
Practical synthesis of quinolone drugs via a novel TsCl-mediated domino reaction sequence†
Jie
Lei
a,
Yong
Ding
a,
Hao-Yi
Zhou
a,
Xin-Yan
Gao
a,
Yi-Hua
Cao
a,
Dian-Yong
Tang
 a,
Hong-yu
Li
*b,
Zhi-Gang
Xu
*a and
Zhong-Zhu
Chen
*a
a,
Hong-yu
Li
*b,
Zhi-Gang
Xu
*a and
Zhong-Zhu
Chen
*a
aCollege of Pharmacy, National & Local Joint Engineering Research Center of Targeted and Innovative Therapeutics, IATTI, Chongqing University of Arts and Sciences, Chongqing 402160, China. E-mail: xzg@cqwu.com.cn; 18883138277@163.com
bDepartment of Pharmaceutical Sciences, College of Pharmacy, University of Arkansas for Medical Sciences, Little Rock, AR 72205, USA. E-mail: HLi2@uams.edu
First published on 8th July 2022
Abstract
A novel TsCl-mediated domino sequence to expeditiously access quinolone-based antibiotics, such as ciprofloxacin, norfloxacin, pefloxacin, oxilinic acid, ivacaftor and the precursor of grepftfloxacin and ozenoxacin, starting from commercially available chromone-3-carboxaldehydes and amines, was developed. The total synthesis of these quinolone-based drugs via this sequence shortens the original seven/eight step synthesis to three/four steps with a high overall yield under environmentally benign conditions. The quinolone-based antibiotic drug analogues could also be efficiently synthesized by varying the starting materials and chemical reagents for discovering and developing new antibiotics.
Quinolone is a privileged scaffold widely found in natural products and biologically active molecules, and an essential heterocyclic motif in antibacterial, anticancer and anti-inflammatory drugs (Fig. 1).1–9 The quinolone antibiotics arose in the early 1960s. After sixty years of effort in the discovery of novel quinolone antibiotics with favourable pharmacokinetic properties, many well-known quinolone drugs have been popularly used as prescription drugs or over-the-counter medicines as shown in Fig. 1, for example, ciprofloxacin, norfloxacin, pefloxacin, oxilinic acid, ivacaftor, grepftfloxacin and ozenoxacin. The effective construction of these drugs is therefore a key subject in organic synthesis. Notable efforts by utilizing the Conrad–Limpach reaction, Knorr quinoline synthesis, and Doebner–von Miller reaction gave the quinolinone core with undesired positions for substituents, thus limiting their applicability in the synthesis of quinolone drugs.10,11
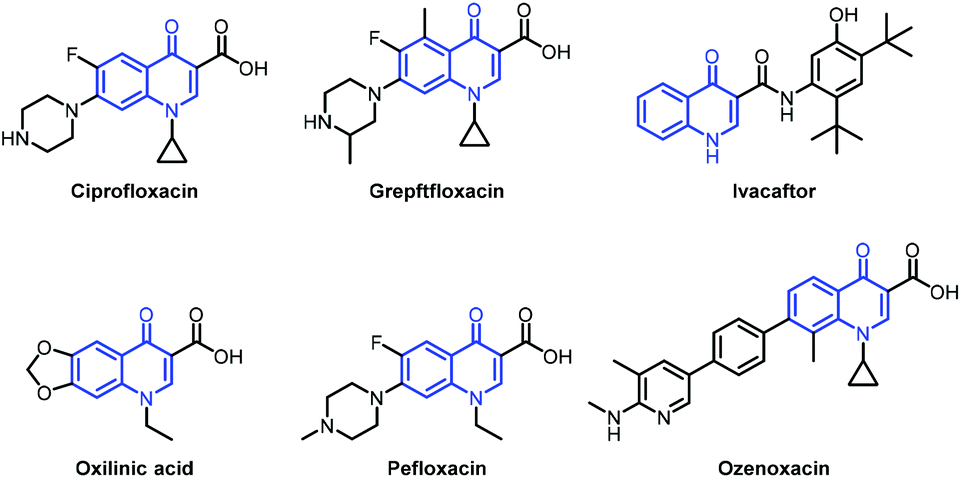 | ||
| Fig. 1 Representative examples of quinolone drugs. | ||
To date, the Gould–Jacobs reaction was extensively studied in accessing the quinolone scaffold, but this classical approach usually requires a multistep sequence with a low overall yield. In detail, anilinomethylene malonates are prepared by the condensation of aniline derivatives with diethyl ethoxymethylenemalonate, which readily undergoes an electrocyclization via a ketene intermediate usually under harsh conditions (Scheme 1a, eqn (1)).12–16 The long reaction time at high temperature leads to decomposition and side-reactions.17–20 To overcome the intrinsic drawbacks of the Gould–Jacobs reaction, functionalized benzoylacetate was used as the starting material via a five-step sequence to furnish the quinolone (Scheme 1a, eqn (2)).21,22 However, the procedure still suffers from high reaction temperature (>250 °C), narrow functional group compatibility, expensive reagents, large excess of additives, and low overall yields.
