
Visible light mediated synthesis of 6H-benzo[c]chromenes: transition-metal-free intramolecular direct C–H arylation†
Micaela D.
Heredia
 ,
Marcelo
Puiatti
,
Roberto A.
Rossi
and
María E.
Budén
*
,
Marcelo
Puiatti
,
Roberto A.
Rossi
and
María E.
Budén
*
INFIQC, Departamento de Química Orgánica, Facultad de Ciencias Químicas, Universidad Nacional de Córdoba, X5000HUA Córdoba, Argentina. E-mail: eugebuden@unc.edu.ar
First published on 22nd November 2021
Abstract
A synthetic approach towards the 6H-benzo[c]chromene ring under visible light and transition-metal-free conditions has been developed. Benzochromenes are synthesized from the corresponding (2-halobenzyl) phenyl ethers or (2-halophenyl) benzyl ethers using KOtBu in dimethyl sulfoxide (DMSO) at room temperature (rt) and blue light-emitting diodes (LEDs) as the light source. This methodology replaces the use of ligands or additives, high temperatures and toxic solvents. The photostimulated reaction exhibits very good tolerance to different functional groups and 5H-dibenzo[c,f]chromenes are also effectively obtained. An electron donor–acceptor complex formed by the dimsyl anion and (2-halobenzyl) phenyl ethers was found and it induces the ET as the initial step in the photocyclization reaction. Furthermore, in order to explain the regiochemical outcome of this reaction, a theoretical analysis was performed using DFT methods.
Introduction
The 6H-benzo[c]chromene ring (Fig. 1, 1) is widely present in pharmaceutical and natural products.1 Cannabinol (2) has an antimicrobial property2 and pulchrol (3) isolated from Bourreria pulchra has an interesting antiprotozoal activity.3 Moreover, heterocyclic compound 4 acts as a G-protein-coupled receptor in the central nervous system.4 | ||
| Fig. 1 The 6H-Benzo[c]chromene ring in bioactive compounds. | ||
Many different synthetic methodologies are described for the construction of 6H-benzo[c]chromenes (Scheme 1). It is known that one of the most used strategies begins from 2-halo aryl benzyl ethers using metal catalysts such as Pd,5–13 Co14 or Au15 by intramolecular dehydrohalide coupling (Scheme 1, route A, Y = H, X = I, Br, Cl or SiMe3). A similar reaction is exemplified via radical intermediates generated from n-Bu3SnH and AIBN.16,17 Zhu et al. also demonstrated the intramolecular Pd-catalyzed decarboxylative coupling of 2-((2-bromobenzyl)oxy) benzoic acid or ethers (route A, Y = COOH or COOR, X = Br) leading to 6H-benzo[c]chromenes.18 Recently, an interesting strategy involving the Pd-catalyzed dehydrogenative coupling of two non-activated aryl C–H bonds to form the key aryl–aryl bond (route A, X = Y = H) has been reported.19 However, the o-(2-pyridyl) sulfonyl group (R1 = O-SO2Py) is needed as a directing group in this synthetic approach. The use of transition metals and toxic reagents or the need to introduce an appropriate directing group into the arene substrate to control the regioselectivity and reactivity are the main disadvantages of these types of transformations.
 | ||
| Scheme 1 Strategies for the synthesis of the 6H-benzo[c]chromene ring. | ||
Furthermore, 6H-benzo[c]-chromenes can be synthesized by Pd- or Ag-catalyzed intramolecular C–H acyloxylation of ortho-aryl benzoic acids followed by a reduction of the carbonyl group (Scheme 1, route B).20,21 In 2018, Sun and co-workers reported the use of iridium catalyst and visible light to initiate an intermolecular radical addition to biaryl vinyl ethers for the synthesis of 6-fluoroalkyl 6H-benzo[c]-chromenes.22 More recently, another study of Sun demonstrated that the addition cascade cyclization of biaryl vinyl ethers can also be promoted using benzoyl peroxide (BPO) at 100 °C (ref. 23) (Scheme 1, route C). Another strategy involves a tandem reduction of various 2′-bromo-biaryl-2-carbaldehydes24 or 2′-phenoxy-biaryl-2-ketones25 to form the corresponding 2′-bromo- or 2′-phenoxy-biaryl-2-alcohol followed by Pd catalysis via Caryl–Oalcoholic coupling, instead of C–C coupling (Scheme 1, route D). Alternatively, a direct C–O bond formation of 2′-bromo-biaryl-2-alcohol could take place in the presence of KOtBu at 100 °C via intramolecular SNAr (route D).26 Fused chromenes are also prepared by cascade hydroarylation/cycloisomerization reactions of polyyne-type aryl propargyl ethers using indium(III) as the catalyst (Scheme 1, route E).27 Starting from 2-(2-(allyloxy)phenyl)furan by the intramolecular Diels–Alder reactions of furan with unactivated alkene in aqueous media under microwave (MW), the benzo[c]chromenes were obtained in very good yields (Scheme 1, route F).28 However, in routes E and F, several steps were necessary to obtain the precursors. Reddy et al. also demonstrated that Zn(II) or Cu(I) chloride promoted a formal [4 + 2] cycloaddition of 4-(phenylethynyl)-2H-chromene-3-carbaldehydes with terminal alkynes at 80 °C using 1,2-dichloroethane (DCE) as the solvent (route G).29,30 Moreover, the access to 6H-benzo[c]chromenes via a three-component cascade reaction by Rh(III)-catalyzed annulation of aryl ketone O-acyloximes, quinones and acetone was reported (Scheme 1, route H).31 Another widely employed approach is to place the leaving group in the aryl moiety instead of the benzyl moiety (route A, Y = N2BF4, X = H). In this regard, 2-phenoxybenzenediazonium or 2-phenoxymethyl phenyl diazonium tetrafluoroborate was efficiently used in the synthesis of highly substituted 6H-benzo[c]chromenes by Pd-catalyzed C–H activation32 or using Ru(bpy)3Cl2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 33 or eosin34 as the photocatalyst. Despite a myriad of available methodologies due to the biological importance of benzo[c]chromenes, it is still necessary to develop new ecological synthetic methods avoiding the use of high temperatures, toxic solvents, expensive or non-commercially available ligands and even the use of transition metals.
33 or eosin34 as the photocatalyst. Despite a myriad of available methodologies due to the biological importance of benzo[c]chromenes, it is still necessary to develop new ecological synthetic methods avoiding the use of high temperatures, toxic solvents, expensive or non-commercially available ligands and even the use of transition metals.
Over the past few years, transition-metal free coupling reactions of haloarenes to obtain biaryls via base-promoted homolytic aromatic substitution (BHAS) have been exhaustively studied.35–37 In 2008, Itami et al.38 showed that KOtBu-induced electron transfer reactions can be used to obtain biaryls using high temperatures or MW irradiation. Following this methodology, many groups reported biaryl synthesis by direct C–H arylation of benzene in the presence of different additives as ligands or super electron donors.39–48 BHAS was also applied for the construction of various types of heterocyclic compounds. Phenanthridinones,49,50 pyrrolophenanthridines,51 dibenzo-azepinones49 and phenanthridines,52,53 among others35,54,55 can be obtained using ligands and high temperatures to initiate the reaction. Particularly, two metal-free strategies have recently been developed to obtain 6H-benzo[c]chromenes via the neocuproine–KOtBu complex in benzene at 100 °C (ref. 56) (Scheme 1, route A, Y = H, X = Br, I) or in the presence of KOtBu in pyridine at 160 °C under MW irradiation57 (Scheme 1, route A, Y = Br, I, X = H). The application of visible light irradiation in organic synthesis is tempting because it helps to avoid the use of ligands and high temperatures. It is known that HAS promoted by a base and light efficiently works in intermolecular coupling reactions for the synthesis of biaryls58 and stylbenes.59 However, no heterocycle has been obtained by using this strategy. Here, we demonstrated that 6H-benzo[c]chromenes can be easily obtained from the corresponding 2-halobenzyl phenyl ethers using just visible light to initiate base (KOtBu) promoted reactions at room temperature.
