
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-pot carboxyl enrichment fosters water-dispersibility of reduced graphene oxide: a combined experimental and theoretical assessment†
Francesco
Amato
 *a,
Alessandro
Motta
ab,
Leonardo
Giaccari
a,
Roberto
Di Pasquale
a,
Francesca Anna
Scaramuzzo
c,
Robertino
Zanoni
a and
Andrea Giacomo
Marrani
*a
*a,
Alessandro
Motta
ab,
Leonardo
Giaccari
a,
Roberto
Di Pasquale
a,
Francesca Anna
Scaramuzzo
c,
Robertino
Zanoni
a and
Andrea Giacomo
Marrani
*a
aDipartimento di Chimica, Università di Roma La Sapienza, p.le A. Moro 5, I-00185 Rome, Italy. E-mail: francesco.amato@uniroma1.it; andrea.marrani@uniroma1.it; Tel: +39 0649913568 Tel: +39 0649913316
bConsorzio INSTM, UdR Roma “La Sapienza”, p.le A. Moro 5, I-00185 Rome, Italy
cDipartimento di Scienze di Base e Applicate per l'Ingegneria (S.B.A.I.), Università di Roma La Sapienza, Via del Castro Laurenziano 7, I-00161 Rome, Italy
First published on 17th January 2023
Abstract
Graphene, one of the allotropic forms of carbon, has attracted enormous interest in the last few years due to its unique properties. Reduced graphene oxide (RGO) is known as the nanomaterial most similar to graphene in terms of electronic, chemical, mechanical, and optical properties. It is prepared from graphene oxide (GO) in the presence of different types of reducing agents. Nevertheless, the application of RGO is still limited, owing to its tendency to irreversibly aggregate in an aqueous medium. Herein, we disclosed the preparation of water-dispersible RGO from GO previously enriched with additional carboxyl functional groups through a one-pot reaction, followed by chemical reduction. This novel and unprecedentedly reported reactivity of GO toward the acylating agent succinic anhydride (SA) was experimentally investigated through XPS, Raman, FT-IR, and UV-Vis, and corroborated by DFT calculations, which have shown a peculiar involvement in the functionalization reaction of both epoxide and hydroxyl functional groups. This proposed synthetic protocol avoids use of sodium cyanide, previously reported for carboxylation of graphene, and focuses on the sustainable and scalable preparation of a water-dispersible RGO, paving the way for its application in many fields where the colloidal stability in aqueous medium is required.
1. Introduction
Graphene is a carbon-based bidimensional material prepared through top–down and bottom–up approaches.1–6 In the first route, graphene is prepared from graphite through its exfoliation, sometimes in presence of intercalating agents and, in the second one, through chemical vapor deposition (CVD) from a mixture of methane/hydrogen. While the first approach is hampered by some drawbacks, such as low yield of production, low purity and time-consuming processes, the second one is limited to the growth of graphene sheets onto transition metals and alloys surfaces, which narrows its bulk production for practical applications. An innovative synthetic method for the preparation of graphene using the flash Joule heating from inexpensive carbon sources was also recently reported with encouraging results, though its synthetic apparatus may not be easily available.7GO, the oxidized form of graphene prepared through the chemical oxidation of graphite,8,9 is a non-stoichiometric bidimensional material, and is constituted prevalently by sp2-hybridized carbon atoms and oxygen-based functional groups. Among these, the most abundant are epoxide and hydroxyl groups, which are mainly localized in the basal plane of the sheet, differently from the less numerous carboxyl groups, confined at the edge. Moreover, both carbonyl and phenol-like hydroxyl groups are present in low percentages and most usually confined at the edges of the layer.10
Owing to the simultaneous presence of aromatic domains and oxygen-based functional groups in the layers, the chemical modifications of GO have opened new applicative horizons and opportunities. In particular, these modifications can be performed through non-covalent and covalent approaches, with the latter undoubtedly more explored.10,11
As reported in the pioneering review of S. Guo et.al., many types of chemoselective covalent functionalizations have been performed, taking advantage of the oxygen-based functional groups (epoxide, hydroxyl and carboxyl) as well as C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds present in the structure of GO. In particular, exhaustively disclosed examples of chemical modifications include silanization, esterification, epoxide ring-opening and radical addition to CC, in addition to multi-functionalization strategies with the relative advantages and limitations of each one.11
C bonds present in the structure of GO. In particular, exhaustively disclosed examples of chemical modifications include silanization, esterification, epoxide ring-opening and radical addition to CC, in addition to multi-functionalization strategies with the relative advantages and limitations of each one.11
Considering the low amount of carboxyl groups in the structure of GO, a route able to effectively increase their number via a carboxylation reaction would be highly desirable to broaden the range of its possible applications.12,13 For example, a carboxyl-rich GO could be used as a platform in metal-ion removal from wastewater, employed in amide coupling with amine-rich nanomaterials, as well as in the preparation of supramolecular hydrogels.14–18
Examples of carboxylation include the one reported by A. Bakandritsos et al.,19,20 who obtained a carboxyl-rich GO from cyanographene hydrolyzed with an aqueous solution of HNO3 at 20% in order to transform the –CN groups into –COOH groups. The content of this functional groups was estimated as 9.3% on the basis of the deconvoluted C 1s XPS spectrum.19–21
Another method of GO carboxylation reported in the literature is based on the selective derivatization of hydroxyls with chloroacetic acid in the presence of sodium hydroxide.22–24 This type of functionalization, although practical, suffers from the reduction of GO induced by the strong base, which changes the physical–chemical properties of GO.25–27 The influence of different amounts of sodium hydroxide in the carboxylation of GO in presence of chloroacetic acid was deeply investigated by S. Guo et al., proving that only in presence of high amounts of base the functionalization occurs quantitatively. In particular, GO previously carboxylated in presence of chloroacetic acid and 3 M NaOH in water, was doubly functionalized combining an epoxide-ring opening with BocNH-PEG10-NH2, followed by amide coupling in presence of EDC/NHS. Since the strong basic conditions necessary for carboxylation lead to a simultaneous loss of oxygenated functionalities, the authors observed a low efficiency of functionalization in comparison to a direct epoxide-ring aperture reaction. This result suggests that, in the experimental conditions of high pH, carboxylation is not an effective approach to functionalize GO.25
A further route to GO carboxylation makes use of maleic anhydride, involving two different and competitive types of reactivity of GO: hydroxyl groups can undergo esterification, and diene groups may react with the dienophile maleic anhydride according to a Diels–Alder (DA) reaction, followed by acid hydrolysis, as presented by Brisebois et al.28–30 In particular, this [4 + 2] pericyclic reaction was deeply investigated through 13C-SS NMR measurements, and a dual reactivity of GO was observed toward maleic anhydride, at the edges and on the basal plane, analogously to 1,3-butadiene and cis-3,5-cyclohexadiene-1,2-diol systems, respectively. It is noteworthy that this work proves the existence of cis diene in the structure of GO. A decrease of oxygenated species at prolonged reaction times was observed, as a consequence of thermal decomposition of the labile oxygenated groups of GO or by a possible retro-DA reaction.29
RGO is the closest nanomaterial to graphene, both from the morphological and physical-chemical point of view. In fact, it is a bidimensional nanomaterial prepared from GO with an atomic thickness that shows, differently from an ideal sheet of graphene, plenty of structural defects resulting from deoxygenation reaction processes, in addition to residual oxygen-based functional groups. Notably, the restoration of the extended π-conjugated network after the chemical reduction of GO makes this material very appealing for optical and electronic applications. On the other hand, its dispersibility in water is strongly reduced, due to loss of the abundant polar functional groups present in the structure of the parent GO.9,31
In this work, we present an unprecedentedly investigated one-pot carboxylation of GO, employing succinic anhydride (SA) followed by chemical reduction of GO in presence of L-ascorbic acid.32,33 Inspired both by the O-acylation and O-alkylation of phenols in the presence of sodium carbonate as a base in anhydrous aprotic polar solvents, we propose the same synthetic protocol on GO.34,35 As a control experiment, we conducted the same functionalization of GO using tetra-fluorosuccinic anhydride (F-SA), benefiting from the high sensitivity of XPS to F atoms. All the experimental results were supported by DFT calculations, which allowed to investigate the energetics of various possible reaction mechanisms. Particular attention was devoted to explore the feasibility of a reaction path involving base-catalyzed epoxide ring-opening in presence of SA.36,37 Considering the absence of both side-reactions and degradation phenomena on GO during the carboxyl-functionalization, we explored the subsequent reduction of the obtained carboxyl-rich GO in presence of the green reducing agent L-ascorbic acid, in order to remove the residual epoxide and hydroxyl functional groups. In the end, this chemical reduction affords a water-dispersible RGO, wherein the numerous carboxyl functional groups ensure an improved colloidal kinetic stability in water.
To the best of our knowledge, this is the first report of such synthetic route for the preparation of water-dispersible RGO.
2. Experimental
Commercial reagents and solvents were purchased from Sigma-Aldrich, Fluka, Alfa Aesar, VWR, TCI and used as received, without further purification, unless otherwise stated.Pristine GO (GO) was synthesized by following a reported procedure with some modifications.38,39 Briefly, 69 mL of H2SO4 96 wt% were added to a mixture of synthetic graphite (3.0 g) and NaNO3 (1.5 g) and the mixture was cooled at 0 °C. Then, KMnO4 (9.0 g) was added in portions and the mixture was stirred at 35 °C for 30 min. Subsequently, distilled water (138 mL) was added slowly and the mixture was heated at 98 °C for 15 min. Next, additional distilled water (420 mL) and H2O2 (3 mL, 30% v/v) were added. For workup, a filtration through a filter paper was performed and the solid obtained was centrifuged at 4000 rpm for 30 min in presence of HCl 1.12 M and distilled water (four times). During this step of purification, the supernatant was whenever discharged away and replaced with distilled water. Next, the solid was transferred in a round-bottom flask, then distilled water was added, and the mixture was stirred for a night to ensure a proper exfoliation. Next, the dispersion was sonicated for 30 min and centrifuged at 3000 rpm for 40 min in order to recover the yellowish supernatant and discharge, as precipitate, the graphite oxide that may be present. This procedure was repeated until obtainment of an almost colorless supernatant. Finally, the dispersion of GO was dried under vacuum by means of a rotavapor at 35.0 °C and the solid powder stored in a desiccator.
Carboxyl-functionalized GO (C-GO), 25.5 mg of GO and 25.5 mg of K2CO3 (0.18 mmol) were added to 30 mL of dry DMF. Then, 130 mg of succinic anhydride (SA, 1.30 mmol) were added, and the mixture was sonicated for 30 min. Subsequently, the mixture was stirred at 50 °C for 24 h under inert atmosphere (N2). After this time, the stirring was stopped and, subsequently, 160 mL of diethyl ether were added in order to precipitate the product. The mixture was then filtered through a filter paper and the solid collected washed with additional 40 mL of diethyl ether. Finally, after the complete solvent evaporation, 76.0 mg of a brown solid were obtained.
For the synthesis of the Fluorinated-GO (F-GO), 13.9 mg of GO and 14.0 mg of K2CO3 (0.10 mmol) were added to 15 mL of dry DMF. Then, 70 μL of tetra-fluorosuccinic anhydride (0.65 mmol), were added and the mixture was sonicated for 30 min. Subsequently, the mixture was stirred at 50 °C for 24 h under inert atmosphere (N2). After this time, the stirring was stopped and 80 mL of diethyl ether were added in order to precipitate the product. The mixture was then filtered through a filter paper and the solid collected washed with additional 20 mL of diethyl ether. Finally, after the complete solvent evaporation, 55.0 mg of a brown solid were collected.
Carboxyl-rich Reduced Graphene Oxide (C-RGO) was synthesized from C-GO. Briefly, 6 mg of C-GO were dispersed in 20 mL of distilled water and the pH was adjusted to ≅7 by adding NaOH 0.5 M. After, 60 mg of L-ascorbic acid were added to the aqueous dispersion and the mixture was stirred at room temperature for 72 h. Finally, after this time, the black dispersion was centrifuged at 4000 rpm for 30 min (five times, four in water and the last one in ethanol) discharging away the supernatant and keeping the solid black-precipitate.
