
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNitrous oxide activation by picoline-derived Ni–CNP hydrides†
José
Bermejo‡
,
Isabel
Ortega-Lepe‡
,
Laura L.
Santos
,
Nuria
Rendón
 ,
Joaquín
López-Serrano
,
Eleuterio
Álvarez
and
Andrés
Suárez
*
,
Joaquín
López-Serrano
,
Eleuterio
Álvarez
and
Andrés
Suárez
*
Instituto de Investigaciones Químicas (IIQ) and Centro de Innovación en Química Avanzada (ORFEO-CINQA), CSIC-Universidad de Sevilla, Avda. Américo Vespucio 49, 41092, Sevilla, Spain. E-mail: andres.suarez@iiq.csic.es
First published on 12th January 2024
Abstract
Oxygen atom transfer (OAT) from N2O to the Ni–H bond of proton-responsive picoline-derived CNP nickel complexes has been investigated both experimentally and theoretically. These Ni–CNP complexes efficiently catalyse the reduction of N2O with pinacolborane (HBpin) under mild conditions.
Nitrous oxide (N2O) is an important contributor to the global climate change due to its high greenhouse impact and ozone-depleting properties.1,2 The increasing concentrations of N2O in the Earth's atmosphere have been chiefly attributed to anthropogenic sources associated with the use of nitrogen-based fertilizers, combustion of biomass and fossil fuels, and the production of industrial chemicals.3 While thermodynamically very favourable, N2O degradation to N2 and O2 is associated with high kinetic barriers, which explains its average long life (118 years) in the atmosphere.4 The known ability of transition metal complexes to react with small molecules, including CO2, CO or H2, make them interesting alternatives for N2O activation and subsequent transformation. However, the reactivity of transition metal complexes towards N2O is critically hampered by its weak σ-donor and π-acceptor properties as a ligand.5 Interestingly, a favourable pathway for N2O activation by transition metal complexes involves the nucleophilic attack of a metal-bound hydride to the terminal nitrogen of N2O, thus generating an O-bound oxyldiazene intermediate (M–ONNH). Subsequent nitrogen extrusion from this derivative produces a hydroxy complex (M–OH), this process representing an overall oxygen atom transfer (OAT) to the complex's M–H bond.6–11 Moreover, the reaction of the hydroxyl derivative with common reductants, such as H2,6,7 alcohols,8 CO9 or silanes,7,10 might lead to the regeneration of the M–H bond, either in a stoichiometric or catalytic process.
With the exception of a hafnium hydride complex,11 metal systems examined for the OAT from N2O to M–H bonds are based on electron-rich precious metals (Rh, Ru, Os, Ir).6–10 In recent years, efficient catalytic systems incorporating Earth-abundant first-row transition metals are in great demand due to their lower economic and environmental costs. As a result of being less hydridic, hydride complexes of first-row late transition metals often lack the high reactivity usually associated with their precious metal counterparts.12 Nickel hydride pincer complexes have been found to be active catalysts in a plethora of reduction reactions,13,14 and have been profusely investigated in the activation of CO2,15–17 an isoelectronic molecule to N2O. Moreover, nickel derivatives have been shown to activate N2O by different modes.18 Herein, we report on the reactivity of Ni–H pincer complexes based on a picoline-derived CNP (C = N-heterocyclic carbene, P = phosphine) ligand towards N2O, including the OAT from N2O to the metal-hydride bond and the catalytic activity of these complexes in the reduction of N2O with pinacolborane (HBpin).