Results and discussion
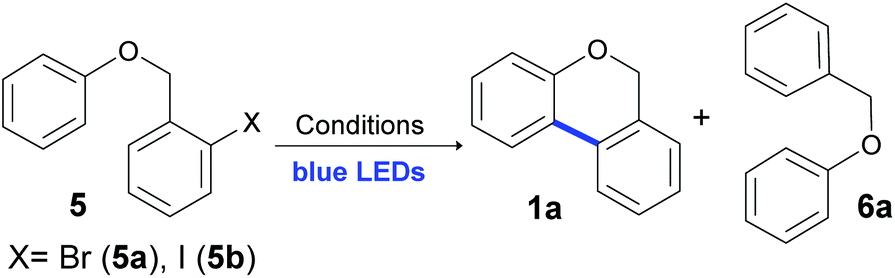
First, the reaction was studied using 2-bromobenzyl phenyl ether 5a as a model substrate. However, the desired product 1a was obtained only in 15% yield when the reaction was carried out in dry dimethyl sulfoxide (DMSO) using 3 equiv. of KOtBu even after 24 hours of irradiation (Table 1, entry 1). Under identical conditions, full conversion was obtained starting from the iodide substrate 5b (the reaction time was not optimized) and 1a was obtained in 70% isolated yield with 25% of the reduction product 6a (Table 1, entry 2). Solvents such as tetrahydrofuran (THF) and acetonitrile (MeCN) were tested and no conversion into products was detected (Table 1, entries 3 and 4). Despite full conversion being achieved, when N,N-dimethylacetamide (DMA) or N,N-dimethyl-formamide (DMF) was tested as a solvent, the main product was 6a instead of 1a, obtained in 79% and 84% yields, respectively (entries 5 and 6). Other bases such as KOH, K2CO3, and Cs2CO3, were examined, and no cyclization product 1a was obtained (entries 7–9). Moreover, when NaOtBu was used as base, 1a was afforded in similar yield as KOtBu (65% yield, entry 10). The treatment of 5b in DMF for 24 h in the presence of K2CO3 (3 equiv.) did not afford the product 1a (entry 11).60 In order to optimize the reaction conditions, after different assays, complete conversion with similar yields of 1a was achieved within just 2 h, decreasing notably the reaction time (Table 1, entry 12). No reaction was observed when no base was used (Table 1, entry 13), indicating that a homolytic C–X cleavage was not possible. Moreover, a lower yield with a low conversion was observed when the reaction was carried out with only 1.5 equiv. of KOtBu (46% of 1a with 23% of 6a, entry 14). Finally, the reaction was carried out in the presence of 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) as a radical trapping agent and no product was obtained, indicating the presence of radicals as intermediates (entry 15).|
|
||||
|---|---|---|---|---|
| Entry | X | Conditions | Yield 1ab (%) | Yield 6ab (%) |
| a Reactions were carried out under a N2 atmosphere using 5 (1 equiv., 0.1 mmol) and a base (3 equiv., 0.3 mmol) in 2 mL of the solvent. Samples were irradiated using blue LEDs (467 nm). b Yields were quantified by 1H NMR using 4-nitroacetopehenone as the standard. Isolated yield is given in parenthesis. c Using 1.5 equiv. of KOtBu. | ||||
| 1 | Br | DMSO, KOtBu, 24 h | 15 | 2 |
| 2 | I | DMSO, KOtBu, 24 h | 67 (70) | 25 |
| 3 | I | THF, KOtBu, 24 h | — | 9 |
| 4 | I | MeCN, KOtBu, 24 h | 10 | 7 |
| 5 | I | DMA, KOtBu, 24 h | 16 | 79 |
| 6 | I | DMF, KOtBu, 24 h | 13 | 84 |
| 7 | I | DMSO, KOH, 24 h | — | — |
| 8 | I | DMSO, K2CO3, 24 h | 1 | — |
| 9 | I | DMSO, Cs2CO3, 24 h | — | — |
| 10 | I | DMSO, NaOtBu, 24 h | 65 | 24 |
| 11 | I | DMF, K2CO3, 24 h | — | — |
| 12 | I | DMSO, KO t Bu, 2 h | 69 | 30 |
| 13 | I | DMSO, 2 h | — | — |
| 14c | I | DMSO, KOtBu, 24 h | 46 | 23 |
| 15 | I | DMSO, KOtBu, TEMPO (1 equiv.), 2 h | — | — |
In order to study the impact of the light properties on the photocyclization reaction, the reaction was conducted using different irradiation sources under the optimal reaction conditions previously found using blue LEDs (Table 2, entry 1). First, no reaction occurred under dark conditions (Table 2, entry 2), indicating that light is a key reactant for the reaction to proceed at room temperature. In addition, green and violet LEDs were also effective and resulted in 57% and 60% yields of 1a, respectively (entries 3 and 4). Moreover, the cyclization reaction was also carried out using 400 W HPIT lamps (λ > 350 nm) and 1a was obtained in a similar yield to using blue LEDs (entry 5). As shown in Table 2, the best results were achieved applying blue LED light with λmax = 467 nm (entry 1) in comparison with other light sources, under the same reaction conditions.
|
|
|||
|---|---|---|---|
| Entry | Light source | Yield 1ab (%) | Yield 6ab (%) |
| a Reactions were carried out under a N2 atmosphere using 5b (1 equiv., 0.1 mmol) and KOtBu (3 equiv., 0.3 mmol) in 2 mL of DMSO. b Yields were quantified by 1H NMR using 4-nitroacetopehenone as the standard. | |||
| 1 | Blue LEDs 3 W, λ = 467 nm | 69 | 30 |
| 2 | Dark | — | — |
| 3 | Green LEDs 3 W, λ = 522 nm | 57 | 29 |
| 4 | Violet LEDs 3 W, λ = 395 nm | 60 | 32 |
| 5 | HPIT 400 W, λ ≥ 350 nm | 70 | 22 |

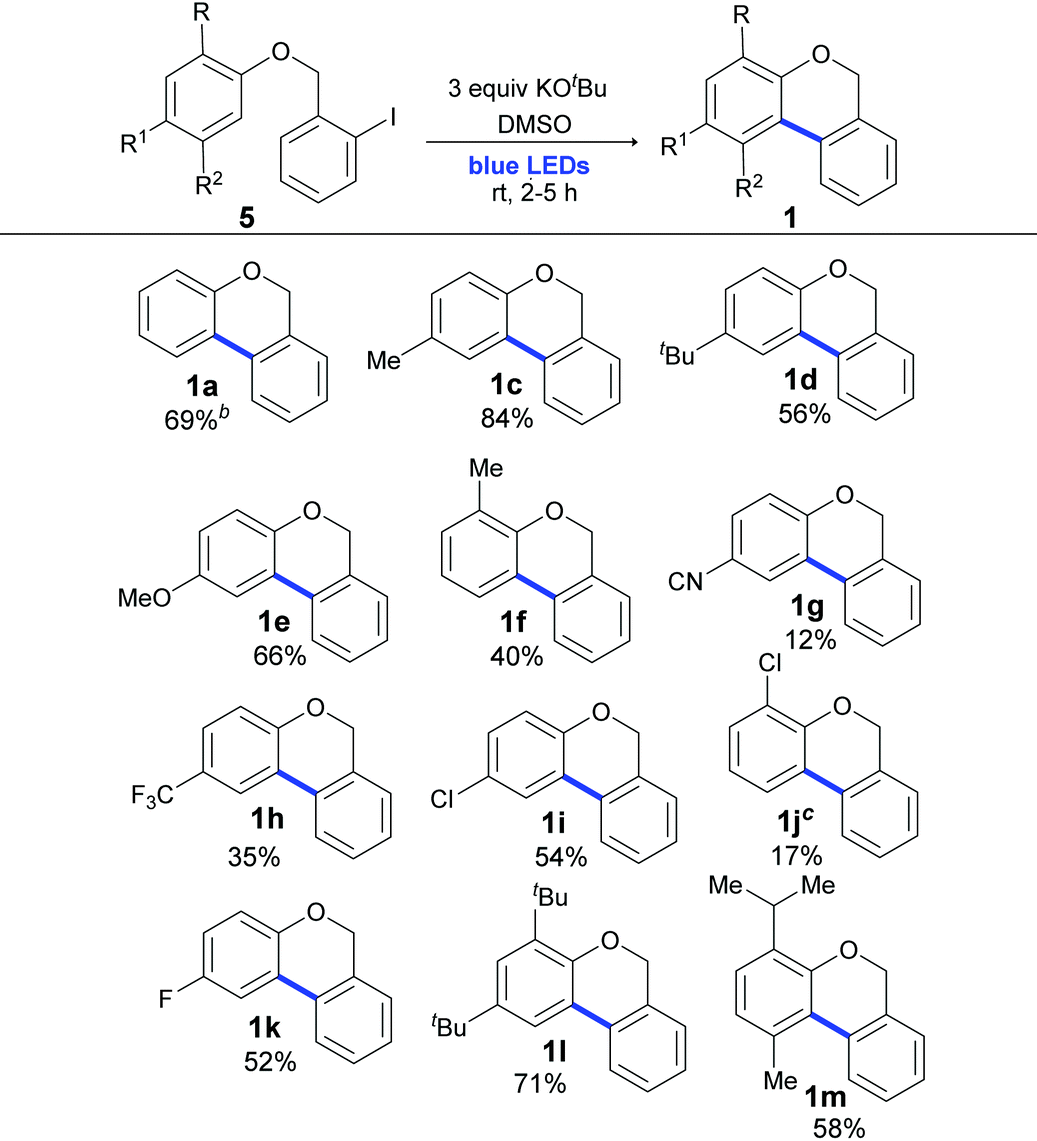
Once the optimal reaction conditions were determined, we focused on the scope of this cyclization methodology. As shown in Table 3, (2-halobenzyl) phenyl ethers with different functional groups (Me, t-Bu, OMe, F, CN, CF3 and Cl) as well as o- or p-substitution were well tolerated and full conversion after 2 or 5 h of irradiation was achieved. As shown in Table 3, similar yields were obtained when light was used instead of the ligand, high temperature and toxic solvent.56 Yields were quantified by 1H NMR (Table 3), and the isolated yields are provided in the ESI (in some cases, the isolated yields were lower than those observed in 1H NMR due to the difficulty in the purification procedures).‡
|
a The reaction was carried out under a N2 atmosphere using 5 (1 equiv., 0.1 mmol) and KOtBu (3 equiv., 0.3 mmol) in DMSO (2 mL) and by irradiating with blue LEDs (467 nm) for 2 h (substrates 5a–5e) or 5 h (substrates 5f–5m). Yields were quantified by 1H NMR using 4-nitroacetophenone as the standard.
