
A double-triggered self-immolative spacer for increased selectivity of molecular release†
Steve
Huvelle
a,
Thomas
Le Saux
b,
Ludovic
Jullien
 *b and
Frédéric
Schmidt
*c
*b and
Frédéric
Schmidt
*c
ai-CleHS, UMR 8060, Chimie ParisTech – PSL, Paris, Île-de-France, France
bPASTEUR, Département de chimie, École normale supérieure, PSL University, Sorbonne Université, CNRS, 24, rue Lhomond, 75005 Paris, France. E-mail: Ludovic.Jullien@ens.psl.eu
cInstitut Curie, PSL University, CNRS UMR 3666 – INSERM U1143, 26 rue d'Ulm, 75248 PARIS CEDEX 05, FRANCE. E-mail: Frederic.Schmidt@curie.fr
First published on 1st December 2021
Abstract
A self-immolative spacer based on dissymmetrical N,N′-bis-carbamate aniline is introduced to liberate a substrate from a precursor after dual activation. The proof of principle of its exclusive selectivity for substrate liberation has been conducted on a profluorophore.
Introduction
Prodrug strategies1,2 are increasingly used in therapy to overcome toxicity issues. In antitumoral chemotherapy, they are considered to circumvent lethal side effects, provided that cytotoxic agents (e.g. auristatins) can be reliably released at the tumor site after activation involving overexpressed enzymes or metabolites.3,4 The search for selectivity in discriminating cancerous and healthy cells is key. Since the former differs from the latter by a plurality of factors, it has been considered to govern the release of the active compound not by one but by several triggering events as it is performed in OR logic gate strategies.5,6 In this report, we aim at using two triggering events to liberate a covalently bound substrate from a precursor of low molecular weight.In relation to the preceding goal, profluorophores requiring dual (e.g. enzymatic and chemical) activation followed by bond breakage to become fluorescent have been interestingly reported.7–10 However, the reported strategies have relied on directly introducing two orthogonal protecting groups on the fluorophore backbone. This constraint has restricted their scope to specific multifunctional substrates and few prodrug examples have been reported at present with these strategies. To overcome this limitation, an AND logic gate strategy recently developed uses a self-immolative spacer that triggers the drug release after two enzymatic reaction steps.11–13 However, this sequential strategy intrinsically carries on a hierarchy: a first enzymatic activation has to precede a second one for the liberation to occur. In order to solve this drawback, Dal Corso, Gennari and co-workers14 developed the first non-sequential dual-responsive self-immolative spacer, considering a protease and a phosphatase sensitive moiety. Both enzymatic steps can occur independently without intrinsic hierarchy but it has to be noted that the protease step is determinant in the self-immolation process. Indeed, the drug release occurs slowly but significantly after the action of the protease even if the phosphatase step did not occur.
In this communication, we report on an alternative AND logic gate strategy, which does not require such a hierarchy for liberating a substrate conditioning it to two trigger. Instead of being sequential, dual activation is parallel so as to confer exactly the same status to both activating triggers.
Results and discussion
Molecular design
Our self-immolative motif is based on an elimination mechanism for self-immolation since this is compatible with liberating alcohols and amines through ether, carbonate, carbamate or ammonium functions.15,16 In order to avoid any possible steric hindrance, we adopted a heteroatom-para-benzyl core. To enable parallel dual activation, we chose nitrogen as the initiating heteroatom since this atom can be further linked to two activatable groups. To avoid any detrimental self-immolation after the first activation step, we linked the nitrogen to two carbonyl groups so as to deactivate its lone pair up to the second activation step. Further envisioning therapeutical applications, we balanced considerations of stability and reactivity and integrated the nitrogen atom in a di-carbamate. To establish a proof of principle, we eventually retained (i) light and Pd(II) as triggers and respectively introduced 4,5-dimethoxy-2-nitrobenzyl alcohol and allyl alcohol in the di-carbamate and (ii) 7-hydroxy-trifluoromethylcoumarin as a model substrate, which becomes bright when it is liberated from its ether-bound precursor.We performed a series of experiments on model compound 7 to establish the orthogonality of the triggers and their effectiveness once combined. The validation of our design of a double-triggered self-immolative spacer is shown in Scheme 1.