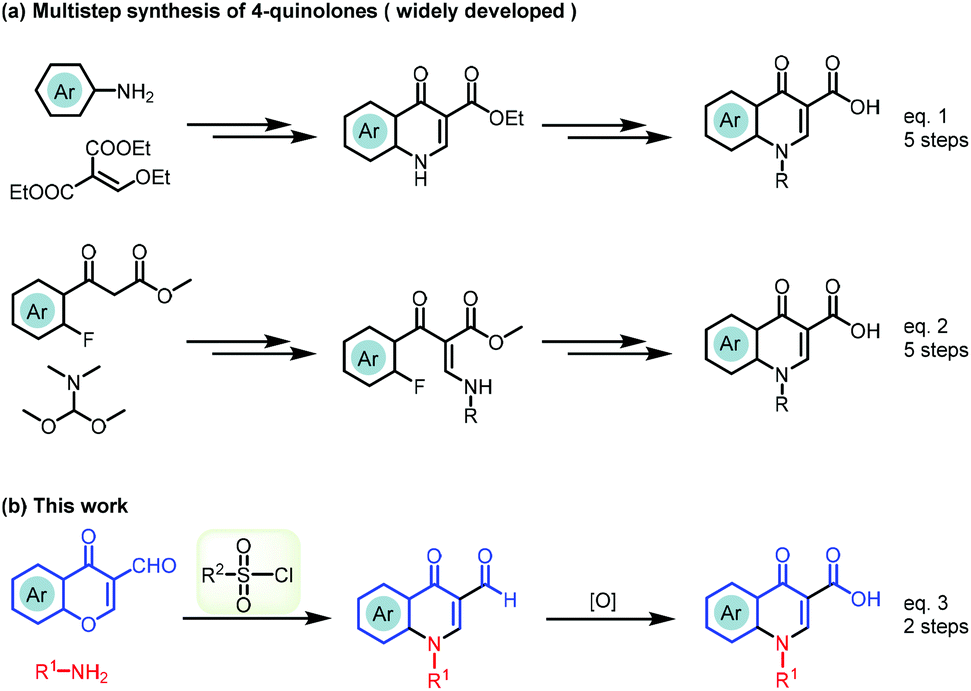 | ||
| Scheme 1 Strategies for accessing 4-quinolones and the current reaction design. | ||
The lack of a green strategy for the efficient synthesis of 4-quinolone drugs opens up an opportunity to develop novel synthetic approaches towards this important class of drugs. Therefore, a method that could be carried out under mild conditions with broad functional group tolerance and atom- and step-economical advantages would be highly desirable. Inspired by literature reports and our recent findings on the ring-opening reaction of chromone with a strong nucleophile,23–27 we assumed that the chromone ring system could be opened with a weak nucleophile, but the intermediate may not be stable and be quickly cyclized back to chromone. Thus, trapping of the active intermediate to drive the equilibrium to the product provided a practical and efficient strategy for the synthesis of drug-like scaffolds.28–30 By utilizing this strategy, we report a novel TsCl-mediated domino sequence for the efficient assembly of quinolone-based drugs (Scheme 1b, eqn (3)). There are three advantages of this methodology: (1) access to 4-quinolones in only two steps avoiding a long multi-step synthesis, high-boiling solvents, high temperature or stoichiometric polyphosphoric acid; (2) construction of quinolone-based drugs starting from commercially available starting materials; and (3) expansion of this convenient and expeditious strategy to promote the discovery of other pharmaceutical molecules with a viable alternative.