The reaction of the carbene ligand precursors 1a–b19 with KHMDS (potassium bis(trimethylsilyl)amide), followed by addition of NiBr2(dme) (dme = 1,2-dimethoxyethane), afforded the isolation of the bromide complexes 2a–b in good yields (83–87%) (Scheme 1). These derivatives were characterised by NMR spectroscopy and elemental analysis. Aiming to access Ni hydride pincer derivatives, the reaction of the bromide complexes 2a–b with NaBH4 was targeted. Upon anion exchange using NaBPh4, the cationic hydride complexes 3a–b were isolated with yields of 68 and 74%, respectively. These derivatives were analytical and spectroscopically characterised. For instance, the hydride region of the 1H NMR spectrum of 3a exhibits a doublet at −18.0 ppm with a coupling constant of 2JHP = 72 Hz. The IR spectra of 3a and 3b include a band at 1901 and 1888 cm−1, respectively, attributable to the stretching of the Ni–H bond. Moreover, the proposed structure of complex 3a was further confirmed in the solid state by single crystal X-ray diffraction analysis (Fig. 1a). This complex shows a square planar coordination geometry (Σ(Ni) = 359.9°), with the pincer ligand adopting the expected κ3-(P,N,C) coordination mode (C–Ni–P angle: 170.3°).
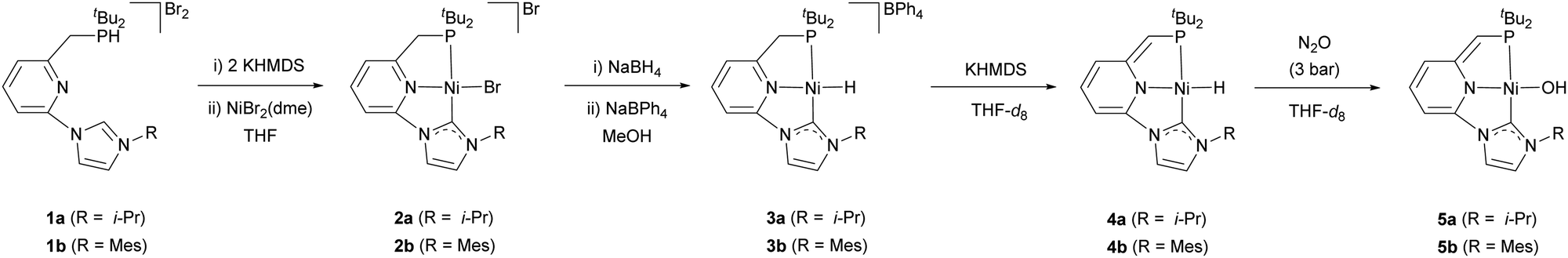 | ||
| Scheme 1 Synthesis of the Ni complexes 2a–b, 3a–b, 4a–b and 5a–b. | ||
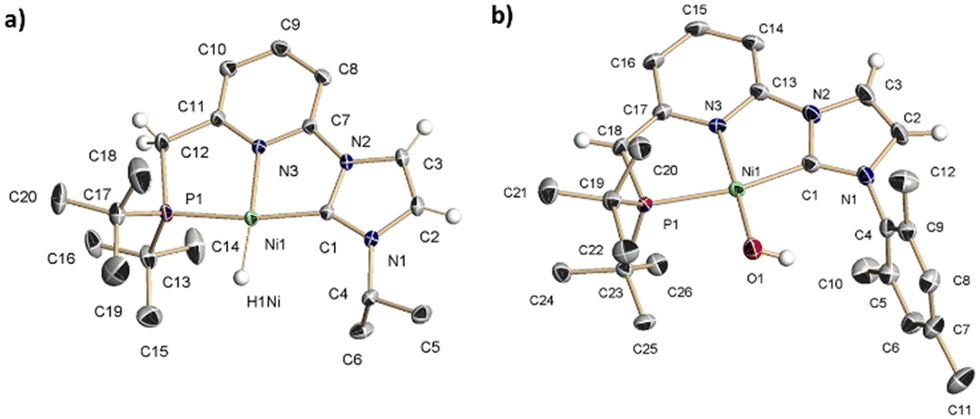 | ||
| Fig. 1 ORTEP drawings at 30% ellipsoid probability of: (a) the cationic fragment of complex 3a, and (b) complex 5b. Most hydrogen atoms have been omitted for the sake of clarity. See ESI† for selected bond lengths and angles. | ||
As result of the expected acidity of the methylene hydrogens of the CNP ligand,20 treatment of complexes 3a–b with a strong base, such as KHMDS, in THF-d8 produced the instantaneous colour change of the initially pale yellow solutions to dark red. The clean formation of the new species 4a–b, deprotonated at the pincer methylene bridge, was evidenced by 1H and 13C{1H} NMR spectroscopies. Attempted isolation of complexes 4a–b only led to significant decomposition, likely due to a poor steric stabilization,14 and consequently they were characterised spectroscopically in solution. In the 1H NMR spectrum of 4a, deprotonation of the methylene arm of the pincer was evidenced by the presence of a singlet appearing at 3.26 ppm, corresponding to the ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHP methyne bridge, and the up-field shift of the resonances of the dearomatized pincer central N-containing ring that appear between 6.41 and 5.34 ppm. In addition, a doublet resonance appearing at ca. −17.5 ppm (2JHP = 67 Hz) in the hydride region was detected in the same experiment, which is attributable to the Ni–H hydrogen. Moreover, the stretching mode of the Ni–H bond produces in the IR spectra of 4a–b absorption bands at lower wavelengths than in their protonated counterparts 3a–b (1841 and 1857 cm−1 for 4a and 4b, respectively), in agreement with the expected larger trans influence of the anionic amide central donor group of the pincer in comparison to a neutral pyridine donor.