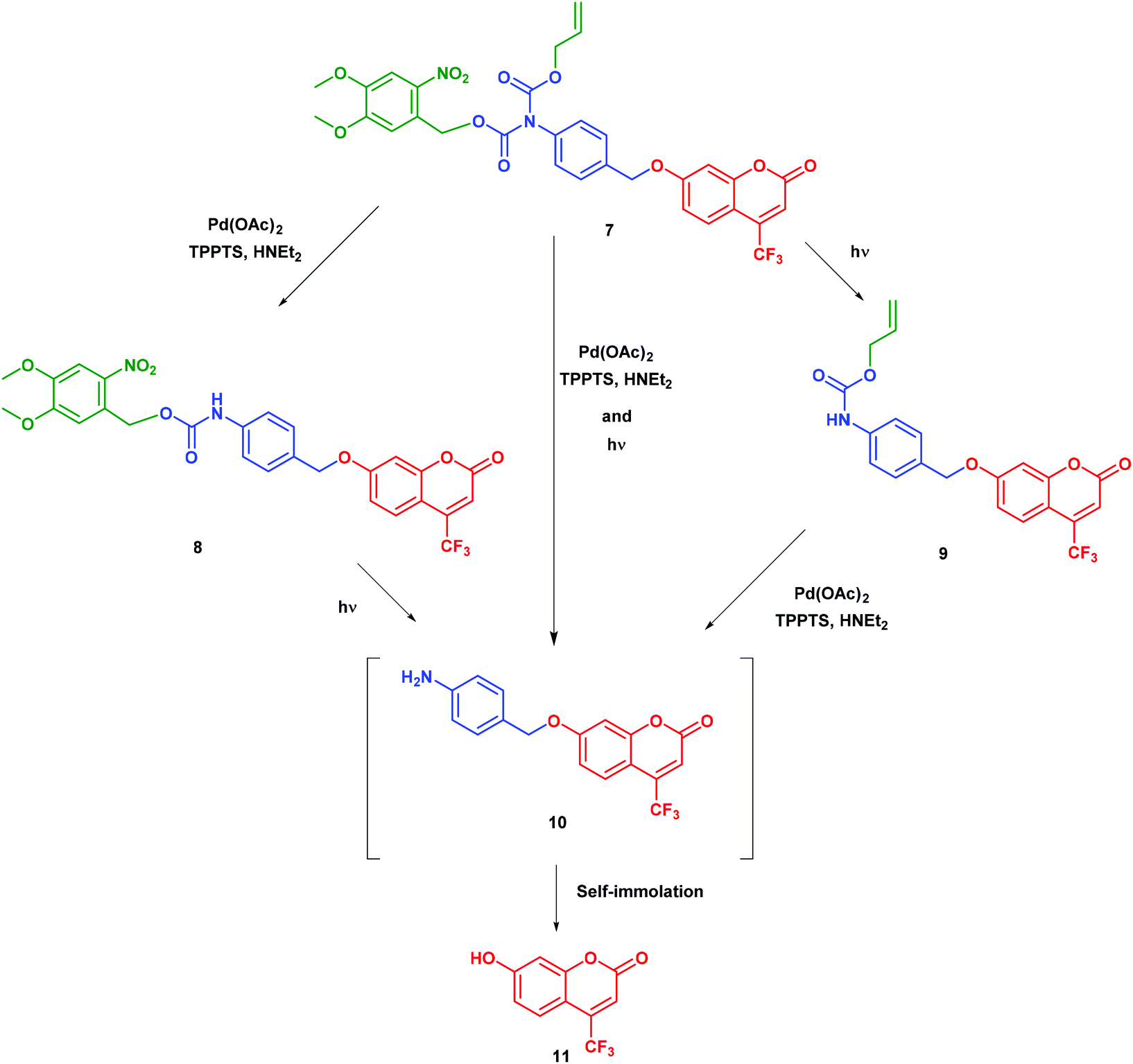 | ||
Scheme 1 Release of the coumarin substrate 11 from its N,N′-biscarbamate precursor 7. Pd(II): Pd(OAc)2 2 mol%, TPPTS 4 mol%, and HNEt2 2.2 eq.; hν: 365 nm. The last step denoted Δ necessitates H2O to occur. Solvent: CH3CN/H2O (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v:v); T = 293 K. :1 v:v); T = 293 K. | ||
Synthesis of bis-carbamate 7
The synthesis of the bis-carbamate spacer 7 is displayed in Scheme 2. It started from 4-amino benzyl alcohol, which was quantitatively protected with the tert-butyldimethylsilyl group to yield the silyl ether 1. To our knowledge, no dissymmetrical N,N′-biscarbamate has been ever reported in the field of self-immolative spacers. Hence, we had to find conditions for synthesizing this new family of compounds. Among the few related compounds,17–19 aniline could yield the corresponding bis-carbamate after reaction with anhydride or chloroformate in basic media, which indicated that the first carbamate could react with strong electrophiles under these conditions. Thus, we quantitatively generated the alloc monocarbamate 2 after the reaction of 1 with the corresponding chloroformate. Then we had to introduce the second nitro-veratryl after the reaction of the carbamate moiety. All attempts with a soft base (Et3N, DIEA, and K2CO3) with or without a catalyst (DMAP, KI) were unsuccessful. Although carbamates were often reported as unstable under strongly basic conditions,20 we achieved deprotonation of 2 with several strong bases (LDA, nBuLi, tBuOK, NaH, and KHMDS) to yield the anion that