b Same as entry 12 of Table 1.
c Compound 1j was obtained as a yellow oil in an inseparable mixture (9:1) with the reductive product 1-(benzyloxy)-2-chlorobenzene.
|
|---|
|
Yields were affected by the electronic properties of the substituents on the aromatic ring and the substrates with electron donating groups (EDG) gave better yields (Table 3, 84–40%, products 1c–1f and 1l and 1m) than those with electron-withdrawing groups (EWG) (Table 3, 54–12%, products 1g–1k). It should also be noted that the EWG moiety on the substrate was ineffective in a similar radical ring closure protocol.57 However, this effect was not observed in the intermolecular coupling of ArX with arenes in the synthesis of biaryls by light induced BHAS reactions.58 Moreover, disubstituted 2-iodobenzyl phenyl ethers smoothly participated in this cyclization reaction and gave diversely substituted 6H-benzo[c]chromenes in very good to moderate yields (products 1l and 1m with 71% and 58% yields, respectively).
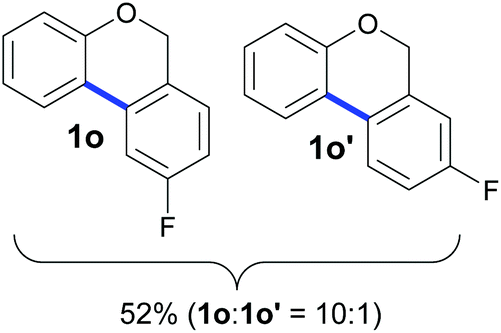
Also, 2-halophenylbenzyl ethers were evaluated as substrates. Contrary to 2-halobenzyl aryl halide, starting from 5o and 5p, a mixture of 6H-benzo[c]chromenes (1o–1o′ and 1p–1p′) was obtained in moderate yields (52% and 57% yields, respectively, Table 4). This behaviour was also found in MW assisted conditions at 160 °C in pyridine, obtaining similar regioselectivity.57
|
|
||
|---|---|---|
| Entry | Substrate | Product (1, yield %)b |
| a The reaction was carried out under a N2 atmosphere using 5 (1 equiv., 0.1 mmol) and KOtBu (3 equiv., 0.3 mmol) in DMSO (2 mL) and by irradiating with blue LEDs (467 nm) for 2 h (substrate 5n) or 5 h (substrates 5o and 5p). b Yields were quantified by NMR 1H using 4-nitroacetophenone as standard. For isolated yields, see ESI.† | ||
| 1 |

|

|
| 2 |
|
|
| 3 |

|

|
The presence of yellow coloration in the reaction mixture led us to test whether an electron donor–acceptor (EDA) complex between the substrate 5b and dimsyl anion was formed.61 UV-visible measurements were performed in order to confirm the formation of the complex and a new peak was observed with a maximum at λ = 319 nm when KOtBu was added to a solution of 5b in DMSO (Fig. 2). Besides, an identical peak was also observed when KOtBu was replaced with other bases such as NaOtBu or NaH.‡ These results enabled us to propose the photoexcitation of an EDA complex which induces the ET (Scheme 2).62
 | ||
| Fig. 2 The UV-visible spectra of CTC between the substrate 5b and dimsyl anion. | ||
 | ||
| Scheme 2 The homolytic aromatic substitution mechanism promoted by a base and light for the synthesis of 6H-benzo[c]chromenes with an EDA complex in the first step of the chain process. | ||
This hypothesis was evaluated with molecular modelling using DFT and TD-DFT approaches. As described in the experimental and theoretical procedures, different DFT functionals were employed.‡ In the first place, the possible formation of the EDA complex was studied for the model substrate 5b. Indeed, a favourable ΔHformation was obtained (approx. −3.8 kcal mol−1 with M062X and −2.5 kcal mol−1 with B3LYP), but since it is unfavourable from the entropic point of view, the ΔGformation is positive, ∼6.2 and 7.4 kcal mol−1 with both methods. Due to the differences between both DFT functionals, the geometries of the complexes are slightly different; however, in both geometries the dimsyl anion is close to 5b by less than 3 Å. Further information about the complex geometries and the procedure employed for the construction and evaluation of the geometries of the complexes is available in the ESI.† Then the excited states of the complex were evaluated using TD-DFT, revealing indeed an excited state with a charge-transfer complex (CTC) character. As shown in Fig. 3, the unpaired spin density indicated the origin, in orange, and the destination of the electron, in green.
 | ||
| Fig. 3 Left: the unpaired electron spin density for the hole, orange, and electron, green, of the excited state of the EDA complex. Right: the geometry of the complex, with the oxygen of the dimsyl anion at ∼2.3 Å of substrate 5b. Obtained at B3LYP/def2tzvp (IEFPCM-DMSO). | ||
The next stage is the computation of the energies involved in the proposed mechanism shown in Scheme 2. It should be noted that with B3LYP and M062X it was not possible to obtain the radical anion of the model substrate 5b since the C–I bond breaks, showing that is not a minimum in the potential energy surface (PES). If we compute the ΔG of the ET from the dimsyl anion to 5b to give radical 7˙ after the C–I fragmentation, it is not energetically favoured. However, if we include the excitation energy of the EDA complex, for the formation of CTC, the reaction turns out to be an exergonic process (Scheme 2). Hence, after this process, formally known as the initiation step, radical 7˙ is formed, starting a chain reaction. The next step is the 6-endo coupling to yield the cyclized intermediate 8˙, and according to our modelling results, it is exergonic (ΔG ∼ −15.1 kcal mol−1 and −16.3 kcal mol−1 for B3LYP and M062X), with a free energy of activation barrier (ΔGact) of ∼9.4 kcal mol−1 and ∼10.8 kcal mol−1 for B3LYP and M062X, respectively. In other ways, the intermediate radical 7˙ can generate the reduction (or dehalogenated) product 6 by removing a hydrogen from the solvent molecule. Once closed radical 8˙ is formed, a very favourable H+ loss in the basic media triggered by the re-aromatization takes place, forming 1a˙− in a very exergonic step, (ΔG ∼ −30.0 kcal mol−1 and −32.6 kcal mol−1 for B3LYP and M062X). Finally, an ET from 1a˙− to 5b gives the final products 1a and 7˙ after the C–I bond fragmentation, restarting the reaction cycle. This final ET is also exergonic for B3LYP (ΔG ∼ −1.5 kcal mol−1) and slightly endergonic for M062X (ΔG ∼ 5.2 kcal mol−1). The cyclic character of the proposed reaction mechanism could be one of the reasons for the relatively short reaction times.