First, the reaction proceeded through a one-pot domino cascade by selecting chromone-3-carboxaldehyde 1a, tert-butylamine 2a and p-toluenesulfonyl chloride 3a as the starting materials with a base (Table 1). As our previous work indicated, a Brønsted acid was capable of affording 1,4-addition adducts under mild conditions.31 Under these conditions, however, no final compound 4a was observed. Thus, we switched our attention to basic conditions. Among the inorganic bases screened, tBuOLi afforded the final product 4a in 58% isolated yield (entry 4). A little lower yield was obtained by the replacement of tBuOLi with tBuONa, K2CO3 or Na2CO3. The effect of organic bases was further evaluated in N,N-dimethylformamide (DMF) (entries 8–10), but with quite lower conversion. After briefly screening common organic solvents (entries 11–18), we found that performing the reaction with toluene as the solvent delivered 2a in an obvious higher 71% yield (entry 14). Prolonging the reaction time from 4 h to 8 h afforded 4a in the highest 86% yield (entry 20). Further prolonging the reaction time or increasing the reaction temperature led to slightly diminished yields (entries 21 and 22). Therefore, the optimized reaction conditions were determined to be: “starting materials with 2.0 equiv. of a base in toluene at 100 °C for 8 h in a sealed system”.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Base | Solvent | Time (h) | Temp. (°C) | Yield (%) |
| a Reaction conditions: 1a (0.2 mmol), 2a (0.22 mmol), 3a (0.2 mmol), 2.0 equiv. of base, solvent (2.0 mL), in a sealed tube. b 2a (0.4 mmol). | |||||
| 1 | MeONa | DMF | 4 | 100 | 26 |
| 2 | KOH | DMF | 4 | 100 | 31 |
| 3 | NaHCO3 | DMF | 4 | 100 | 39 |
| 4 | t BuOLi | DMF | 4 | 100 | 58 |
| 5 | t BuONa | DMF | 4 | 100 | 49 |
| 6 | K2CO3 | DMF | 4 | 100 | 43 |
| 7 | Na2CO3 | DMF | 4 | 100 | 41 |
| 8 | Et3N | DMF | 4 | 100 | 14 |
| 9 | DBU | DMF | 4 | 100 | 23 |
| 10 | DABCO | DMF | 4 | 100 | 19 |
| 11b | — | DMF | 4 | 100 | 17 |
| 12 | t BuOLi | THF | 4 | 100 | 13 |
| 13 | t BuOLi | DMSO | 4 | 100 | 33 |
| 14 | t BuOLi | Toluene | 4 | 100 | 71 |
| 15 | t BuOLi | MeCN | 4 | 100 | 40 |
| 16 | t BuOLi | DCE | 4 | 100 | 44 |
| 17 | t BuOLi | Dioxane | 4 | 100 | 36 |
| 18 | t BuOLi | NMP | 4 | 100 | 53 |
| 19 | t BuOLi | EtOH | 4 | 100 | 18 |
| 20 | t BuOLi | Toluene | 8 | 100 | 86 |
| 21 | t BuOLi | Toluene | 8 | 120 | 75 |
| 22 | t BuOLi | Toluene | 10 | 100 | 82 |
As we expected, p-toluenesulfonyl chloride likely drove the reaction to the desired product by sulfonating the transient hydroxyl group from chromone ring-opening. To obtain the optimal sulfonyl chloride for this transformation, a series of sulfonyl chlorides were investigated (Scheme 2). Me, MeO, or tBu substituted benzenesulfonyl chloride led to slightly diminished yields. Similarly, ethanesulfonyl chloride did not improve the yield either. However, benzenesulfonyl chloride containing an electron-withdrawing group, such as CF3, NO2 and Cl, gave 4a in a slightly higher yield. We then decided to employ cost-effective p-toluenesulfonyl chloride for further investigations.
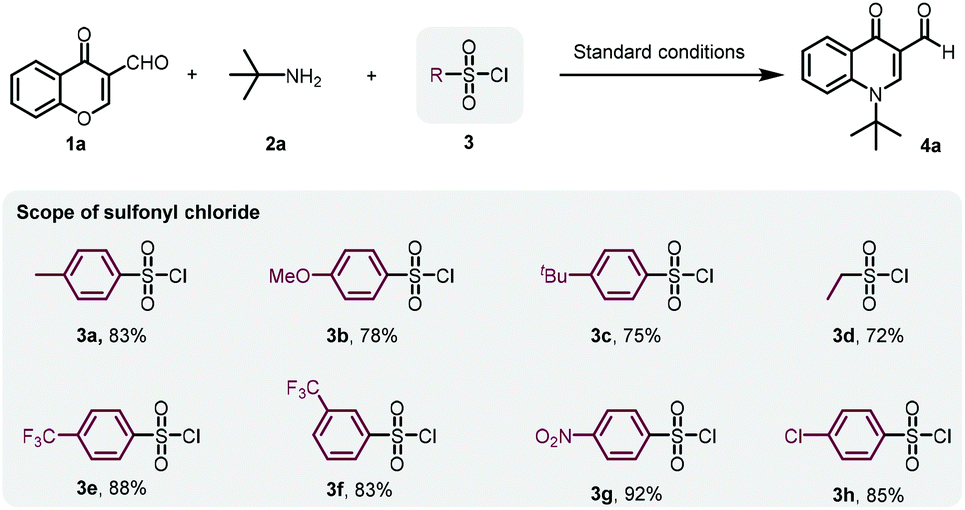 | ||
| Scheme 2 Scope of sulfonyl chlorides in the reaction. | ||
With the optimal conditions in hand, chromone-3-carboxaldehyde, aliphatic amine and p-toluenesulfonyl chloride were subjected to this domino process for the generation of a library of quinolinone heterocycles, as shown in Scheme 3. The bulky amine of tert-butylamine afforded the desired compound 4a with a yield of 86%. Specifically, while 4-methylbenzylamine was selected as the amine source, compound 4d was obtained with 77% yield. As the scope for amines depicted, not only general aliphatic amines could be tolerated, but also aromatic amines provided the final products in good yields. Various chromone-3-carboxaldehydes were then investigated to expand the scope of quinoline-3-aldehydes (4h–4s) as shown in Scheme 3. To our great delight, the reaction worked well with electron-donating and electron-withdrawing groups on chromone-3-carboxaldehyde with yields from 73% to 90%. It is worth noting that 4h could be directly converted to oxilinic acid (Scheme 4) via the oxidation reaction. Also, 6-methyl and 6-bromo-substituted chromone-3-carboxaldehyde led to the final compounds 4n and 4r, in yields of 85% and 82%, respectively. Importantly, the precursors of many 4-quinolone-based drugs could be furnished in one step with 73% to 85% yields (4aa–4ah). The reactive Michael acceptor was introduced to provide 4ai in 73% yield. The modification of an erlotinib analogue was carried out with the corresponding aldehyde to give 4aj with 76% yield (see the ESI† for details).
