
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEvaluation of histone deacetylase inhibitor substituted zinc and indium phthalocyanines for chemo- and photodynamic therapy†
Başak Arua,
Aysel Günayb,
Gülderen Yanıkkaya Demirel*a,
Ayşe Gül Gürek *b and
Devrim Atilla*b
*b and
Devrim Atilla*b
aFaculty of Medicine, Immunology Department, Yeditepe University, 34755 Ataşehir, İstanbul, Turkey. E-mail: gulderen.ydemirel@yeditepe.edu.tr
bDepartment of Chemistry, Gebze Technical University, 41400 Gebze, Kocaeli, Turkey. E-mail: gurek@gtu.edu.tr; datilla@gtu.edu.tr
First published on 28th October 2021
Abstract
In this study, we synthesized and characterized 3-hydroxypyridin-2-thione (3-HPT) bearing zinc (ZnPc-1 and ZnPc-2) and indium (InPc-1 and InPc-2) phthalocyanine (Pc) derivatives, either non-peripherally or peripherally substituted as photosensitizer (PS) agents and evaluated their anti-cancer efficacy on two breast cancer cell lines, MDA-MB-231 and MCF-7 as well as a human endothelial cell line, HUVEC. Our results indicated different localization patterns between ZnPcs and InPcs in addition to enhanced effects on the mitochondrial network for InPcs. Moreover, peripheral or non-peripheral substitution of HDACi moieties altered cellular localization between ZnPc-1 and ZnPc-2, leading to increased IC50 values along with decreased anti-cancer activity for non-peripheral substitution. When considering the compounds' differential effects in vitro, our data indicates that further research is required to determine the ideal Pcs for anti-cancer PDT treatments since the core metals of the compounds have affected the cellular localization, and positioning of the chemotherapeutic residues may inhibit cellular penetrance.
Introduction
Consisting of a photosensitizer (PS), light, and oxygen; photodynamic therapy (PDT) aims to destroy malignant tissue via irradiating the PS by a light source at a wavelength matching the compound's absorption maximum.1,2 Irradiation promotes chemical reactions that are induced by electron or energy transfer; in turn, reactive oxygen and nitrogen species are generated. PDT exerts its anti-cancer effects mainly by a highly reactive state of oxygen, so-called singlet oxygen (1O2),3 and targets tumours via three distinct mechanisms. Irradiation of the PS promotes accumulation of reactive oxygen species (ROS) that directly kill tumour cells by activating programmed cell death mechanisms.4,5 PDT can impair tumour associated vascularization and leads to nutrient and oxygen deprivation, moreover, PDT-induced stress signals also enable the immune system to recognise and destroy tumour cells.6,7 The role of each mechanism depends on the type and dose of the PS administered, incubation duration of the target tissue with the PS, optical dose of irradiation and tumour oxygen levels,6 and all three mechanisms mentioned above are important for tumour regression8,9 while the importance of each in tumour eradication needs further research. Among other PSs such as tetrapyrrolic porphyrins and chlorins, thanks to their high extinction coefficients and longer absorption wavelengths (due to additional benzene rings connected to each pyrrolic subunit which causes more electron delocalization),10,11 phthalocyanines (Pcs) are regarded as potent second-generation PS compounds in cancer treatment. The efficacy and specificity of the treatment can be further increased by introducing chemotherapeutic moieties or tumour-specific markers to the PS for targeted anti-cancer therapy.12 Introduction of functional groups, axially or peripherally to Pcs also decreases their aggregation in aqueous interfaces while alleviating their hydrophobicity.13 Histone deacetylases (HDACs), regulators of reversible histone acetylation play a role in carcinogenesis, and are potential targets for anti-cancer treatments.14–16 Inhibition of HDACs is shown to induce cell cycle arrest, promote cell death, modulate immune responses, and inhibit angiogenesis.17 Histone deacetylase inhibitor (HDACi) treatments are mainly approved for haematological malignancies.18,19 Examples include vorinostat, belinostat, panobinstat and romidepsin for cutaneous T-cell lymphoma, peripheral T-cell lymphoma, multiple myeloma and cutaneous T-cell lymphoma, respectively.17,18 Considering the high HDAC expression observed in certain solid tumours, (such as high HDAC6 expression in breast cancer),17 the success of HDACi treatments in haematological cancers introduced them into clinical trials for targeting solid tumours. However, the results were controversial: HDACi monotherapies in solid tumours remained ineffective while leading to toxic side effects,20 in contrast, combinatorial administration of HDACi and chemotherapy provided beneficial results, both in animal studies and clinical trials.21–23Being the most common malignancy among women, every year 1.5 million patients are being diagnosed with breast cancer (which accounts for 25% of all women diagnosed with cancer), and it is estimated that 12% of all women will be diagnosed with breast cancer during their lifetimes.24,25 Survival rates differ between countries: the estimated 5-year survival rate is ∼80% in developed countries whereas it remains below 40% in developing countries.24 Due to its heterogenic nature as well as variable morphological and biological features, different clinical approaches are adopted for the treatment of the disease.26 Various studies have underlined the involvement of HDAC6 and HDAC8 activities in the breast cancer, and reported that their inhibition may be a beneficial approach for treatment.27–30
Reports involving combinatorial applications of PDT and HDACi suggested enhanced anti-cancer activity, yet, none of them have evaluated efficacy of a Pc derivative as the PS agent.31–33 Previously, we reported 3-hydroxypyridin-2-thione (3-HPT), a HDACi substituted silicon phthalocyanine derivative (SiPc-HDACi) exerts anti-cancer properties on breast cancer cell lines by activating programmed cell death pathways and inducing cell cycle arrest.12 Herein, we synthesized and characterized either non-peripherally or peripherally 3-HPT bearing zinc (Zn) and indium (In) Pc derivatives, and evaluated their efficacy on breast cancer cell lines (MCF-7 and MDA-MB-231) in addition to the healthy endothelial cell line HUVEC.
Experimental section
Materials and methods
IR spectra were recorded between 4000 and 500 cm−1 using a PerkinElmer Spectrum 100 FT-IR spectrometer with an attenuated total reflection (ATR) accessory featuring a zinc selenide (ZnSe) crystal. MALDI-TOF mass spectrometry analyses were carried out on Bruker micro flex LT MALDI-TOF MS spectrometer using dihydroxybenzoic acid as a matrix. 1H NMR and 13C NMR spectra were recorded in DMSO-d6 solutions on a Varian 500 MHz spectrometer. TMS standart was used as internal standart for 1H-NMR measurements. Elemental analysis was carried out using Thermo Finnigan Flash 1112 Instrument. Singlet oxygen quantum yield (ΦΔ) measurements were done by Horiba Jobin–Yvon Fluorometer with Hamamatsu NIR PMT 5509 by using direct method. All solvents and chemicals were of reagent-grade quality, purchased from Sigma Aldrich Chemical Co. and Merck.Starting compounds, 3-nitrophthalonitrile (2a),34 4-nitrophthalonitrile (2b)35 and HDACi (1-(2-methyl-(1,1′-biphenylmethyl))-3-hydroxyoxypyridin-2-thione)36 (1) were synthesized according to the literature.