Since an important substituent effect was found in the yields of the cyclization reaction, we carried out a DFT analysis in order to explain these reactivity differences. This substituent effect was also observed in similar reactions with radical anions as intermediates,57 and it could be related to differences in either the initiation or the propagation of the reaction cycle. These two steps could be related to the balance between the electron affinity (EA) of the substrate and the product (or the ionization potential of its radical anion).63,64 This balance affects the length of the reaction chain and directly affects the reaction yields. In Table 5, the results of the computed ΔEAs of (2-iodobenzyl) phenyl ethers 5b–m and 1a–m are presented. In the list, there are two groups, in one group, compounds with EDG (5b–f/1a–f and 5l and 5m/1l and 1m), and in the other, compounds with EWG (5g–k/1g–k). In this table, a tendency is observed: for those compounds where the ΔEA is lower (ΔEA ∼ 0.4 eV), good to very good yields were obtained (1a–f and 1l and 1m), whereas when the ΔEA is higher (ΔEA ∼ 0.6 eV), the yields are not higher than 55%. Moreover, the differences in the EAs of the substrates could be ascribed to slight variances in the nature of the radical anion, which could be observed comparing their unpaired spin density or the Lowest Unoccupied Molecular Orbital (LUMO) distribution.‡
| Cpd. 5/1 | Substituent | ΔEAa,b (eV) |
|---|---|---|
| a Calculations were carried out at B3LYP/def2tzvp (SCRF:IEFPCM/DMSO). b Considering that the radical anions of most of the substrates are not stable, vertical electron affinities are used for these molecules; ΔEA is calculated as EAvert5x − EAad1x. | ||
| 5b/1a | p-H | 0.47 |
| 5c/1c | p-CH3 | 0.49 |
| 5d/1d | p-tBu | 0.43 |
| 5e/1e | p-OCH3 | 0.50 |
| 5f/1f | o-CH3 | 0.43 |
| 5g/1g | p-CN | 0.61 |
| 5h/1h | p-CF3 | 0.66 |
| 5i/1i | p-Cl | 0.63 |
| 5j/1j | o-Cl | 0.60 |
| 5k/1k | p-F | 0.62 |
| 5l/1l | o,p-tBu | 0.35 |
| 5m/1m | o,iProp; m-CH3 | 0.40 |
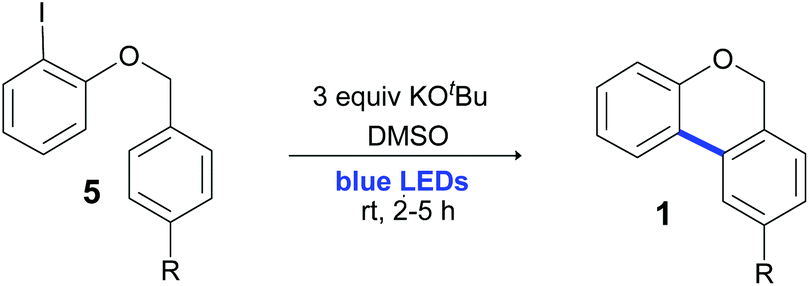
When 2-iodophenyl benzyl ethers (5o and 5p) were used as substrates, a mixture of regioisomers was only observed; the cyclization of the aryl radicals 7˙ was also studied by molecular modeling calculations with the DFT methodology, and B3LYP as the functional (Fig. 4). A previously proposed mechanism for this radical cyclization is shown in Fig. 4A.57,60 Upon the formation of radicals 7o and 7p, a spirocyclohexadienyl radical 916,64 is generated by a 5-exo-trig ipso attack and a concerted ring expansion65 ocurrs to give the thermodynamically more stable radical 10˙. This radical finally affords products 1o′ and 1p′ after re-aromatization. Alternatively, aryl radical 9˙ can form the fused-cyclic system 11˙via a 3-exo closure followed by a successive neophyl rearrangement,66 giving the isomers 1o and 1p. The free activation energy and ΔG for 5-exo and 6-endo cyclization of 7p˙ (R = CH3) were calculated (Fig. 4B). On one hand, a low energy difference between both free energy of activation barriers (ΔGact = 1.5 kcal mol−1) was found (ΔGact 6-endo = 9.6 kcal mol−1vs. ΔGact 5-exo = 8.1 kcal mol−1). These results could help to explain the regioisomeric mixture of products experimentally obtained (a mixture of thermodynamic and kinetic control products). On the other hand, as a comparison, radicals 7c–m derived from 2-iodobenzyl aryl ethers 5c–m only give products derived from 6-endo cyclization (the kinetic control products). In the case of radical 7c for example (Fig. 4C), a higher energy difference between both free energy of activation barriers (ΔΔGact = 4 kcal mol−1) was found, but it favours 6-endo over 5-exo cyclization (ΔGact 6-endo = 11.1 kcal mol−1vs. ΔGact 5-exo = 15.1 kcal mol−1), in agreement with the regiochemistry presented in Table 4.
 | ||
| Fig. 4 (A) The proposed radical pathway for the intramolecular photocyclization reaction of 7o and 7p. (B) 6-endo vs. 5-exo cyclization coupling reaction of radical 7p by B3LYP/def2-tzvp in the presence of a continuum solvent (DMSO). (C) 6-endo vs. 5-exo cyclization coupling reaction of radical 7c by B3LYP/def2-tzvp in the presence of a continuum solvent (DMSO). | ||
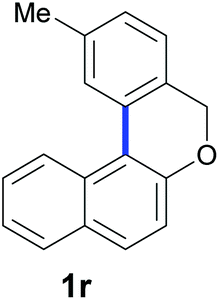
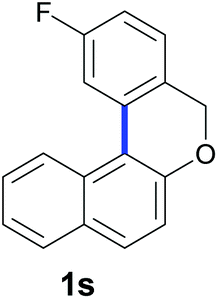
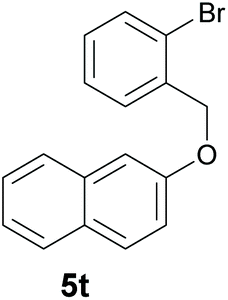
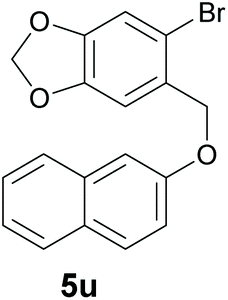
Finally, in order to extend the scope of our reaction, 5H-dibenzo[c,f]chromenes were also effectively synthetized by using this metal-free photoinduced strategy. As shown in Table 6, products 1q–u derived from naphthols were successfully obtained under visible light irradiation starting from the corresponding bromide substrates in very good yields (56–91% yields).
|
|
||
|---|---|---|
| Substrate | Product | Yieldb |
|
a The reaction was carried out under a N2 atmosphere using 5 (1 equiv., 0.1 mmol) and KOtBu (3 equiv., 0.3 mmol) in DMSO (2 mL) and by irradiating with blue LEDs (467 nm) for 2 h.
b Yields were quantified by 1H NMR using 4-nitroacetopehenone as the standard. For isolated yields, see the ESI.†
c A mixture of regioisomers 1s–1s′ was detected by GC-MS.
d The C–H coupling at the C1 of the naphthyl ring was favoured over the C3 coupling product (C1:C3 = 15:1). The C3 coupling product (1u′) was detected by GC-MS, but it was not possible to isolate it.