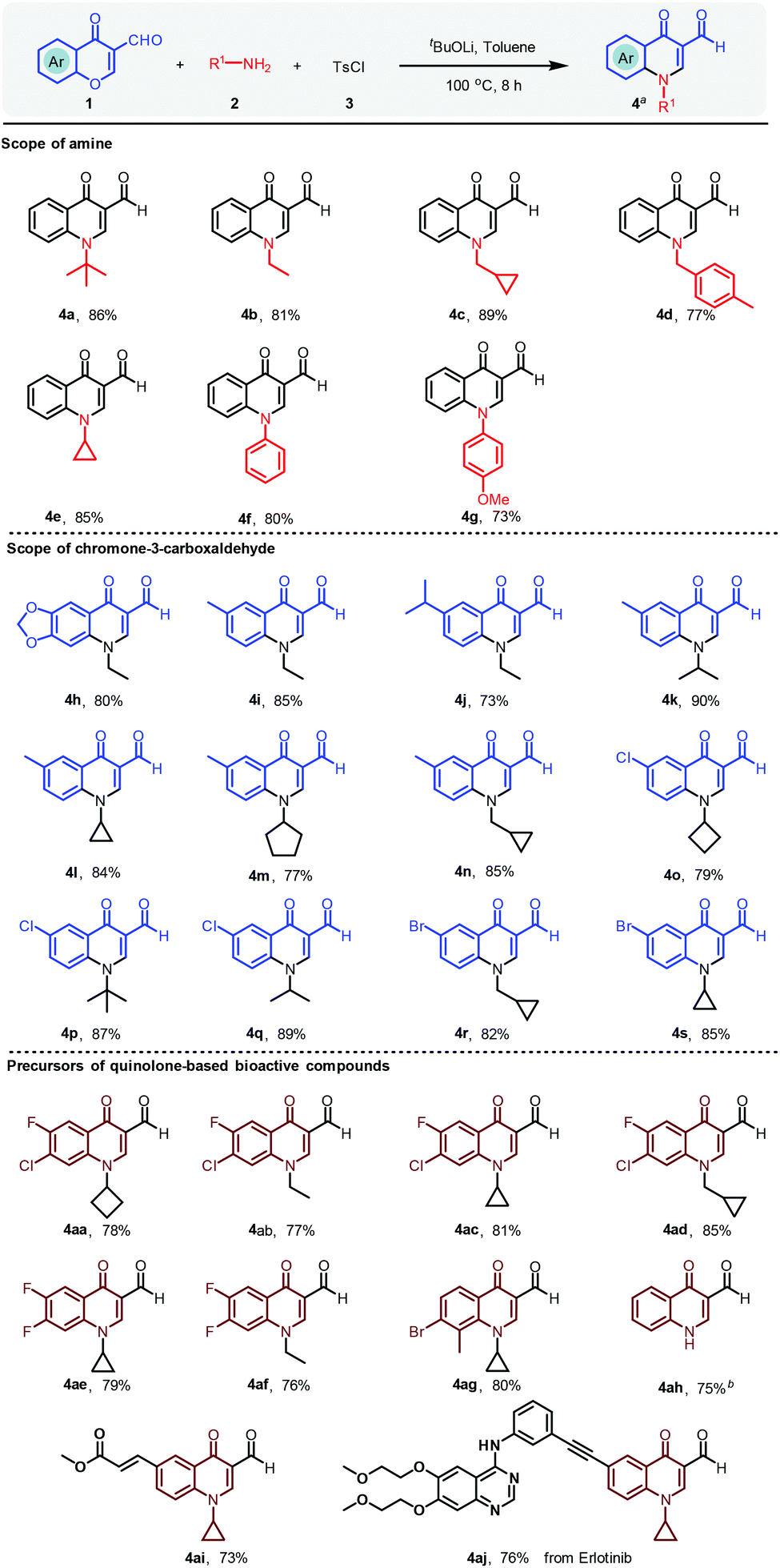 | ||
Scheme 3 Scope of 4-quinolinones in the TsCl-mediated reaction. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Reaction conditions: 1 (0.2 mmol), 2 (0.22 mmol), 3 (0.2 mmol), tBuOLi (0.4 mmol), toluene (2.0 mL), 100 °C, 8 h. Isolated yields are reported. bDeprotection reaction of 4a: 1 mol L−1 of HCl (2.0 equiv.), MeCN, 80 °C, 6 h. Two-step yield is given. See the ESI† for details. Reaction conditions: 1 (0.2 mmol), 2 (0.22 mmol), 3 (0.2 mmol), tBuOLi (0.4 mmol), toluene (2.0 mL), 100 °C, 8 h. Isolated yields are reported. bDeprotection reaction of 4a: 1 mol L−1 of HCl (2.0 equiv.), MeCN, 80 °C, 6 h. Two-step yield is given. See the ESI† for details. | ||
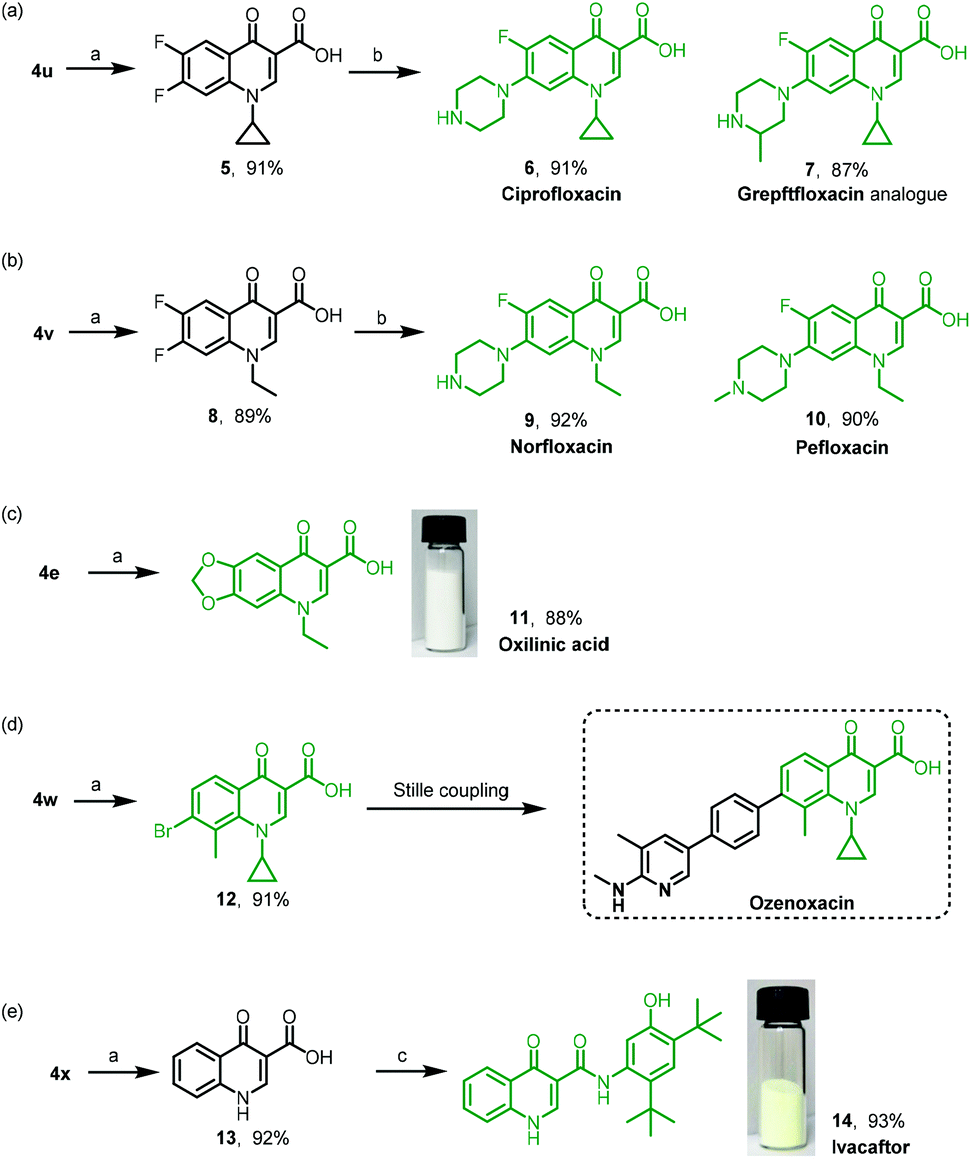 | ||
| Scheme 4 Applications of the strategy for quinolone drugs and their precursors. Reaction conditions: (a) NH2SO3H (4.0 equiv.), NaClO2 (5.0 equiv.), DCM, r.t., 4 h. (b) Secondary amine (2.5 equiv.), microwave irradiation, 120 °C, 30 min. (c) HBTU (1.2 equiv.), Et3N (3.0 equiv.), DCM, r.t., 12 h. See the ESI† for details. | ||
Quinolone is an important motif in synthetic antibacterial agents due to their excellent safety profile, favourable pharmacokinetic characteristics, and broad antibacterial effectiveness.32–34 We then applied our strategy of the TsCl-mediated domino sequence to construct quinolone-based drugs. As shown in Scheme 4, 4u was synthesized by mixing 6,7-difluoro chromone-3-carboxaldehyde, cyclopropylamine and TsCl, followed by oxidation as depicted in Scheme 3 to give the corresponding