
Tetrahedral DNA-directed core-satellite assembly as SERS sensor for mercury ions at the single-particle level†
Ning
Feng
a,
Jingjing
Shen
a,
Chang
Li
a,
Qianqian
Zhao
a,
Essy Kouadio
Fodjo
b,
Lei
Zhang
 *a,
Shufen
Chen
a,
Quli
Fan
a and
Lianhui
Wang
*a
*a,
Shufen
Chen
a,
Quli
Fan
a and
Lianhui
Wang
*a
aState Key Laboratory of Organic Electronics and Information Displays & Institute of Advanced Materials(IAM), Nanjing University of Posts and Telecommunications, 9 Wenyuan Road, Nanjing 210023, China. E-mail: iamlzhang@njupt.edu.cn; iamlhwang@njupt.edu.cn
bLaboratory of constitution and reaction of matter, University of Felix Houphouet-Boigny, 22 BP 582, Abidjan 22, Cote d’Ivoire
First published on 30th March 2022
Abstract
To monitor the deteriorating mercury emissions, it is imperative to propose methods for detecting mercury ions (Hg2+) with sensitivity and selectivity. The SERS spectral-resolved single-particle detection approach can be carried out using dark-field optical microscopy (DFM) combined with Raman spectroscopy. Herein, we have designed a novel yet convenient single-particle detection assay for quantifying Hg2+ using DFM-correlated Raman spectroscopy. In the assay, a tetrahedral DNA-directed core-satellite nanostructure is used as the SERS probe. Especially, one edge of the tetrahedron is made up of a single-stranded DNA containing a Hg2+ aptamer, which reconfigures upon the specific recognition of Hg2+. As a result, the interparticle distance reduces from 4.5 to 1.2 nm, thus generating Raman signal enhancement. As a proof of concept, Hg2+ was detected in a linear range from 1 to 100 nM based on the variation in SERS intensity. Furthermore, the experimental results were supported by the finite difference time domain (FDTD) calculations. Owing to its high sensitivity and selectivity, this method was further employed to detect Hg2+ in practical tap water and lake water samples, revealing that the single-particle detection strategy holds great promise for Hg2+ analysis in real environment analysis.
Introduction
Mercury ions (Hg2+) can gradually accumulate in the human body through drinking water and food chains, which cause irreversible damage to the central nervous system, digestive system, kidneys, and other organs.1,2 The damage caused by mercury is related to the amount of mercury accumulated in the body. Thus, this fact prompted us to look for a sensitive and selective method for Hg2+ detection. Until now, a series of methods for Hg2+ measurement have been developed, including fluorescence,3 electrochemistry,4 Raman scattering,5 surface plasma resonance,6 and colorimetric sensing.7 Among these methods, surface-enhanced Raman scattering (SERS) is a powerful tool for Hg2+ detection with sensitivity and efficiency.8 SERS techniques for Hg2+ detection are mainly based on indirect SERS since Hg2+ has no Raman activity, which usually requires additional Raman reporters. The main assay strategy is tightly associated with the regulation of the “hotspot” structure. Hotspots serve as signal transducers to produce SERS signals. The Raman signal of the hotspot structure changes with the addition of the target molecule. However, it is hard to achieve a well-defined and controllable interparticle distance, which governs the SERS signal of hotspot structures. Besides, during the recognition process of Hg2+, the auto-aggregation of nanoparticles is caused by external interferences, such as pH, temperature, and salt concentration, which usually lead to a false positive signal readout. Therefore, there is a need to develop a SERS-based biosensor with high reliability and precision.Recently, the spectral-resolved single-particle detection strategy has received great attention for its advantageous capability to analyze the target object at the single-particle level by dark-field microscopy (DFM).9 For example, Xiao and coworkers10 designed a single-particle detection assay for the quantitative detection of alkaline phosphatase activity in human serum by using a single MnO2-modified gold nanoparticle (AuNP) as the sensor. The SERS spectra of a single plasmonic nanoparticle can be collected by using DFM combined with Raman spectroscopy,11 which opens up unprecedented opportunities for SERS biosensors in detecting target molecules at the single-particle level.12 Compared with the traditional approaches, single-particle detection employs a single particle to generate an independent signal output, which effectively avoids the ensemble-averaged signal effect and greatly improves sensitivity and specificity.13
DNA nanotechnology has emerged as structural and functional building blocks in various plasmonic architectures owing to its excellent molecular recognition controllability and highly controllable length.14,15 Especially, the tetrahedral DNA structure, with exceptional mechanical rigidity and structural stability, has greatly advanced the accurate fabrication of plasmonic architectures. Tetrahedral DNA-directed assembly is proven to be advantageous for the precise control of interparticle distance.16,17 More importantly, the shape of the tetrahedron can be changed by the mechanical reconfiguration of the DNA strand at the arm of the tetrahedron using external stimuli.18 Such reconfigurations may be exploited for the development of plasmonic probes based on the surface plasmon coupling effect. DNA tetrahedron-directed plasmonic architectures have displayed great potential as plasmonic probes in the biosensing field.19–21
In this study, we designed a single-particle detection method based on tetrahedral DNA-directed core-satellite assemblies for Hg2+ detection. The core-satellite nanostructures were composed of satellite AuNPs tethered by tetrahedral DNA structures to a core AuNP. One edge of the tetrahedron consisted of single-stranded DNA containing a Hg2+ aptamer. The Hg2+ aptamer folded to form a hairpin structure in the presence of Hg2+. As a consequence, the configuration of the tetrahedron changed from a relaxed state to a taut state (Scheme 1), leading to a decrease in the interparticle distance and thus producing Raman signal enhancement. A remarkable magnification in SERS intensity was observed with the increase in Hg2+ at the single-particle level. The lowest detection limit of Hg2+ was 0.36 nM. Besides, the finite difference time domain (FDTD) calculation verified the strong linear correlation between the variation in SERS intensity with Hg2+ concentration. Moreover, this SERS probe showed high specificity. Finally, this SERS probe successfully demonstrated Hg2+ detection in tap water and lake water. Taking advantage of its excellent sensitivity and specificity, this proposed method can be useful for Hg2+ analysis in natural water.