Reduced Graphene Oxide (RGO) was synthesized in the same experimental conditions of C-RGO from pristine GO, rather than C-GO.
For the acid/base back-titration, a known amount of GO (about 10 mg) was dispersed in 10 mL of distilled water. Subsequently, 200 μL of HCl 0.1 M (Normex) was added and then the resulting solution was titrated with a 0.5 M solution of NaOH (Normex). For the quantification of acid sites, a Gran Plot analysis was performed. By plotting the μmol of H+ and OH−vs. the volume (μL) of titrant, two linear regions were identified. The resulting amounts of titrant at the equivalent point (μmoleq1 and μmoleq2) were extrapolated through a linear fitting. Finally, the total number of acid active sites, expressed in μmol g−1, were calculated by subtracting μmoleq2 from μmoleq1 and dividing the resulting number by the amount of GO analyzed. Back-titrations and Gran plot analyses were repeated at least three times.
For the XPS characterization, an H-terminated Si(100) surface was used as a support for the drop-casting of 50 μL of 0.06 mg mL−1 graphene derivatives dispersions.
UV-Vis measurements were carried out with a Cary 500 UV-Vis-NIR spectrophotometer. All the spectra were recorded at room temperature using 10 mm path-length quartz cuvettes.
Atomic Force Microscopy (AFM) measurements were performed in tapping mode on a Veeco AFM Multimode equipped with Nanoscope IIIa and using a RTESP Bruker tip with nominal parameters r = 8 nm, f = 300 kHz, k = 40 N m−1. The images were recorded with a 512 × 512 pixels resolution and corrected by polynomial background filters using the software Gwyddion 2.31. The measurements were performed on an H-terminated Si(100) surface.
Infrared spectra were recorded on a Varian FT-IR 660 instrument in the range of 4000–400 cm−1 with a spectral resolution of 4 cm−1 and 32 scans in KBr pellets.
Raman spectra were run at room temperature in backscattering geometry with an inVia Renishaw micro-Raman spectrometer equipped with an air-cooled CCD detector and super-Notch filters. An Ar+ ion laser (λlaser = 514 nm) was used, coupled to a Leica DLML microscope with a 20× objective. The resolution was 2 cm−1 and spectra were calibrated using the 520.5 cm−1 line of a silicon wafer. Raman spectra were acquired in several (6–10) different spots on the surface of the samples. For GO and its derived-materials, each spectrum was acquired with 1% of power, 10 seconds of spectral acquisition, and 20 scans. Raman spectrum of succinic anhydride was obtained with 1% of power, 10 seconds of spectral acquisition, and 5 scans.
XPS measurements were carried out using a modified Omicron NanoTechnology MXPS system equipped with a monochromatic Al Kα (hν = 1486.7 eV) X-ray source (Omicron XM-1000), operating the anode at 14 kV and 16 mA. The C 1s photoionization region was acquired using an analyzer pass energy of 20 eV, while the survey spectra were acquired with a pass energy of 50 eV. A take-off angle of 21° with respect to the sample surface normal was adopted. The experimental spectra were theoretically reconstructed by fitting the secondary electrons background to a Shirley function and the elastic peaks to pseudo-Voigt functions described by a common set of parameters: position, full-width at half-maximum (FWHM), Gaussian–Lorentzian ratio. The relative amount of the different oxygenated functional groups was determined through the area of the peaks within the curve fitting envelope of the C 1s region, with an uncertainty of ±10%. For the sake of comparison between different samples, all intensities (areas) of the peaks were normalized to the one of the contribution from sp2 C atoms in the CC network (peak at 284.8 eV).
3. Computational methods
Geometry optimizations of all reactants, products, intermediates, and transition states were carried out along the entire reaction paths. Calculations were performed adopting the B3LYP hybrid GGA functional.40 The standard all-electron 6-311G** triple zeta basis set41 plus polarization was used for all the atoms. Unrestricted calculations were always performed owing to a RHF/UHF instability verified in some cases. Molecular geometry optimization of stationary points was carried out without symmetry constrains and using analytical gradient techniques. The transition states were searched with the synchronous, transit-guided quasi-Newton (STQN) method.42 Frequency analysis was performed in order to verify stationary and saddle points and to obtain thermochemical information at standard conditions (298.15 K and 1 atm). Owing to the presence of anionic species among the intermediates, a single point energy calculation with the addition of diffuse functions in the basis set was performed on the optimized geometry of all the investigated species. The force constants were determined analytically with the harmonic approximation. In order to evaluate ΔG of solvation, the SMD variation43 of the Polarized Continuum formalism (PCM)44 was adopted. All calculations were performed using the G16 code45 on Linux cluster systems. Molecular graphics were produced by the CHEMCRAFT graphical package.46Pristine and oxidized graphene sheets were modeled adopting a hydrogen passivated graphene cluster (Fig. 1) of 48 carbon atoms (C48) as a starting point.47
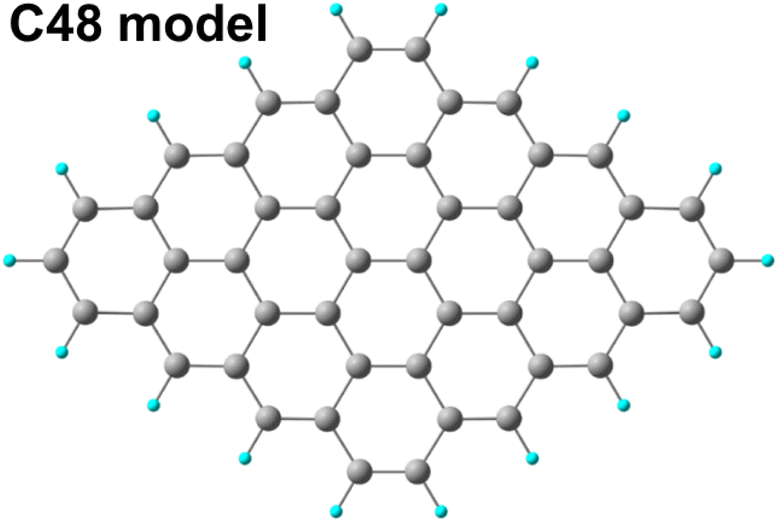 | ||
| Fig. 1 Graphene cluster C48 used as model in the DFT calculations. | ||
Oxidized species were then appended on the basal plane and on the edges to model local environment on the graphene oxide sheet.
4. Results and discussion
4.1. AFM morphological characterization
The pristine GO material obtained following a modified Hummers' method (see Experimental section) was studied with AFM for its morphological characterization. This revealed the presence of isolated and large bi-layers of GO (Fig. S1a†) with lateral size in the order of micrometers with an average thickness of 1.65 ± 0.15 nm (Fig. S1b†). The presence of bi-layers in the AFM micrographs ensures that the starting material used for the further chemical functionalizations is effectively well-exfoliated.4.2. UV-Vis, FTIR and Raman spectroscopy characterization of carboxyl-rich GO
Subsequently, GO and its derivatives were characterized with routine spectroscopic techniques. The powder of GO is easily dispersed in water and its UV-Vis spectrum (Fig. 2, black line) shows a broad absorption in the full range of visible light, with a tail up to 800 nm in addition to the main peak at 232 nm, and a shoulder at 300 nm due to π–π* and n–π* electron transitions, respectively. The carboxylation reaction of GO with SA yielded a powder (C-GO) well dispersible in water (roughly up to 1 mg mL−1), yet less than GO. Aqueous dispersions of these samples can be visually inspected in Fig. 3. The UV-Vis spectrum of the C-GO water dispersion shows an increased absorbance compared to GO, particularly below 232 nm and above 300 nm (Fig. 2, red line), calling for the occurrence of chemical modifications upon reaction of GO with SA. More specifically, the increased absorbance above 300 nm hints at an increased similarity with reduced graphene oxide, suggesting a probable modification of the oxygen-based functional groups.48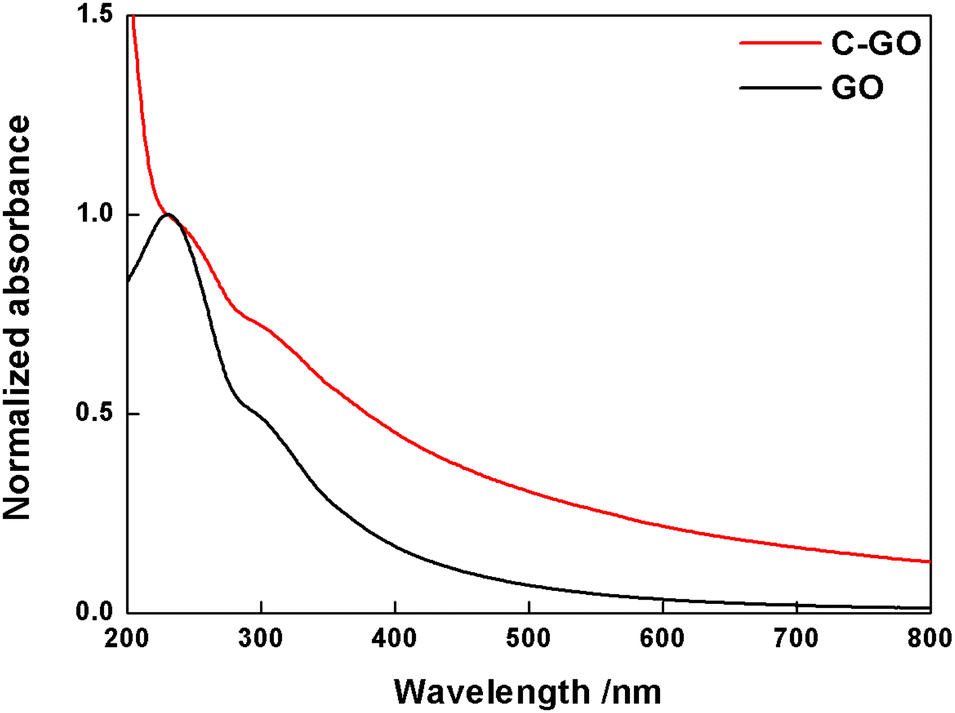 | ||
| Fig. 2 Normalized UV-Vis spectra of GO (black line) and C-GO (red line) recorded in water at room temperature. | ||
 | ||
| Fig. 3 Aqueous dispersions of the samples addressed in this work: pristine GO (A), C-GO (B), C-RGO (C) and RGO (D). | ||
The FT-IR spectrum of GO (Fig. 4a, black line) shows the typical aspect, with a broad and intense band observed at 3380 cm−1 due to the stretching modes of O–H bond, arising from residual adsorbed water molecules, a CO feature at 1723 cm−1, belonging to aldehydes, ketones and carboxyl groups, a further one at 1614 cm−1, assigned to the bending modes of water molecules, and a last one at 1218 cm−1, assigned to the C–O stretching mode.11 The FT-IR spectrum of C-GO (Fig. 4a, red line) clearly shows the asymmetrical and symmetrical stretching vibrations of the methylene (–CH2–) groups localized at 2945 and 2918 cm−1, respectively.