Synthesis
Phthalonitrile derivatives (3a and 3b)
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:0.2)) gave the target product 3a (597 mg, 1.72 mmol) in 89% yield as yellow solid. 1H NMR (500 MHz, CDCl3) δ (ppm): 7.79–7.73 (m, 1H), 7.52 (dd, J = 10.0, 6.3 Hz, 1H), 7.46–7.40 (m, 1H), 7.36 (d, J = 11.0 Hz, 4H), 7.26 (s, 1H), 7.19 (d, J = 7.0 Hz, 1H), 6.97 (dd, J = 8.9, 3.8 Hz, 1H), 6.68 (dd, J = 8.7, 5.5 Hz, 1H), 5.87 (d, J = 4.2 Hz, 2H), 2.26 (s, 2H). 13C NMR (125 MHz, CDCl3) δ (ppm): 174.6, 159.3, 153.5, 141.1–125.9, 120.1, 117.0, 115.3, 112.8, 110.8, 105.6, 58.9, 20.6. IR (ATR-thin film) νmax/cm−1; 3025, 2900–2800, 2230, 1621, 1575, 1456, 1398, 1266. MALDI-TOF (m/z) calc. for C27H19N3OS: 433.53; found: 433.34 [M]+.:1:0.2)) gave the target product 3b (617 mg, 1.78 mmol) in 89% as yellow solid. 1H NMR (500 MHz, CDCl3) δ (ppm): δ 7.80 (dd, J = 6.7, 1.8 Hz, 1H), 7.77–7.71 (m, 1H), 7.43–7.18 (m, 10H), 6.75–6.68 (m, 1H), 5.91 (s, 2H), 2.29 (s, 3H). 13C NMR (125 MHz, CDCl3) δ (ppm): 174.9, 160.1, 153.6, 141.1–126.0, 121.1, 117.5, 115.6, 115.3, 110.9, 109.2, 105.6, 59.2, 20.6. IR (ATR-thin film) νmax/cm−1; 3040, 2900–2800, 2232, 1620, 1570, 1455, 1398, 1265. MALDI-TOF (m/z) calc. for C27H19N3OS: 433.53; found: 433.33 [M]+.
:1:0.2)) gave the target product 3a (597 mg, 1.72 mmol) in 89% yield as yellow solid. 1H NMR (500 MHz, CDCl3) δ (ppm): 7.79–7.73 (m, 1H), 7.52 (dd, J = 10.0, 6.3 Hz, 1H), 7.46–7.40 (m, 1H), 7.36 (d, J = 11.0 Hz, 4H), 7.26 (s, 1H), 7.19 (d, J = 7.0 Hz, 1H), 6.97 (dd, J = 8.9, 3.8 Hz, 1H), 6.68 (dd, J = 8.7, 5.5 Hz, 1H), 5.87 (d, J = 4.2 Hz, 2H), 2.26 (s, 2H). 13C NMR (125 MHz, CDCl3) δ (ppm): 174.6, 159.3, 153.5, 141.1–125.9, 120.1, 117.0, 115.3, 112.8, 110.8, 105.6, 58.9, 20.6. IR (ATR-thin film) νmax/cm−1; 3025, 2900–2800, 2230, 1621, 1575, 1456, 1398, 1266. MALDI-TOF (m/z) calc. for C27H19N3OS: 433.53; found: 433.34 [M]+.:1:0.2)) gave the target product 3b (617 mg, 1.78 mmol) in 89% as yellow solid. 1H NMR (500 MHz, CDCl3) δ (ppm): δ 7.80 (dd, J = 6.7, 1.8 Hz, 1H), 7.77–7.71 (m, 1H), 7.43–7.18 (m, 10H), 6.75–6.68 (m, 1H), 5.91 (s, 2H), 2.29 (s, 3H). 13C NMR (125 MHz, CDCl3) δ (ppm): 174.9, 160.1, 153.6, 141.1–126.0, 121.1, 117.5, 115.6, 115.3, 110.9, 109.2, 105.6, 59.2, 20.6. IR (ATR-thin film) νmax/cm−1; 3040, 2900–2800, 2232, 1620, 1570, 1455, 1398, 1265. MALDI-TOF (m/z) calc. for C27H19N3OS: 433.53; found: 433.33 [M]+.Phthalocyanine derivatives (ZnPc-1, ZnPc-2, InPc-1 and InPc-1)
:C2H5OH (30:1)) gave ZnPc-1 (121 mg, 0.067 mmol) in 15% yield as green color solid. 1H NMR (500 MHz, DMSO-d6) δ (ppm): δ 8.57 (bs, 5H, ArH), 8.09–6.94 (m, ArH, 51H), 6.32–6.10 (m, CH2, 8H), 2.34–2.16 (m, CH3, 12H). 