acid 5 in 91% yield. The sequential nucleophilic substitution on the C–F bond with piperazine or 2-methylpiperazine produced 6 (ciprofloxacin, 91%) and 7 (grepftfloxacin analogue, 87%), respectively (Scheme 4a). Similarly, the oxidized product 8 from 4v underwent the nucleophilic substitution reaction to afford 9 (norfloxacin, 92%) and 10 (pefloxacin, 90%), respectively (Scheme 4b). It is worth mentioning that the synthesis of each of these two drugs took only three steps via our TsCl-mediated domino sequence. Oxilinic acid and ozenoxacin are famous antibiotics. The oxidation reaction of 4e and 4w gave oxalinic acid (11, 88%) and the precursor (12, 91%) of ozenoxacin (Scheme 4c and d), respectively. As indicated in the literature, ozenoxacin was synthesized directly by the Stille coupling reaction from 12 in a high yield. An efficient and short-step synthesis of ivacaftor was then designed by changing the starting materials, aldehyde and amine. Compound 13 resulting from the TsCl-mediated domino sequence and oxidation was sequentially conjugated with 5-amino-2,4-di-tert-butylphenol to give ivacaftor (14, 93%, Scheme 4e). Subsequently, the gram-scale synthesis of oxilinic acid and ivacaftor was carried out in high overall yields; see the ESI† for details. Each synthesis could be completed by a graduate student in three working days.