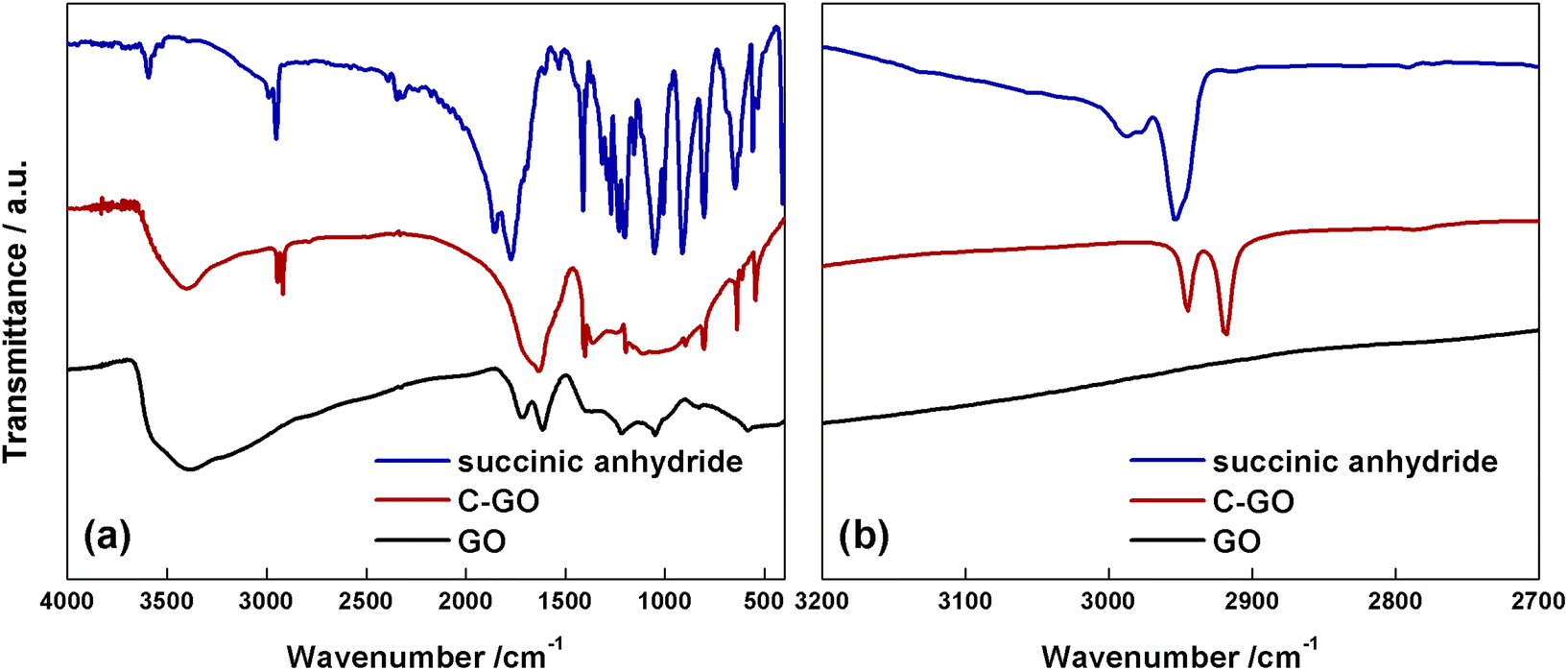 | ||
| Fig. 4 (a) FT-IR spectra of succinic anhydride (blue line), C-GO (red line) and GO (black line). (b) Magnified spectra in the methylene stretching region. | ||
It is noteworthy that the comparison with the spectrum of pure SA (Fig. 4a and b, blue line) shows that the wavenumber of methylene stretching increases when this group is part of a strained ring, as in SA, supporting the hypothesized ring aperture of the anhydride as a consequence of the esterification reaction on GO.49 Furthermore, in C-GO, differently to the case of pure SA which displays two carbonyl stretching bands localized at 1855 and 1773 cm−1, a wide band between 1630–1730 cm−1 is observed. This feature calls for the presence of both ester and carboxyl/carboxylate functional groups.49
The Raman spectrum of pristine GO shows the prominent D and G bands centered at ≈1362 cm−1 and ≈1600 cm−1 (Fig. 5a, black line). Notably, after the carboxyl-functionalization reactions the intensity of the D band, which is associated to the ring-breathing mode from sp2 carbon-rings adjacent to an edge or defect, increases. Starting from GO, characterized by an ID/IG = 0.83, this ratio increases to 0.87 in C-GO (Fig. 5a, red line). Moreover, the D band is affected not only in terms of intensity, but also in its position, which is red-shifted of ≈9 cm−1 (Fig. 5a). Furthermore, the Raman spectra in the high energy region (2400–3200 cm−1, Fig. 5b), where the 2D and D + G bands of GO can typically be found, show the presence in C-GO of narrow features at 2933 and 2949 cm−1 (Fig. 5b, red line) which can be traced back to the methylene stretching of pure SA (Fig. 5b, black line). These contributions, as already detected in FT-IR spectra (Fig. 4b), are found to redshift of ≈39 cm−1 also in Raman spectra, as expected if the ring aperture of anhydride is taken into account. On the other hand, the presence of unreacted SA within the C-GO sample can confidently be ruled out according to the Raman spectrum of pure SA (Fig. S2†). Overall, both FT-IR and Raman spectroscopy experimental observations provide a robust evidence of the presence of new signals to be associated to reacted SA, which confirm the occurrence of functionalization.50 At the same time, the occurrence of a slight reduction can be envisaged on the basis of the increment of the ID/IG ratio in the Raman spectra of C-GO (Fig. 5a, red line).51,52
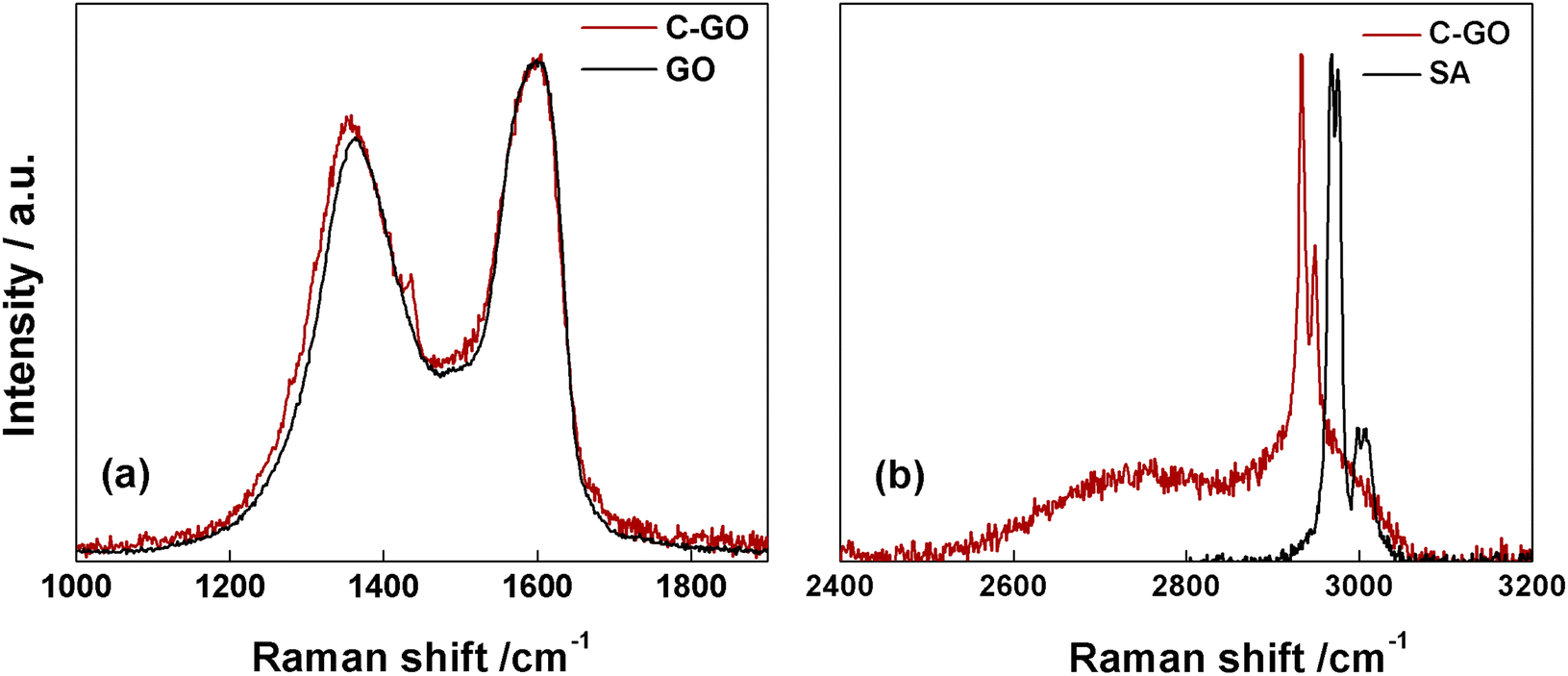 | ||
| Fig. 5 (a) Raman spectra of C-GO (red line) and GO (black line). (b) Raman spectra of C-GO (red line) and SA (black line) in the high-energy region. | ||
4.3. Back-titration of acidic groups on carboxyl-rich GO
It is well known that the bidimensional material GO is rich in oxygen-based functional groups. In particular, epoxide and hydroxyl groups are mainly localized in the basal plane, whereas the less numerous carboxyl groups can be found at the edges of the layers. The native pH of a freshly prepared aqueous dispersion of GO at concentration 0.76 mg mL−1 was 2.8, proving the presence of acidic groups. To evaluate the number of acidic groups in the structure of GO, a back-titration was performed (see Experimental section for details). As reported in Fig. 6a, GO was titred in the range of pH 4–9. Notably, three different pKa values were observed: (i) 4.3, typically attributed to carboxyl groups in ortho-position with respect to hydroxyl ones; (ii) 6.6, to be assigned to isolated carboxyl groups, and (iii) 9.0, from phenol-like hydroxyls, in accordance to the literature.53 The linearization of the titration curve of GO (Fig. 6b), by means of the Gran Plot analysis, afforded a value of 2200 ± 100 μmol g−1 of acidic sites.54,55 The same experiment was performed on the functionalized C-GO, providing a value of 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 ± 2600 μmol g−1 of acidic sites (Fig. 6c and d), and a clearly visible pKa of ≈4.8 (associated to the most abundant acidic species), close to that of propionic acid. The detected increment of acidic sites is most probably due an increased concentration of carboxyl groups, strongly pointing at a well-behaved functionalization reaction.
300 ± 2600 μmol g−1 of acidic sites (Fig. 6c and d), and a clearly visible pKa of ≈4.8 (associated to the most abundant acidic species), close to that of propionic acid. The detected increment of acidic sites is most probably due an increased concentration of carboxyl groups, strongly pointing at a well-behaved functionalization reaction.