|
||

|
|
84 |
|
|
56 |

|
|
63c |
|

|
56d |
|

|
91d |

Experimental
General remarks
Purification of the desired compounds was carried out by column chromatography on silica gel or by preparative High Performance Liquid Chromatography (HPLC). Gas chromatographic (GC) analysis was performed using a flame-ionization detector on a 30 m capillary column of 0.32 mm × 0.25 μm film thickness, with a 5% phenylpolysiloxane phase. Gas chromatography-mass spectroscopy (GC-MS) analysis was performed employing an electronic impact (EI) ionization method and a 25 m × 0.2 mm × 0.33 μm column with a 5% phenylpolysiloxane phase. 1H NMR and 13C NMR{1H} spectra were recorded on 400 and 500 MHz on spectrometers with CDCl3 or DMSO-d6 as solvents with TMS as the internal standard. Coupling constants are given in Hz and chemical shifts are reported in δ values in ppm. Data are reported as follows: chemical shift, multiplicity (s = singlet, s br = broad singlet, d = doublet, t = triplet, dd = double doublet, dt = double triplet, ddd = double double doublet and m = multiplet), coupling constants (Hz), and integration.Computational procedure
The experimental behavior of the system was investigated by molecular modeling calculations with the DFT methodology, the B3LYP,67 M062X68 and ωB97XD69 functionals, the def2-tzvp basis set70 with ECP for iodine71 downloaded from the BSE website,72 and the PCM continuum solvent model (DMSO)73 as implemented in Gaussian16 (Rev. C.01). Models of the reaction profiles were explored by using the distinguished reaction coordinate scan. The characterization of the stationary points was carried out by Hessian matrix calculations. The energy recorded for the TSs, anions and radical anions includes zero-point and thermochemical corrections and corresponds to free energy differences at 298 K.Materials
Phenol, 2-iodophenol, 2-chlorophenol, 2-methylphenol, 4-methylphenol, 4-methoxyphenol, 4-tert-butylphenol, 4-fluorophenol, 4-cyanophenol, 4-chlorophenol, 4-(trifluoromethyl)phenol, 2,4-di-tert-butylphenol, 2-isopropyl-5-methylphenol, 1-bromonaphthol, 1-naphthol, 2-naphthol, benzyl chloride, benzyl bromide, 2-bromobenzyl bromide, 2-iodobenzyl chloride, 2-iodobenzyl bromide, 4-methylbenzyl chloride, 4-fluorobenzyl chloride, 5-bromo-6-(bromomethyl)benzo[d][1,3]dioxole, KOtBu, NaOtBu, K2CO3, Cs2CO3 KOH, NaH, NH4NO3, NH4Cl, Na2SO4, 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO), 18-crown-6, 4-nitroacetophenone and 2,4,6-trinitroanisole were purchased from commercial suppliers and used without further purification. Acetone was previously distilled and DMSO, DMF, DMA, THF and MeCN were distilled and dried under molecular sieves (3 Å). MeOH HPLC was previously filtered. All solvents were of analytical grade. The silica used in column chromatography corresponds to silica gel 60 (0.063–0.200 mm).General procedures for the synthesis of compounds 5
Method A: employing the previously described conditions,10,56 to a solution of the corresponding phenol (1 equiv., 2 mmol) with K2CO3 (1 equiv., 2 mmol, 276 mg) in acetone (7.2 mL), the required benzyl bromide (1 equiv., 2 mmol) was added. The mixture was stirred followed by heating to 50 °C for 24 h. The reaction mixture was then cooled to room temperature and water was added and the layers were separated. The organic layer was washed with water (3 × 20 mL), dried over Na2SO4 and concentrated under reduced pressure to afford the reaction crude, which was analyzed by TLC and GC and isolated by column chromatography over silica gel.Method B: to a solution of the corresponding phenol (1 equiv., 2 mmol) with K2CO3 (1 equiv., 2 mmol, 276 mg) in DMF (7.2 mL), 18-crown-6 (1 equiv., 2 mmol, 426 μL) and the required benzyl chloride (1 equiv., 2 mmol) were added. The mixture was stirred followed by heating to 120 °C for 24 h. The reaction mixture was then cooled to room temperature and water was added and the layers were separated. The organic layer was washed with water (3 × 20 mL), dried over Na2SO4 and concentrated under reduced pressure to afford the reaction crude, which was analyzed by TLC and GC and isolated by column chromatography over silica gel.
Characterization data of compounds 5
:0→80:20). A white solid was isolated in 79% yield (1.57 mmol, 528 mg). 1H NMR (400 MHz, CDCl3) δ 7.89 (dd, J = 7.9, 1.2 Hz, 1H), 7.64–7.59 (m, 2H), 7.45 (dd, J = 7.7, 1.8 Hz, 1H), 7.38 (td, J = 7.5, 1.3 Hz, 1H), 7.09–7.01 (m, 3H), 5.10 (s, 2H). 13C NMR{1H} (101 MHz, CDCl3) δ 161.7, 139.6, 138.0, 134.2, 130.1, 128.8, 128.7, 119.2, 115.8, 104.7, 97.4, 74.3. GC/MS EI m/z 335 (M+, 9), 217 (100), 90 (86), 89 (41), 63 (17), 50 (4). FTIR (cm−1): 3064, 2912, 2222, 1307, 1261, 1168, 1038, 1012, 837, 826, 751, 545. HRMS (ESI-TOF+) m/z: [M + Na]+ calcd for C14H10lNNaO 357.9699; found 357.9696.
:0→90:10). A white solid was isolated in 46% yield (0.95 mmol, 233 mg). 1H NMR (400 MHz CDCl3) δ 7.85 (d, J = 8 Hz, 1H), 7.63 (d, J = 7.6 Hz, 1H), 7.39 (td, J = 8.1, 1.4 Hz, 2H), 7.21 (td, J = 7.9, 1.7 Hz, 1H), 7.03 (td, J = 7.7, 1.7 Hz, 1H), 6.98–6.90 (m, 2H), 5.10 (s, 2H). 13C NMR{1H} (101 MHz, CDCl3) δ 154.0, 139.2, 138.8, 130.6, 129.6, 128.6, 128.5, 127.9, 123.4, 122.1, 114.2, 96.6, 74.8. GC/MS EI m/z 344 (M+, 9), 217 (100), 90 (60), 89 (31), 63 (13). FTIR (cm−1): 3064, 2912, 1253, 1064, 1041, 1010, 847, 746, 423. HRMS (ESI-TOF+) m/z: [M + Na]+ calcd for C13H10ClINaO 366.9357; found 366.9337.
:20). A white solid was isolated in 46% yield (0.91 mmol, 333.7 mg). 1H NMR (400 MHz, CDCl3) δ 7.87 (dd, J = 7.9, 1.2 Hz, 1H), 7.56 (ddd, J = 7.7, 1.8, 0.8 Hz, 1H), 7.39 (td, J = 7.5, 1.2 Hz, 1H), 7.14 (d, J = 7.7 Hz, 1H), 7.03 (td, J = 7.7, 1.7 Hz, 1H), 6.79 (dd, J = 7.7, 1.6 Hz, 1H), 6.72 (dd, J = 1.6, 1H), 5.01 (s, 2H), 3.40 (m, 1H), 2.33 (s, 3H), 1.24 (d, J = 6.8, 6H). 13C NMR{1H} (101 MHz, CDCl3) δ 155.5, 139.9, 139.3, 136.6, 134.5, 129.4, 128.5, 128.5, 126.2, 121.9, 112.9, 97.1, 74.2, 26.8, 23.0, 21.5. GC/MS EI m/z 366 (M+, 73), 217 (100), 197 (20), 148 (12), 91 (17), 90 (46), 89 (20). FTIR (cm−1): 3052, 2959, 1255, 1162, 1010, 746, 808, 594. HRMS (ESI-TOF+) m/z: [M + Na]+ calcd for C17H19INaO 389.0373; found 389.0389.
:0→97:3). Yellow oil was isolated in 37% yield (0.71 mmol, 233 mg). 1H NMR (500 MHz CDCl3) δ 7.81 (dd, J = 7.8, 1.6 Hz, 1H), 7.48 (dd, J = 8.7, 5.4 Hz, 2H), 7.29 (ddd, J = 8.2, 7.5, 1.6 Hz, 1H), 7.09 (ddd, J = 9.6, 5.8, 2.5 Hz, 2H), 6.86 (dd, J = 8.2, 1.2 Hz, 1H), 6.74 (td, J = 7.6, 1.3 Hz, 1H), 5.11 (s, 2H). 13C NMR{1H} (101 MHz, CDCl3) δ 162.6 (J = 246 Hz), 157.2, 139.8, 132.4 (J = 3 Hz), 129.6, 129.0 (J = 8 Hz), 123.1, 115.4 (J = 8 Hz), 113.0, 87.0, 70.4. GC/MS EI m/z 328 (M+, 4), 109 (100), 50 (1). FTIR (cm−1): 3062, 2925, 1274, 1224, 1051, 1018, 821, 749, 648, 599, 493. HRMS (ESI-TOF+) m/z: [M + Na]+ calcd for C13H10FINaO 350.09653; found 350.9659.