subsequently reacted with the nitro-veratryl chloroformate to afford the dicarbamate 3. After optimisation, we could obtain a 60% yield by using KHMDS as a base, concentration in the 0.3–0.5 M range, and temperature evolving overnight from −78 °C to room temperature. The next step was the deprotection of the silyl ether. Fluoride salts such as TBAF generated significant degradation. In contrast, acidic treatment with Amberlyst®15 cleanly gave the deprotected alcohol (89% yield). The resulting benzylic alcohol 4 was engaged in chlorination with MsCl to give the benzylic chloride 5, which was subsequently converted into benzylic iodide 6 by the Finkelstein reaction. The final compound 7 was eventually obtained by coupling 6 with the coumarin phenol 11 in 49% yield by using Ag2O in acetonitrile at room temperature. Overall, 7 was obtained in 7 steps with a 20% yield.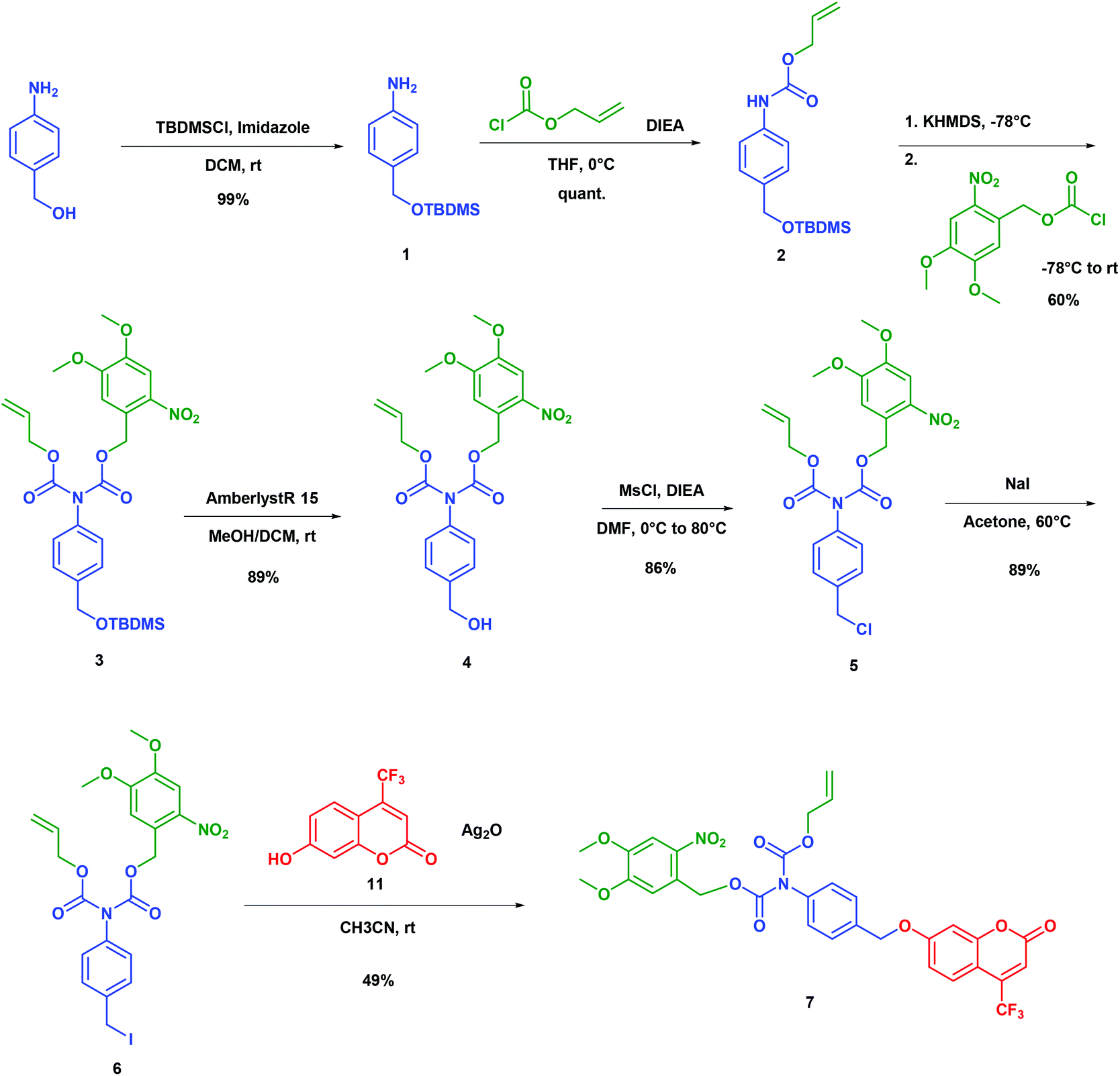 | ||
| Scheme 2 Synthesis of model 7 to establish the proof-of-principle of a double-triggered self-immolative spacer. | ||
Self-immolation experiments
In the first set of preparative experiments (see the ESI†), we first subjected 7 to Pd(II) in a water/acetonitrile mixture in order to remove the alloc carbamate moiety. After 1 h treatment (no more starting material 7 detected by UPLC/MS), we could isolate the nitro-veratryle monocarbamate 8 in 91% yield. We similarly subjected 7 to UV light (365 nm) to remove the nitro-veratryl photolabile protecting group and isolated the alloc monocarbamate 9 in 86% yield after 48 h illumination (disappearance of 7). We further evaluated