13C NMR (125 MHz, DMSO-d6) δ (ppm): 182.14, 159.81, 156.11, 154.81, 142.30, 149.73, 145.91, 144.83, 141.92, 139.20, 137.84, 135.11, 126.18, 122.45, 122.10, 120.05, 114.82, 106.71, 59.81, 21.12. IR (ATR-thin film) νmax/cm−1; 3078, 2900, 1600, 1640, 1485, 1455, 1360, 1093, 880, 744. MALDI-TOF (m/z) calc. for C108H76N12O4S4Zn: 1799.50, found: 1799.35 [M]+. Elemental analysis (%) calc. for C108H76N12O4S4Zn: C, 72.09; H, 4.26; N, 9.34; found C, 72.20; H, 4.35; N, 9.10. UV-vis (DMSO): λmax nm (logε) 335 (5.10), 638 (4.56), 706 (5.28).ε) 354 (5.04), 608 (4.58), 672 (5.26).:C2H5OH (40:1)) gave InPc-1 (100 mg, 0.053 mmol) in 12% yield as dark green color solid. 1H NMR (500 MHz, DMSO-d6) δ (ppm): δ 9.32–6.48(m, ArH, 56H), 5.90 (s, CH2, 8H), 2.27–1.92 (m, CH3, 12H). 13C NMR (125 MHz, DMSO-d6) δ (ppm): 178.4, 142.90, 135.65, 131.05, 130.09, 129.80, 126.90, 112.56, 104.3, 57.2, 20.1. IR (ATR-thin film) νmax/cm−1; 3074, 2921–2851, 1610, 1483, 1450, 1350. MALDI-TOF (m/z) calc. for C108H76ClInN12O4S4: 1884.38, found: 1884.54 [M]+, 1853.14 [M − 2(CH3)]+. Elemental analysis (%) calc. for C108H76ClInN12O4S4Zn: C, 68.84; H, 4.07; N, 8.92; found C, 69.10; H, 4.20; N, 8.80. UV-vis (DMSO): λmax nm (logε) 356 (4.59), 650 (3.15), 722 (5.55).ε) 352 (4.62), 645 (4.25), 695 (5.42).In vitro studies
Cell culture conditions
MCF-7 and MDA-MB-231 cells were cultured in high glucose (4.5 g L−1) Dulbecco's Modified Eagle's Medium (DMEM), while HUVECs were cultured in DMEM/F12 medium; both supplemented with 10% fetal bovine serum (FBS) and antibiotic solution (100 IU penicillin and 0.1 mg streptomycin) (all purchased from Gibco, Thermo Fisher Scientific, USA). HUVECs used in this study were between 8th and 10th passages whereas MCF-7 cells were at 24th and MDA-MB-231 cells were 34th passages. For evaluating compounds' cytotoxic properties under dark and light conditions, cells were seeded as 5000 cells per well into 96-well test plates as triplicates and incubated overnight in 5% CO2 containing cell culture incubator with humid environment for attachment. All compounds were dissolved in dimethyl sulfoxide (DMSO) and diluted in culture medium in order to prepare 1, 5, 25, 50 and 100 μM solutions. The final concentration of DMSO did not exceed 0.1%, which is considered as safe in terms of cytotoxicity. Untreated cells were used as control.For flow cytometric analyses and western blotting, 500.000 cells were seeded into 60 mm tissue culture dishes as triplicates; for confocal microscopy, cells were seeded on Millicell® EZ Slides (Merck Millipore, USA) as 20.000 cell per well and incubated overnight for attachment. Cells were treated with respective IC50 doses and were irradiated 24 hours later as indicated above. After an additional 24 hours, cells were detached with trypsin–EDTA (w/phenol red, 0.25%, Gibco, Thermo Fisher Scientific, USA); for flow cytometric analyses, cells were washed twice with DPBS containing 0.1% sodium azide as a preservative, for western blotting, protein extraction was performed, followed by measuring protein concentration with Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific, USA).