To get insight into this domino sequence mechanism, a series of control experiments were performed as depicted in Scheme 5. At room temperature, the reaction with chromone-3-carboxaldehyde 1a, tert-butylamine 2a and p-toluenesulfonyl chloride 3a gave β-enamino dicarbonyl compound (15, 92%). On increasing the reaction temperature to 100 °C, 15 was smoothly transformed to compound 4a in 95% yield. Accordingly,35 the Schiff base was the key intermediate in the transformation, and was synthesized via the condensation reaction of 3-formylchromone 1a with tert-butylamine 2a in ethanol. As we expected, compound 4a was converted from the Schiff base in 88% yield under the optimized conditions (Scheme 5b). In the absence of either TsCl or K2CO3, no desired compounds were detected (Scheme 5c and d). Therefore, our synthesis of 4-quinolone scaffolds occurred in two key steps: the formation of β-enamino dicarbonyl and Schiff base intermediates.
 | ||
| Scheme 5 Control experiments. | ||
Based on the literature information27,31 and control experiments, we postulated the reaction mechanism as shown in Scheme 6. First, the synthesis of 4-quinolone was initiated by the condensation of Schiff base I with the loss of one molar water. p-Toluenesulfonyl chloride in the presence of water led to the generation of a small amount of acids (TsOH and HCl). The nucleophilic attack of a water molecule on the protonated Schiff base II gave enamine III. The sulfonation of the reactive oxygen connected to the phenyl ring would provide the key intermediate sulfonate. Then the tautomerization of enol IV would give β-enamino dicarbonyl compound V. As a good leaving group, sulfonate would drive the intramolecular nucleophilic substitution to form a new C–N bond to give the final product 4.