the stabilities against hydrolysis of the monocarbamates 8 and 9 by UV absorption. Hence, we could evidence the absence of release of coumarin 11 over 24 h, which confirmed that monodeprotection could occur and stop at this stage. In the second series of experiments analyzed by UPLC/MS (see the ESI†), we showed that the exposure of (i) the nitro-veratryl monocarbamate 8 to UV light (365 nm) for 24 h removed the nitro-veratryle photolabile protecting group and liberated coumarin 11 in 86% yield; (ii) the alloc monocarbamate 9 to Pd(II) in a H2O/CH3CN mixture removed the alloc carbamate moiety and quantitatively yielded coumarin 11 after 30 min. We eventually demonstrated that coumarin 11 could be quantitatively obtained from 7 upon combining both deprotection conditions (UV illumination + Pd(II)).To further analyze the liberation of coumarin 11 from the dual activation of 7, we then recorded the temporal evolution of the fluorescence intensity from a 7 solution subjected to both Pd(II) and continuous illumination at 365 nm. Fig. 1 displays the temporal evolution of the fluorescence emission reporting on the release of coumarin 11 upon continuously illuminating a 20 μM solution of precursor 7 containing Pd(II) at 298 K. The complete kinetic analysis is reported in the ESI.† The experiment was reproduced for various incident light intensities to rely on a global fit to extract the kinetic constants associated with the photochemically- and thermally-driven steps. Starting from a poorly fluorescent 7 solution, fluorescence emission increases as anticipated upon releasing 11.