CHP methyne bridge, and the up-field shift of the resonances of the dearomatized pincer central N-containing ring that appear between 6.41 and 5.34 ppm. In addition, a doublet resonance appearing at ca. −17.5 ppm (2JHP = 67 Hz) in the hydride region was detected in the same experiment, which is attributable to the Ni–H hydrogen. Moreover, the stretching mode of the Ni–H bond produces in the IR spectra of 4a–b absorption bands at lower wavelengths than in their protonated counterparts 3a–b (1841 and 1857 cm−1 for 4a and 4b, respectively), in agreement with the expected larger trans influence of the anionic amide central donor group of the pincer in comparison to a neutral pyridine donor.
Next, having access to the Ni hydride complexes 3 and 4, we examined their reactions with N2O. Pressurisation with N2O (3 bar) of solutions of complexes 3a–b in THF-d8 did not produced observable changes in their NMR spectra. Conversely, complexes 4a–b, formed in situ by the reaction of 3a–b with KHMDS, gradually react with N2O (3 bar) (reaction half-life of 4b: 36 h at r.t. and 5.5 h at 55 °C) (Scheme 1). The 1H NMR spectrum of the resulting hydroxy derivative 5a includes a doublet signal attributable to the hydroxo moiety appearing at −3.85 ppm with a P–H coupling constant of 3JHP = 7.4 Hz; meanwhile, significant changes in the deprotonated CNP* pincer ligand were not observed. Similar spectroscopic data were obtained for complex 5b. Analysis by X-ray diffraction of a single crystal of 5b revealed a square-planar coordination geometry (Σ(Ni) = 360.1°, C–Ni–P angle: 166.65(7)°), with quite similar metric parameters to the Ni-pincer framework of complex 3a (Fig. 1b). The Ni atom in 5b resides in a distorted square-planar environment with the hydroxyl oxygen displaced 0.17 Å out of the least-square plane defined by the P1, C1, N3 and Ni atoms. Finally, the Ni–O bond length, 1.8272(18) Å, lies at the lowest extreme of the range observed for related nickel square-planar hydroxo complexes based on anionic pincer ligands (Ni–O distances: 1.83–1.93 Å).21 It is remarkable that, to our knowledge, this is the first example of an OAT from N2O to a base metal hydride complex.