Determining cytotoxicity under dark and light conditions
Dose interval (1–100 μM) was determined according to a study previously published.37 Compounds' cytotoxic properties under dark conditions were evaluated by measuring viability after incubating cells with compounds for 24 hours. Light toxicity was evaluated by irradiating treated cells with red light at 5 mA for 34 minutes to obtain 500 mJ optical dose37 after 24 hours of incubation at dark. Viability was measured after an additional 24 hours and IC50 doses for each compound were calculated. Tetrakis-(4,7,10-trioxaundecan-1-sulfanyl)phthalocyaninato zinc (ZnPc-C) was used as a positive control in this study, applied, irradiated and analysed in the same manner.37 Alamar Blue assay was used for determining viability (Sigma Aldrich, USA) which relies on the reduction in response to metabolic activity.38 Absorbances were measured at 570 nm and 600 nm with a spectrophotometer (Epoch, BioTek Instruments, USA). Viability percentages were calculated according to the equation given in equation (S1).†Annexin V/propidium iodide staining
Apoptosis, necrosis and viability were evaluated with annexin V/propidium iodide staining, which is considered as the gold standard for apoptosis detection.39 Detached cells were washed twice and then suspended in annexin V binding buffer (BioVision Inc., USA), followed by staining with annexin V-FITC reagent (1 μL/1 × 105 cells) (BioVision Inc., USA) and propidium iodide (0.25 μg/1 × 105 cells) (Thermo Fisher Scientific) by incubating cells on ice for 15 minutes. Analyses were immediately performed on Beckman Coulter CytoFLEX flow cytometry system. CytExpert software was used for evaluating data.DNA content analysis
DNA content analysis relies on the difference DNA amount cells have through cell division stages G0/1 pre-replicative cells, S (dividing cells) and G2/M (post replicative plus mitotic cells).40 Cells with fragmented DNA can also be identified and this fraction is named as “sub-G1” population. In this study, Cell Cycle Kit (Beckman Coulter) was used. Detached cells were fixed in 70% ethanol by incubating at 4 °C for an hour. Ethanol was discarded, cells were washed with PBS for one time and stained with the reagent supplied by incubating at room temperature for 40 minutes at dark. Samples were kept at 4 °C until analysis and read with Navios EX flow cytometry system (Beckman Coulter). Analyses were performed on ModFit Software (Version 5).Evaluation of HDAC6 and HDAC8 expressions by western blotting
Anti-HDAC6 (cat. no. 7558, Cell Signaling Technologies, USA) and anti-HDAC8 (cat. no. 685502, BioLegend Inc., USA) primary antibodies were prepared at 1:1000 dilutions while anti-β actin antibody (4970, Cell Signaling Technologies, USA) was prepared at 1:2000 dilution in tris buffered saline-Tween 20 solution (TBS-T) with 3% Blocker™ BSA (cat. no. 37520, Thermo Fisher Scientific, USA). 30 μg denaturated protein samples were separated on 10% bis-acrylamide gels by running gels at 80 V for 3.5 hours at room temperature. Wet protein transfer was performed at 300 mA for 1 h to 0.45 μm PVDF membranes (cat. no. IPVH00010, Merck Millipore, USA). Membranes were blocked by incubating with 5% skimmed milk powder in TBS-T at room temperature for one hour; respective proteins were labelled by incubating membranes with primary antibody solutions overnight at 4 °C on shaker platform, followed by probing with either HRP-conjugated anti-rabbit or anti-mouse IgG secondary antibodies (7074 and 7076, respectively, both from Cell Signaling Technologies, USA) at room temperature for 1 h. Images were acquired on Vilber Fusion Pulse imaging system (Vilber, Germany).