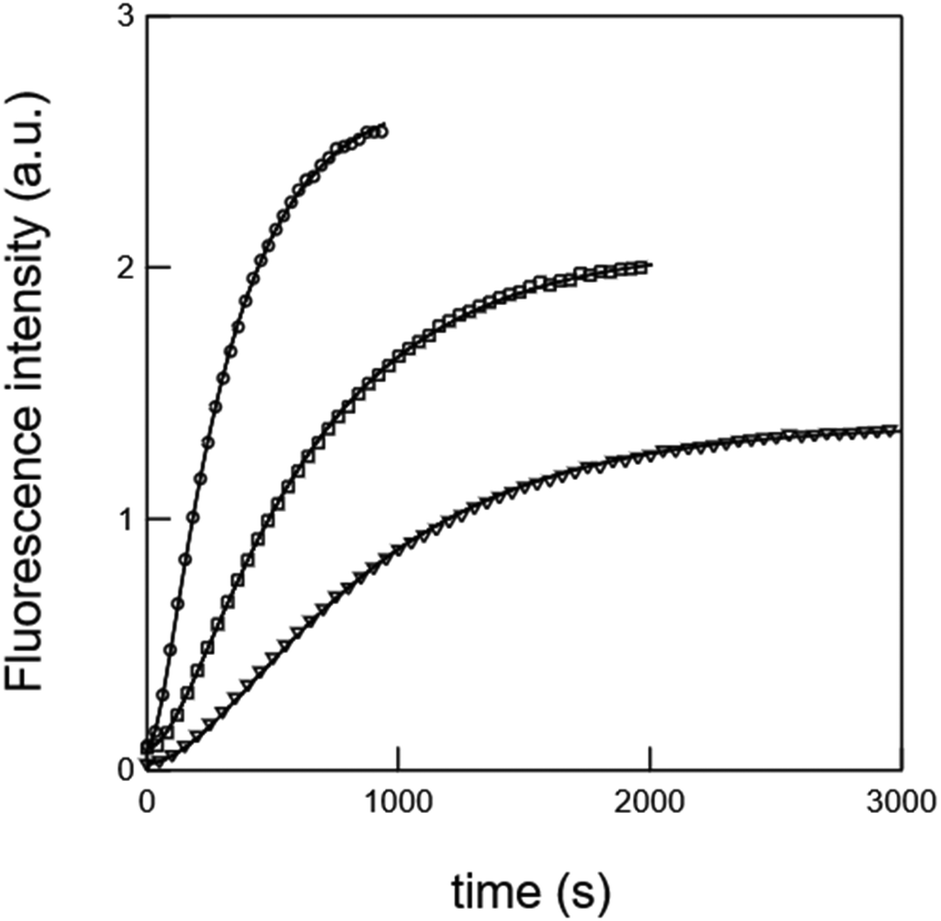 | ||
| Fig. 1 Temporal evolution of the fluorescence emission (λem = 500 nm) upon illuminating at λexc = 365 nm a 20 μM 7 solution containing Pd(OAc)2 2 mol%, TPPTS 4 mol%, and HNEt2 2.2 eq. at various light intensities in the ratios 1 (circles), 0.4 (squares), and 0.2 (triangles). Markers: experimental data; solid line: fits with eqn (8) in the ESI.† Solvent: CH3CN:H2O (4:1 v:v). T = 298 K. | ||
The time evolution of the fluorescence signal was analyzed by adopting the reduced kinetic scheme shown in Scheme 1S (see the ESI†). In relation to Scheme 2 and after the second series of experiments analyzed by UPLC/MS, the precursor 7 yields the intermediate 10 (rate constant klow), which subsequently releases coumarin 11 (rate constant k3). We obtained satisfactory fits by constraining klow to be proportional to the light intensity (as expected from a photochemical step) and k3 = 2.2 × 10−3 ± 1 × 10−3 s−1. The same experiment was reproduced at 310 K (see the ESI†). In addition, we obtained satisfactory fits by constraining klow to be proportional to the light intensity (as expected from a photochemical step) and k3 = 8.4 × 10−3 ± 1 × 10−3 s−1.