To attain further insight into the observed dissimilar reactivity of complexes 3 and 4 towards N2O, the mechanism of the N2O oxygen atom transfer to the Ni–H bonds of 3a and 4a was investigated by performing DFT calculations (B3LYP-D3/def2TZVP) (Fig. 2). In the case of 3a, initial outer-sphere transfer of the hydrido ligand to the terminal nitrogen atom of N2O leads to the endergonic formation of intermediate A, with an associated barrier TS(3a→A) of 29.2 kcal mol−1.22 Subsequent N2 release from A to yield the corresponding hydroxo complex B was found to be largely exergonic, having a relatively high energy barrier of 27.8 kcal mol−1. Interestingly, in the case of 4a, the transition state TS(4a→A*) associated to the transfer of the Ni–H hydride to N2O has an energy (ΔG‡) of 26.0 kcal mol−1, and leads to the formation of A* with an energy return of 4.0 kcal mol−1. The lower barrier for the hydride transfer to N2O for 4a and the thermodynamically favourable formation of A* resemble reported results regarding the insertion of CO2 into Ni–H bonds of square-planar pincer nickel complexes.16,17 These studies have shown lower energy barriers and higher stability of the insertion products as the ligand trans to the hydride becomes a stronger donor. In our case, the different behaviour of the hydrides 3 and 4 towards N2O can be attributed to the expected larger trans influence of the deprotonated pincer ligand, which produces a weakening of the Ni–H bond and increases the nucleophilic character of the hydride ligand.16,17 In fact, the calculated Ni–H distances and NBO (natural bond orbital) charges on the hydride for 3a and 4a are in good agreement with a less strong Ni–H bond and a higher nucleophilicity of the Ni–H group in the case of 4a. The Ni–H bond is slightly longer for 4a [d(Ni–H) = 1.48 Å (3a), 1.51 Å (4a)]; whereas the hydride ligand bears a larger negative charge [NBO charges on the Ni: 0.41 (3a) and 0.42 (4a); NBO charges on the hydride: –0.24 (3a), –0.31 (4a)].
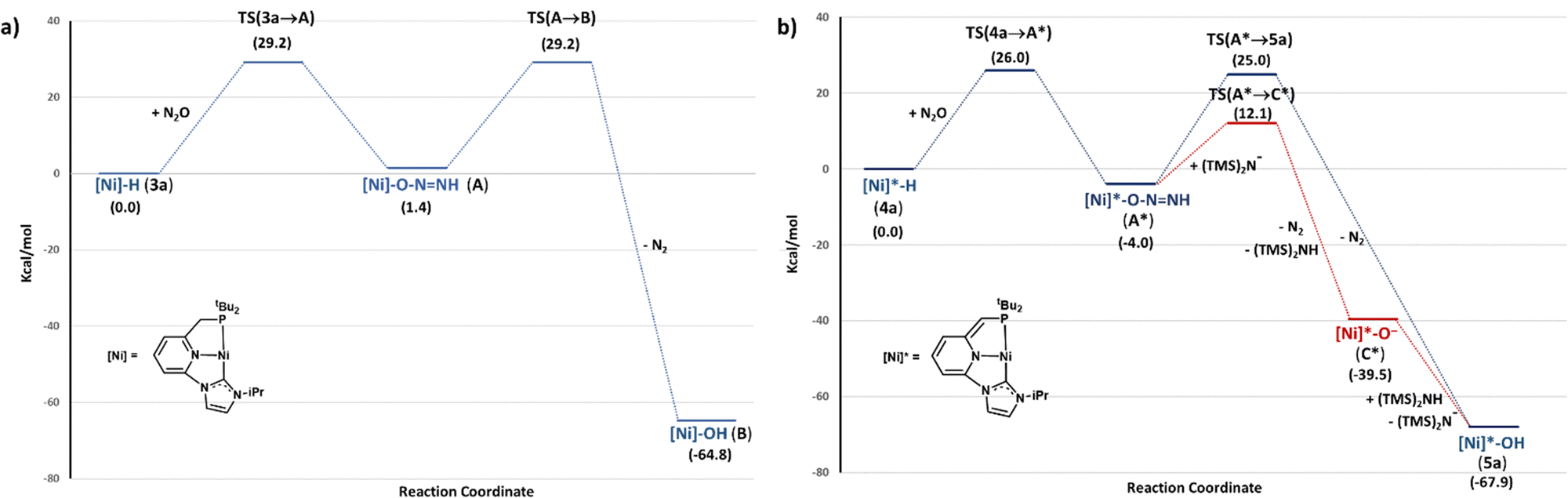 | ||
| Fig. 2 DFT calculated free energy (ΔG in THF, kcal mol−1) profile of the N2O insertion into the Ni–H bond of complexes 3a (a) and 4a (b). | ||
N2 extrusion from A* to form the corresponding hydroxo derivative 5a was found to be highly exergonic, having an energy barrier of 29.0 kcal mol−1. However, since complex 4a is generated in situ from 3a in the presence of a slight excess of KHMDS, the participation of the base in the N2 release step was also investigated. Deprotonation of the Ni–ONNH moiety yields the unusual species C* through a transition state located at 16.1 kcal mol−1. This intermediate can be readily protonated by (Me3Si)2NH leading to the hydroxo complex 5a. This last step is exergonic by 28.4 kcal mol−1 from C*, and takes place without energy barrier, as indicated by relaxed potential energy (PES) scans (see ESI†). The overall energy return for the formation of 5a and N2 from 4a and N2O is 67.9 kcal mol−1.