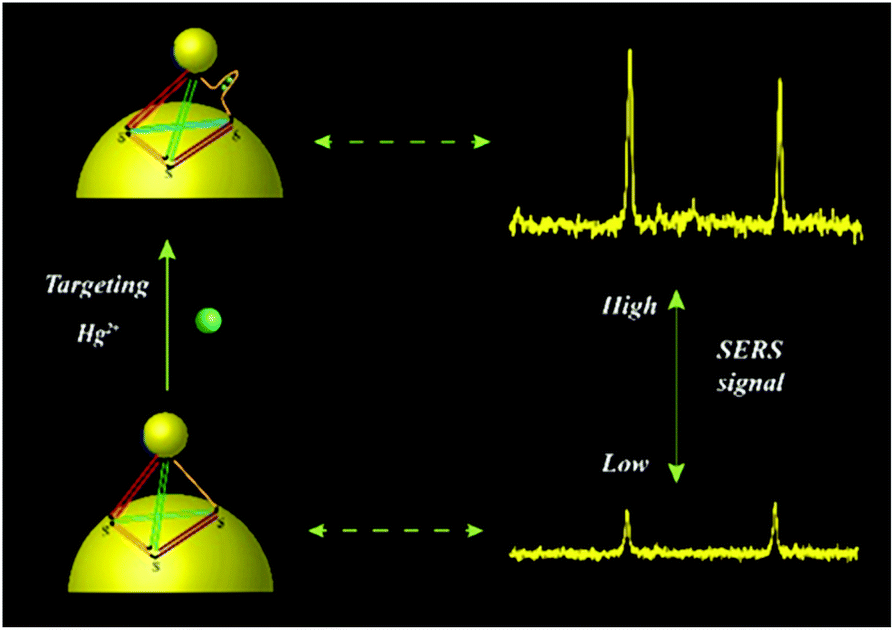 | ||
| Scheme 1 Schematic of the mechanism of Hg2+ detection. | ||
Experimental synthesis of AuNPs
Small satellite AuNPs were prepared by following a published protocol with slight modifications,22,23 and core AuNPs were synthesized according to a previous report using the seed-growth method.21 For more details, see ESI.†Preparation of the substrate
Briefly, indium tin oxide (ITO) glass slides were selected as the substrates. The surfaces of the substrates were cleaned by using a cleaning agent, acetone, ethanol, and ultrapure water. The substrates were cleaned in each solvent for 0.5 hours. After intense flushing with water, the cleaned substrates were modified with thiol by immersing them in 3-mercaptopropyltriethoxysilane (MPTES) (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) dissolved in anhydrous ethanol and incubating for 3 hours. Finally, the silanized substrates were washed with ethanol to remove excess MPTES.
:1 v/v) dissolved in anhydrous ethanol and incubating for 3 hours. Finally, the silanized substrates were washed with ethanol to remove excess MPTES.