Conclusion
Through the synthetic pathway developed in this article, an original asymmetric N,N′-bis-carbamate linker has been obtained with 20% overall yield in 7 steps. These new synthetic conditions open the way to the synthesis of a family of new molecules. The potential use of this structure as a new molecular delivery system, according to the “double gate” strategy, has been established. The structure performed the quantitative release leaving a phenol in a non-sequential activation pathway upon two independent triggers. Moreover, intermediates 8 and 9 generated after single activations were found to be stable under physiological-like conditions, meaning that the second activation was mandatory to initiate the self-immolation process. Despite 6-nitroveratryl and the alloc protecting groups being used in a medicinal chemistry or chemical biology context21,22 and considering that any carbamate-linked leaving group is supposed to give similar results, these groups could be replaced by biological relevant moieties (enzymatic or metabolic substrates)23–25 and cytotoxic compounds could replace the coumarin derivative. The use of bis-carbamate was validated on a model and therefore opens the way to applications in medicinal chemistry or chemical biology strategies based on self-immolative spacers.Experimental
General information
Commercially available chemicals were used without further purification. Anhydrous solvents were freshly distilled before use or obtained from a Pure SolvTM solvent purificator. Low actinic glassware and aluminium film were used for all experiments involving compounds bearing the nitroveratryl moiety. Column chromatography (CC) was led on silica gel 60 (0.040–0.063 mm) (Merck). Analytical and thin layer chromatography (TLC) was performed on Merck silica gel 60 F-254 precoated plates; detection by UV (254 and 365 nm). 1H NMR spectra were recorded at 300 MHz. 13C NMR spectra were recorded at 75 MHz with complete proton decoupling; chemical shifts (δ) in ppm related to protonated solvent as an internal reference (1H: CHCl3 in CDCl3, 7.26 ppm; 13C: 13CDCl3 in CDCl3, 77.16 ppm; 1H: CH3OH in CD3OD, 3.31 ppm; 13C: 13CD3OD in CD3OD, 49.00 ppm; 1H: CH3CN in CD3CN, 1.94 ppm; 13C: 13CD313CN in CD3CN, 1.32, 118.26 ppm); coupling constants J in Hz; current notations are used for multiplicity (s: singlet; bs: broad singlet; d: doublet; dd: double doublet; t: triplet; q: quadruplet; m: multiplet).Experimental procedures
:1 (v:v) mixture of acetonitrile and water before palladium(II) acetate (0.1 mg, 0.0004 mmol, 0.02 eq.), triphenylphosphine m-trisulfonate (0.4 mg, 0.0008 mmol, 0.04 eq.) and diethylamine (4 μL, 0.04 mmol, 2.2 eq.) were added. The solution was stirred for 1 h at room temperature, then diluted with DCM and washed with water and brine. After drying on MgSO4, it was evaporated and the residue was purified on silica gel (0 to 100% DCM/EtOAc). The monocarbamate 8 was obtained as a slightly yellow solid (9.3 mg, 91%). For the physicochemical experiments, a further purification was done by HPLC using 80 to 90% gradient of CH3CN/water/0.1% TFA and the Waters XBridge® Prep C18 5 μm OBD™ 30 × 150 mm Column. Rf = 0.29 (40% EtOAc/Cx). δH (CDCl3) 7.65 (s, 1H), 7.56 (dd, 1H, J = 9.0, 1.5 Hz), 7.39 (d, 2H, J = 8.6 Hz), 7.35 (d, 2H, J = 8.5 Hz), 6.97 (s, 3H), 6.91 (dd, 1H, J = 9.0, 2.4 Hz), 6.86 (d, 1H, J = 2.4 Hz), 6.76 (bs, 1H), 6.55 (s, 1H), 5.53 (s, 2H), 5.04 (s, 2H), 3.90 (s, 3H), 3.89 (s, 3H). δC (CDCl3) 162.4, 159.3, 156.3, 153.4, 152.8, 148.5, 141.8, 141.4, 140.2, 137.8, 130.7, 128.7, 126.4, 119.0, 114.0, 112.4, 110.0, 108.3, 107.2, 102.4, 70.3, 64.1, 56.5, 56.4, 48.4. HRMS [M + H]+ calcd for C27H22F3N2O9 575.1277, found 575.1269, [M + Na]+ calcd for C27H21F3N2NaO9 597.1097, found 597.1095.
