinze, M. Gottschaldt and U. S. Schubert, Macromolecules, 2009, 42, 2387 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc05452g |
| This journal is © The Royal Society of Chemistry 2024 |