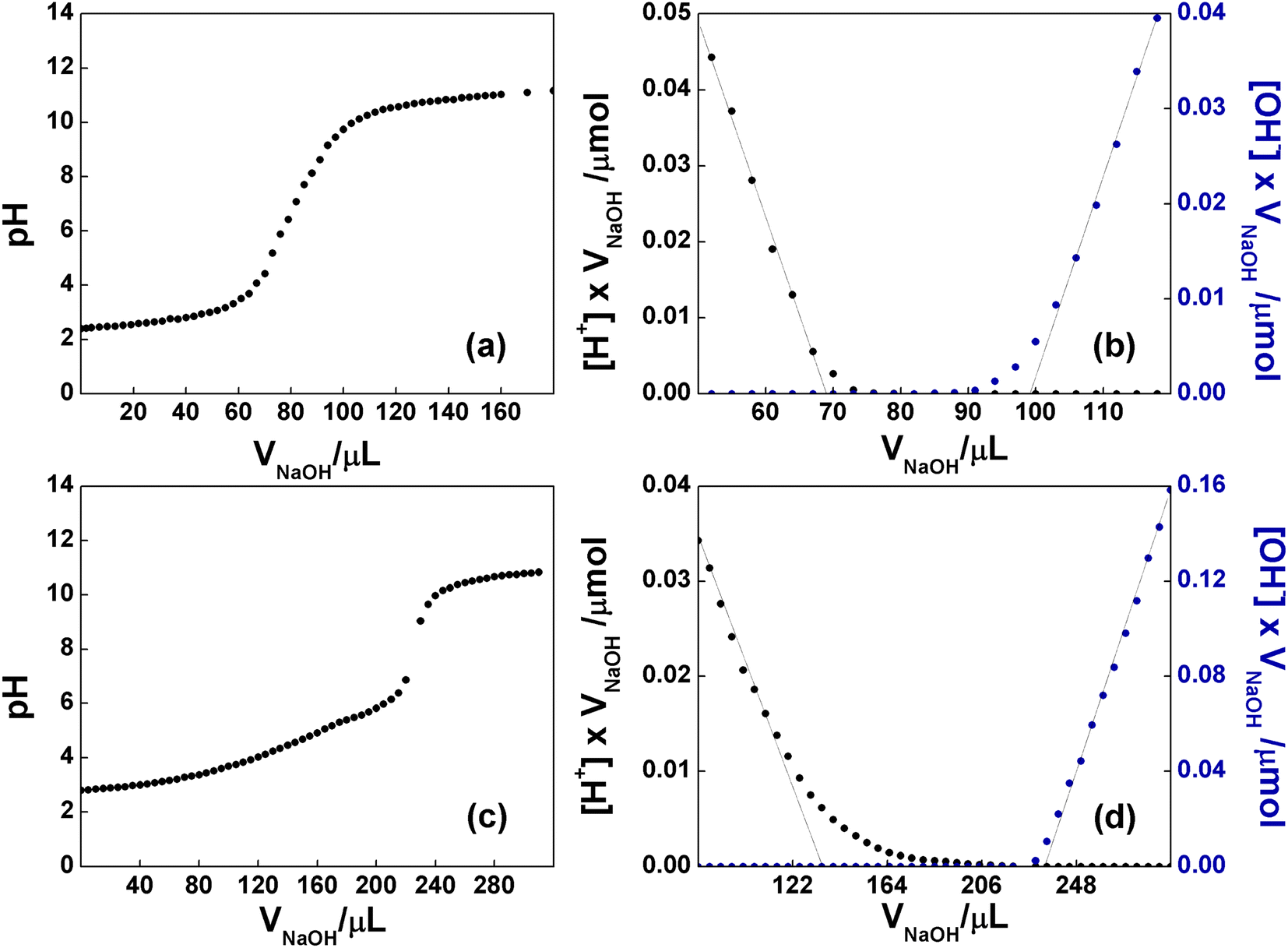 | ||
| Fig. 6 (a) pH curve observed upon backtitration of GO (0.76 mg mL−1) with NaOH, after the addition of 20 equiv of HCl. (b) Gran plot obtained from the data of (a). (c) pH curve observed upon backtitration of C-GO (0.27 mg mL−1) with NaOH, after the addition of 30 equiv of HCl. (d) Gran plot obtained from the data of (c). In the plateau regions, the acid/base sites of GO, are being deprotonated. | ||
4.4. X-ray photoelectron spectroscopy of carboxyl-rich GO
XPS spectroscopy is a fundamental tool for the investigation of the surface functionalities of GO-based materials.33,48,51,52,56,57 The deconvoluted C 1s spectra of GO and its carboxylated derivatives are reported in Fig. 7 (see Table S1† for details). The C 1s spectrum of the pristine GO displays the two main peaks ascribable to the sp2 C network at 284.8 eV (red shaded curve in Fig. 7a) and a convolution of C–O bonds from hydroxyl and epoxide groups at 286.9 eV (blue and green curves in Fig. 7a).33,56,58 The shoulder at higher binding energy (BE) is deconvoluted into two components, assigned to carbonyl/carboxylate groups, at 288.1 eV (magenta curves in Fig. 7), and to the less abundant carboxyl moieties at 289.2 eV (yellow curves in Fig. 7), whose normalized area (see Experimental section) is about 0.06 (Table 1).58 After the carboxylation reaction with SA in the presence of K2CO3 as a base, the C 1s XPS spectrum of C-GO sensibly changes (Fig. 7b). The first apparent difference is the significant abatement of the epoxide component at 287.1 eV, whose area contribution dramatically drops from 0.75 to 0.28 (see Table 1), which strongly points at a participation of these functional groups in the reaction of functionalization, as anticipated before.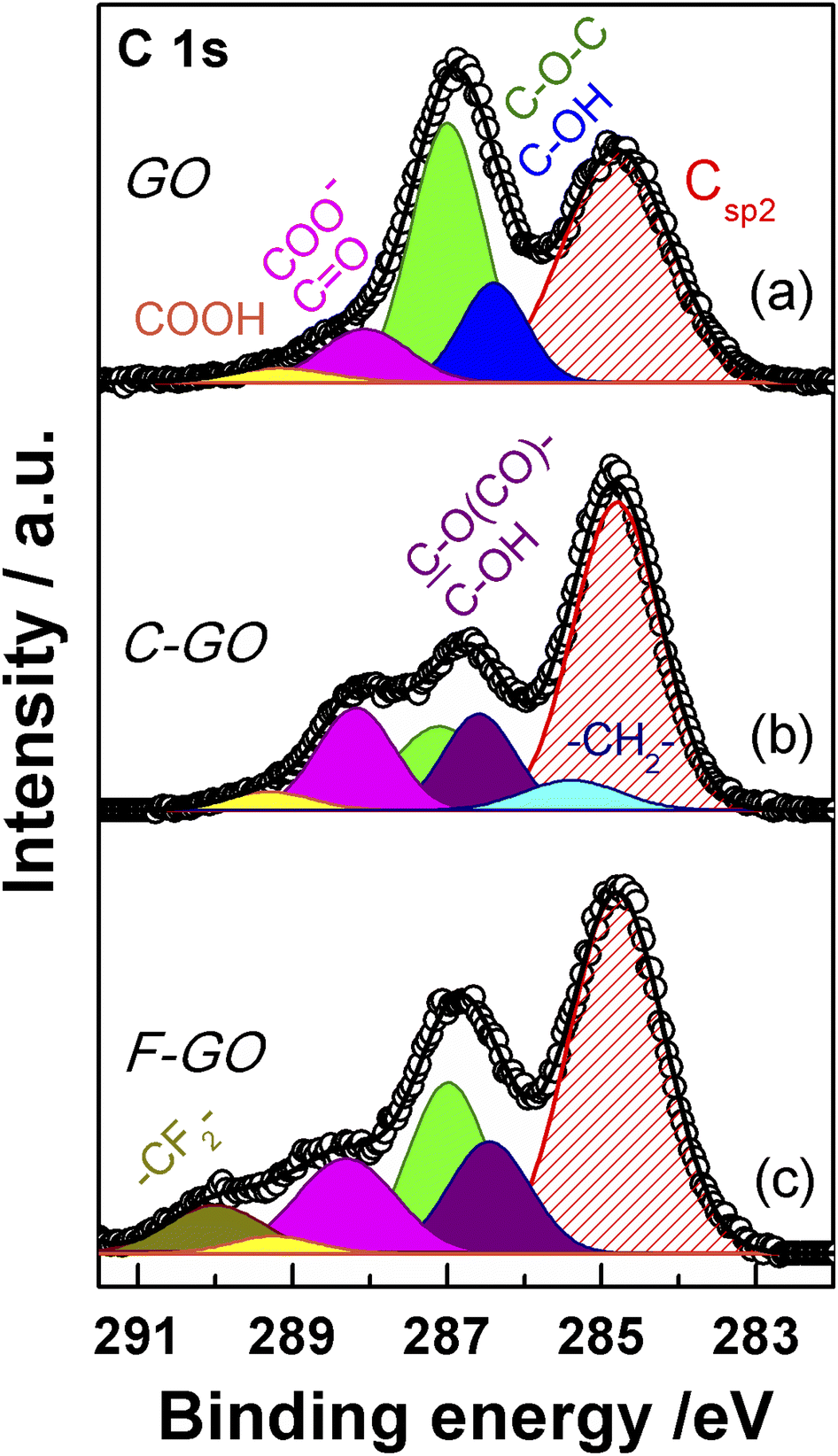 | ||
| Fig. 7 XPS spectra in the region C 1s of GO (a), C-GO (b), and F-GO (c). Experimental data are reported in dots, while fitting reconstruction is represented with a continuous line and color-filled curves corresponding to the various functional groups. Color code: red: sp2 CC; blue: hydroxyl; green: epoxy; magenta: carbonyl/carboxylate; yellow: carboxyl; light cyan: methylene; violet: ester/hydroxyl (see text); dark yellow: di-fluoromethylene. | ||
C (Csp2) from C 1s XPS spectra of GO, C-GO, F-GO, C-RGO and RGO
| Sample | C–OH/Csp2 | C–O–C/Csp2 | CO + COO−/Csp2 |
COOH/Csp2 | –CH2–/Csp2 | –CF2–/Csp2 |
|---|---|---|---|---|---|---|
|
a Associated error is ±10%.
b In C-GO, F-GO and C-RGO the C–OH signal is also contributed by the estereal C–O(CO)– atoms of graphene oxide bound to the succinate moiety, which display the same BE as C–OH.
|
||||||
| GO | 0.26 | 0.75 | 0.19 | 0.06 | — | — |
| C-GO | 0.25b | 0.28 | 0.30 | 0.06 | 0.11 | — |
| F-GO | 0.26b | 0.40 | 0.28 | 0.04 | — | 0.14 |
| C-RGO | 0.07b | 0.01 | 0.17 | 0.03 | 0.03 | — |
| RGO | 0.28 | 0.21 | 0.12 | 0.06 | — | — |
Moreover, an additional small component is needed for a proper reconstruction of the experimental profile at 285.4 eV (light cyan curve in Fig. 7b). This contribution can be assigned to the methylene groups within the succinate moiety, supposedly bound to GO after functionalization (vide infra).58,59 Furthermore, in the spectrum of C-GO, the component at 286.6 eV (violet curve in Fig. 7b), typically assigned to hydroxyl groups in GO, may receive contribution also from estereal C–O(CO)– atoms of graphene oxide, should this be bound to the succinate moiety after reaction with SA (vide infra).58,59 As to the intensity of this peak, it is found that its normalized area remains nearly constant compared to pristine GO (0.25 vs. 0.26, Table 1). This suggests that a diminution of the reactive hydroxyl groups is equally counterbalanced by the build-up of estereal functionalities upon carboxylation reaction with SA, as expected considering a direct O-acylation mechanism. Another striking feature of the C 1s XPS spectrum of C-GO is the combined increase of the signal of both carbonyl/carboxylate groups at 288.2 eV and carboxyls at 289.3 eV compared to pristine GO (see Table 1), which strongly supports the accomplishment of its chemical modification via carboxyl enrichment. The increment of the carbonyl/carboxylate signal at 288.2 eV also suggests an increased number of carbonyl groups, which may derive, through resonance effect, from edge enolate groups close to epoxides reactive towards carboxylation with SA (vide infra).
Overall, besides the obvious increase in carboxyl features, the decrease of epoxides, coupled to the perfect counterbalance between hydroxyls and estereal carbon atoms, and the increase of carbonyl groups, nicely matches with a picture of the carboxylation reaction where both edge enol-like and basal epoxide groups are reactive towards SA (vide infra, theoretical modelling).