Fabrication of core-satellite nanostructures on the substrate
The silanized substrates were immersed in the 100 nm AuNPs solution for 10 min. The AuNP-functionalized substrates were washed with water and dried under nitrogen flow. Subsequently, a 100 μL of tetrahedron-structured DNA solution (1 pM) was dropped onto the surface of the AuNP-modified substrate, followed by incubation in the shaker at room temperature for 3 hours. Then, the excess tetrahedron-structured DNA solution on the surface of the substrate was removed by thorough rinsing with ultrapure water and dried under a nitrogen flow. After that, 100 μL of satellite AuNPs was dropped onto the substrate containing tetrahedral DNA and incubated for 4 h at room temperature with slow shaking. Finally, the core-satellite-nanostructure-decorated ITO substrates were washed with ultrapure water and dried with nitrogen.SERS spectrum of the core-satellite nanostructure and Hg2+ detection
To obtain the SERS scattering spectrum, the core-satellite-nanostructure-functionalized substrates were immobilized on an automatic stage. Then, the target Hg2+ solution was pipetted on the surface of the substrate. For the detection of Hg2+, the SERS spectrum of a single core-satellite nanostructure was collected for 20 seconds before and after adding solutions containing various concentrations of Hg2+.Results and discussion
Fabrication and characterization of the DNA-mediated core-satellite nanostructure
The synthesized SERS probes were made of core-satellite nanostructures. The core-satellite nanostructure was fabricated with several 20 nm-sized AuNPs connected by a tetrahedral DNA structure to 100 nm-sized AuNPs, as shown schematically in Fig. 1A. The tetrahedral DNA structure was constructed from a single DNA strand and three thiol-modified single DNA strands (listed in Table S1†). Each thread of the tetrahedral DNA structure contained 26 base pairs. The tetrahedron-structured DNA carried a polyA sequence at one vertex and three thiol groups at the other three vertices. It is worth noting that one edge of the tetrahedron-structured DNA contained a single-stranded DNA that could specifically recognize Hg2+. To confirm the successful assembly of the tetrahedron-structured DNA, polyacrylamide gel electrophoresis (PAGE) was performed. As displayed in Fig. S1,† the tetrahedron moved more slowly than both the single-strand DNA and other combinations of two or three DNA strands, demonstrating the successful formation of a tetrahedral DNA structure.24 To prove that the Hg2+ aptamer does not hybridize with polyA, PAGE was performed. Tetra 3 and tetra 4 strands contained the Hg2+ aptamer and polyA, respectively (Table S1†). As expected, in Fig. S1,† there are two bands in column 15. The result proved that tetra 3 could not hybridize with tetra 4, indicating that polyA could not hybridize with the Hg2+ aptamer. The AuNPs were characterized by transmission electron microscopy (TEM) and extinction spectra. As displayed in their TEM images (Fig. S2B and D†), the acquired 20 nm- and 100 nm-sized AuNPs were both spherical and presented high monodispersity. The as-prepared 20 nm- and 100 nm-sized AuNPs showed the main absorbance peaks at 520 and 568 nm, respectively (Fig. S2A and D†). The core-satellite nanostructure was fabricated by following our protocol previously reported.21 The tetrahedral DNA structures were immobilized onto the surface of core AuNP via the strong gold–thiol bonds, and the residual polyA section that served as an effective anchoring block was introduced for connecting the 20 nm-sized AuNPs.25,26 | ||
| Fig. 1 Fabrication of DNA-mediated core-satellite nanostructures. (A) Synthetic scheme of the core-satellite nanostructure. (B) Typical DFM images and the corresponding scattering spectra (C) of (I) core AuNPs, (II) tetrahedral DNA structure-decorated core AuNPs, and (III) core-satellite nanostructures. (D) Typical SEM image of the core-satellite nanostructures (Inset: the enlarged SEM image). | ||
The self-assembly process was monitored by the DFM images and scattering spectra (Fig. 1B and C). The scattering peak of the individual bare 100 nm-sized AuNPs was found at 560 nm. The corresponding scattered light showed yellow color. When the tetrahedral DNA structure was attached to a 100 nm-sized AuNP, the scattering spectra shifted from 560 to 570 nm. Simultaneously, the color of the scattered spot evolved from yellow to yellow-red due to the change in the reflective index of the gold surface microenvironment.27 After adding the satellite AuNP solution, the decoration of the satellite AuNPs on the core AuNP led to an extraordinary red-shift in the scattering spectra, accompanied by the variation of the scattering light from yellow-red to red. We attribute this phenomenon to the plasmon coupling effect.28 Moreover, the successful formation of the core-satellite nanostructure was confirmed by the scanning electron microscopic (SEM) image showing that the surface of the core AuNP was decorated with the satellite AuNPs. (Fig. 1D). Besides, we noted that the satellite AuNPs were also deposited on the ITO glass substrate due to nonspecific adsorption (Fig. 1D). Because of the limit of CCD sensitivity, the image resolution of DFM was limited. The scattered light from AuNPs with a size of at least 40 nm could not be detected by DFM microscopy (Fig. 1B).29
With respect to the SERS test, the SERS spectrum of a single particle was recorded by using DFM combined with the Raman spectroscopy setup (Fig. S3†). The results are depicted in Fig. S4,† which shows the DFM image of the core-satellite nanostructure (Fig. S4B†) and the corresponding SERS spectrum (Fig. S4†). A sufficiently intense Raman signal from a single particle could be observed, confirming the single-particle test. The most prominent bands at 1072 and 1583 cm−1 were assigned to the v8a and v12 aromatic ring vibrations, respectively.30 After the optical measurement, we characterized the morphology of individual nanoparticles via in situ SEM. The results are displayed in Fig. S4C,† which shows the surface topography of the core-satellite nanostructure and demonstrates that the SERS spectrum was captured from a single particle. Besides, to rule out the influence of small AuNPs nonspecifically adsorbed on the substrates, SERS tests were performed at 10 random blank positions near or far from the scattering light spot, and almost no SERS signal could be obtained (Fig. S5†). This is because the small AuNPs offer a relatively low electromagnetic field within these regions. Therefore, the small AuNPs nonspecifically adsorbed on the substrates would have a negligible effect in the following experiments.