Next, the catalytic performance of the Ni(II) hydride derivatives in the reduction of N2O using HBpin was examined (Table 1).23 Whilst no reaction was observed in the absence of the Ni–CNP catalysts, HBpin was fully consumed using 2.0 mol% of the cationic complexes 3a–b, leading to the formation of pinBOH and (pinB)2O, in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8 ratio (entries 1 and 2); moreover, generation of N2 and H2 was detected, although it could not be quantified (see ESI† for details). By employing in situ formed complexes 4a and 4b, somewhat faster reactions using lower catalyst loadings (0.5 mol%) than for the corresponding complexes 3 were observed (entries 3 and 4). Finally, THF-d8 solutions of complex 5b, formed in situ, were also found to catalyse the reduction of N2O with HBpin (entry 5).
:8 ratio (entries 1 and 2); moreover, generation of N2 and H2 was detected, although it could not be quantified (see ESI† for details). By employing in situ formed complexes 4a and 4b, somewhat faster reactions using lower catalyst loadings (0.5 mol%) than for the corresponding complexes 3 were observed (entries 3 and 4). Finally, THF-d8 solutions of complex 5b, formed in situ, were also found to catalyse the reduction of N2O with HBpin (entry 5).
| Entry | Cat. | Cat. loading [mol%] | Conv. [%] (time, [h]) | pinBOH: (pinB)2O ratio |
|---|---|---|---|---|
| a Reaction conditions, unless otherwise noted: 2 bar N2O, r.t., THF-d8, [HBpin] = 0.4 M. Complexes 4a–b were formed in situ from 3a–b with KHMDS. Complex 5b was formed in situ from 3b and KHMDS under N2O pressure. Conversion and selectivity were determined by 1H and 11B{1H} NMR spectroscopies using mesitylene as internal standard. N2 and H2 formation were detected by GC-MS analysis of the headspace gas and 1H NMR spectroscopy, respectively (see ESI). | ||||
| 1 | 3a | 2.0 | >99 (6.5) | 2:8 |
| 2 | 3b | 2.0 | >99 (2.0) | 2:8 |
| 3 | 4a | 0.5 | >99 (3.0) | 1:9 |
| 4 | 4b | 0.5 | >99 (2.0) | 2:8 |
| 5 | 5b | 2.0 | >99 (2.0) | 2:8 |
The observed fast kinetics for the catalytic reaction are at odds with the relatively slow N2O insertion in complexes 4 and the lack of reactivity of derivatives 3. In an attempt to obtain further information, a series of control experiments were performed. First, no reaction was observed by NMR spectroscopy between HBpin (ca. 4 equiv.) and the hydride complexes 3 and 4 in the temperature range between −60 °C and 55 °C.24 Moreover, a solution of the in situ formed complex 5b in THF-d8 was treated with HBpin (0.9 equiv.) at −65 °C. After approximately 0.25 h, 82% conversion of 5b to a mixture of the hydride derivatives 3b and 4b (5:1 ratio) was observed, demonstrating that Ni–H bond regeneration from 5 is facile with HBpin. In the 11B{1H} NMR spectrum of this experiment, a new broad signal was observed at 4.5 ppm. Unfortunately, attempts to isolate and characterise this boron species were unsuccessful. The presence of borane Lewis acids has been shown to impact on both the reaction kinetics and product distribution in the CO2 hydroboration,25 and has been found to play a role in the CO2 activation by Ni-PBP hydride complexes. Therefore, we hypothesise that Lewis acid borane species might catalyse or co-catalyse the hydroboration of N2O in the presence of complexes 3 and 4.26 In fact, complete catalyst inhibition was observed when the reaction catalysed by 3b was performed in the presence of 20 mol% Et3N, acting as a Lewis acid scavenger.27
To conclude, the OAT from N2O to hydride Ni complexes based on CNP ligands has been found to be dependent on the trans influence of the pincer. Thus, the reactivity of complexes 3a–b towards N2O is triggered by a strong base (KHMDS), leading to the formation of the deprotonated Ni–CNP* species 4a–b, which are capable of performing N2O activation. Moreover, the use of a strong base promotes the N2 release from the Ni–ONNH intermediate resulting from the N2O insertion into the Ni–H bond. Finally, both complexes 3 and 4 catalyse the reduction of N2O with HBpin, in a process that is proposed to be catalysed or co-catalysed by the presence of boron Lewis acids generated under catalytic conditions.