:0→90:10). A light brown solid was isolated in 34% yield (0.65 mmol, 212 mg). 1H NMR (400 MHz, CDCl3) δ 8.23 (d, J = 8.6 Hz, 1H), 7.73 (t, J = 6.8 Hz, 2H), 7.54 (t, J = 7.7 Hz, 1H), 7.38 (t, J = 5.5 Hz, 3H), 7.24 (d, J = 9.0 Hz, 1H), 7.18 (d, J = 7.6 Hz, 2H), 5.24 (s, 2H), 2.34 (s, 3H). 13C NMR{1H} (101 MHz, CDCl3) δ 153.1, 137.9, 133.8, 133.3, 130.1, 129.4, 128.9, 128.1, 127.8, 127.4, 126.6, 124.6, 115.8, 110.1, 71.9, 21.4. GC/MS EI m/z 328 (M++ 2, 1), 326 (M+, 2), 105 (100), 79 (10), 77 (10). FTIR (cm−1): 3049, 2915, 1266, 1067, 1018, 806, 764, 744, 483. HRMS (ESI-TOF+) m/z: [M + Na]+ calcd for C18H15BrNaO 349.0174; found 349.0163.
:0→80:20). A white solid was isolated in 51% yield (1.27 mmol, 420.5 mg). 1H NMR (400 MHz, CDCl3) δ 8.23 (d, J = 8.5 Hz, 1H), 7.76 (d, J = 8.8 Hz, 2H), 7.56 (t, J = 7.6 Hz, 1H), 7.51–7.46 (m, 2H), 7.40 (t, J = 7.3 Hz, 1H), 7.26–7.22 (m, 1H), 7.07 (t, J = 8.4 Hz, 2H), 5.23 (s, 2H). 13C NMR{1H} (101 MHz, CDCl3) δ 162.7 (J = 247 Hz), 152.9, 133.3, 132.6 (J = 3 Hz), 130.3, 129.3 (J = 8 Hz), 129.0, 128.2, 127.9, 126.5, 124.8, 115.8 (J = 4 Hz), 115.6, 110.3, 71.4. GC/MS EI m/z 332 (M+ + 2, 4), 330 (M+, 3), 114 (11), 109 (100), 83 (16). FTIR (cm−1): 3065, 2933, 1261, 1053, 826, 795, 745, 410. HRMS (ESI-TOF+) m/z: [M + Na]+ calcd for C17H12BrFNaO 352.9948; found 352.9926.
:0→90:10). A white solid was isolated in 53% yield (1.05 mmol, 338 mg). 1H NMR (400 MHz, CDCl3) δ 7.77 (d, J = 8.7 Hz, 2H), 7.73 (d, J = 8.3 Hz, 1H), 7.61 (d, J = 8.0 Hz, 2H), 7.45–7.42 (t, J = 6.8 Hz, 1H), 7.34 (t, J = 7.6 Hz, 2H), 7.28–7.17 (m, 3H), 5.25 (s, 2H). 13C NMR{1H} (101 MHz, CDCl3) δ 156.6, 136.4, 134.7, 132.8, 129.7, 129.4, 129.4, 129.1, 127.8, 127.7, 127.0, 126.6, 124.0, 122.6, 119.0, 107.6, 69.6. GC/MS EI m/z 314 (M+ + 2, 1), 312 (M+, 1), 233 (51), 171 (95), 169 (100), 115 (35), 90 (67), 89 (51), 63 (18). FTIR (cm−1): 3052, 2930, 1268, 1240, 1095, 1064, 788, 770, 682. HRMS (ESI-TOF+) m/z: [M + Na]+ calcd for C17H13BrNaO 335.0042; found 335.0034.
:0→90:10). A brown solid was isolated in 43% yield (0.83 mmol, 295 mg). 1H NMR (400 MHz, CDCl3) δ 7.78–7.72 (m, 3H), 7.43 (ddd, J = 8.2, 6.8, 1.4 Hz, 1H), 7.34 (ddd, J = 8.1, 6.8, 1.3 Hz, 1H), 7.25–7.18 (m, 2H), 7.07 (s, 1H), 7.05 (s, 1H), 5.97 (s, 2H), 5.15 (s, 2H). 13C NMR{1H} (101 MHz, CDCl3) δ 156.4, 148.2, 147.8, 134.6, 129.7, 129.6, 129.3, 127.8, 127.0, 126.6, 124.0, 119.0, 113.2, 112.9, 109.2, 107.5, 102.0, 69.5. GC/MS EI m/z 356 (M+, 5), 277 (22), 215 (97), 213 (100), 207 (24), 157 (11), 115 (20), 78 (43), 77 (16), 76 (33), 75 (34), 50 (11). FTIR (cm−1): 3059, 2902, 1475, 1498, 1237, 1036, 930, 811, 744. HRMS (ESI-TOF+) m/z: [M + Na]+ calcd for C18H13BrNaO3 378.9940; found 378.9933.
General procedures for photoinduced BHAS reactions under visible light irradiation
The reactions were carried out in a vial at rt and under a N2 atmosphere and irradiated with a blue LED (3 W) using 1 equiv. (0.1 mmol) of 2-halobenzyl phenyl ether and 3 equiv. of KOtBu (0.3 mmol, 33.7 mg) in DMSO (2 mL previously dried and deoxygenated). After the reaction was complete, it was quenched with NH4NO3 or NH4Cl and excess water and the residue was extracted with EtOAc (3 × 30 mL). The organic layers extracted were combined, washed with water, dried with anhydrous Na2SO4 and concentrated under reduced pressure to leave the crude products. Yields were quantified by 1H NMR using an internal method employing 2,4,6-trinitro anisole as the standard. Isolated yields were obtained by column chromatography on silica gel or preparative HPLC.Characterization data of 6H-benzo[c]chromenes and 5H-dibenzo[c,f]chromenes
Compounds 1o and 1o′9 were obtained as a yellow oil by preparative HPLC, eluting with MeOH in 28% isolated yield (7 mg, 0.028 mmol), a mixture of inseparable isomers (1o:1o′ = 10:1). 9-Fluoro-6H-benzo[c]chromene (1o): 1H NMR (400 MHz, CDCl3) δ 7.66 (dd, J = 7.8, 1.0 Hz, 1H), 7.38 (dd, J = 9.9, 2.4 Hz, 1H), 7.30–7.20 (m, 1H), 7.10 (m, 2H), 7.02–6.96 (m, 2H), 5.09 (s, 2H). 13C NMR{1H} (101 MHz, CDCl3) δ 163.2 (J = 246 Hz), 155.0, 132.5, 132.4, 130.3, 127.2 (J = 3 Hz), 126.4 (J = 8 Hz), 123.6, 122.4, 117.6, 114.5 (J = 22 Hz), 109.2 (J = 23 Hz), 68.1. GC/MS EI m/z 201 (M+ + 1, 7), 200 (M+, 56), 199 (100), 171 (12), 170 (30), 100 (11), 85 (17). FTIR (cm−1): 3041, 2922, 1274, 1243, 1044, 1038, 806, 754, 534. HRMS (ESI-TOF+) m/z: [M − H]+ calcd for C13H8FO 199.0565; found 199.0551.