The principle of Hg2+ detection
For the construction of the SERS sensor, the core-satellite nanostructures used as SERS probes were prepared with several satellite AuNPs connected by the tetrahedral DNA structure to core AuNPs. The sequence for Hg2+ recognition was designed at one edge of the tetrahedron. According to previous reports, it is well-known that Hg2+ ions induce the conjunction of the thymine–thymine (T–T) base pair.31,32 The thorough principle for Hg2+ detection is described in Scheme 1. Upon binding the target Hg2+, the Hg2+ aptamer is converted to a hairpin structure owing to the formation of the T–Hg2+–T bond, which induces the transformation of the tetrahedra from a relaxed state to a taut state.18 Consequently, the transformation of the tetrahedra draws the satellite AuNP closer to the core AuNP, producing strong SERS intensity due to the surface plasmon coupling effect. Benefiting from the specific coordination between the Hg2+ aptamer and Hg2+ ions, this can be an ideal SERS probe for Hg2+ detection with high sensitivity and specificity. To verify the working principle of Hg2+ detection at the single-particle level, the core-satellite nanostructures immobilized on a substrate were exposed to 100 nM Hg2+ and incubated for 10 min. The variation in the SERS spectra in the absence and presence of Hg2+ was tested. As shown in Fig. 2A, the SERS intensity enhanced remarkably, illustrating that the detection platform can be used for the qualitative analysis of Hg2+ based on the SERS spectral changes. The above principle was further verified by using scattering spectra. Upon adding Hg2+, the Hg2+ aptamer combined with Hg2+ to form a hairpin structure, resulting in a remarkable red-shift of the scattering spectra (Fig. 2B). This result indicated that the addition of Hg2+ could decrease the interparticle distance, leading to an obvious increase in the plasmon coupling effect. Thus, it can be concluded that the qualitative detection of Hg2+ is feasible with our proposed principle. To further demonstrate the Hg2+ stimulus-responsive enhancement in the SERS signal of the SERS sensor, I− ions were used as the inhibitor of the SERS sensor after Hg2+ ion stimulation. When I− ions are introduced, the complex of HgI2 is preferably formed because it is an energetically stabilized product, which would lead to the structural separation of T–Hg2+–T, recovering to the initial level.33 Therefore, I− ions can work as the inhibitor for surface recycling. As expected, the SERS signal observably increased on adding 100 nM Hg2+ ions; comparatively, the intensity of the Raman scattering peaks significantly decreased, with the structure being restored to its initial state upon the addition of I− ions (Fig. S7†). This result indicates that the changes in the SERS signal were derived in response to the Hg2+ stimulus. | ||
| Fig. 2 Detection mechanism. (A) Raman spectra and (B) the corresponding scattering spectra of the individual core-satellite assembly in the absence and presence of 100 nM Hg2+. Typical TEM image of the isolated gold dimer (D) before and (E) after treatment with excess Hg2+. Statistical analysis of gap width in the (C) absence and (F) presence of excess Hg2+. | ||
The TEM images demonstrated the Hg2+ stimuli-responsive variation in the interparticle distance. However, the core-satellite nanostructures in the TEM images appeared as two-dimensional structures owing to the capillary forces and drying effect during sample preparation. This fact led to difficulty in measuring the interparticle distance from the TEM images.34,35 To precisely characterize the interparticle distance, a dimer of two 20 nm-sized AuNPs was synthesized by using the same tetrahedral DNA structure to replace the core-satellite nanostructure. Because the two AuNPs of the dimer were located on the same plane of the TEM grid during sample preparation, the spacing between the dimers should be close to the real situation in the experiment. The interparticle distance between the dimer in the absence and presence of excess Hg2+ was characterized by TEM (Fig. 2D and E). Statistical analysis showed that the average distance between the dimers decreased from 4.5 to 1.2 nm upon exposure to the Hg2+ solution (Fig. 2C and F). Additionally, in the presence of excess Hg2+, the intensity of the Raman scattering peaks significantly increased (Fig. S8†). This result is in good agreement with the foregoing SERS spectra in Fig. 2A.