:1 (v:v) mixture of acetonitrile and water. It was subsequently stirred and irradiated for 48 h at room temperature by using 3 TLC 6 W lamps emitting at 365 nm. The solution became red/orange over the time. After checking that the starting material disappeared by TLC, the solution was diluted with DCM and then washed with water and brine. After drying on MgSO4, it was evaporated and the residue was purified on silica gel (0 to 40% EtOAc/Cx). The mono carbamate 9 was obtained as an off-white solid (13 mg, 86%). For the physicochemical experiments, another purification was done by HPLC using 80 to 90% gradient of CH3CN/water/0.1% TFA and the Waters XBridge® Prep C18 5 μm OBD™ 30 × 150 mm Column. Rf = 0.29 (40% EtOAc/Cx). δH (CD3CN) 7.63 (bs, 1H), 7.43 (dd, 1H, J = 7.8, 1.7 Hz), 7.27 (d, 2H, J = 8.5 Hz), 7.18 (d, 2H, J = 8.5 Hz), 6.80 (dd, 2H, J = 6.9, 2.4 Hz), 5.77 (m, 1H), 6.44 (s, 1H), 5.14 (dd, 1H, J = 17.3, 1.5 Hz), 5.01 (d, 1H, J = 10.5 Hz), 4.91 (s, 2H), 4.40 (d, 2H, J = 5.3 Hz). δC (CD3CN) 163.3, 159.9, 157.0, 154.1, 140.6–141.9 (q, CF3), 139.8, 133.9, 131.2, 129.6, 126.9, 124.5, 120.8, 119.3, 117.6, 114.4, 113.9, 113.8, 107.8, 103.3, 71.0, 65.9. HRMS [M + H]+ calcd for C21H17F3NO5 420.1059, found 420.1051.
Author contributions
Conceptualization, F. S. and L. J.; methodology, S. H., F. S., T. L. S., and L. J.; software, T. L. S.; formal analysis, S. H., T. L. S, and L. J.; investigation, S. H. and T. L. S.; resources, S. H. and T. L. S.; writing original draft, S. H., T. L. S., L. J., and F. S.; writing-review & editing, S. H., L. J., and F. S.; funding acquisition, L. J., and F. S.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The ComUE Paris Sciences et Lettres (PSL) is acknowledged for financial support to SH. Christine Gaillet is acknowledged for NMR measurements and UPLC experiments.Notes and references
- F. Kratz, I. A. Muller, C. Ryppa and A. Warnecke, ChemMedChem, 2008, 3, 20 CrossRef CAS PubMed.
- A. Dal Corso, L. Pignataro, L. Belvisi and C. Gennari, Chem. – Eur. J., 2019, 25, 14740 CrossRef CAS PubMed.
- Y. L. Leu, C. S. Chen, Y. J. Wu and J. W. Chern, J. Med. Chem., 2008, 51, 1740 CrossRef CAS PubMed.
- M. Grinda, B. Renoux, I. Tranoy-Opalinski and S. Papot, MedChemComm, 2012, 3, 68 RSC.
- B. A. Badeau, M. P. Comerford, C. K. Arakawa, J. A. Shadish and C. A. DeForrest, Nat. Chem., 2018, 10, 251 CrossRef CAS PubMed.