Compounds 1s and 1s′ were obtained as a yellow oil by preparative HPLC, eluting with MeOH in 60% isolated yield (0.12 mmol, 30 mg), a mixture of inseparable isomers (1s:1s′ = 10:1). 2-Fluoro-5H-dibenzo[c,f]chromene (1s): 1H NMR (400 MHz, CDCl3) δ 8.49 (d, J = 8.6 Hz, 1H), 7.86 (d, J = 2 Hz, 1H), 7.79–7.71 (m, 2H), 7.57 (ddd, J = 8.4, 6.9, 1.4 Hz, 1H), 7.43 (ddd, J = 8.0, 6.9, 1.1 Hz, 1H), 7.28 (dd, J = 8.3, 5.8 Hz, 1H), 7.23 (d, J = 8.8 Hz, 1H), 7.04 (td, J = 8.4, 2.5 Hz, 1H), 5.02 (s, 2H). 13C NMR{1H} (101 MHz, CDCl3) δ 162.8 (J = 245 Hz), 154.5, 133.8 (J = 4 Hz) 131.0, 130.7, 130.1, 129.1, 128.4 (J = 3 Hz), 127.5, 127.02, 126.7 (J = 4 Hz), 124.3 (×2), 122.6, 118.3, 113.7 (J = 23 Hz), 113.2 (J = 22 Hz), 68.9. GC/MS EI m/z 251 (M+ + 1, 22), 250 (M+, 100), 249 (77), 233 (14), 222 (17), 221 (49), 220 (61), 218 (25), 207 (22), 201 (10), 125 (11), 110 (31), 109 (12), 101 (13), 100 (10), 97 (20), 85 (10), 81 (11), 73 (19), 71 (11), 69 (13), 57 (22), 55 (18). FTIR (cm−1): 3057, 2922, 1240, 1222, 1022, 824, 803, 589. HRMS (ESI-TOF+) m/z: [M − H]+ calcd for C17H10FO 249.0721; found 249.0730.
Compounds 1u and 1u′ were obtained as a yellow oil by preparative HPLC in 85% isolated yield (46 mg, 0.17 mmol), a mixture of inseparable isomers (1u:1u′ = 15:1). 8H-[1,3]dioxolo [4′,5′:4,5]benzo[1,2-c]benzo[f]chromene (1u): 1H NMR (400 MHz, CDCl3) δ 8.45 (d, J = 8.5 Hz, 1H), 7.84 (d, J = 8.1 Hz, 1H), 7.70 (d, J = 8.8 Hz, 1H), 7.59–7.48 (m, 2H), 7.40 (m, 1H), 7.22 (d, J = 8.8 Hz, 1H), 6.81 (s, 1H), 6.03 (s, 2H), 4.94 (s, 2H). 13C NMR{1H} (101 MHz, CDCl3) δ 153.5, 147.7, 146.7, 130.8, 130.1, 129.6, 129.0, 128.0, 126.9, 126.4, 124.7, 124.0, 121.8, 118.2, 107.3, 106.3, 101.4, 69.4. GC/MS EI m/z 276 (M+, 33), 275 (100), 209 (12), 208 (16), 207 (86), 191 (19), 180 (20), 177 (11), 163 (10), 137 (13), 133 (12), 101 (13), 96 (24), 94 (11), 88 (11), 87 (12), 75 (16), 73 (76), 61 (10). FTIR (cm−1): 3057, 2922, 1240, 1222, 1012, 816, 751, 589. HRMS (ESI-TOF+) m/z: [M + Na]+ calcd for C18H12NaO3 299.0671; found 299.0671.
Conclusions
In conclusion, a green methodology involving a visible-light-promoted method has been developed to synthesize 6H-benzo[c]chromenes. In this protocol, benzochromenes were obtained from the corresponding (2-iodobenzyl) phenyl ethers by utilizing only KOtBu in DMSO and blue LEDs as a light source at room temperature. The reaction was highly tolerant to a variety of functional groups and was further expanded to obtain several 5H-dibenzo[c,f]chromenes. A photoinduced base-promoted homolytic aromatic substitution reaction (photo-BHAS) is proposed as a plausible mechanism for the intramolecular reaction. The photoexcitation of an EDA complex (formed by the dimsyl anion and substrate 5) starts the reaction with a chain mechanism. The computational calculations helped to explain or understand the reactivity of the halogenated substrates in the propagation of this mechanism, as well as the regiochemistry of the radical coupling.Notable features of the above processes include:
(1) transition-metal and ligand free methodology;
(2) room temperature reactions without conventional heating or MW assistance;
(3) environmentally friendly conditions.
With the advantages described above, the synthetic methods developed in this work are expected to be used as valuable alternative protocols for the preparation of synthetically and biologically interesting 6H-benzo[c]chromenes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported partly by Agencia Córdoba Ciencia, Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Secretaría de Ciencia y Tecnología, Universidad Nacional de Córdoba (SECyT) and Agencia Nacional de Promoción Científica y Técnica (ANPCyT). M. D. H. gratefully acknowledges the receipt of the fellowship from CONICET. This work used computational resources from CCAD – Universidad Nacional de Córdoba (https://ccad.unc.edu.ar/), which are part of SNCAD – MinCyT, República Argentina.Notes and references
- R. Pratap and V. J. Ram, Chem. Rev., 2014, 114, 10476–10526 CrossRef CAS PubMed.
- G. Appendino, S. Gibbons, A. Giana, A. Pagani, G. Grassi, M. Stavri, E. Smith and M. M. Rahman, J. Nat. Prod., 2008, 71, 1427–1430 CrossRef CAS.
- D. Killander and O. Sterner, Eur. J. Org. Chem., 2014, 1594–1596 CrossRef CAS.
- G. A. Thakur, S. Bajaj, C. Paronis, Y. Peng, A. L. Bowman, L. S. Barak, M. G. Caron, D. Parrish, R. Deschamps and A. Makriyannis, J. Med. Chem., 2013, 56, 3904–3921 CrossRef CAS.
- L. Campeau, M. Parisien, A. Jean and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 581–590 CrossRef CAS.
- Y. Tanji, N. Mitsutake, T. Fujihara and Y. Tsuji, Angew. Chem., Int. Ed., 2018, 57, 10314–10317 CrossRef CAS PubMed.
- R. Cano, J. M. Pérez, D. J. Ramón and G. P. McGlacken, Tetrahedron, 2016, 72, 1043–1050 CrossRef CAS.
- T. Korenaga, N. Suzuki, M. Sueda and K. Shimada, J. Organomet. Chem., 2015, 780, 63–69 CrossRef CAS.
- M. C. Ortiz Villamizar, F. I. Zubkov, C. E. Puerto Galvis, L. Y. Vargas Méndez and V. V. Kouznetsov, Org. Chem. Front., 2017, 4, 1736–1744 RSC.
- L. Campeau, M. Parisien, M. Leblanc and K. Fagnou, J. Am. Chem. Soc., 2004, 126, 9186–9187 CrossRef CAS.
- Y. An, B. S. Zhang, Z. Zhang, C. Liu, X. Y. Gou, Y. N. Ding and Y. M. Liang, Chem. Commun., 2020, 56, 5933–5936 RSC.
- M. Parisien, D. Valette and K. Fagnou, J. Org. Chem., 2005, 70, 7578–7584 CrossRef CAS.
- M. Lafrance, D. Lapointe and K. Fagnou, Tetrahedron, 2008, 64, 6015–6020 CrossRef CAS.
- W. Liu, H. Cao, J. Xin, L. Jin and A. Lei, Chem. – Eur. J., 2011, 17, 3588–3592 CrossRef CAS.
- T. J. A. Corrie, L. T. Ball, C. A. Russell and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2017, 139, 245–254 CrossRef CAS.
- W. R. Bowman, E. Mann and J. Parr, J. Chem. Soc., Perkin Trans. 1, 2000, 2991–2999 RSC.
- A. M. Rosa, A. M. Lobo, P. S. Branco and P. Sundaresan, Tetrahedron, 1997, 53, 285–298 CrossRef CAS.
- Z. Shen, Z. Ni, S. Mo, J. Wang and Y. Zhu, Chem. – Eur. J., 2012, 18, 4859–4865 CrossRef CAS.
- D.-D. Guo, B. Li, D.-Y. Wang, Y.-R. Gao, S.-H. Guo, G.-F. Pan and Y.-Q. Wang, Org. Lett., 2017, 19, 798–801 CrossRef CAS.
- Y. Li, Y. Ding, J. Wang, Y. Su and X. Wang, Org. Lett., 2013, 11, 2574–2577 CrossRef PubMed.
- J. Dai, W. Xu, Y. Wu, W. Zhang, Y. Gong, X. He, X. Zhang and H. Xu, J. Org. Chem., 2015, 80, 911–919 CrossRef CAS.
- Q. Deng, L. Tan, Y. Xu, P. Liu and P. Sun, J. Org. Chem., 2018, 83, 6151–6161 CrossRef CAS.