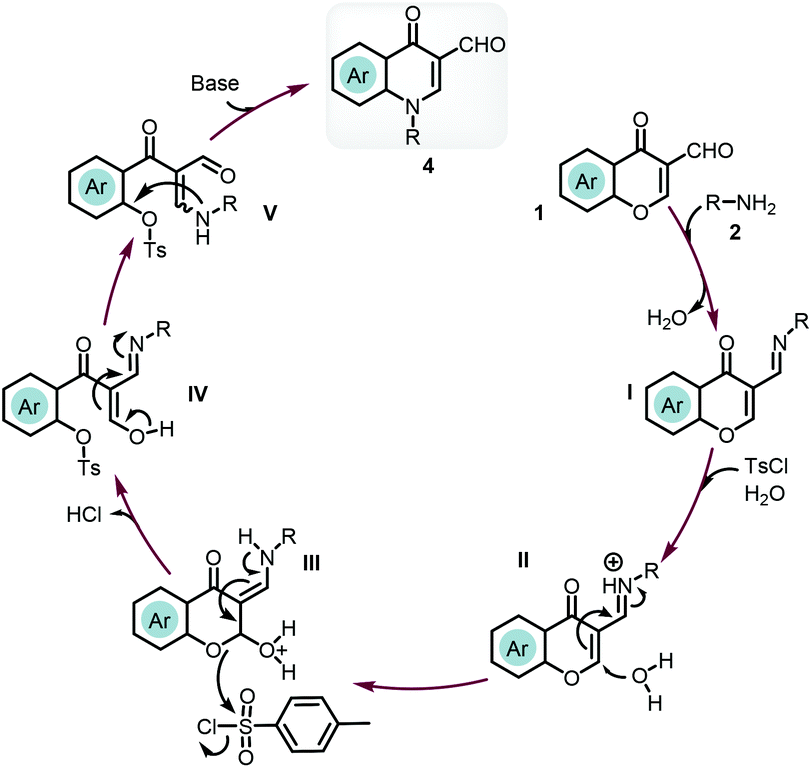 | ||
| Scheme 6 Proposed mechanism for the synthesis of 4-quinolone 4. | ||
In summary, we have uncovered a TsCl-mediated reaction for the construction of 4-quinolone scaffolds with commercially available chromone-3-carboxaldehydes and amines. Importantly, the strategy was successfully expanded to synthesize market drugs, such as ciprofloxacin, norfloxacin, pefloxacin, oxilinic acid, ivacaftor and the precursor of grepftfloxacin and ozenoxacin. The total synthesis of these quinolone-based drugs via this sequence shortens the original seven/eight step synthesis to three/four steps with a high overall yield under environmentally benign conditions. Further exploration of this green domino strategy to synthesize fourth-generation antibiotic drugs and develop novel antibacterial reagents is currently under way in our laboratory.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the Science and Technology Research Program of Chongqing Municipal Education Commission (KJQN202001338, KJQN201801321, KJQN202101340 and KJZD-M201801301), the Natural Science Foundation Project of CQ CSTSC (cstc2020jcyj-msxmX0733, cstc2019jcyj-msxmX0012 cstc2018jszx-cyzdX0023 and cstc2021jcyj-bsh0233) and the Chongqing University of Arts and Sciences: Program for Talents Introduction (R2021FYX05). We would also like to thank Ms H. Z. Liu and J. Xu for obtaining the LC/MS, HRMS and NMR data.Notes and references
- S. Pasquini, M. De Rosa, A. Ligresti, C. Mugnaini, A. Brizzi, N. P. Caradonna, M. G. Cascio, D. Bolognini, R. G. Pertwee, V. D. Marzo and F. Corelli, Eur. J. Med. Chem., 2012, 58, 30–43 CrossRef CAS PubMed.
- A. Gaurav, M. R. Yadav, R. Giridhar, V. Gautam and R. Singh, Med. Chem. Res., 2011, 20, 192–199 CrossRef CAS.
- V. Cecchetti, C. Parolin, S. Moro, T. Pecere, E. Filipponi, A. Calistri, O. Tabarrini, B. Gatto, M. Palumbo, A. Fravolini and G. Palu, J. Med. Chem., 2000, 43, 3799–3802 CrossRef CAS PubMed.
- M. Llinas-Brunet, M. D. Bailey, E. Ghiro, V. Ghorys, T. Halmos, M. Poirier, J. Rancourt and N. Goudreau, J. Med. Chem., 2004, 47, 6584–6594 CrossRef CAS PubMed.
- R. M. Cross, A. Monastyrskyi, T. S. Mutka, J. N. Burrows, D. E. Kyle and R. Manetsch, J. Med. Chem., 2010, 53, 7076–7094 CrossRef CAS PubMed.
- M. Hadjeri, E.-L. Peiller, C. Beney, N. Deka, M. A. Lawson, C. Dumontet and A. Boumendjel, J. Med. Chem., 2004, 47, 4964–4970 CrossRef CAS PubMed.
- Y. Xia, Z.-Y. Yang, P. Xia, T. Hackl, E. Hamel, A. Mauger, J.-H. Wu and K.-H. Lee, J. Med. Chem., 2001, 44, 3932–3936 CrossRef CAS PubMed.
- M. Sato, T. Motomura, H. Aramaki, T. Matsuda, M. Yamashita, Y. Ito, H. Kawakami, Y. Matsuzaki, W. Watanabe, K. Yamataka, S. Ikeda, E. Kodama, M. Matsuoka and H. Shinkai, J. Med. Chem., 2006, 49, 1506–1508 CrossRef CAS PubMed.
- R. Musiol, Expert Opin. Drug Discovery, 2017, 12, 583–597 CrossRef CAS PubMed.
- Y. Kimura, S. Atarashi, K. Kawakami, K. Sato and I. Hayakawa, J. Med. Chem., 1994, 37, 3344–3352 CrossRef CAS PubMed.
- L. A. Mitscher, P. N. Sharma, T. Daniel and W. Chu, J. Med. Chem., 1987, 30, 2283–2286 CrossRef CAS PubMed.
- R. Gould and W. Jacobs, J. Am. Chem. Soc., 1939, 61, 2890–2895 CrossRef CAS.