Financial support from MCIN/AEI/10.13039/501100011033/FEDER,UE (PID2019-104159GB-I00, TED2021-129181B-I00, PID2022-136570OB-I00) and CSIC (COOPB20604), and the use of the computational facilities of the Supercomputing Center of Galicia (CESGA) is gratefully acknowledged. J. B. thanks CSIC, Junta de Andalucía and Fondo Social Europeo (FSE) for a Sistema Nacional de Garantía Juvenil contract.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. Hansen and M. Sato, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16109 CrossRef CAS PubMed; S. A. Montzka, E. J. Dlugokencky and J. H. Butler, Nature, 2011, 476, 43 CrossRef PubMed.
- M. J. Prather, Science, 1998, 279, 1339 CrossRef CAS PubMed; A. R. Ravishankara, J. S. Daniel and R. W. Portmann, Science, 2009, 326, 123 CrossRef PubMed.
- E. A. Davidson and D. Kanter, Environ. Res. Lett., 2014, 9, 105012 CrossRef.
- S. I. Gorelsky, S. Ghosh and E. I. Solomon, J. Am. Chem. Soc., 2006, 128, 278 CrossRef CAS PubMed; S. Javoy, R. Mevel and C. E. Paillard, Int. J. Chem. Kinet., 2009, 41, 357 CrossRef.
- W. B. Tolman, Angew. Chem., Int. Ed., 2010, 49, 1018 CrossRef CAS PubMed.
- A. W. Kaplan and R. G. Bergman, Organometallics, 1997, 16, 1106 CrossRef CAS; A. W. Kaplan and R. G. Bergman, Organometallics, 1998, 17, 5072 CrossRef; J.-H. Lee, M. Pink, J. Tomaszewski, H. Fan and K. G. Caulton, J. Am. Chem. Soc., 2007, 129, 8706 CrossRef PubMed; L. E. Doyle, W. E. Piers and J. Borau-Garcia, J. Am. Chem. Soc., 2015, 137, 2187 CrossRef PubMed; J. D. Smith, E. Chih, W. E. Piers and D. M. Spasyuk, Polyhedron, 2018, 155, 281 CrossRef; P. Jurt, A. S. Abels, J. J. Gamboa-Carballo, I. Fernández, G. Le Corre, M. Aebli, M. G. Baker, F. Eiler, F. Müller, M. Wörle, R. Verel, S. Gauthier, M. Trincado, T. L. Gianetti and H. Grützmacher, Angew. Chem., Int. Ed., 2021, 60, 25372 CrossRef PubMed; I. Ortega-Lepe, P. Sánchez, L. L. Santos, P. Lara, N. Rendón, J. López-Serrano, V. Salazar-Pereda, E. Álvarez, M. Paneque and A. Suárez, Inorg. Chem., 2022, 61, 18590 CrossRef PubMed.
- R. Zeng, M. Feller, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2017, 139, 5720 CrossRef CAS PubMed.