Optimization of conditions
To achieve the best detection effect, we investigated the optimized conditions, such as the number of satellite AuNPs around the core AuNP and reaction time. Firstly, the effect of the satellite AuNPs number on the core AuNP was investigated. The number of satellite AuNPs attached to the core AuNP surface was controlled by tuning the immersion time in the satellite AuNP solution. As we all know, the localized surface plasmon resonance (LSPR) property heavily depends on the number of satellite AuNPs per core AuNP.31 Therefore, the DFM and scattering spectra techniques were applied to monitor the assembly process. As the immersion time increased, the color of the scattering spot gradually changed from yellow-red to red under the dark-field microscope (Fig. 3A). Simultaneously, the scattering peak increased gradually along with the increase in immersion time (Fig. 3B and C). This result is mainly due to the strong plasmon coupling effect. The red-shift of scattering spectral peak achieved the max value and then remained constant after 4 h, implying that the surface coverage reached saturation. To understand the assembly process in-depth, the surface coverage of the core AuNP was characterized by using SEM at different time points. The typical SEM images of a single isolated core-satellite nanostructure at different immersion times were observed (Fig. 3D, inset). Moreover, a statistical analysis of the satellite AuNP number was performed. As the immersion time increased, an increasing number of satellite AuNPs were adsorbed to the surface of the core AuNPs (Fig. 3D). When the immersion time was more than 4 h, the surface coverage of the core AuNP had reached saturation. This result is in excellent agreement with the behavior observed in the scattering spectra. When the incubation time exceeded 4 h, the number of satellite AuNPs remained almost constant. In contrast, more satellite AuNPs were nonspecifically adsorbed on the surface of the substrate, making it difficult to distinguish the scattering light spot of the individual particles in the DFM image. Therefore, 4 h was considered the optimum immersion time for the surface coverage to reach saturation. Subsequently, the influence of reaction time was also studied. Reaction time is a critical factor influencing the affinity between Hg2+ and the Hg2+ aptamer, which determines the analytical performance. To monitor this reaction in real-time, the time-dependent SERS spectra of a single core-satellite assembly were recorded over 10 min at a certain time interval upon treatment with 100 nM Hg2+ (Fig. 3E and F). With the increase in reaction time, the Raman intensity ascended. The reaction achieved equilibrium in 5 min, showing the maximum intensity. To ensure the accuracy of data, 7 min was taken as the optimal reaction time. | ||
| Fig. 3 Optimization of the assay conditions. (A) DFM images and (B) scattering spectra of the core-satellite nanostructures at various incubation times. (C) Plot of the SPR wavelength shift with incubation time. (D) Statistical distribution of the satellite AuNP number attached per core AuNP. Inset: typical SEM images of the core-satellite nanostructures treated for various immersion times (scale bars, 100 nm). (E) Time-resolved Raman spectra of an individual core-satellite assembly in the presence of 100 nM Hg2+. (F) Time-resolved SERS intensity of a single core-satellite nanostructure exposed to 100 nM Hg2+. | ||
Detection of Hg2+
Under the optimized conditions, the sensitivity of this sensor was evaluated based on SERS signals after introducing different concentrations of Hg2+ (1 to 100 nM). Fig. 4A presents the representative SERS spectra under different Hg2+ concentrations. According to the literature,21 the Raman scattering peak at 1583 cm−1, which belongs to the aromatic ring breathing mode, suffers from relevant interference due to the attachment of Hg2+ to the carboxyl group of 4-mercaptobenzoic acid (4-MBA). The reason might be the redistribution of charges between the ring breathing vibrational band upon the adsorption of Hg2+ on the carboxyl group of 4-MBA.36,37 On the contrary, the Raman scattering peak at 1073 cm−1 exhibited no frequency shift, indicating that it did not suffer from any interference upon the coordination of Hg2+ to the carboxyl group. Therefore, for the accuracy of quantitative Hg2+ detection, the characteristic SERS peak of 4-MBA at 1072 cm−1 was employed. To demonstrate the stability of the SERS probe for Hg2+ detection at the single-particle level, lots of SERS spectral experiments were repeated at the same Hg2+ concentration for statistical analyses. The thirty SERS intensity variations at 1072 cm−1 (ΔI) were distributed as a Gaussian curve (Fig. S9†). The SERS intensity increased linearly with the Hg2+ concentration in the range of 10 to 100 nM. Moreover, the linear regression equation (ΔI = 0.3494C(Hg2+) + 4.4314) for Hg2+ detection was obtained for the SERS signals at 1072 cm−1 (Fig. 4B). The corresponding correlation R2 was 0.9953. The detection limit (LOD) for Hg2+ was 0.36 nM at a signal-to-noise ratio of 3. The lowest concentration for quantitative detection of Hg2+ by this method corresponded to 1 nM, which is lower than the allowable maximum residue limit of Hg2+ (10 nM) in drinkable water as per the United States Environmental Protection Agency (USEPA).33,38 Compared with the previously reported methods for Hg2+ measurement,39–43 the proposed strategy for Hg2+ shows much higher detection sensitivity (Table S2†).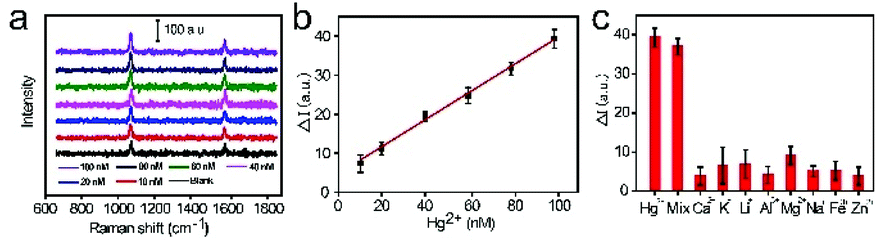 | ||
| Fig. 4 Detection of Hg2+. (A) The Hg2+ concentration-dependent SERS spectra ranging from 100 to 1 nM and the blank control. (B) The linear relationship between the variation in the SERS intensity of 4-MBA at 1072 cm−1 and Hg2+ concentration. (C) Selectivity of the method for Hg2+. | ||
To further demonstrate the sensing mechanism based on the SERS intensity variation with the concentration of Hg2+, FDTD simulation was carried out to investigate the variation in the electromagnetic intensity of the core-satellite nanostructures during the compression of the particles. The SERS intensity was dominated by the enhancement in the local electromagnetic field, which is directly proportional to the second power of the electromagnetic field. Five structural models with a core AuNP of size 100 nm and four satellite AuNPs of size 20 nm were used in the simulation (Fig. S6†). The interparticle distance between the core AuNP and the satellite AuNPs varied from 4.5 to 1.2 nm. For simplicity, the contributions of the DNA strands, 4-MBA and glass slides were ignored.44Fig. 5A–E vividly displays the electric-field distribution of the different models with four satellite AuNPs distributed around the surface of a core AuNP. The electromagnetic enhancement is highly localized in the gap regions. The intensity of the electromagnetic field in the gap is enhanced as the interparticle distance between the satellite and core AuNP decreases from 4.5 to 1.2 nm. As shown in Fig. 5F, with the increase in the compressed satellite AuNP number, a linear rise in SERS intensity happens. These calculated results are in accordance with the experimental results, which demonstrate that the increase in SERS intensity observed in our experiments is due to the increase in the compressed satellite AuNP number.
 | ||
| Fig. 5 FDTD simulations of the core-satellite nanostructure as a function of the increasing number of satellites. (A–E) The local electric field distribution of core AuNPs assembled with different numbers of compressed satellite AuNPs: 0–4, respectively. The labels 0–4 correspond to the number of compressed satellite AuNPs. (F) The linear range between the calculated SERS intensity and the compressed satellite AuNP number. | ||
To prove the selectivity of detection by this SERS biosensor, several similar metal ions, such as Hg2+, Ca2+, K+, Li+, Al3+, Mg2+, Na+, Fe3+, Zn2+ and the mixture containing all the above metal ions, were further employed under optimized conditions. The SERS signal of the individual core-satellite assembly in solutions containing 100 nM Hg2+ and 100-fold other metal ions, respectively, are described in Fig. 4C. The SERS intensity remained unchanged after treatment with the other metal ions except for Hg2+ and the mixture of metal ions. The high selectivity of this detection is due to the specific coordination between the Hg2+ aptamer and Hg2+ ions. This comparison demonstrated that the proposed SERS sensor possesses excellent selectivity, making it suitable for working in a complex ecological system.
To prove the ability of the method in the measurement of Hg2+ ions in practical applications, tap water and lake water collected from our laboratory and a lake in our university were tested. Before analysis, samples with three different Hg2+ concentrations (10, 50 and 100 nM) were prepared with diluted tap water and lake water. Fig. S10† depicts the SERS spectra of the Hg2+ detection in tap water and lake water. To prove the accuracy and reliability of this method, the spiked recovery was calculated and is summarized in Table 1. It was found that the relative standard deviation (RSD) ranged from 1.29% to 2.07%. We attribute this to the favorable affinity of the Hg2+ aptamer and Hg2+. The results indicate that our SERS sensor can be used for detecting Hg2+ ions in complex biological samples.
| Water sample | Spilled Hg2+ (nM) | Measured Hg2+ (nM) | RSD (%) |
|---|---|---|---|
| Tap water | 10 | 9.59 | 1.82 |
| 50 | 49.8 | 2.01 | |
| 100 | 101.6 | 1.68 | |
| Lake water | 10 | 10.9 | 1.29 |
| 50 | 48.1 | 2.07 | |
| 100 | 102.3 | 1.84 |
Conclusions
In summary, we designed a novel single-particle detection assay for the sensitive and selective quantitative analysis of Hg2+ by using DFM-correlated Raman spectroscopy. After the addition of Hg2+, the Hg2+ aptamer folded up and formed a hairpin structure, which led to a variation in the tetrahedral DNA configuration. Consequently, the interparticle distance reduced from 4.5 to 1.2 nm, which tremendously increased the SERS signal. Based on this principle, a detection range from 1 to 100 nM, with a low LOD of 0.36 nM, was obtained. Furthermore, the FDTD simulation results revealed that the variation in SERS intensity had a linear response to the concentration of Hg2+. In addition, the proposed Hg2+ detection process presented highly favorable specificity in the presence of a series of similar metal ions. Thus, the SERS probe was applied for the detection of Hg2+ ions in real water samples. The proposed SERS probe exhibits great potential in Hg2+ detection in real water samples.Author contributions
N. Feng performed all the experiments. N. Feng, J. J. Shen, C. Li, Q. Q Zhao, E. K. Fodjo, L. Zhang, S. F. Chen, Q. L. Fan and L. H Wang analyzed the data. N. Feng, L. Zhang, and L. H Wang wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Key Research and Development Program of China (2017 YFA0205300), the National Natural Science Foundation of China (62075103, 21674048), Program for Changjiang Scholars and Innovative Research Team in University (IRT_15R37), the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD, YX030003), the Key Research and Development Program of Jiangsu (BE2018732), the Natural Science Foundation of Jiangsu Province (BK20211271).Notes and references
- Z. C. Castilhos, S. Rodrigues-Filho, A. P. C. Rodrigues, R. C. Villas-Bôas, S. Siegel, M. M. Veiga and C. Beinhoff, Sci. Total Environ., 2006, 368, 320–325 CrossRef CAS PubMed.
- E. M. Nolan and S. J. Lippard, Chem. Rev., 2008, 108, 3443–3480 CrossRef CAS PubMed.
- H. Chu, D. Yao, J. Chen, M. Yu and L. Su, ACS Omega, 2020, 5, 9558–9565 CrossRef CAS PubMed.
- C. Lai, S. Liu, C. Zhang, G. Zeng, D. Huang, L. Qin, X. Liu, H. Yi, R. Wang, F. Huang, B. Li and T. Hu, ACS Sens., 2018, 3, 2566–2573 CrossRef CAS PubMed.
- Y. Cui, Y. Tang, S. Fan, Y. Ge, M. Han, J. Liu, M. Li and J. Hu, J. Phys. Chem. C, 2020, 124, 19267–19272 CrossRef CAS.
- J. R. Crockett, H. Win-Piazza, J. E. Doebler, T. Luan and Y. Bao, ACS Appl. Nano Mater., 2021, 4, 1654–1663 CrossRef CAS.
- W.-R. Cui, C.-R. Zhang, W. Jiang, R.-P. Liang, S.-H. Wen, D. Peng and J.-D. Qiu, ACS Sustainable Chem. Eng., 2019, 7, 9408–9415 CrossRef CAS.
- C. Song, B. Yang, Y. Yang and L. Wang, Sci. China: Chem., 2016, 59, 16–29 CrossRef CAS.
- W. Wang, Chem. Soc. Rev., 2018, 47, 2485–2508 RSC.
- S. Wang, Z. Ye, X. Wang and L. Xiao, ACS Appl. Nano Mater., 2019, 2, 6646–6654 CrossRef CAS.
- L.-L. Qu, D.-W. Li, L.-X. Qin, J. Mu, J. S. Fossey and Y.-T. Long, Anal. Chem., 2013, 85, 11675–11675 CrossRef CAS.
- K. Ock, W. I. Jeon, E. O. Ganbold, M. Kim, J. Park, J. H. Seo, K. Cho, S.-W. Joo and S. Y. Lee, Anal. Chem., 2012, 84, 2172–2178 CrossRef CAS PubMed.
- M. Tian, Z. Yuan, Y. Liu, C. Lu, Z. Ye and L. Xiao, Analyst, 2020, 145, 4737–4752 RSC.
- N. Liu and T. Liedl, Chem. Rev., 2018, 118, 3032–3053 CrossRef CAS PubMed.
- N. Li, Y. Shang, Z. Han, T. Wang, Z.-G. Wang and B. Ding, ACS Appl. Mater. Interfaces, 2019, 11, 13835–13852 CrossRef CAS PubMed.
- T. Peng, X. Li, K. Li, Z. Nie and W. Tan, ACS Appl. Mater. Interfaces, 2020, 12, 14741–14760 CrossRef CAS PubMed.
- X. Zhang, C. Liu, Y. Pei, W. Song and S. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 28671–28680 CrossRef CAS PubMed.
- H. Pei, L. Liang, G. Yao, J. Li, Q. Huang and C. Fan, Angew. Chem., Int. Ed., 2012, 51, 9020–9024 CrossRef CAS PubMed.
- J. C. Fraire, V. N. Sueldo Ocello, L. G. Allende, A. V. Veglia and E. A. Coronado, J. Phys. Chem. C, 2015, 119, 8876–8888 CrossRef CAS.
- W. Ma, H. Yin, L. Xu, X. Wu, H. Kuang, L. Wang and C. Xu, Chem. Commun., 2014, 50, 9737–9740 RSC.
- N. Feng, L. Zhang, J. Shen, Y. Hu, W. Wu, E. K. Fodjo, S. Chen, W. Huang and L. Wang, Chem. Eng. J., 2022, 433, 133666 CrossRef CAS.
- Y. Tian, Z. Shuai, J. Shen, L. Zhang, S. Chen, C. Song, B. Zhao, Q. Fan and L. Wang, Small, 2018, 14, 1800669 CrossRef PubMed.
- X. Lin, Y.-l. Hu, C. Zhang, J. Yin, R. Cui, D.-l. Yang and B. Chen, J. Cent. South Univ., 2021, 28, 3642–3653 CrossRef CAS.
- H. Pei, N. Lu, Y. Wen, S. Song, Y. Liu, H. Yan and C. Fan, Adv. Mater., 2010, 22, 4754–4758 CrossRef CAS PubMed.
- J.-W. Oh, D.-K. Lim, G.-H. Kim, Y. D. Suh and J.-M. Nam, J. Am. Chem. Soc., 2014, 136, 14052–14059 CrossRef CAS PubMed.
- D. Zhu, P. Song, J. Shen, S. Su, J. Chao, A. Aldalbahi, Z. Zhou, S. Song, C. Fan, X. Zuo, Y. Tian, L. Wang and H. Pei, Anal. Chem., 2016, 88, 4949–4954 CrossRef CAS PubMed.
- Y. Zhang, Z. Shuai, H. Zhou, Z. Luo, B. Liu, Y. Zhang, L. Zhang, S. Chen, J. Chao, L. Weng, Q. Fan, C. Fan, W. Huang and L. Wang, J. Am. Chem. Soc., 2018, 140, 3988–3993 CrossRef CAS PubMed.
- M.-X. Li, C.-H. Xu, N. Zhang, G.-S. Qian, W. Zhao, J.-J. Xu and H.-Y. Chen, ACS Nano, 2018, 12, 3341–3350 CrossRef CAS PubMed.
- P. L. Truong, X. Ma and S. J. Sim, Nanoscale, 2014, 6, 2307–2315 RSC.
- A. Michota and J. Bukowska, J. Raman Spectrosc., 2003, 34, 21–25 CrossRef CAS.
- Z. Duan, X. Zhang, T. Ye, X. Zhang, S. Dong, J. Liu, X. Xiao and C. Jiang, ACS Appl. Mater. Interfaces, 2018, 10, 25737–25743 CrossRef CAS PubMed.
- N. Ma, X. Ren, H. Wang, X. Kuang, D. Fan, D. Wu and Q. Wei, Anal. Chem., 2020, 92, 14069–14075 CrossRef CAS PubMed.
- B. Sun, X. Jiang, H. Wang, B. Song, Y. Zhu, H. Wang, Y. Su and Y. He, Anal. Chem., 2015, 87, 1250–1256 CrossRef CAS PubMed.
- T.-T. Zhai, D. Ye, Y. Shi, Q.-W. Zhang, X. Qin, C. Wang and X.-H. Xia, ACS Appl. Mater. Interfaces, 2018, 10, 33966–33975 CrossRef CAS PubMed.
- K. Li, K. Wang, W. Qin, S. Deng, D. Li, J. Shi, Q. Huang and C. Fan, J. Am. Chem. Soc., 2015, 137, 4292–4295 CrossRef CAS PubMed.
- P. L. Yap, S. Kabiri, D. N. H. Tran and D. Losic, ACS Appl. Mater. Interfaces, 2019, 11, 6350–6362 CrossRef CAS PubMed.
- S. J. Lee and M. Moskovits, Nano Lett., 2011, 11, 145–150 CrossRef CAS PubMed.
- T. Kang, S. M. Yoo, I. Yoon, S. Lee, J. Choo, S. Y. Lee and B. Kim, Chem. – Eur. J., 2011, 17, 2211–2214 CrossRef CAS PubMed.
- P. Veerakumar, G. Jaysiva, S.-M. Chen and K.-C. Lin, ACS Appl. Mater. Interfaces, 2022, 14, 5908–5920 CrossRef CAS PubMed.
- Y. Zhang, S. Guo, Z. Jiang, G. Mao, X. Ji and Z. He, Anal. Chem., 2018, 90, 9796–9804 CrossRef CAS PubMed.
- X. Guo, J. Huang, Y. Wei, Q. Zeng and L. Wang, J. Hazard. Mater., 2020, 381, 120969 CrossRef CAS PubMed.
- X. Zhao, S. Campbell, P. Z. El-Khoury, Y. Jia, G. Q. Wallace, A. Claing, C. G. Bazuin and J.-F. Masson, ACS Sens., 2021, 6, 1649–1662 CrossRef CAS PubMed.
- D. S. Franciscato, T. A. Matias, J. Shinohara, J. M. Gonçalves, N. P. Coelho, C. S. Fernandes, E. A. Basso, H. S. Nakatani, K. Araki, H. E. Toma and V. R. de Souza, Spectrochim. Acta, Part A, 2018, 204, 174–179 CrossRef CAS PubMed.
- X. Lan, Z. Chen, B.-J. Liu, B. Ren, J. Henzie and Q. Wang, Small, 2013, 9, 2308–2315 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary statement, figures and data. See DOI: https://doi.org/10.1039/d2an00402j |
| This journal is © The Royal Society of Chemistry 2022 |