- R. J. Amir, M. Popkov, R. A. Lerner, C. F. Barbas III. and D. Shabat, Angew. Chem., Int. Ed., 2005, 44, 4378 CrossRef CAS PubMed.
- S. Debieu and A. Romieu, Org. Biomol. Chem., 2015, 13, 10348 RSC.
- S. Y. Li, L. H. Liu, H. Cheng, B. Li, W. X. Qiu and X. Z. Zhang, Chem. Commun., 2015, 51, 14520 RSC.
- J. B. Grimm, T. D. Gruber, G. Ortiz, T. A. Brown and L. D. Lavis, Bioconjugate Chem., 2016, 27, 474 CrossRef CAS PubMed.
- For selected reviews, see: (a) A. Romieu, Org. Biomol. Chem., 2015, 13, 1294 RSC; (b) J. L. Kolanowski, F. Liu and E. J. New, Chem. Soc. Rev., 2018, 47, 195 RSC; (c) H.-W. Liu, L. Chen, C. Xu, Z. Li, H. Zhang, X.-B. Zhang and W. Tan, Chem. Soc. Rev., 2018, 47, 7140 RSC.
- G. C. Van de Bittner, C. R. Bertozzi and C. J. Chang, J. Am. Chem. Soc., 2013, 135, 1783 CrossRef CAS PubMed.
- M. Prost and J. Hasserodt, Chem. Commun., 2014, 50, 14896 RSC.
- J. D. Bargh, S. J. Walsh, N. Ashman, A. Isidro-Llobet, J. S. Carroll and D. R. Spring, Chem. Commun., 2021, 57, 3457 RSC.
- A. Dal Corso, S. Arosio, N. Arrighetti, P. Perego, L. Belvisi, L. Pignataro and C. Gennari, Chem. Commun., 2021, 57, 7778 RSC.
- A. Alouane, R. Labruere, T. Le Saux, F. Schmidt and L. Jullien, Angew. Chem., Int. Ed., 2015, 54, 7492 CrossRef CAS PubMed.
- P. J. Burke, J. Z. Hamilton, T. A. Pires, J. R. Setter, J. H. Hunter, J. H. Cochran, A. B. Waight, K. A. Gordon, B. E. Toki, K. K. Emmerton, W. Zeng, I. J. Stone, P. D. Senter, R. P. Lyon and S. C. Jeffrey, Mol. Cancer Ther., 2016, 15, 938 CrossRef CAS PubMed.
- T. J. Thornton and M. Jarman, Synthesis, 1990, 295 CrossRef CAS.
- M. Nowak, Z. Malinowski, A. Jozwiak, E. Fornal, A. Blaszczyk and R. Kontek, Tetrahedron, 2014, 70, 5153 CrossRef CAS.
- Y. Basel and A. Hassner, J. Org. Chem., 2000, 65, 6368 CrossRef CAS PubMed.
- J. E. C. Hutchins and T. H. Fife, J. Am. Chem. Soc., 1973, 95, 2282 CrossRef CAS.
- P. Murat, M. V. Gormally, D. Sanders, M. Di Antonio and S. Balasubramanian, Chem. Commun., 2013, 49, 8453 RSC.
- E. Latocheski, G. M. Dal Forno, T. M. Ferreira, B. L. Oliveira, G. J. L. Bernardes and J. B. Domingos, Chem. Soc. Rev., 2020, 49, 7710 RSC.
- E. W. P. Damen, T. J. Nevalainen, T. J. M. Van der Bergh, F. M. H. de Groot and H. W. Scheeren, Bioorg. Med. Chem., 2002, 10, 71 CrossRef CAS PubMed.
- J. L. M. Jourden and S. M. Cohen, Angew. Chem., Int. Ed., 2010, 49, 6795 CrossRef PubMed.
- B. Huang, W. Chen, Y. Q. Kuang, W. Liu, X. J. Liu, L. J. Tang and J. H. Jiang, Org. Biomol. Chem., 2017, 15, 4383 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ob02124a |
| This journal is © The Royal Society of Chemistry 2022 |