- A. Mermer, N. Demirbas, Y. Sirin, H. Uslu, Z. Özdemir and A. Demirbaş, Bioorg. Chem., 2018, 78, 236–248 CrossRef CAS PubMed.
- E. Stern, G. G. Muccioli, R. Millet, J. Goossens, A. Farce, P. Chavatte, J. H. Poupaert, D. M. Lambert, P. Depreux and J. P. Hénichart, J. Med. Chem., 2006, 49, 70–79 CrossRef CAS PubMed.
- P. E. Harrington, M. D. Croghan, C. Fotsch, M. Frohn, B. A. Lanman, L. D. Pennington, A. J. Pickrell, A. B. Reed, K. K. C. Sham, A. Tasker, H. A. Arnett, M. Fiorino, M. R. Lee, M. McElvain, H. G. Morrison, H. Xu, Y. Xu, X. Zhang, M. Wong and V. J. Cee, ACS Med. Chem. Lett., 2012, 3, 74–78 CrossRef CAS PubMed.
- A. C. Flick, H. X. Ding, C. A. Leverett, R. E. Kyne Jr., K. K.-C. Liu, S. J. Fink and C. J. O'Donnell, J. Med. Chem., 2017, 60, 6480–6515 CrossRef CAS PubMed.
- D. Lecoq, B. A. Chalmers, R. N. Veedu, D. Kvaskoff, P. V. Bernhardt and C. Wentrup, Aust. J. Chem., 2009, 62, 1631–1638 CrossRef CAS.
- G. R. Lappin, J. Am. Chem. Soc., 1948, 70, 3348–3350 CrossRef CAS PubMed.
- R. B. Smith, H. Faki and R. Leslie, Synth. Commun., 2011, 41, 1492–1499 CrossRef CAS.
- Z. Mészáros and I. Hermecz, Tetrahedron Lett., 1975, 12, 1019–1020 CrossRef.
- R. A. Bunce, E. J. Lee and M. T. Grant, J. Heterocycl. Chem., 2011, 48, 620–625 CrossRef CAS.
- R. Song, Y. Wang, M. Wang, R. Gao, T. Yang, S. Yang, C. Yang, Y. Jin, S. Zou, J. Cai, R. Fan and Q. He, Bioorg. Chem., 2020, 103, 104176 CrossRef CAS PubMed.
- F. A. Carrey and R. J. Sundberg, Advanced Organic Chemistry: Part A: Structure and Mechanisms, Springer Science, 5th edn, 2007, ch. 3 Search PubMed.
- N. Poomathi, P. T. Perumala and S. Ramakrishna, Green Chem., 2017, 19, 2524–2529 RSC.
- T. Lepitre, R. L. Biannic, M. Othman, A. M. Lawson and A. Daïch, Org. Lett., 2017, 19, 1978–1981 CrossRef CAS PubMed.
- J. Y. Liao, W. J. Yap, J. E. Wu, M. W. Wong and Y. Zhao, Chem. Commun., 2017, 53, 9067–9070 RSC.
- J. Lei, J. Xu, D.-Y. Tang, J.-W. Shao, H. Li, Z.-Z. Chen and Z.-G. Xu, Org. Chem. Front., 2020, 7, 2657–2663 RSC.
- S. Liu, W. Yao, Y. Liu, Q. Wei, J. Chen, X. Wu, F. Xia and W. Hu, Sci. Adv., 2017, 3, e1602467 CrossRef PubMed.
- X. Guo and W. Hu, Acc. Chem. Res., 2013, 46, 2427–2440 CrossRef CAS PubMed.
- J. He, A. G. K. Reddy, L. Niu, D. Xing and W. Hu, Org. Lett., 2019, 21, 4571–4574 CrossRef PubMed.
- J. Lei, Y. Li, J. Xu, D.-Y. Tang, J.-W. Shao, H. Li, Z.-Z. Chen and Z.-G. Xu, Green Chem., 2020, 22, 3716–3720 RSC.
- S. Sabatini, F. Gosetto, G. Manfroni, O. Tabarrini, G. W. Kaatz, D. Patel and V. Cecchetti, J. Med. Chem., 2011, 54, 5722–5736 CrossRef CAS PubMed.
- S. Srinivasan, R. M. B. Shafreen, P. Nithyanand, P. Manisankar and S. K. Pandian, Eur. J. Med. Chem., 2010, 45, 6101–6105 CrossRef CAS PubMed.
- Y. Wang, G. L. V. Damu, J.-S. Lv, R.-X. Geng, D.-C. Yang and C.-H. Zhou, Bioorg. Med. Chem. Lett., 2012, 22, 5363–5366 CrossRef CAS PubMed.
- K. M. Khan, N. Ambreen, S. Hussain, S. Perveen and M. I. Choudhary, Bioorg. Med. Chem., 2009, 17, 2983–2988 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and spectroscopic characterization information. See DOI: https://doi.org/10.1039/d2gc01689c |
| This journal is © The Royal Society of Chemistry 2022 |