- T. L. Gianetti, S. P. Annen, G. Santiso-Quinones, M. Reiher, M. Driess and H. Grützmacher, Angew. Chem., Int. Ed., 2016, 55, 1854 CrossRef CAS PubMed; J. Bösken, R. E. Rodríguez-Lugo, S. Nappen, M. Trincado and H. Grützmacher, Chem. – Eur. J., 2023, 29, e202203632 CrossRef PubMed.
- R. Zeng, M. Feller, Y. Diskin-Posner, L. J. W. Shimon, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2018, 140, 7061 CrossRef CAS PubMed.
- P. Molinillo, B. Lacroix, F. Vattier, N. Rendón, A. Suárez and P. Lara, Chem. Commun., 2022, 58, 7176 RSC.
- G. A. Vaughan, P. B. Rupert and G. L. Hillhouse, J. Am. Chem. Soc., 1987, 109, 5538 CrossRef CAS.
- E. S. Wiedner, M. B. Chambers, C. L. Pitman, R. M. Bullock, A. J. M. Miller and A. M. Appel, Chem. Rev., 2016, 116, 8655 CrossRef CAS PubMed.
- V. Arora, H. Narjinari, P. G. Nandia and A. Kumar, Dalton Trans., 2021, 50, 3394 RSC.
- N. A. Eberhardt and H. Guan, Chem. Rev., 2016, 116, 8373 CrossRef CAS PubMed.
- T. J. Schmeier, N. Hazari, C. D. Incarvito and J. A. Raskatov, Chem. Commun., 2011, 47, 1824 RSC; J. E. Heimann, W. H. Bernskoetter, J. A. Guthrie, N. Hazari and J. M. Mayer, Organometallics, 2018, 37, 3649 CrossRef CAS; C. Yoo, J. Kim and Y. Lee, Organometallics, 2013, 32, 7195 CrossRef; K. J. Jonasson and O. F. Wendt, Chem. – Eur. J., 2014, 20, 11894 CrossRef PubMed; L. J. Murphy, H. Hollenhorst, R. McDonald, M. Ferguson, M. D. Lumsden and L. Turculet, Organometallics, 2017, 36, 3709 CrossRef.
- H.-W. Suh, T. J. Schmeier, N. Hazari, R. A. Kemp and M. K. Takase, Organometallics, 2012, 31, 8225 CrossRef CAS.
- P. Ríos, N. Curado, J. López-Serrano and A. Rodríguez, Chem. Commun., 2016, 52, 2114 RSC; P. Ríos, A. Rodríguez and J. López-Serrano, ACS Catal., 2016, 6, 5715 CrossRef CAS; J. E. Heimann, W. H. Bernskoetter, N. Hazari and J. M. Mayer, Chem. Sci., 2018, 9, 6629 RSC; T. J. Schmeier, A. Nova, N. Hazari and F. Maseras, Chem. – Eur. J., 2012, 18, 6915 CrossRef PubMed.
- T. Yamada, K. Suzuki, K. Hashimoto and T. Ikeno, Chem. Lett., 1999, 1043 CrossRef CAS; P. T. Matsunaga and G. L. Hillhouse, J. Am. Chem. Soc., 1993, 115, 2075 CrossRef; N. D. Harrold, R. Waterman, G. L. Hillhouse and T. R. Cundari, J. Am. Chem. Soc., 2009, 131, 12872 CrossRef PubMed; B. Horn, C. Limberg, C. Herwig, M. Feist and S. Mebs, Chem. Commun., 2012, 48, 8243 RSC; F. Le Vaillant, A. Mateos Calbet, S. González-Pelayo, E. J. Reijerse, S. Ni, J. Busch and J. Cornella, Nature, 2022, 604, 677 CrossRef PubMed; J. Gwak, S. Ahn, M.-H. Baik and Y. Lee, Chem. Sci., 2019, 10, 4767 RSC.
- T. Simler, A. A. Danopoulos and P. Braunstein, Chem. Commun., 2015, 51, 10699 RSC.