- L. Ji, Q. Deng, P. Liu and P. Sun, Org. Biomol. Chem., 2019, 17, 7715–7722 RSC.
- A. Ahmed, S. Dhara and J. K. Ray, Tetrahedron Lett., 2013, 54, 1673–1676 CrossRef CAS.
- F. Wang, Y. Liu, F. Chen, M. Qu and M. Shi, Tetrahedron Lett., 2015, 56, 2393–2396 CrossRef CAS.
- R. Singha, A. Ahmed, Y. Nuree, M. Ghosh and J. K. Ray, RSC Adv., 2015, 5, 50174–50177 RSC.
- L. Alonso-Marañón, L. A. Sarandeses, M. M. Martínez and J. Pérez Sestelo, Org. Chem. Front., 2018, 5, 2308–2312 RSC.
- X. F. Yan He, X. Zhang, L. Cui and J. Wang, Green Chem., 2012, 14, 3429–3435 RSC.
- B. Muralidhar and S. R. Reddy, ChemistrySelect, 2017, 2, 2539–2543 CrossRef CAS.
- B. Muralidhar and S. R. Reddy, Bull. Chem. Soc. Jpn., 2018, 91, 65–70 CrossRef CAS.
- W. Yang, J. Wang, H. Wang, L. Li, Y. Guan, X. Xu and D. Yu, Org. Biomol. Chem., 2018, 16, 6865–6869 RSC.
- J. Zhou, L. Z. Huang, Y. Q. Li and Z. T. Du, Tetrahedron Lett., 2012, 53, 7036–7039 CrossRef CAS.
- Q. Bai, J. He, X. Zhu, G. Feng and C. Jin, Chin. J. Org. Chem., 2019, 39, 527–531 CrossRef CAS.
- J.-Y. He, Qi.-F. Bai, C. Jin and G. Feng, Synlett, 2018, 29, 2311–2315 CrossRef CAS.
- C. Sun and Z. Shi, Chem. Rev., 2014, 114, 9219–9280 CrossRef CAS PubMed.
- I. Hussain and T. Singh, Adv. Synth. Catal., 2014, 356, 1661–1696 CrossRef CAS.
- E. Shirakawa and T. Hayashi, Chem. Lett., 2012, 41, 130–134 CrossRef CAS.
- S. Yanagisawa, K. Ueda, T. Taniguchi and K. Itami, Org. Lett., 2008, 10, 4673–4676 CrossRef CAS.
- C. Sun, H. Li, D. Yu, M. Yu, X. Zhou, X. Lu and K. Huang, Nat. Chem., 2010, 2, 1044–1049 CrossRef CAS PubMed.
- E. Shirakawa, K. Itoh, T. Higashino and T. Hayashi, J. Am. Chem. Soc., 2010, 132, 15537–15539 CrossRef CAS.
- W. Liu, H. Cao, H. Zhang, H. Zhang, K. H. Chung, C. He and H. Wang, J. Am. Chem. Soc., 2010, 132, 16737–16740 CrossRef CAS PubMed.
- W. Liu and L. Xu, Tetrahedron, 2015, 71, 4974–4981 CrossRef CAS.
- K. Masters, S. Lindner and S. Bra, J. Fluor. Chem., 2015, 179, 102–105 CrossRef.
- W. Liu, R. Liu and Y. Bi, Tetrahedron, 2015, 71, 2622–2628 CrossRef CAS.
- D. Ghosh, J. Lee, C. Liu, Y. Chiang and H. Man, Adv. Synth. Catal., 2014, 356, 406–410 CrossRef CAS.
- Q. Song, D. Zhang, Q. Zhu and Y. Xu, Org. Lett., 2014, 16, 5272–5274 CrossRef CAS.
- Z. Xue, Y. Ying and K. Shing, Tetrahedron Lett., 2014, 55, 6180–6183 CrossRef CAS.
- A. Dewanji, S. Murarka, D. P. Curran and A. Studer, Org. Lett., 2013, 15, 6102–6105 CrossRef CAS PubMed.
- B. S. Bhakuni, A. Kumar, S. J. Balkrishna, J. A. Sheikh, S. Konar and S. Kumar, Org. Lett., 2012, 14, 2838–2841 CrossRef CAS PubMed.
- S. Sharma, M. Kumar, S. Sharma, O. S. Nayal, N. Kumar, B. Singh and U. Sharma, Org. Biomol. Chem., 2016, 14, 8536–8544 RSC.
- S. De, S. Ghosh, S. Bhunia, J. A. Sheikh and A. Bisai, Org. Lett., 2012, 14, 4466–4469 CrossRef CAS.
- S. De, S. Mishra, B. N. Kakde, D. Dey and A. Bisai, J. Org. Chem., 2013, 78, 7823–7844 CrossRef CAS.
- Y. Wu, S. M. Wong, F. Mao, T. L. Chan and F. Y. Kwong, Org. Lett., 2012, 14, 5306–5309 CrossRef CAS PubMed.
- B. S. Bhakuni, A. Yadav, S. Kumar, S. Patel, S. Sharma and S. Kumar, J. Org. Chem., 2014, 79, 2944–2954 CrossRef CAS.
- S.-B. Lin, W.-W. Wang, J.-P. Meng, X.-W. Li, J. Wu and X.-L. Sun, Tetrahedron Lett., 2019, 60, 151309 CrossRef CAS.
- C. L. Sun, Y. F. Gu, W. P. Huang and Z. J. Shi, Chem. Commun., 2011, 47, 9813–9815 RSC.
- D. S. Roman, Y. Takahashi and A. B. Charette, Org. Lett., 2011, 13, 3242–3245 CrossRef CAS.
- M. E. Budén, J. F. Guastavino and R. A. Rossi, Org. Lett., 2013, 15, 1174–1177 CrossRef.
- J. F. Guastavino, M. E. Budén and R. A. Rossi, J. Org. Chem., 2014, 79, 9104–9111 CrossRef CAS.
- P. D. Q. Dao, S. L. Ho, H. J. Lim and C. S. Cho, J. Org. Chem., 2018, 83, 4140–4146 CrossRef CAS.
- M. E. Budén, J. I. Bardagí, M. Puiatti and R. A. Rossi, J. Org. Chem., 2017, 82, 8325–8333 CrossRef PubMed.
- J. P. Barham, G. Coulthard, K. J. Emery, E. Doni, F. Cumine, G. Nocera, M. P. John, L. E. A. Berlouis, T. Mcguire, T. Tuttle and J. A. Murphy, J. Am. Chem. Soc., 2016, 138, 7402–7410 CrossRef CAS PubMed.
- D. M. A. Vera and A. B. Pierini, Phys. Chem. Chem. Phys., 2004, 6, 2899–2903 RSC.
- J. L. Borioni, M. Puiatti, D. M. A. Vera and A. B. Pierini, Phys. Chem. Chem. Phys., 2017, 19, 9189–9198 RSC.
- L. Benati, G. Calestani, R. Leardini, M. Minozzi, D. Nanni, P. Spagnolo, S. Strazzari and G. Zanardi, J. Org. Chem., 2003, 68, 3454–3464 CrossRef CAS.
- A. Studer and M. Bossart, Tetrahedron, 2001, 57, 9649–9667 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1998, 98, 5648–5662 CrossRef.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- J. Chai and M. Head-gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- F. Weigend, R. Ahlrichs and F. K. Gmbh, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- K. A. Peterson, D. Figgen, E. Goll, H. Stoll, M. Dolg, D. Figgen, E. Goll and H. Stoll, J. Chem. Phys., 2003, 119, 11113–11123 CrossRef CAS.
- B. P. Pritchard, D. Altarawy, B. Didier, T. D. Gibson and T. L. Windus, J. Chem. Inf. Model., 2019, 59, 4814–4820 CrossRef CAS.
- G. Scalmani, M. J. Frisch, G. Scalmani and M. J. Frisch, J. Chem. Phys., 2010, 132, 114110–114115 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ob01673c |
| ‡ Two DFT functionals were employed in the molecular modelling studies: M062X, and B3LYP. The results in the main text correspond to the ones with B3LYP. The results obtained with M062X are also included in the ESI.† All the calculations were carried out with the def2tzvp basis set and a pseudopotential for iodine in DMSO modelled within the IEFPCM approximation. |
| This journal is © The Royal Society of Chemistry 2022 |