In order to support these findings, XPS spectra were recorded also from the GO sample reacted with the fluorinated derivative of SA (F-SA). In this case, the fluorine atoms of the opened anhydride possibly appended to the GO sheet constitute an easily detectable tag in the XPS spectrum, both in the F 1s (see Fig. S3†) and in the C 1s region, which may render easier the identification of the reaction products. The C 1s XPS spectrum of F-GO (Fig. 7c) displays, in comparison to the pristine GO, a substantial intensity decrease of epoxide groups (0.40 vs. 0.75), with the hydroxyl/estereal area contribution constant at 0.26. The amount of carbonyl/carboxylate moieties rises up with a normalized area of 0.28, while undissociated carboxyl groups at 289.2 eV show an area of only 0.04. In this case, the fluorinated methylene groups, –CF2–, of the appended succinate moiety display an intense chemical shift which make them clearly visible at 290.0 eV (dark yellow curve in Fig. 7c). These results, together with the F 1s spectrum (Fig. S3†), further support the validity of the applied method for a carboxyl enrichment of GO.59
4.5. Theoretical modelling of the carboxylation reaction mechanism
On the basis of the obtained experimental results, a two-fold reaction path can be envisaged for the reaction of GO with SA in aprotic solvent in the presence of K2CO3. These hypothesized routes are the direct O-acylation at edge phenolates and the ring-opening of basal epoxide, as schematized in Fig. 8 (see Fig. S4† for the corresponding scheme in the presence of F-SA). The obtained C-GO product is expected to be functionalized with linear succinate moieties.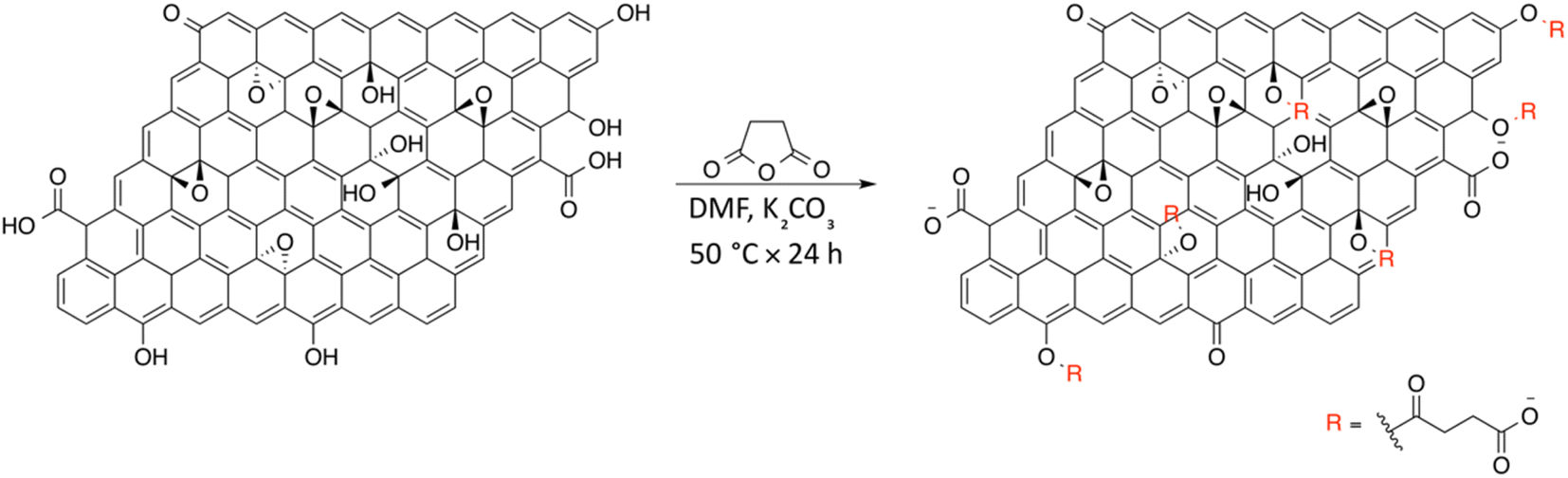 | ||
| Fig. 8 Reaction scheme for the synthesis of C-GO. | ||
With these premises, the reaction of SA with GO to form the carboxyl-functionalized GO (C-GO) was modeled into the DFT framework to acquire useful information on the mechanisms underlying the carboxylation process. Both investigated reaction paths involve a nucleophilic attack of the GO oxygenated species on the SA carbonyl group, and both require an acid–base activation, in order to enhance the nucleophilic nature of the GO functionalities. The acid–base activation is promoted by the carbonate ion (see Experimental section), which extracts a proton from an acidic site on the GO. Beside the carboxyl groups, also enol/phenol groups can be involved in this proton transfer process in presence of a strong enough base. Therefore, the first modelled reaction path involves the approach of SA carbonyl to the enolate/phenolate group located at the GO edge (Fig. 9, path 1).
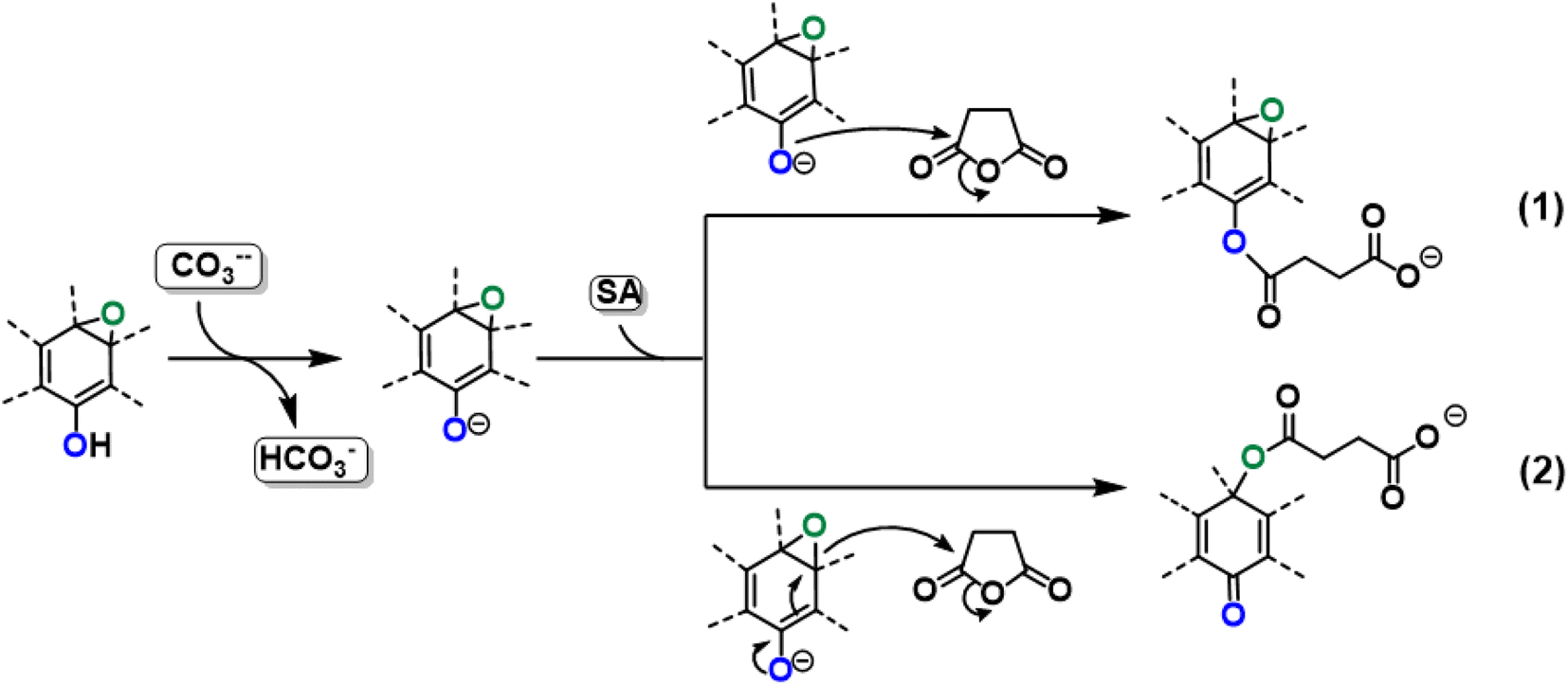 | ||
| Fig. 9 Reactivity of GO toward SA in presence of K2CO3 with two different reaction paths. | ||
The second path considered involves the approach of a SA carbonyl group to an epoxy group located on the GO basal plane, which turns an edge phenolate/enolate group into a carbonyl, while promoting the epoxide opening (Fig. 9, path 2). Our model starts by considering the epoxide group located in position 3,4 with respect to the hydroxyl. The activation step is shared by both mechanisms and it is slightly esoergonic (ΔG = −4.3 kcal mol−1). Geometrical analysis of the system before and after the acid–base proton transfer reveals that the C–OH distance (1.37 Å) is strongly reduced after the proton transfer (C–O = 1.24 Å), while charge analysis shows that the negative charge increases on passing from enol to enolate (−0.3 vs. −0.5, respectively). It is worthy of note that also the negative charge on the epoxide group slightly increases after the proton transfer (0.00 vs. −0.1, respectively) as a consequence of the partial delocalization of the negative charge on both oxygen species. At this point, the approach of SA can occur towards either the edge enolate group, leading to mechanism (1), or to the basal epoxide group, leading to the epoxide ring-opening (2). Computed energy profiles reveal that mechanism (2) is preferred over mechanism (1), with an evaluated Gibbs free energy barrier of 26.8 kcal mol−1 and 30.7 kcal mol−1, respectively (Fig. 10). Notably, without the initial base activation, the carboxylation step does not occur.
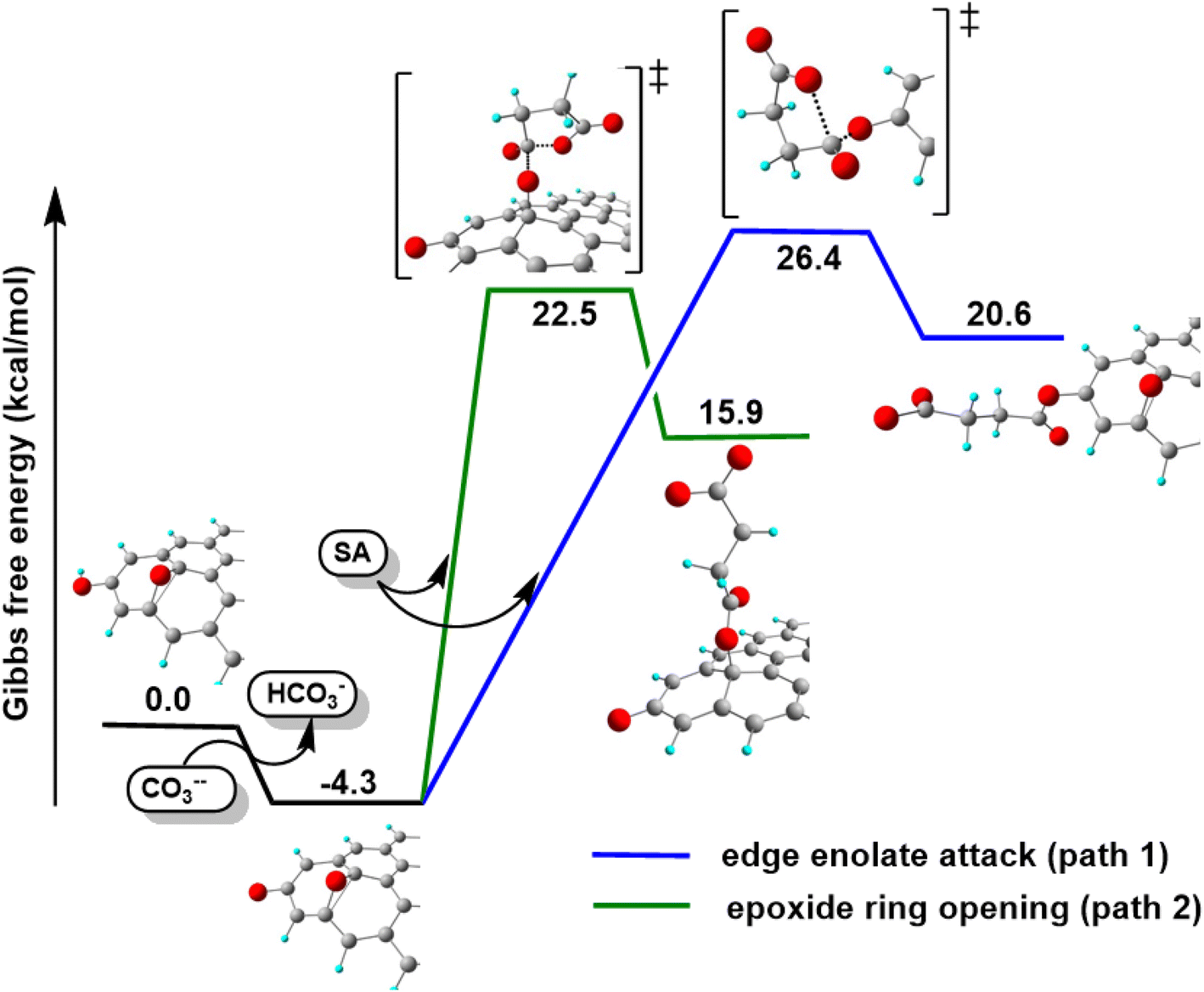 | ||
| Fig. 10 Energy profiles for the proposed mechanisms of the O-acylation of GO promoted by SA. | ||
Moreover, it was found that the epoxide ring-opening mechanism occurs preferentially when enol and epoxide groups are closely located within the carbon network. In fact, moving the epoxide far from the enol brings about a parallel increase in the energy barrier for its ring-opening mechanism (Table 2).
|
|
|
|
|---|---|---|
| Enolate–epoxide distance (Å) | 4.0 | 13.7 |
| Path 1, ΔG‡ (kcal mol−1) | 30.7 | 25.3 |
| Path 2, ΔG‡ (kcal mol−1) | 26.8 | 39.8 |
| Charge on enolate | −0.749 | −0.784 |
| Charge on epoxide | −0.607 | −0.565 |
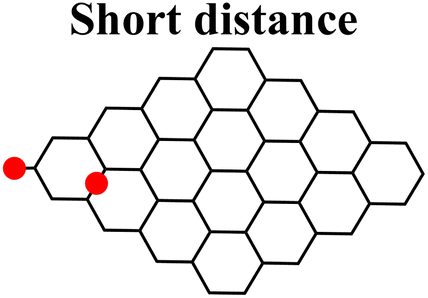
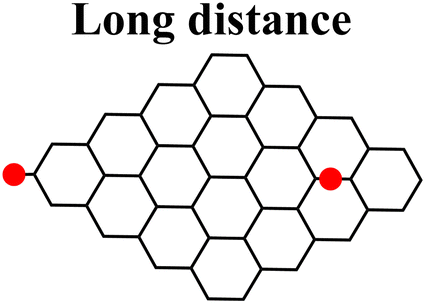
By contrast, the energy barrier for the acylation of the enolate, simultaneously decreases with the epoxide distance. This inverted behavior of the two mechanisms with respect to the distance between enolate and epoxide groups can be explained by their different nucleophilic character. By following the charge population on both the enolate and epoxide O atoms, it appears evident that moving the epoxide away from the enolate, the negative charge increases on the enolate O atom, while decreases on the epoxide (Table 2). As a consequence, at short distance the nucleophilic character of epoxide is slightly enhanced compared to long distance. This effect favors the SA approach to the epoxide group, rather than to the enolate, in those regions of the GO sheet where they are both present and close to each other.
From the above results, it emerges that the carboxylation process occurs preferentially in proximity (epoxide ring-opening mechanism) or at the edge (O-acylation on the enolate) of the GO sheet, with the latter process being less probable. The activation step, consisting in the proton transfer from the GO surface to the carbonate, is fundamental for the carboxylation process. The negative charge thereby resulting can be delocalized on an epoxide group in the close vicinity, promoting the ring-opening mechanism, in keeping with the depression of the epoxide signal observed by XPS in the C-GO sample.
4.6. Spectroscopical characterization of chemically reduced carboxyl-rich GO
In order to obtain a water dispersible reduced graphene oxide, the effect of using ascorbic acid (AA) as a reducing agent on C-GO was investigated (Fig. 11). The expected reduced graphene oxide derivative will be referred to as C-RGO.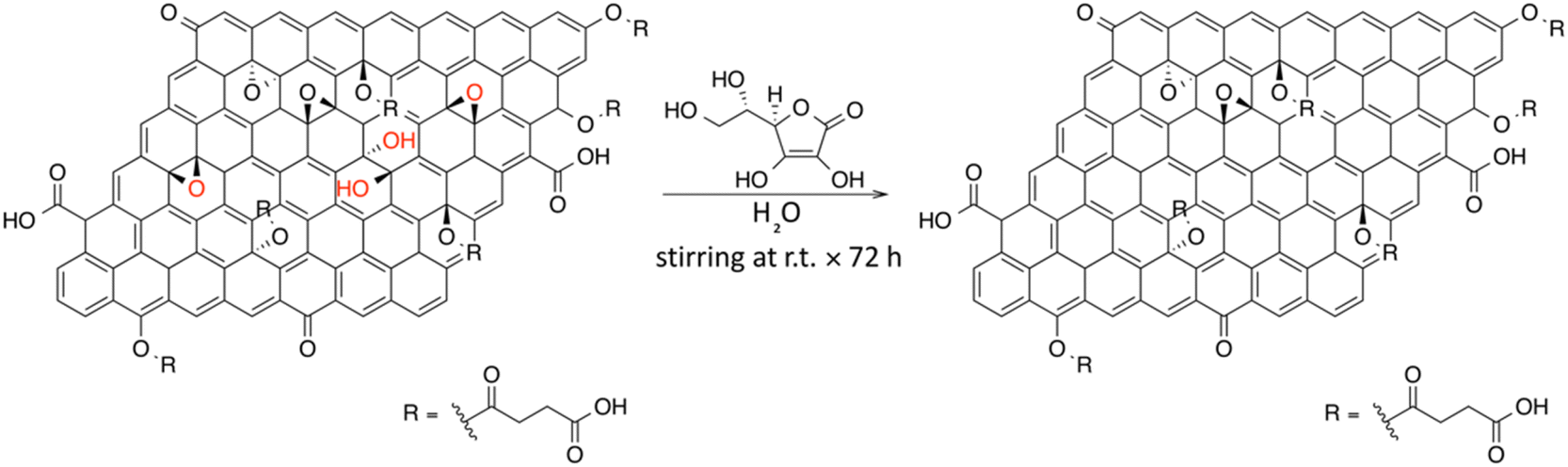 | ||
| Fig. 11 Possible reaction scheme for the synthesis of C-RGO. | ||
AA is a known reducing agent toward the epoxide and diol functional groups of GO, with no reported effects on the carboxyl functionalities.32,33,51,60
The black-powder of C-RGO collected after the purification turned out to be easily dispersible in water (see Fig. 2), up about 0.06 mg mL−1, strongly suggesting that the purpose of the reaction was successfully reached. The UV-Vis spectrum shows that the shoulder localized at 232 nm in C-GO is red-shifted to 255 nm in C-RGO, whereas the other shoulder at 300 nm associated to the n–π* electron transitions disappears (Fig. 12a). Furthermore, in C-RGO, absorption in the whole range of visible light is dramatically enhanced, as a consequence of the restoration of the π-conjugated C texture, in accordance to the literature.32,61
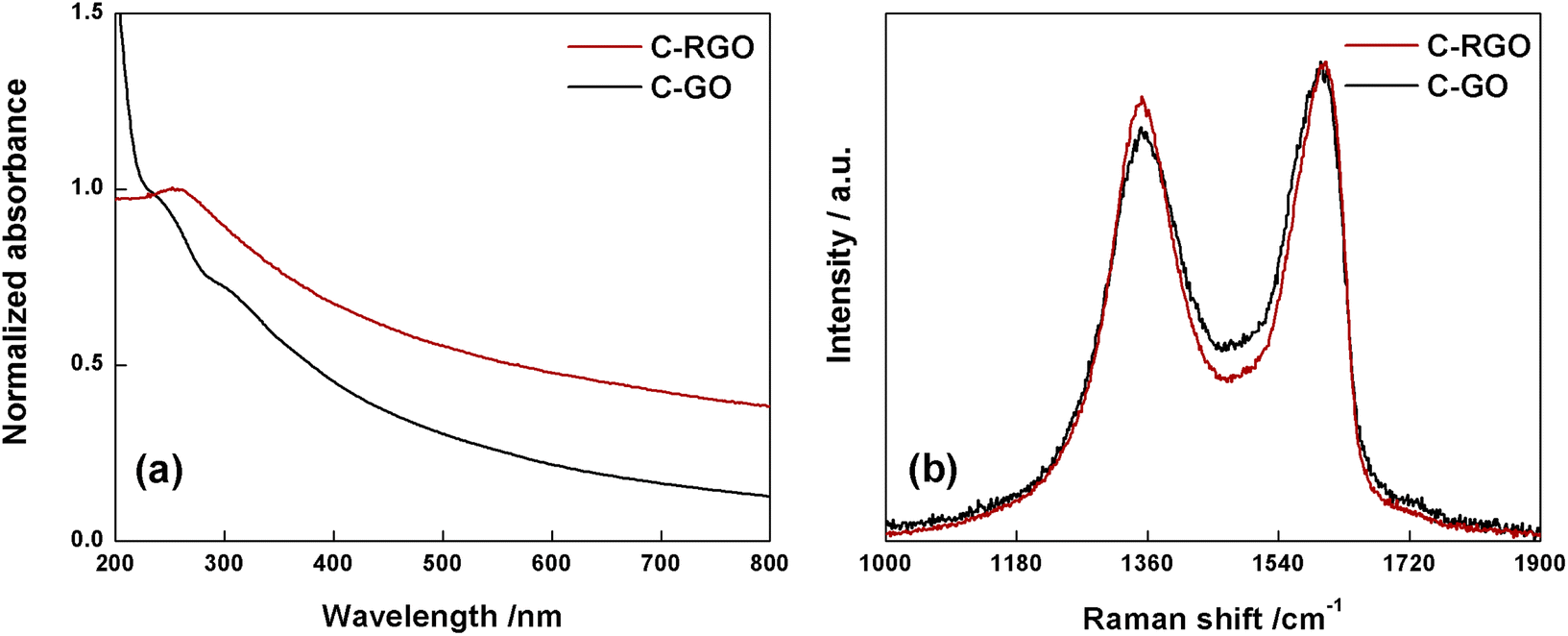 | ||
| Fig. 12 (a) Normalized UV-Vis spectra of C-RGO (red line) and C-GO (black line) recorded in water at room temperature. (b) Raman spectra of C-GO (black line) and C-RGO (red line). | ||
The Raman spectrum (Fig. 12b) of C-RGO in the region of D and G bands, shows an increased ID/IG ratio (0.92) compared to C-GO (0.86), a trend which is fully coherent with reduction of a graphene oxide based material.33,51,52
The C 1s XPS spectrum of C-RGO (Fig. 13a) shows a dramatic decrease of the hydroxyl and epoxide groups related features (see Table 1) compared to C-GO, as a consequence of the further elimination of these moieties operated by AA. In particular, the peak due to the residual epoxide groups almost disappears (normalized area: 0.01 vs. 0.28 in C-GO). In addition, the hydroxyl/ester signal at 286.3 eV also decreases (normalized area: 0.07 vs. 0.25 in C-GO), with a residual intensity most likely contributed by the ester groups alone, deriving from the carboxylation reaction. Finally, the XPS spectrum of C-RGO displays, in comparison to its starting material C-GO, a lower, yet significant, content of carbonyl/carboxylate groups (normalized area: 0.17 vs. 0.30), and of undissociated carboxyl groups (normalized area: 0.03 vs. 0.06). This clearly demonstrates that the carboxyl enrichment is preserved after reduction with AA. To further support the interpretation of C 1s XPS results, an additional control experiment was performed, by applying the same reduction protocol with AA to the pristine GO (see Experimental section). The obtained product (RGO) turned out to be hardly soluble in water, and its C 1s XPS spectrum (Fig. 13b) shows, in comparison to C-RGO, a much more prominent intensity in the OH/epoxide region (yet much lower than pristine GO), and a lower intensity of the carbonyl/carboxylate related features.
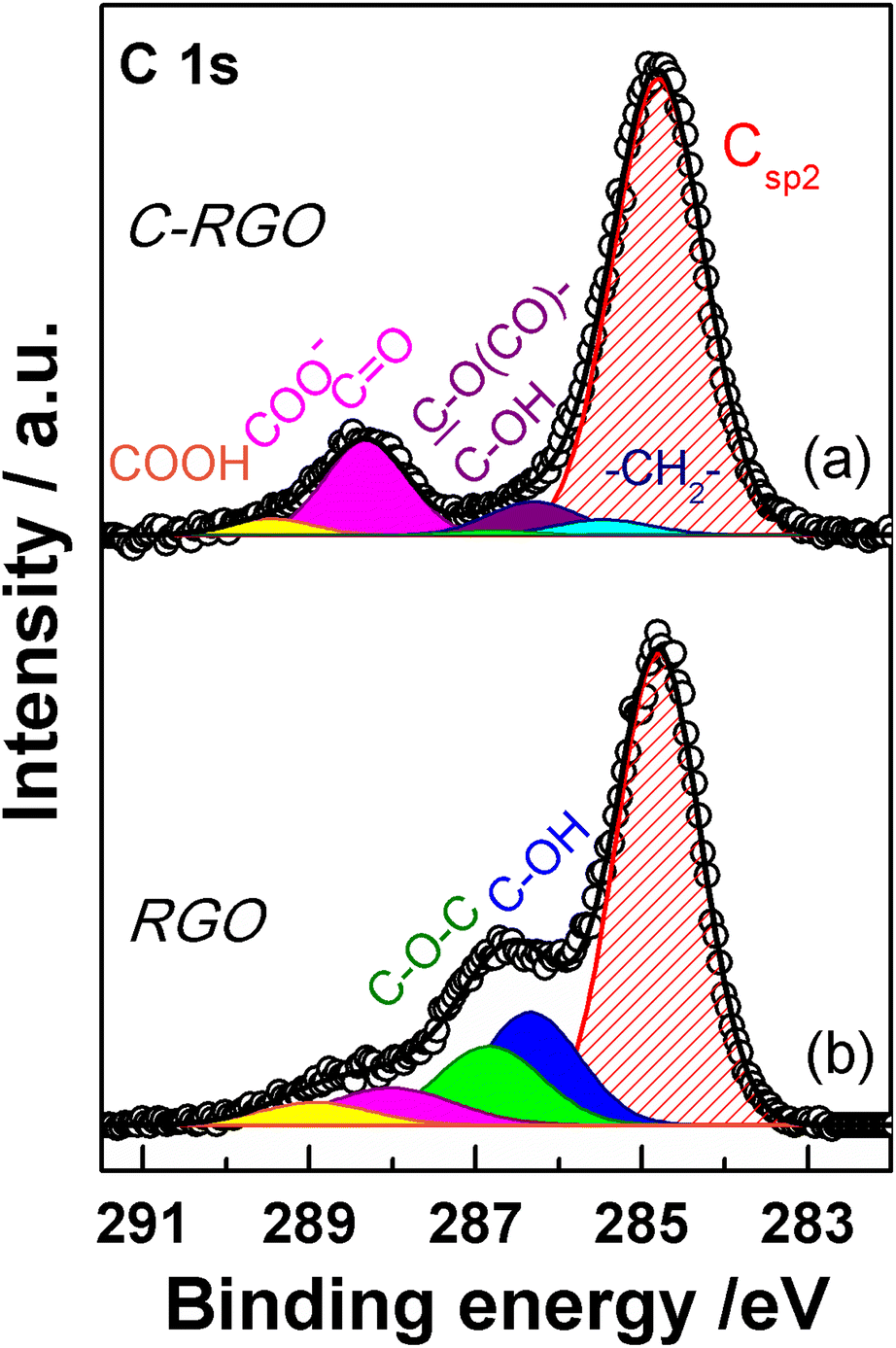 | ||
| Fig. 13 XPS spectra of the C 1s region of (a) C-RGO and (b) RGO. Experimental data are reported in dots, while fitting reconstruction is represented with a continuous line and color-filled curves corresponding to the various functional groups. Color code: red: sp2 CC; blue: hydroxyl; green: epoxy; magenta: carbonyl/carboxylate; yellow: carboxyl; light cyan: methylene; violet: ester/hydroxyl (see text). | ||
5. Conclusions
In summary, we investigated a one-pot carboxylation reaction of GO through a novel sustainable synthetic procedure, which employs succinic anhydride. The obtained novel functionalized material was characterized by means of spectroscopic and analytical methods, which evidenced the nature and extent of the chemical modifications, confirming the occurrence of a carboxyl-enrichment of the GO sheet. DFT calculations were conducted to support the interpretation of the experimental results, and put in evidence a twofold reactivity of GO in presence of succinic anhydride and a strong enough base, such as potassium carbonate, which provides deprotonation of edge hydroxyl groups. In particular, an (unprecedented) ring-opening based reactivity of epoxide groups towards succinic anhydride, more favored when proximal to hydroxyls at the edges of the sheet, was demonstrated. This reaction route was found to be more probable than that directly involving deprotonated edge hydroxyls (direct O-acylation), which may however partially contribute to the overall process. This synthetic route to carboxyl-rich GO constitutes an improvement over use of hazardous chemicals, such as cyanides, and may be particularly desirable in those applications were a high concentration of carboxyl groups is required, e.g. preparation of supramolecular hydrogels, catalysis and heavy metal ions removal from wastewater.The obtained carboxyl-enrichment of GO was then employed to achieve a reduced graphene oxide material with the advantage of being water-dispersible, through reaction with L-ascorbic acid, a mild and green reducing agent. According to the spectroscopical results, we demonstrated that the carboxyl functionalities preliminary appended to the structure of GO are not involved in the subsequent reduction process, which selectively operates on epoxide groups, ensuring a significantly improved colloidal kinetic stability in water, compared to reduced graphene oxide material obtained with traditional methods. The chemical reduction of a formerly carboxylated GO is more advantageous than the carboxyl-functionalization of a RGO sample, since, in this latter case, the amount of oxygen-based functional groups available to react with SA would be very low.
Improvements in water-dispersibility of reduced graphene oxide materials may result prominently beneficial in fields like smart electronic-textiles, environmental chemistry, biomedicine and nano-scaffolding. In particular, considering the known dependence of biocompatibility of the graphene-based nanomaterials on the amount and type of oxygen groups, C-RGO could be beneficial in the numerous biological applications where cells growth and differentiation require the presence of acidic functions.
Author contributions
Francesco Amato: conceptualization, data curation, investigation, methodology, writing – original draft, writing – review & editing. Alessandro Motta: conceptualization, data curation, formal Analysis, funding acquisition, resources, software, writing – original draft, writing – review & editing. Leonardo Giaccari: data curation, investigation, formal analysis. Roberto Di Pasquale: data curation, investigation, formal analysis. Francesca Anna Scaramuzzo: data curation, investigation, formal analysis. Robertino Zanoni: conceptualization, funding acquisition, resources, supervision, writing – review & editing. Andrea Giacomo Marrani: conceptualization, data curation, investigation, methodology, funding acquisition, writing – original draft, writing – review & editing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
F. A. thanks: Sapienza University of Rome for financial support through the Ateneo Project AR2221814CBEA5BC. A. G. M. thanks: Sapienza University of Rome for financial support through the Ateneo Project RM120172B80059DA and RM11916B88D8E044; Italian Ministry of Health for financial support through the project Ricerca Finalizzata 2019 GR-2019-12370086. RZ gratefully acknowledges support by the PRIN 2017 program of Italian Ministry of Research and University (MIUR, Grant No. 2017RSAFK7). Computational resources were provided by CINECA Computing Cluster under the ISCRA initiative (award no. HP10C0T7WT 2022). Prof. D. Gazzoli is kindly acknowledged for assistance in Raman spectroscopy and Prof. M. P. Donzello is kindly acknowledged for assistance in UV-Vis and FT-IR spectroscopy.References
- N. Kumar, R. Salehiyan, V. Chauke, O. Joseph Botlhoko, K. Setshedi, M. Scriba, M. Masukume and S. Sinha Ray, Top-down synthesis of graphene: A comprehensive review, FlatChem, 2021, 27, 100224, DOI:10.1016/j.flatc.2021.100224.
- B. R. Muñoz and C. Gómez-aleixandre, Review of CVD Synthesis of Graphene, Chem. Vap. Depos., 2013, 19, 297–322, DOI:10.1002/cvde.201300051.
- W. Zheng and L. Y. S. Lee, Beyond sonication: Advanced exfoliation methods for scalable production of 2D materials, Matter, 2022, 5, 515–545, DOI:10.1016/j.matt.2021.12.010.
- L. Niu, J. N. Coleman, H. Zhang, H. Shin, M. Chhowalla and Z. Zheng, Production of Two-Dimensional Nanomaterials via Liquid-Based Direct Exfoliation, Small, 2016, 12, 272–293, DOI:10.1002/smll.201502207.
- I. Ferrari, A. Motta, R. Zanoni, F. A. Scaramuzzo, F. Amato, E. A. Dalchiele and A. G. Marrani, Understanding the nature of graphene oxide functional groups by modulation of the electrochemical reduction: A combined experimental and theoretical approach, Carbon, 2023, 203, 29–38, DOI:10.1016/j.carbon.2022.11.052.
- A. Ambrosi, C. K. Chua, N. M. Latiff, A. H. Loo, C. H. A. Wong, A. Y. S. Eng, A. Bonanni and M. Pumera, Graphene and its electrochemistry-an update, Chem. Soc. Rev., 2016, 45, 2458–2493, 10.1039/c6cs00136j.
- D. X. Luong, K. V. Bets, W. A. Algozeeb, M. G. Stanford, C. Kittrell, W. Chen, R. V. Salvatierra, M. Ren, E. A. McHugh, P. A. Advincula, Z. Wang, M. Bhatt, H. Guo, V. Mancevski, R. Shahsavari, B. I. Yakobson and J. M. Tour, Gram-scale bottom-up flash graphene synthesis, Nature, 2020, 577, 647–651, DOI:10.1038/s41586-020-1938-0.
- R. K. Singh, R. Kumar and D. P. Singh, Graphene oxide: Strategies for synthesis, reduction and frontier applications, RSC Adv., 2016, 6, 64993–65011, 10.1039/c6ra07626b.
- S. Eigler and A. Hirsch, Chemistry with graphene and graphene oxide - Challenges for synthetic chemists, Angew. Chem., Int. Ed., 2014, 53, 7720–7738, DOI:10.1002/anie.201402780.
- V. Georgakilas, M. Otyepka, A. B. Bourlinos, V. Chandra, N. Kim, K. C. Kemp, P. Hobza, R. Zboril and K. S. Kim, Functionalization of graphene: Covalent and non-covalent approaches, derivatives and applications, Chem. Rev., 2012, 112, 6156–6214, DOI:10.1021/cr3000412.
- S. Guo, S. Garaj, A. Bianco and C. Ménard-Moyon, Controlling covalent chemistry on graphene oxide, Nat. Rev. Phys., 2022, 4, 247–262, DOI:10.1038/s42254-022-00422-w.
- Y. Heng Cheong, M. Z. M. Nasir, A. Bakandritsos, M. Pykal, P. Jakubec, R. Zbořil, M. Otyepka and M. Pumera, Cyanographene and Graphene Acid: The Functional Group of Graphene Derivative Determines the Application in Electrochemical Sensing and Capacitors, ChemElectroChem, 2019, 6, 229–234, DOI:10.1002/celc.201800675.
- O. Jankovský, M. Nováček, J. Luxa, D. Sedmidubský, V. Fila, M. Pumera and Z. Sofer, A New Member of the Graphene Family: Graphene Acid, Chem. - A Eur. J., 2016, 22, 17416–17424, DOI:10.1002/chem.201603766.
- B. Adhikari, A. Biswas and A. Banerjee, Graphene oxide-based supramolecular hydrogels for making nanohybrid systems with Au nanoparticles, Langmuir, 2012, 28, 1460–1469, DOI:10.1021/la203498j.
- S. Sandoval, A. Fuertes and G. Tobias, Solvent-free functionalisation of graphene oxide with amide and amine groups at room temperature, Chem. Commun., 2019, 55, 12196–12199, 10.1039/c9cc05693a.
- F. Khan, M. S. Khan, S. Kamal, M. Arshad, S. I. Ahmad and S. A. A. Nami, Recent advances in graphene oxide and reduced graphene oxide based nanocomposites for the photodegradation of dyes, J. Mater. Chem. C, 2020, 8, 15940–15955, 10.1039/d0tc03684f.
- R. Allgayer, N. Yousefi and N. Tufenkji, Graphene oxide sponge as adsorbent for organic contaminants: comparison with granular activated carbon and influence of water chemistry, Environ. Sci. Nano., 2020, 7, 2669–2680, 10.1039/d0en00193g.
- W. Zhang, H. Xu, F. Xie, X. Ma, B. Niu, M. Chen, H. Zhang, Y. Zhang and D. Long, General synthesis of ultrafine metal oxide/reduced graphene oxide nanocomposites for ultrahigh-flux nanofiltration membrane, Nat. Commun., 2022, 13, 1–10, DOI:10.1038/s41467-022-28180-4.
- A. Bakandritsos, M. Pykal, P. Boński, P. Jakubec, D. D. Chronopoulos, K. Poláková, V. Georgakilas, K. Čépe, O. Tomanec, V. Ranc, A. B. Bourlinos, R. Zbořil and M. Otyepka, Cyanographene and Graphene Acid: Emerging Derivatives Enabling High-Yield and Selective Functionalization of Graphene, ACS Nano, 2017, 11, 2982–2991, DOI:10.1021/acsnano.6b08449.
- M. Blanco, D. Mosconi, M. Otyepka, M. Medveď, A. Bakandritsos, S. Agnoli and G. Granozzi, Combined high degree of carboxylation and electronic conduction in graphene acid sets new limits for metal free catalysis in alcohol oxidation, Chem. Sci., 2019, 10, 9438–9445, 10.1039/c9sc02954k.
- M. Pumera and Z. Sofer, Towards stoichiometric analogues of graphene: Graphane, fluorographene, graphol, graphene acid and others, Chem. Soc. Rev., 2017, 46, 4450–4463, 10.1039/c7cs00215g.
- X. Sun, Z. Liu, K. Welsher, J. T. Robinson, A. Goodwin, S. Zaric and H. Dai, Nano-graphene oxide for cellular imaging and drug delivery, Nano Res., 2008, 1, 203–212, DOI:10.1007/s12274-008-8021-8.
- T. Jiang, W. Sun, Q. Zhu, N. A. Burns, S. A. Khan, R. Mo and Z. Gu, Furin-mediated sequential delivery of anticancer cytokine and small-molecule drug shuttled by graphene, Adv. Mater., 2015, 27, 1021–1028, DOI:10.1002/adma.201404498.
- T. Yin, J. Liu, Z. Zhao, Y. Zhao, L. Dong, M. Yang, J. Zhou and M. Huo, Redox Sensitive Hyaluronic Acid-Decorated Graphene Oxide for Photothermally Controlled Tumor-Cytoplasm-Selective Rapid Drug Delivery, Adv. Funct. Mater., 2017, 27 DOI:10.1002/adfm.201604620.
- S. Guo, J. Raya, D. Ji, Y. Nishina, C. Ménard-Moyon and A. Bianco, Is carboxylation an efficient method for graphene oxide functionalization?, Nanoscale Adv., 2020, 2, 4085–4092, 10.1039/d0na00561d.
- C. Chen, W. Kong, H. M. Duan and J. Zhang, Theoretical simulation of reduction mechanism of graphene oxide in sodium hydroxide solution, Phys. Chem. Chem. Phys., 2014, 16, 12858–12864, 10.1039/c4cp01031k.
- A. M. Dimiev, L. B. Alemany and J. M. Tour, Graphene oxide. Origin of acidity, its instability in water, and a new dynamic structural model, ACS Nano, 2013, 7, 576–588, DOI:10.1021/nn3047378.
- W. Li, M. Wang, Y. Yue, W. Ji and R. Ren, Enhanced mechanical and thermal properties of bismaleimide composites with covalent functionalized graphene oxide, RSC Adv., 2016, 6, 54410–54417, 10.1039/c6ra09260h.
- P. P. Brisebois, C. Kuss, S. B. Schougaard, R. Izquierdo and M. Siaj, New Insights into the Diels-Alder Reaction of Graphene Oxide, Chem.–Eur. J., 2016, 22, 5849–5852, DOI:10.1002/chem.201504984.
- Y. Cao, S. Osuna, Y. Liang, R. C. Haddon and K. N. Houk, Diels-Alder reactions of graphene: Computational predictions of products and sites of reaction, J. Am. Chem. Soc., 2013, 135, 17643–17649, DOI:10.1021/ja410225u.
- O. C. Compton and S. T. Nguyen, Graphene oxide, highly reduced graphene oxide, and graphene: Versatile building blocks for carbon-based materials, Small, 2010, 6, 711–723, DOI:10.1002/smll.200901934.
- J. Zhang, H. Yang, G. Shen, P. Cheng, J. Zhang and S. Guo, Reduction of graphene oxide vial-ascorbic acid, Chem. Commun., 2010, 46, 1112–1114, 10.1039/b917705a.
- A. G. Marrani, A. Motta, V. Palmieri, G. Perini, M. Papi, E. A. Dalchiele, R. Schrebler and R. Zanoni, A comparative experimental and theoretical study of the mechanism of graphene oxide mild reduction by ascorbic acid and: N-acetyl cysteine for biomedical applications, Mater. Adv., 2020, 1, 2745–2754, 10.1039/d0ma00456a.
- G. Brieger, D. Hachey and T. Nestrick, Convenient O-alkylation of phenols, J. Chem. Eng. Data, 1968, 13, 581–582, DOI:10.1021/je60039a048.
- R. Sekiya, Y. Uemura, H. Murakami and T. Haino, White-light-emitting edge-functionalized graphene quantum dots, Angew. Chem., Int. Ed., 2014, 53, 5619–5623, DOI:10.1002/anie.201311248.
- H. Sehaqui, K. Kulasinski, N. Pfenninger, T. Zimmermann and P. Tingaut, Highly Carboxylated Cellulose Nanofibers via Succinic Anhydride Esterification of Wheat Fibers and Facile Mechanical Disintegration, Biomacromolecules, 2017, 18, 242–248, DOI:10.1021/acs.biomac.6b01548.
- V. Thakur, A. Guleria, S. Kumar, S. Sharma and K. Singh, Recent advances in nanocellulose processing, functionalization and applications: A review, Mater. Adv., 2021, 2, 1872–1895, 10.1039/d1ma00049g.
- D. C. Marcano, D. V Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, Improved Synthesis of Graphene Oxide, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- J. Chen, Y. Li, L. Huang, C. Li and G. Shi, High-yield preparation of graphene oxide from small graphite flakes via an improved Hummers method with a simple purification process, Carbon, 2015, 81, 826–834, DOI:10.1016/j.carbon.2014.10.033.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652, DOI:10.1063/1.464913.
- A. D. McLean and G. S. Chandler, Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z=11-18, J. Chem. Phys., 1980, 72, 5639–5648, DOI:10.1063/1.438980.
- C. Peng, P. Y. Ayala, H. B. Schlegel and M. J. Frisch, Using Redundant Internal Coordinates to Optimize Equilibrium Geometries and Transition States, J. Comput. Chem., 1996, 17, 49–56 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions, J. Phys. Chem. B, 2009, 113, 6378–6396, DOI:10.1021/JP810292N/SUPPL_FILE/JP810292N_SI_003.PDF.
- J. Tomasi, B. Mennucci and R. Cammi, Quantum mechanical continuum solvation models, Chem. Rev., 2005, 105, 2999–3093, DOI:10.1021/CR9904009/ASSET/IMAGES/MEDIUM/CR9904009E00090.GIF.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Revision B.01, 2016 Search PubMed.
- Chemcraft – graphical software for visualization of quantum chemistry computations, https://www.chemcraftprog.com Search PubMed.
- D. Baniya, The Hydrogen Passivated Graphene Cluster and its Stability - First Principle DFT (B3LYP) Levels of Approximation with the Basis Set 3-21G, Kathford J. Eng. Manag., 2018, 1, 5–10, DOI:10.3126/KJEM.V1I1.22013.
- V. Palmieri, E. A. Dalchiele, G. Perini, A. Motta, M. De Spirito, R. Zanoni, A. G. Marrani and M. Papi, Biocompatible: N -acetyl cysteine reduces graphene oxide and persists at the surface as a green radical scavenger, Chem. Commun., 2019, 55, 4186–4189, 10.1039/c9cc00429g.
- R. M. Silverstein, F. X. Webster and D. Kiemle, Spectrometric Identification of Organic Compounds, John Wiley & Sons, 7th edn, 2005 Search PubMed.
- K. N. Kudin, B. Ozbas, H. C. Schniepp, R. K. Prud’homme, I. A. Aksay and R. Car, Raman spectra of graphite oxide and functionalized graphene sheets, Nano Lett., 2008, 8, 36–41, DOI:10.1021/nl071822y.
- A. G. Marrani, A. Motta, F. Amato, R. Schrebler, R. Zanoni and E. A. Dalchiele, Effect of electrolytic medium on the electrochemical reduction of graphene oxide on Si(111) as probed by XPS, Nanomaterials, 2022, 12, 1–21, DOI:10.3390/nano12010043.
- A. G. Marrani, A. C. Coico, D. Giacco, R. Zanoni, F. A. Scaramuzzo, R. Schrebler, D. Dini, M. Bonomo and E. A. Dalchiele, Integration of graphene onto silicon through electrochemical reduction of graphene oxide layers in non-aqueous medium, Appl. Surf. Sci., 2018, 445, 404–414, DOI:10.1016/j.apsusc.2018.03.147.
- B. Konkena and S. Vasudevan, Understanding aqueous dispersibility of graphene oxide and reduced graphene oxide through p K a measurements, J. Phys. Chem. Lett., 2012, 3, 867–872, DOI:10.1021/jz300236w.
- J. A. Boiani, The gran plot analysis of an acid mixture :An undergraduate experiment to highlight this alternate method, J. Chem. Educ., 1986, 63, 724–726, DOI:10.1021/ed063p724.
- G. Filippini, F. Amato, C. Rosso, G. Ragazzon, A. Vega-Peñaloza, X. Companyó, L. Dell'Amico, M. Bonchio and M. Prato, Mapping the Surface Groups of Amine-Rich Carbon Dots Enables Covalent Catalysis in Aqueous Media, Chem, 2020, 6, 3022–3037, DOI:10.1016/j.chempr.2020.08.009.
- A. G. Marrani, R. Zanoni, R. Schrebler and E. A. Dalchiele, Toward Graphene/Silicon Interface via Controlled Electrochemical Reduction of Graphene Oxide, J. Phys. Chem. C, 2017, 121, 5675–5683, DOI:10.1021/acs.jpcc.7b00749.
- A. Kovtun, D. Jones, S. Dell'Elce, E. Treossi, A. Liscio and V. Palermo, Accurate chemical analysis of oxygenated graphene-based materials using X-ray photoelectron spectroscopy, Carbon, 2019, 143, 268–275, DOI:10.1016/j.carbon.2018.11.012.
- J. E. Castle, Practical Surface Analysis by Auger and X-Ray Photoelectron Spectroscopy, ed. D. Briggs and M. P. Seah, John Wiley and Sons Ltd, Chichester, 1983, p. 533 Search PubMed.
- H. Hantsche, High Resolution XPS of Organic Polymers, the Scienta ESCA300 Database, ed. G. Beamson and D. Briggs, Wiley, Chichester, 1992, p. 295 Search PubMed.
- J. Gao, F. Liu, Y. Liu, N. Ma, Z. Wang and X. Zhang, Environment-friendly method to produce graphene that employs vitamin C and amino acid, Chem. Mater., 2010, 22, 2213–2218, DOI:10.1021/cm902635j.
- C. K. Chua and M. Pumera, Chemical reduction of graphene oxide: A synthetic chemistry viewpoint, Chem. Soc. Rev., 2014, 43, 291–312, 10.1039/c3cs60303b.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2na00771a |
| This journal is © The Royal Society of Chemistry 2023 |