- J. I. van der Vlugt, M. Lutz, E. A. Pidko, D. Vogt and A. L. Spek, Dalton Trans., 2009, 1016 RSC; D. Oren, Y. Diskin-Posner, L. Avram, M. Feller and D. Milstein, Organometallics, 2018, 37, 2217 CrossRef CAS PubMed.
- T. J. Schmeier, A. Nova, N. Hazari and F. Maseras, Chem. – Eur. J., 2012, 18, 6915 CrossRef CAS PubMed; D. Adhikari, S. Mossin, F. Basuli, B. R. Dible, M. Chipara, H. Fan, J. C. Huffman, K. Meyer and D. J. Mindiola, Inorg. Chem., 2008, 47, 10479 CrossRef PubMed; D. V. Gutsulyak, W. E. Piers, J. Borau-Garcia and M. Parvez, J. Am. Chem. Soc., 2013, 135, 11776 CrossRef PubMed; J. Cámpora, P. Palma, D. del Río and E. Álvarez, Organometallics, 2004, 23, 1652 CrossRef; A. Castonguay, A. L. Beauchamp and D. Zargarian, Inorg. Chem., 2009, 48, 3177 CrossRef PubMed; C. Yao, P. Chakraborty, E. Aresu, H. Li, C. Guan, C. Zhou, L.-C. Liang and K.-W. Huang, Dalton Trans., 2018, 47, 16057 RSC; K. J. Jonasson, A. H. Mousa and O. F. Wendt, Polyhedron, 2018, 143, 132 CrossRef; N. H. Anderson, J. M. Boncella and A. M. Tondreau, Organometallics, 2018, 37, 4675 CrossRef.
- H. Yu, G. Jia and Z. Lin, Organometallics, 2008, 27, 3825 CrossRef CAS; J. A. Luque-Urrutia and A. Poater, Inorg. Chem., 2017, 56, 14383 CrossRef PubMed; L. Yao, Y. Li, L. Huang, K. Guo, G. Ren, Z. Wu, Q. Lei, W. Fang and H. Xie, Comput. Theor. Chem., 2018, 1128, 48 CrossRef; H. Xie, C. Liu, Y. Yuan, T. Zhou, T. Fan, Q. Lei and W. Fang, Dalton Trans., 2016, 45, 1152 RSC; S. Escayola, M. Solà and A. Poater, Inorg. Chem., 2020, 59, 9374 CrossRef PubMed; H. Xie, Y. Zhang, C. Xiang, Y. Li, T. Fan, Q. Lei and W. Fang, Dalton Trans., 2018, 47, 15324 RSC; G. Liang, M. Zhang and C. E. Webster, Inorganics, 2022, 10, 69 CrossRef; H. Xie, Y. Li, L. Huang, F. Nong, G. Ren, T. Fan, Q. Lei and W. Fang, Dalton Trans., 2016, 45, 16485 RSC.
- Y. Pang, M. Leutzsch, N. Nöthling and J. Cornella, J. Am. Chem. Soc., 2020, 142, 19473 CrossRef CAS PubMed; X. Chen, H. Wang, S. Du, M. Driess and Z. Mo, Angew. Chem., Int. Ed., 2022, 61, e202114598 CrossRef PubMed.
- D. Adhikari, J. C. Huffman and D. J. Mindiola, Chem. Commun., 2007, 4489 RSC; A. Rossin, M. Peruzzini and F. Zanobini, Dalton Trans., 2011, 40, 4447 RSC; S. Chakraborty, J. Zhang, Y. J. Patel, J. A. Krause and H. Guan, Inorg. Chem., 2013, 52, 37 CrossRef CAS PubMed.
- M. R. Espinosa, D. J. Charboneau, A. Garcia de Oliveira and N. Hazari, ACS Catal., 2019, 9, 301 CrossRef CAS.
- S. J. Geier, C. M. Vogels, J. A. Melanson and S. A. Westcott, Chem. Soc. Rev., 2022, 51, 8877 RSC.
- A. D. Bage, K. Nicholson, T. A. Hunt, T. Langer and S. P. Thomas, ACS Catal., 2020, 10, 13479 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2299058 and 2299059. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc05455a |
| ‡ These authors have contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |