
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAustalide derivative from marine-derived Aspergillus sp. and evaluation of its cytotoxic and ADME/TOPKAT properties†
Mohamed S. Elnaggar‡
 ac,
Ahmed M. Elissawy‡*ab,
Fadia S. Youssefa,
Máté Kicsákd,
Tibor Kurtánd,
Abdel Nasser B. Singab*ab and
Rainer Kalscheuerc
ac,
Ahmed M. Elissawy‡*ab,
Fadia S. Youssefa,
Máté Kicsákd,
Tibor Kurtánd,
Abdel Nasser B. Singab*ab and
Rainer Kalscheuerc
aDepartment of Pharmacognosy, Faculty of Pharmacy, Ain Shams University Abbassia, 11566, Cairo, Egypt. E-mail: aelissawy@pharma.asu.edu.eg; dean@pharma.asu.edu.eg
bCenter of Drug Discovery Research and Development, Ain Shams University Abbassia, 11566, Cairo, Egypt
cInstitute of Pharmaceutical Biology and Biotechnology, Heinrich-Heine-Universität Düsseldorf, 40225, Germany
dDepartment of Organic Chemistry, University of Debrecen, Debrecen 4032, Hungary
First published on 1st June 2023
Abstract
In-depth chemical investigation of an ethyl acetate extract of Aspergillus sp. isolated from the soft coral Sinularia species resulted in the isolation of one new meroterpenoid, austalide Z (1), one known austalide W (2), six known prenylated indole diketopiperazine alkaloids (3–8), and phthalic acid and its ethyl derivative (9–10). The structures were established by means of 1D and 2D NMR (one- and two-dimensional nuclear magnetic resonance) experiments supported by UV analysis and ESI-MS (electrospray ionization mass spectrometry). In vitro cytotoxic evaluation was performed against the Caco-2 cancer cell line using the MTT assay, which showed that the examined compounds had weak to moderate activities, with the new meroterpenoid austalide Z (1) displaying an IC50 value of 51.6 μg mL−1. ADME/TOPKAT (absorption, distribution, metabolism, excretion, and toxicity) predication performed in silico showed that most of the isolated compounds possessed reasonable pharmacokinetic, pharmacodynamic, and toxicity properties. Thus, it can be concluded that Aspergillus sp. could act as a source of drug leads for cancer prevention with promising pharmacokinetic and pharmacodynamic properties and thus could be incorporated in pharmaceutical dosage forms.
1 Introduction
The fungi kingdom constitutes a rich source of bioactive compounds, and a plethora of drug candidates has been developed from fungal metabolites since the discovery of penicillin from Penicillium notatum by Sir Alexander Fleming in 1928. These drugs comprise the antihyperlipidemic agents atorvastatin and simvastatin, the antifungal drug griseofulvin, the anti-migraine drug ergotamine, and the immunosuppressant drug cyclosporine.1Basically, endosymbiotic fungi colonize the inner tissues of a host, either plant or animal,2 exhibiting mutual benefits, wherein the host organism provides a proper medium for the growth of the fungus and in turn the growing fungal species provide the host with bioactive compounds enabling it to survive unfavorable conditions3 as well as mediating different ecological interactions.4
Endosymbiotic fungi derived from marine ecosystems have received much attention in the last two decades with the isolation of more than 1000 fungal species from marine habitats.5 Cephalosporin C was the first bioactive molecule isolated from the marine-derived fungus Cephalosporium sp.,6 and since then endosymbiotic fungi have proven to be a fabulous source of bioactive secondary metabolites belonging to different classes, such as terpenoids,7,8 diketopiperazines,9 polyketides,10,11 benzophenones,12 peptides,13 and butyrolactones.14
Meroterpenoids represent a major class of fungal terpenoids,7,15–17 and austalides represent a unique subclass of polycyclic (tetra-, penta-, hexa-, or heptacyclic) meroterpenoids with an isobenzofuranone moiety. Thus far, 38 members of austalides have been mainly isolated from different Penicillium and Aspergillus species and demonstrated potential activities, including anti-osteoporosis, antibacterial, antiviral, cytotoxic, and α-glucosidase inhibitory activities.18–24 Moreover, many synthetic approaches have been reported for different members of the austalide family.25–27
It is noteworthy to highlight that cancer still represents a major impendence in the progress of therapeutic approaches to improve public health. Thus, there is an urgent search for effective naturally occurring drug candidates to combat these challenges that are less expensive and possess less adverse effects compared to the current synthetic agents.28
In continuation of our ongoing search for new secondary metabolites from marine-derived fungi, we herein report the chemical investigation of an ethyl acetate extract of the fungus Aspergillus sp. isolated from the inner tissues of the soft coral Sinularia sp. collected at the coast of Sharm El-Sheikh, Egypt. Herein, the isolation and characterization of a new meroterpenoid, named austalide Z (1), the known austalide W (2), and six known prenylated indole diketopiperazine alkaloids (3–8), along with phthalic acid and its ethyl derivatives (9–10) are reported. All the isolated compounds were assessed for their cytotoxic effect against Caco-2 cells using the MTT assay. Additionally, prediction of the pharmacokinetic and pharmacodynamic potentials as well as the toxicity properties of the isolated compounds were performed in silico using the ADME/TOPKAT protocol in Discovery Studio 4.5 (Accelrys Inc., San Diego, CA, USA).
2 Results and discussion
2.1. Chemical investigation of an ethyl acetate extract of the fungus Aspergillus sp.
In-depth chemical investigation of the ethyl acetate extract of the fungus Aspergillus sp. isolated from the inner tissues of the soft coral Sinularia sp. collected from the coast of Sharm El-Sheikh, Egypt, resulted in the isolation and characterization of one new meroterpenoid austalide Z (1), and one known austalide W (2),19 in addition to six known prenylated indole diketopiperazine alkaloids (3–8),29,30 along with phthalic acid and its ethyl derivative (9–10).Compound 1 was isolated as a pale-yellow amorphous powder. LC-ESI-MS revealed pseudomolecular ion peaks at m/z 505 and 503 representing [M + H]+ and [M − H]−, respectively, whereas HR-ESI-MS revealed a peak at m/z 505.2068 representing [M + H]+ (calculated. for m/z C26H33O10, 505.2090), which corresponded to the molecular formula C26H32O10 (ESI Fig. S1A & B†). The UV spectrum of 1 showed a characteristic pattern of austalides with λmax at 270 nm. Its chemical structure was elucidated through 1D and 2D NMR spectroscopic analyses (ESI Fig. S2–S7†), alongside the reported data for the related analogues austalides V and W.19 Analysis of the 1H and APT-NMR spectra displayed a close similarity between compound 1 and austalide W (2), where the APT spectrum (Table 1) revealed the presence of 26 carbons, including one carbonyl group, six olefinic or aromatic carbons and thirteen aliphatic carbons, one tri-oxygenated quaternary carbon, one di-oxygenated quaternary carbon, three oxygenated quaternary carbons, one quaternary aliphatic carbon, one oxygenated methine carbon, one aliphatic methine carbon, two oxygenated methylene carbons, three aliphatic methylenes, two methoxyl carbons, and four methyls.
| Position | δC, type | δH (J in Hz) |
|---|---|---|
| 1 | 68.2, CH2 | 5.14, s |
| 2 | ||
| 3 | 169.7, C | |
| 4 | 108.3, C | |
| 5 | 155.3, C | |
| 6 | 115.9, C | |
| 7 | 157.1, C | |
| 8 | 114.3, C | |
| 9 | 145.5, C | |
| 10 | ||
| 11 | 79.2, C | |
| 12 | 44.5, CH2 | 2.64, d (14.5) |
| 2.39, d (14.5) | ||
| 13 | 102.0, C | |
| 14 | 92.2, C | |
| 15 | 86.7, C | |
| 16 | ||
| 17 | 118.5, C | |
| 18 | 69.2, CH | 3.80, dd (5.5, 0.9) |
| 19 | 37.7, CH2 | 2.15, dd (15.9, 0.9) |
| 1.85, dd (15.9, 5.5) | ||
| 20 | 39.2, C | |
| 21 | 38.2, CH | 3.05, dd (7.5, 1.8) |
| 22 | 17.8, CH2 | 2.95, m |
| 23 | 10.9, CH3 | 2.06, s |
| 24 | 27.3, CH3 | 1.35, s |
| 25 | 78.2, CH2 | 4.16, d (9.7) |
| 3.95, d (9.7) | ||
| 26 | 23.0, CH3 | 1.69, s |
| 27 | 19.6, CH3 | 0.87, s |
| 28 | 49.1, CH3 | 3.53, s |
| 29 | 62.2, CH3 | 4.16, s |
Meanwhile, the 1H NMR spectrum of 1 (Table 1) displayed two methine protons at δH (ppm) 3.80 (1H, dd, J = 5.5, 0.9 Hz, H-18) and 3.05 (1H, dd, J = 7.5, 1.8 Hz, H-21), in which the former was oxygenated, in addition to five methylene protons at δH (ppm) 5.14 (2H, s, H-1), 4.16, 3.95 (2H, d, J = 9.7 Hz, H-25), 2.95 (2H, m, H-22), 2.64, 2.39 (2H, d, J = 14.5 Hz, H-12), and 2.15, 1.85 (2H, dd, J = 15.9, 0.9 Hz; 15.9, 5.5 Hz, H19), in which the former two methylenes were oxygenated, in addition to the presence of two methoxy protons at δH (ppm) 4.16 (3H, s, OMe-29) and 3.53 (3H, s, OMe-28). Moreover, four methyl protons at δH (ppm) 2.06 (3H, s, Me-23), 1.69 (3H, s, Me-26), 1.35 (3H, s, Me-24), and 0.87 (3H, s, Me-27) were observed.
The above-mentioned data were found to be in complete accordance with the reported data for the austalides class,31–33 in which one known analogue of this group of compounds was also isolated here in our study from the same crude extract, namely austalide W (2).31 Compound 2 was found to exhibit close similarity to the structure of 1. Moreover, it is worthy to highlight that 2 was previously reported to be the first austalide derivative having a 5/6/6/6/6/5/5 heptacyclic ring system, including a tetrahydrofuranyl ring.31 In addition, this ring system was also confirmed in 1 to be in alignment with all previously reported austalides, as well as the affirmation of the presence of ring G through the existence of the down-fielded CH2 group (C-25) at δH (ppm) 4.16, 3.95 (2H, d, J = 9.7 Hz) and δC (ppm) 78.2, alongside the existence of a characteristic down-field carbon shift of C-13 at δC (ppm) 102.0, in which all those characteristic shifts were found to be similar to 2,31 affirming the presence of the unusual tetrahydrofuranyl ring (ring G) in the structure of 1.
By inspection of the NMR data mentioned above for 1, it was clear that it differed from austalide W (2) only by the presence of a newly added hydroxyl group at C-18. This hydroxylation could be unambiguously deduced through the difference of the molecular weight of both compounds by only 16 units in the molecular weight of 1 compared to 2. Moreover, in the COSY spectrum, there was a distinct correlation between H-18 and H2-19, confirming the adjacent positioning of both groups. This suggestion was further confirmed through inspection of the reported data for 2 in comparison to the data of the newly isolated 1, in which instead of the methylene group at C-18 in 2, a clearly observed oxygenated methine proton was detected at δH (ppm) 3.8 on C-18. Hence, there was a low-field chemical shift from δH (ppm) 1.81–1.94 in 2 (ref. 31) to δH (ppm) 3.8 in 1, as well as a low-field shift of carbon from δC (ppm) 30.92 in 2 (ref. 31) to δC (ppm) 69.2 in 1. In addition, a low-field shift was also detected in APT NMR for C-19 from δC (ppm) 30.83 in 2 (ref. 31) to δC (ppm) 37.7 in 2, side by side with the confirmatory HMBC correlations from the newly observed methine proton H-18 to δC 39.2 (C-20) and δC 118.5 (C-17), as well as with the correlations from δH (ppm) 0.87 (H3-27) and δH (ppm) 2.15, 1.85 (H2-19) toward δC (ppm) 69.2 (C-18). It is noteworthy to mention that Antipova et al. isolated an acetylated derivative, austalide V, where the oxygenated methine was placed at C-19.34 In our case, HMBC correlations confirmed the presence of the oxygenated methine at C-18 instead, as revealed by the absence of strong correlations between the methyl protons of C-27 and the oxygenated methine carbon C-18. Thus, 1 possessed a heptacyclic skeleton the same as other austalide analogues and the planar structure was elucidated as shown. The trivial name austalide Z is proposed.
The relative configuration of the eight chirality centers of 1 was determined based on NOESY correlations (Fig. 2). A NOESY correlation between 26-H and 27-H methyl protons suggested the cis diaxial orientation of the two methyl groups and determined the relative configuration of the C-14 and C-17 bridge-head carbons, similarly to other previously reported austalide analogues.31 Moreover, the NOE correlations of 24-H methyl protons with the 21-H methine proton indicated that the C-11 and C-21 bridge-head carbons had cis relative configurations. In addition, the observed NOE correlations between the oxygenated 18-H methine proton with the 21-H methine proton alongside with the absence of any cross-peaks or correlations of the former proton with either C-26 or C-27, revealed the cis orientations of 21-H and 18-H. Thus, the relative configuration of 1 was determined as (11S*, 13S*, 14S*,15S*,17S*,18S*,20R*,21R*).
In addition, the biosynthesis of all austalide derivatives likely followed the same biosynthetic pathway, suggesting that they all originated from the parent 6-[(2E,6E)-farnesyl]-5,7-dihydroxy-4-methylphthalide followed by cyclization and oxidative modification.31 An additional pathway for austalide derivatives was proposed based on the reported total synthesis of austalide B.35,36 Thus, those two reported biosynthetic pathways suggested that the skeleton had the same configuration for all the austalide group members.
The ECD spectrum of austalide Z (Fig. 3) showed negative cotton effects (CEs) at 266 and 213 nm and a positive one at 229 nm, which were quite similar to those of the tetracyclic austalides P and Q (Fig. 1).23 The absolute configuration of austalide P was previously determined by one of the authors,23 using the solution TDDFT-ECD calculation protocol.37 In the absence of the C-22 benzylic chirality center, the ECD spectra of austalides Z, P, and Q are governed by the absolute configuration of the C-11 and C-21 bridge-head chirality centers, which have the same (11S,21R) absolute configuration. Moreover, the ECD spectrum of austalide Z showed a similar pattern to those of austalides V and W. With knowledge of the (11S*,13S*,14S*,15S*,17S*,18S*,20R*,21R*) relative configuration, the ECD pattern of austalide Z was derived from the (11S,13S,14S,15S,17S,18S,20R,21R) absolute configuration.
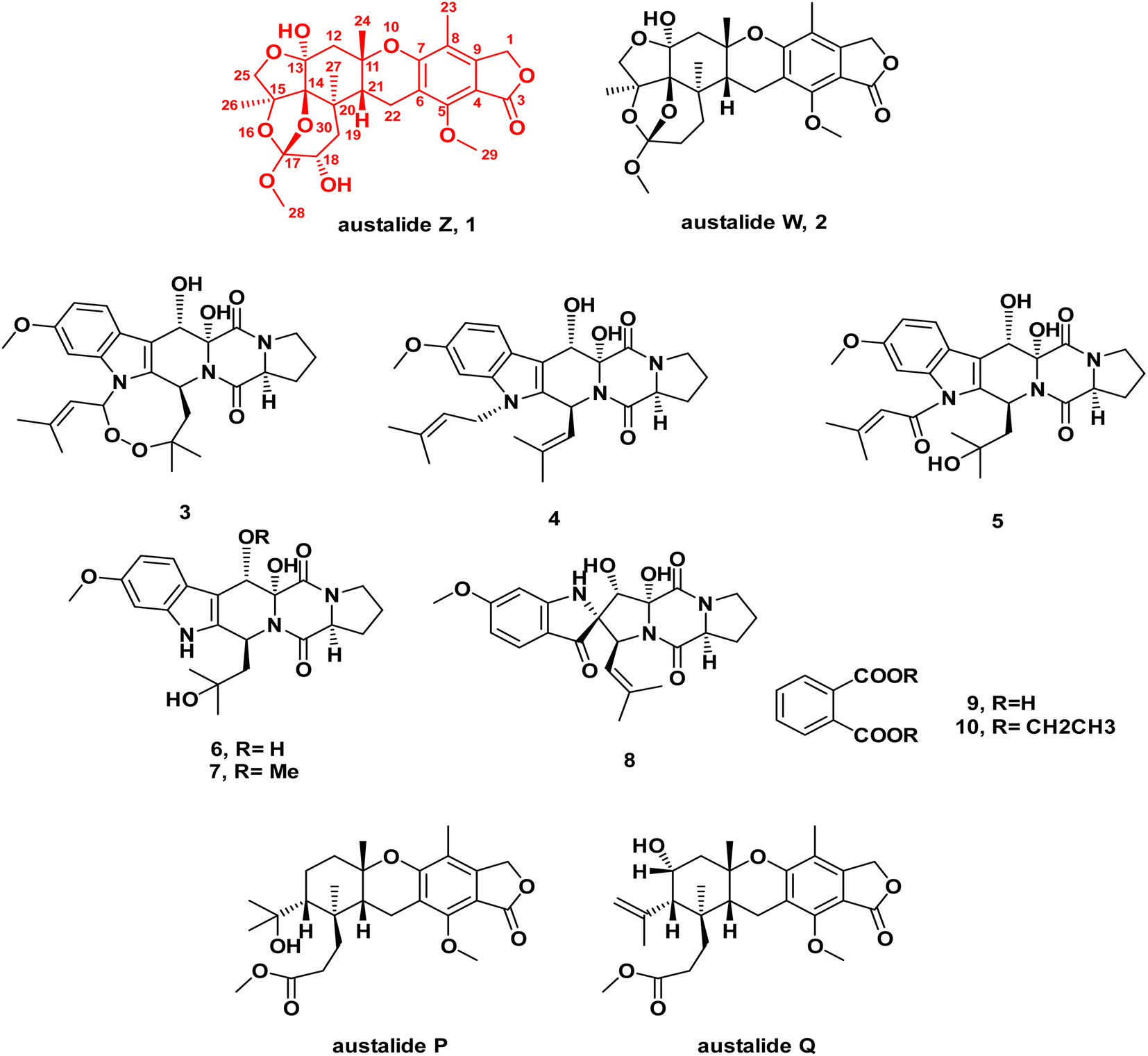 | ||
| Fig. 1 Chemical structures of compounds 1–10 isolated from the ethyl acetate extract of the fungus Aspergillus sp. obtained from the soft coral Sinularia sp. | ||
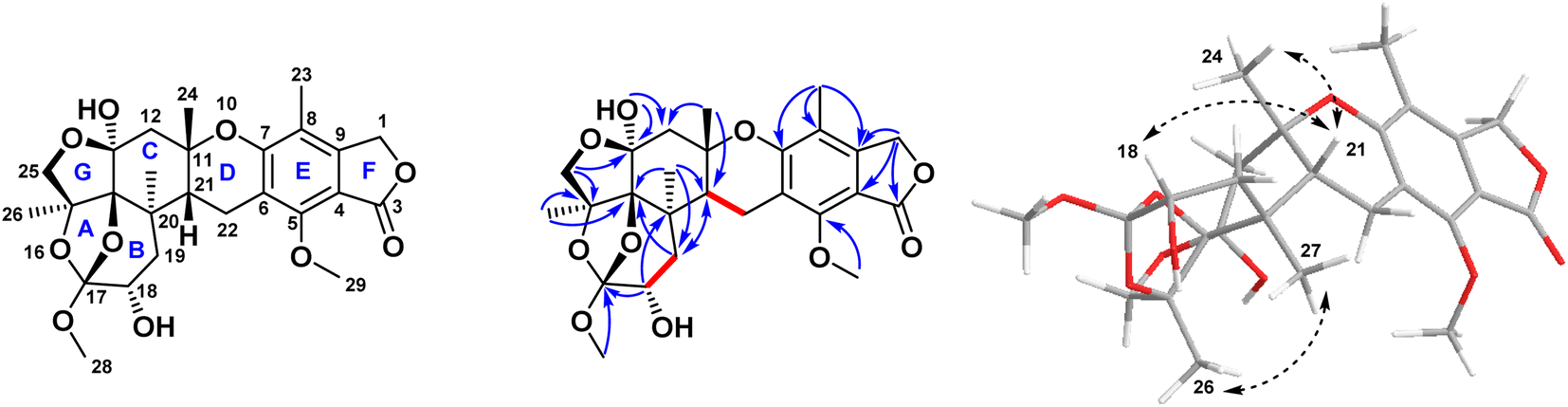 | ||
| Fig. 2 Key COSY, HMBC, and NOESY correlations of compound 1 isolated from the ethyl acetate extract of the fungus Aspergillus sp. obtained from the soft coral Sinularia sp. | ||
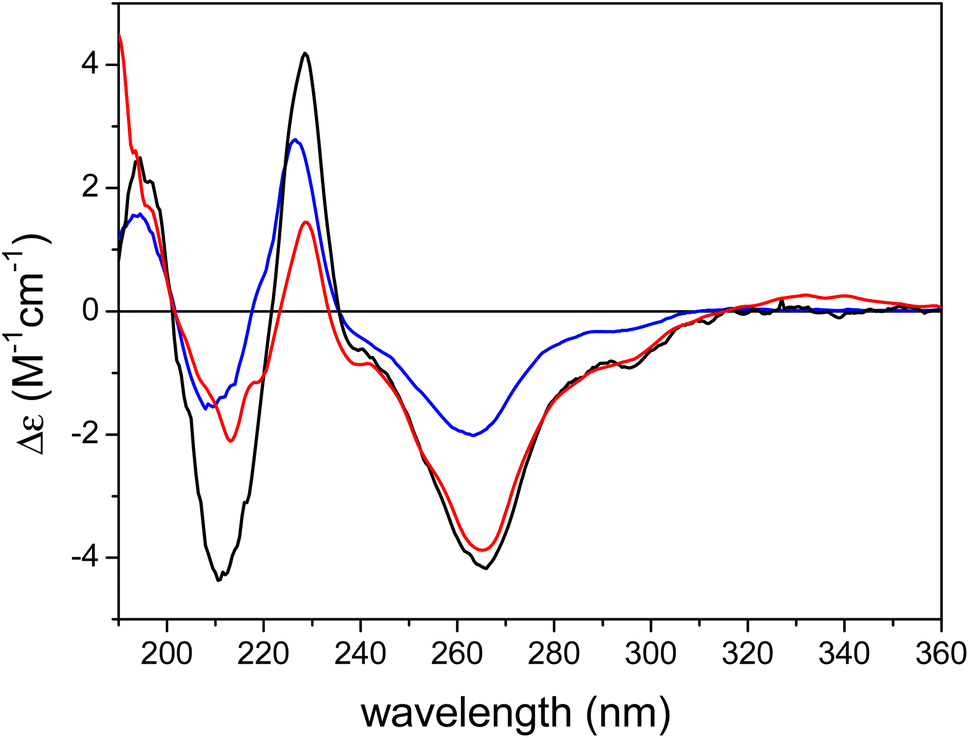 | ||
| Fig. 3 Experimental ECD spectra of austalide Q (X, blue), austalide P (Y, black) versus austalide Z (1, red) in acetonitrile; Copyright Wiley-VCH GmbH. Reproduced with permission. Y. Zhou, A. Mándi, A. Debbab, V. Wray, B. Schulz, W. E. Müller, W. Lin, P. Proksch, T. Kurtán, A. H. Aly, New Austalides from the Sponge-Associated Fungus Aspergillus sp., Eur. J. Org. Chem., 2011 (2011) 6009.23 | ||
2.2. In vitro cytotoxic evaluation of the isolated compounds using the MTT assay
Cytotoxic evaluations of 3, 4, 6, 8 (diketopiperazine derivatives) as well as 1 and 2 (meroterpenoid derivatives) against Caco-2 (human colorectal carcinoma cell lines) revealed weak to moderate activities, displaying IC50 values ranging between 32.5 and 126 μg mL−1, where the new meroterpenoid austalide Z (1) displayed a cytotoxic effect, with an IC50 of 51.6 μg mL−1, as illustrated in Table 2.2.3. ADME/TOPKAT prediction
Prediction of the pharmacokinetic and pharmacodynamic potential as well as the toxicity properties of the isolated compounds was performed in silico using the ADME/TOPKAT protocol in Discovery Studio 4.5 (Accelrys Inc., San Diego, CA, USA). Regarding human intestinal descriptor, 1, 2, 4, and 7–10 showed good human intestinal absorption levels and thus lay within the 99% absorption ellipse as displayed in the ADMET plot (Fig. 4). Concerning the solubility level, 10 showed optimal solubility, while the rest of the compounds revealed possible or good solubility levels. Also, 4, 9, and 10 revealed medium to low blood–brain barrier (BBB) penetration levels and thus lay inside the 99% BBB confidence eclipse in the ADMET plot (Fig. 4). On the contrary, the rest of the compounds had undefined BBB penetration levels, taking level 4 and thus lay outside the 99% BBB confidence eclipse. Regarding the binding of the compounds with plasma protein, 3–8 and 10 displayed less than 90% plasma protein binding (PPB), while the rest of the compounds showed more than 90% PPB. Additionally, all the isolated compounds showed no inhibition to CYP2D6, but some of the compounds showed certain hepatotoxic effects, such as 3–8 and 10 (Table 3).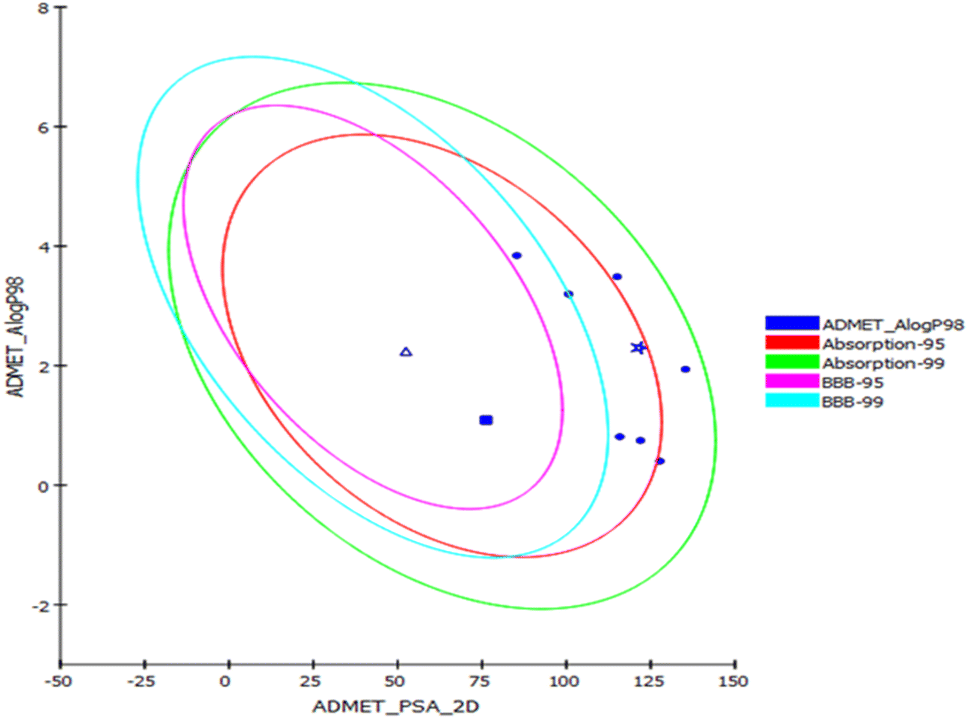 | ||
| Fig. 4 ADMET plot for compounds identified from Aspergillus sp. displaying the 95% and 99% confidence limit ellipses corresponding to the blood–brain barrier (BBB) and the human intestinal absorption. Austalide Z (1) (star); compound 9 (triangle); compound 10 (filled square). | ||
| Compound | Absorption level | Solubility level | BBB level | PPB level | CPY2D6 | Hepatotoxic | PSA-2D | Alog p98 |
|---|---|---|---|---|---|---|---|---|
| a 0, 1, 2, and 3 indicates good, moderate, low, and very low absorption, respectively; 0, 1, 2, 3, 4, and 5 indicates extremely low, very low but possible, low, good, optimal, and too soluble, respectively; 0, 1, 2, 3, and 4 denote very high, high, medium, low, and undefined, penetration via BBB respectively. PBB, plasma protein binding, FALSE means less than 90%, TRUE means more than 90%. NI: non-inhibitor. NT: non-toxic; Tox., toxic. PSA 2D: 2D polar surface area. Alog P98: logarithm of the partition coefficient between n-octanol and water. | ||||||||
| 1 | 0 | 2 | 4 | True | NI | NT | 2.30 | 121.44 |
| 2 | 0 | 1 | 3 | True | NI | NT | 3.20 | 100.63 |
| 3 | 1 | 2 | 4 | False | NI | Tox | 3.49 | 115.08 |
| 4 | 0 | 2 | 2 | False | NI | Tox | 3.84 | 85.33 |
| 5 | 1 | 3 | 4 | False | NI | Tox | 1.94 | 135.33 |
| 6 | 1 | 3 | 4 | False | NI | Tox | 0.40 | 127.74 |
| 7 | 0 | 3 | 4 | False | NI | Tox | 0.81 | 115.85 |
| 8 | 0 | 3 | 4 | False | NI | Tox | 0.75 | 121.98 |
| 9 | 0 | 3 | 2 | True | NI | NT | 2.24 | 52.46 |
| 10 | 0 | 4 | 3 | False | NI | Tox | 1.09 | 76.23 |
Regarding TOPKAT prediction, all the examined isolated compounds displayed no mutagenicity as predicted by the chemical Ames mutagenicity protocol done in silico. Furthermore, 3–5 and 7 showed no carcinogenic effect to both male and female rats (NTP), whereas 9–10 exhibited some carcinogenic potential for female rats and 1–2 and 6–8 for male rats. They displayed rat oral LD50 values between 0.16–9.68 g kg−1-body-weight, with 9–10 showing the highest LD50s of 9.68 and 4.05 g kg−1-body-weight, respectively. Similarly for the rat chronic LOAEL level, all the isolated compounds displayed values between 0.0017–1.6549 g kg−1-body-weight, with 9–10 showing the highest LOAELs of 0.9889 and 1.6549 g kg−1-body-weight, respectively. Regarding skin irritancy, most of the isolated compounds showed no to mild skin irritation. For ocular irritation, most of the isolated compounds showed no to mild irritation except for 6–8 and 10, which showed moderate eye irritation. Thus, it can be concluded that most of the isolated compounds showed reasonable in silico pharmacokinetic, pharmacodynamic, and toxicity properties, and thus could potentially be incorporated in pharmaceutical dosage forms to prevent cancer. Additionally, 9 and 10 that showed the highest fitting scores revealed the best pharmacokinetic and pharmacodynamic properties with slight toxicity, which could be controlled by the given doses when formulated in pharmaceutical preparations (Table 4).
| Compounds | Ames prediction | Rat oral LD50 | Rat chronic LOAEL | Skin irritancy | Ocular irritancy | Rat female NTP | Rat male NTP |
|---|---|---|---|---|---|---|---|
| a Both rat oral LD5 and rat chronic LOAEL are expressed in g kg−1-body-weight. | |||||||
| 1 | Non-mutagen | 0.16 | 0.0038 | Mild | None | Non-carcinogen | Carcinogen |
| 2 | Non-mutagen | 0.29 | 0.0033 | Mild | None | Non-carcinogen | Carcinogen |
| 3 | Non-mutagen | 1.14 | 0.0043 | Mild | Mild | Non-carcinogen | Non-carcinogen |
| 4 | Non-mutagen | 1.43 | 0.0026 | Mild | Mild | Non-carcinogen | Non-carcinogen |
| 5 | Non-mutagen | 1.34 | 0.0036 | Mild | Mild | Non-carcinogen | Non-carcinogen |
| 6 | Non-mutagen | 0.82 | 0.0145 | None | Moderate | Non-carcinogen | Carcinogen |
| 7 | Non-mutagen | 0.89 | 0.0076 | Mild | Moderate | Non-carcinogen | Carcinogen |
| 8 | Non-mutagen | 0.56 | 0.0218 | None | Moderate | Non-carcinogen | Carcinogen |
| 9 | Non-mutagen | 9.68 | 0.9891 | Mild | None | Carcinogen | Non-carcinogen |
| 10 | Non-mutagen | 4.05 | 1.6549 | None | Moderate | Carcinogen | Non-carcinogen |
3 Experimental section
3.1. General experimental procedures
NMR spectra (chemical shifts in ppm) were recorded on a Bruker AVANCE HD III 400 MHz spectrometer (Switzerland). NMR samples were dissolved in methanol-d4 or chloroform-d3, (Sigma Aldrich, Germany) and transferred to 3 mm NMR tubes (Bruker). ECD spectra were recorded on a J-810 spectropolarimeter. HR-ESI-MS analyses were performed on an FTHRMS-Orbitrap (Thermo Finnigan) mass spectrometer. LC-PDA-MS analyses were performed using a Shimadzu LCMS 8045 system coupled with a photodiode array detector (LC-2030/2040) with detection wavelengths of 235, 254, and 280 nm with λmax absorption at 220–400 nm. The LC-PDA-MS system was equipped with an RP-C18 UPLC column (shimpack 2 mm × 150 mm) possessing 2.7 μm particle size using the following gradient elution (acetonitrile (ACN), 0.1% HCOOH in H2O) 0–2 min (10% MeOH); 2–35 min (10% ACN-100% ACN), and 35–40 (100% ACN) with a 0.2 mL min−1 flow rate. The final purification steps were performed using preparative HPLC (Shimadzu, Japan) using a Kromasil C-18RP semi-preparative column (10 mm × 250 mm) at a flow rate of 5 mL min−1 and UV detection at wavelength of 254 nm with a λmax absorption at 220–400 nm. Medium pressure liquid chromatography was performed on a Puriflash 4125 system equipped with a PDA detector. Normal phase column chromatography was performed using silica gel 60 (0.04–0.063, Merck, Germany). TLC analyses were performed using normal phase silica gel pre-coated plates F254 (Merck, Germany). All the consumed solvents were of analytical grade, while those used for HPLC analyses were of HPLC grade.3.2. Fungal material
The fungus Aspergillus sp. was isolated from the inner tissues of the marine soft coral Sinularia sp. The soft coral was collected from the Red Sea close to Sharm El-Sheikh, Egypt, in September 2018 at 5–7 meters depth. For isolation of the fungal strain, the soft coral was rinsed with distilled water, and then surface sterilization was performed using 70% ethanol for 2 min. Small samples from the inner tissues of the soft coral were aseptically cut using a sterilized blade and pressed onto malt agar plate (15 g L−1 malt extract, 15 g L−1 agar, 0.2 g L−1 chloramphenicol to suppress bacterial growth, with the pH adjusted to 7.4–7.8 using 10% NaOH). After incubation at 25 °C, the fungal strain under investigation was found to grow out of the sponge tissue. Pure fungal strains were grown by repeated re-inoculation on fresh culture media.3.3. Identification of the fungal strain
The isolated fungal strain was identified as Aspergillus sp. using a molecular biological protocol by DNA amplification and by sequencing of the ITS region as previously reported.38 The obtained data of sequencing were submitted to GenBank with the accession number OK035701.3.4. Cultivation, extraction, and isolation
Small scale fermentation of the fungal strain was performed on solid rice culture media (100 g rice in 110 mL distilled water, autoclaved for 20 min at 121 °C) in a 1 L Erlenmeyer flask (2 flasks were used). The culture was incubated for 30 days at 25 °C under static conditions. Large-scale fermentation was performed on solid rice medium using 12 Erlenmeyer flasks (1 L each). Fungal cultures were extracted using ethyl acetate (EtOAc), and the combined extracts were then evaporated in vacuum and the residue was partitioned between n-hexane and 90% aqueous methanol yielding about 3 g of reddish-brown solid residue.The defatted extract was then applied to vacuum liquid chromatography (VLC) packed with normal phase silica gel as the stationary phase and applying a stepwise gradient as follows: 80% n-hexane in ethyl acetate to 100% ethyl acetate, followed by dichloromethane in methanol from 95% to 50%. Eluted fractions were analyzed using TLC and similar fractions were pooled together yielding 7 major fractions (FR1–7). Fraction FR3 (500 mg) was fractionated using normal phase medium pressure liquid chromatography eluted with a linear gradient elution from 80% n-hexane: EtOAc to 30% n-hexane: EtOAc. Final purification was performed using RP-semi-preparative HPLC, eluted with a linear gradient elution from 70% ACN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O to 95% ACN:H2O with the final purification of two compounds 3 (25 mg) and 4 (30 mg). Fraction FR4 (350 mg) was directly applied to semi-preparative HPLC and subsequently eluted with a linear gradient elution from 50% ACN:H2O to 90% ACN:H2O yielding 1 (9 mg), 5 (15 mg), and 10 (30 mg). Fraction FR5 (700 mg) was fractionated using normal phase medium pressure liquid chromatography (Puriflash®) eluted with a linear gradient elution from dichloromethane: methanol gradient (from 100% to 80%) yielding three major sub-fractions FR5-V1–FR5-V3. Fraction FR5-V1 was purified using RP-semi-prep-HPLC-PDA eluted with a linear gradient elution from 20% ACN:H2O to 60% ACN:H2O yielding 2 (12 mg) and 9 (20 mg). Similarly, fraction FR5-V3 was purified using semi-prep HPLC and eluted using 30% ACN:H2O to 60% ACN:H2O yielding 6 (11 mg), 7 (7 mg), and 8 (4 mg).
:H2O to 95% ACN:H2O with the final purification of two compounds 3 (25 mg) and 4 (30 mg). Fraction FR4 (350 mg) was directly applied to semi-preparative HPLC and subsequently eluted with a linear gradient elution from 50% ACN:H2O to 90% ACN:H2O yielding 1 (9 mg), 5 (15 mg), and 10 (30 mg). Fraction FR5 (700 mg) was fractionated using normal phase medium pressure liquid chromatography (Puriflash®) eluted with a linear gradient elution from dichloromethane: methanol gradient (from 100% to 80%) yielding three major sub-fractions FR5-V1–FR5-V3. Fraction FR5-V1 was purified using RP-semi-prep-HPLC-PDA eluted with a linear gradient elution from 20% ACN:H2O to 60% ACN:H2O yielding 2 (12 mg) and 9 (20 mg). Similarly, fraction FR5-V3 was purified using semi-prep HPLC and eluted using 30% ACN:H2O to 60% ACN:H2O yielding 6 (11 mg), 7 (7 mg), and 8 (4 mg).
3.5. Compound characterization
Austalide Z (1): pale-yellow amorphous powder. 1H (400 MHz) and 13C (100 MHz) data in CDCl3, as illustrated in Table 1. ECD: (c = 5.5 × 10−5 M, MeCN) λ [nm], (Δε) = 332 (0.26), 296sh (−0.79), 266 (−3.88), 240sh (−0.87), 229 (1.45), 219sh (−1.16), 213 (−2.11); [α]20D = (c 0.10, CDCl3); LC-ESI-MS revealed a pseudomolecular ion peak at m/z 505 and 503 [M + H]+ and [M − H]−, respectively, whereas HR-ESI-MS [M + H]+ showed a peak at m/z 505.2068 (calculated. for m/z C26H33O10, 505.2090) corresponding to the molecular formula C26H32O10. Different spectra are available in the ESI (Fig. S1†).3.6. Cytotoxicity assay
MTT assay was carried out as previously described39 with some modifications. Basically, 100 μL of the tested compound was dissolved in 5% DMSO and RPMI (Roswell Park Memorial Institute) tissue culture medium was then added to each well to reach a final concentration of X μg mL−1. Control wells contained two aliquots of 100 μL of tissue culture medium (MEM + fetal bovine serum in a ratio 9:1), 100 μL sterile DMSO (5%), and RPMI tissue culture medium. Following the specified incubation period (24 h) at 37 °C, the wells were washed with PBS (phosphate Buffered Saline) and the cells were incubated with MTT solution (2 mg mL−1) at 100 μL per each well for 1 h at 37 °C. The supernatants were then removed by decantation and the cells were treated with 100 μL of DMSO per each well to dissolve the formazan crystals formed in the viable metabolically active cells. Elutes of the 8 wells of each test isolate were collected and the absorbance was measured at 540 nm. Control wells were similarly treated and the percentage cytotoxicity was calculated employing the following formula;39,40 cytotoxicity% = 1 − {A540 of test culture/A540 of control culture} × 100.
3.7. ADME/TOPKAT prediction
ADME/TOPKAT (absorption, distribution, metabolism, excretion, and toxicity) prediction was performed for the isolates using Discovery Studio 4.5 (Accelrys Inc., San Diego, CA, USA). The aqueous solubility, plasma protein binding prediction (PPB), human intestinal absorption, blood–brain barrier penetration (BBB), cytochrome P450 (2D6), and hepatotoxicity level were chosen as the ADME descriptors. Meanwhile, the carcinogenic effect on male and female rat FDA, ocular and skin irritation, rat oral LD50, Ames mutagenicity, and rat chronic LOAEL were selected as the toxicity parameters.41,424 Conclusions
In-depth chemical investigation of the ethyl acetate extract of marine fungus Aspergillus sp. isolated from the inner tissues of the soft coral Sinularia sp. resulted in the isolation of one new meroterpenoid austalide Z in addition to nine known compounds belonging to the austalide class, prenylated indole diketopiperazine alkaloids, and phthalic acid derivatives. In vitro cytotoxic evaluation using the MTT assay revealed that most of the examined compounds showed weak to moderate activities, with austalide Z displaying substantial cytotoxic activity. ADME/TOPKAT showed that most of the isolated compounds displayed reasonable pharmacokinetic, pharmacodynamic, and toxicity properties. Thus, it can be concluded that Aspergillus sp. could act as a source of drug leads that could alleviate cancer with reasonable pharmacokinetic and pharmacodynamic properties that could be further improved via various treatments and hence could be incorporated in pharmaceutical dosage forms. However, additional in vivo studies followed by preclinical trials are highly recommended to be conducted on the isolated compounds to confirm the obtained results.Author contributions
Conceptualization, A. S and M. E.; methodology, A. E, M. E., T. K., M. K.; software, F. Y.; validation, R. K., A. S and T. K.; investigation, A. E.; F. S., M. E., M. K. and T. K.; resources, A. S and R. K.; writing—original draft preparation, M. E., A. E., F. Y. And M. K.; writing—review and editing, A. S., T. K., R. K.; supervision, A. S. and R. K.; project administration, A. E. funding acquisition, A. S. and R. K. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research work of M. E, A. E and A. S. was funded by The Science, Technology, and Innovation Funding Authority; STIFA through Joint Egyptian Japanese Scientific Cooperation (JEJSC) grant no. 28925 entitled “Chemical biology on innovative biologically active metabolites from endosymbionts in Egyptian marine invertebrates”. The research work of T. K. and M. K. was supported by the National Research, Development and Innovation Office (K138672). M. S. Elnaggar thanks the Ministry of Higher Education of the Arab Republic of Egypt for fully funding postdoctoral mission. The authors would like thank the CeMSA@HHU (Center for Molecular and Structural Analytics @ Heinrich Heine University) for recording the mass-spectrometric and the NMR spectroscopic data.Notes and references
- N. G. Gomes, F. Lefranc, A. Kijjoa and R. Kiss, Mar. Drugs, 2015, 13, 3950–3991 CrossRef CAS PubMed.
- R. X. Tan and W. X. Zou, Nat. Prod. Rep., 2001, 18, 448–459 RSC.
- M. C. Leal, C. Sheridan, R. Osinga, G. Dionísio, R. J. M. Rocha, B. Silva, R. Rosa and R. Calado, Mar. Drugs, 2014, 12, 3929–3952 CrossRef CAS PubMed.
- V. J. Paul and M. P. Puglisi, Nat. Prod. Rep., 2004, 21, 189–209 RSC.
- J. F. Imhoff, Mar. Drugs, 2016, 14, 19 CrossRef PubMed.
- J. Silber, A. Kramer, A. Labes and D. Tasdemir, Mar. Drugs, 2016, 14, 137 CrossRef PubMed.
- A. M. Elissawy, M. El-Shazly, S. S. Ebada, A. B. Singab and P. Proksch, Mar. Drugs, 2015, 13, 1966–1992 CrossRef CAS PubMed.
- M. S. Elnaggar, S. S. Ebada, M. L. Ashour, W. Ebrahim, A. Singab, W. Lin, Z. Liu and P. Proksch, Fitoterapia, 2017, 116, 126–130 CrossRef CAS PubMed.
- A. M. Elissawy, S. S. Ebada, M. L. Ashour, M. El-Neketi, W. Ebrahim and A. B. Singab, Phytochem. Lett., 2019, 29, 1–5 CrossRef CAS.
- M. S. Elnaggar, S. S. Ebada, M. L. Ashour, W. Ebrahim, W. E. Müller, A. Mándi, T. Kurtán, A. Singab, W. Lin and Z. Liu, Tetrahedron, 2016, 72, 2411–2419 CrossRef CAS.
- M. S. Elnaggar, W. Ebrahim, A. Mándi, T. Kurtán, W. E. G. Müller, R. Kalscheuer, A. Singab, W. Lin, Z. Liu and P. Proksch, RSC Adv., 2017, 7, 30640–30649 RSC.
- M. Bai, C. H. Gao, K. Liu, L. Y. Zhao, Z. Z. Tang and Y. H. Liu, J. Antibiot., 2021, 74, 821–824 CrossRef CAS PubMed.
- F. S. Youssef, M. L. Ashour, A. N. B. Singab and M. Wink, Mar. Drugs, 2019, 17, 559 CrossRef CAS PubMed.
- R. Zhang, W. He, Y. Wang, J. Zhao, R. Zhou, L. Li, Y. He, S. Cen and L. Yu, J. Antibiot., 2021, 74, 225–232 CrossRef CAS PubMed.
- A. M. Elissawy, S. S. Ebada, M. L. Ashour, F. C. Özkaya, W. Ebrahim, A. B. Singab and P. Proksch, Phytochem. Lett., 2017, 20, 246–251 CrossRef CAS.
- Y. Matsuda and I. Abe, Nat. Prod. Rep., 2016, 33, 26–53 RSC.
- B. Sontag, N. Arnold, W. Steglich and T. Anke, J. Nat. Prod., 1999, 62, 1425–1426 CrossRef CAS.
- l. liu and h. ruan, Phytochem. Lett., 2022, 47, 12 Search PubMed.
- T. V. Antipova, K. V. Zaitsev, Y. F. Oprunenko, A. Y. Zherebker, G. K. Rystsov, M. Y. Zemskova, V. P. Zhelifonova, N. E. Ivanushkina and A. G. Kozlovsky, Bioorg. Med. Chem. Lett., 2019, 29, 6 CrossRef PubMed.
- K. J. Kim, J. Lee, W. Wang, Y. Lee, E. Oh, K. H. Park, C. Park, G. E. Woo, Y. J. Son and H. Kang, Int. J. Mol. Sci., 2021, 22, 12 Search PubMed.
- J. X. Peng, X. M. Zhang, W. Wang, T. J. Zhu, Q. Q. Gu and D. H. Li, Mar. Drugs, 2016, 14, 9 CrossRef PubMed.
- J. Wang, Y. N. Sang, S. Q. Tang and P. Zhang, Chem. Nat. Compd., 2020, 56, 670–673 CrossRef CAS.
- Y. Zhou, A. Mándi, A. Debbab, V. Wray, B. Schulz, W. E. Müller, W. Lin, P. Proksch, T. Kurtán and A. H. Aly, Eur. J. Org. Chem., 2011, 2011, 6009 CrossRef CAS.
- A. E. Dejesus, R. M. Horak, P. S. Steyn and R. Vleggaar, J. Chem. Soc., Chem. Commun., 1983, 716–718, 10.1039/c39830000716.
- T. K. Ma, P. J. Parsons and A. G. M. Barrett, J. Org. Chem., 2019, 84, 4961–4970 CrossRef CAS PubMed.
- L. A. Paquette and M. M. Schulze, Heterocycles, 1993, 35, 585–589 CrossRef CAS PubMed.
- L. A. Paquette, T. Z. Wang and M. R. Sivik, J. Am. Chem. Soc., 1994, 116, 2665–2666 CrossRef CAS.
- F. S. Youssef, E. Alshammari and M. L. Ashour, Int. J. Mol. Sci., 2021, 22, 1866 CrossRef CAS PubMed.
- F. Wang, Y. Fang, T. Zhu, M. Zhang, A. Lin, Q. Gu and W. Zhu, Tetrahedron, 2008, 64, 7986–7991 CrossRef CAS.
- F. Xie, X. B. Li, J. C. Zhou, Q. Q. Xu, X. N. Wang, H. Q. Yuan and H. X. Lou, Chem. Biodiversity, 2015, 12, 1313–1321 CrossRef CAS PubMed.
- T. V. Antipova, K. V. Zaitsev, Y. F. Oprunenko, A. Ya. Zherebker, G. K. Rystsov, M. Y. Zemskova, V. P. Zhelifonova, N. E. Ivanushkina and A. G. Kozlovsky, Bioorg. Med. Chem. Lett., 2019, 29, 126708 CrossRef CAS PubMed.
- J. Peng, X. Zhang, W. Wang, T. Zhu, Q. Gu and D. Li, Mar. Drugs, 2016, 14, 131 CrossRef PubMed.
- Y. Zhou, A. Debbab, V. Wray, W. Lin, B. Schulz, R. Trepos, C. Pile, C. Hellio, P. Proksch and A. H. Aly, Tetrahedron Lett., 2014, 55, 2789–2792 CrossRef CAS.
- T. V. Antipova, K. V. Zaitsev, Y. F. Oprunenko, A. Y. Zherebker, G. K. Rystsov, M. Y. Zemskova, V. P. Zhelifonova, N. E. Ivanushkina and A. G. Kozlovsky, Bioorg. Med. Chem. Lett., 2019, 29, 126708 CrossRef CAS PubMed.
- L. A. Paquette, T.-Z. Wang and M. R. Sivik, J. Am. Chem. Soc., 1994, 116, 2665–2666 CrossRef CAS.
- L. A. Paquette, T.-Z. Wang and M. R. Sivik, J. Am. Chem. Soc., 1994, 116, 11323–11334 CrossRef CAS.
- A. Mándi and T. Kurtán, Nat. Prod. Rep., 2019, 36, 889–918 RSC.
- J. Kjer, A. Debbab, A. H. Aly and P. Proksch, Nat. Protoc., 2010, 5, 479–490 CrossRef CAS.
- A. M. Saliba, A. Filloux, G. Ball, A. S. Silva, M.-C. Assis and M.-C. Plotkowski, Microb. Pathog., 2002, 33, 153–166 CrossRef CAS PubMed.
- J. Murakami, K. Kishi, K. Hirai, K. Hiramatsu, T. Yamasaki and M. Nasu, Int. J. Antimicrob. Agents, 2000, 15, 103–109 CrossRef CAS PubMed.
- A. Mollica, G. Zengin, S. Durdagi, R. Ekhteiari Salmas, G. Macedonio, A. Stefanucci, M. P. Dimmito and E. Novellino, J. Biomol. Struct. Dyn., 2019, 37, 726–740 CrossRef CAS PubMed.
- F. S. Youssef, E. Ovidi, N. M. A. Musayeib and M. L. Ashour, Plants, 2021, 10, 1953 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1–S38 comprising 1H NMR, APT, 2D NMR and ESI-MS data of the isolated compounds. See DOI: https://doi.org/10.1039/d3ra02632a |
| ‡ These authors contributed equally to the manuscript. |
| This journal is © The Royal Society of Chemistry 2023 |