Fluorescence imaging
Confocal microscopy was used to determine Pc derivatives' cellular localizations and their impact on autophagy. Cells were fixed with 2% glutaraldehyde solution at 4 °C for 15 minutes. Residual aldehydes were quenched with 0.1 M glycine solution by incubating slides for one hour at room temperature, followed by permeabilization with 0.25% Triton X-100 in PBS at room temperature for an hour. Cells were blocked with TBS containing 1% Blocker™ BSA for an hour at room temperature prior to antibody incubation. Slides were washed trice between each step. For labelling mitochondria, cells were stained with an Alexa Fluor 488 conjugated anti-mitochondria antibody (Clone 113-1, Merck Millipore, USA) at 1:500 dilution by incubating overnight. For imaging autophagosome formation, slides were incubated overnight with LC3A/B antibody (Cell Signaling Technologies, polyclonali USA) at 1:200 dilution, followed by incubating with an Alexa Fluor® 488 conjugated donkey anti-rabbit secondary antibody (Abcam, polyclonal) at 1:100 dilution for an hour at room temperature. Antibody solutions were prepared in TBS containing 3% Blocker™ BSA. Slides were mounted with Dianova Immunoselect Antifading Mounting Medium with DAPI and visualized via Zeiss LSM 880 Airyscan inverted confocal microscope. Photos were taken by Zeiss Zen Black software (Black edition).
Evaluation of mitochondrial activity
Mitochondrial activity was measured by dihydrorhodamine 123 (DHR123), a non-fluorescent substance which gains its fluorescent properties under oxidizing conditions and shown to identify cells with disrupted mitochondrial respiratory chain function.41 In this study, 2.5 × 105 cells were incubated with 5 μM DHR123 at room temperature for 25 minutes and immediately analysed by Beckman Coulter CytoFLEX system. Analyses were performed on CytExpert software.Evaluation of protein expression by flow cytometry
For analysing protein expressions, 2.5 × 105 cells were washed twice with DPBS and suspended in 100 μL DPBS containing 0.1% sodium azide. Cells were labelled with Anti-CD44 PE, anti-CXCR4 PerCP/Cy5.5 and anti-CCR7 PE/Cy7 antibodies (all purchased from BioLegend Inc., Clones BJ18, 12G5 and G043H7 respectively) by incubating at room temperature for 20 minutes under dark. Each antibody was used as 5 μL per test, according to the manufacturer's instructions. Cells were washed twice, resuspended in PBS and analysed on CytoFLEX flow cytometry system. Data analysis was performed on CytExpert software.Statistical analyses
GraphPad Prism Software (Version 8) was used for statistical analysis. Two-way analysis of variance (ANOVA) followed by Tukey's multiple comparison test was used for comparing treatment efficiency between cell lines, while effects of the treatment within the cell lines compared to respective controls were compared by using two-way ANOVA followed by Sidak's multiple comparison test in annexin V/PI staining, western blotting and DHR123 assay. Two-way ANOVA followed by Bonferroni's multiple comparisons test was performed to investigate the significant differences in DNA content analysis. When comparing CD44, CXCR4 and CCR7 protein levels after treatment, comparisons between treatment and control groups were done with two-way ANOVA followed by Sidak's multiple comparison test. P values lower than 0.05 was considered as statistically significant.Results and discussion
Synthesis and characterization
The nucleophilic aromatic substitution reaction of HDACi (1) with 3-nitrophthalonitrile (2a) or 4-nitrophthalonitrile (2b) in dimethylformamide produced the intermediate phthalonitrile derivatives 3a and 3b, successively in high yields. The synthetic pathway and structures of 3a and 3b were given in Scheme 1. The structures of compound 3a and 3b were determined through by FT-IR, 1H and 13C NMR spectroscopies, ESI-MS and MALDI-TOF MS as given in Experimental section. | ||
| Scheme 1 Synthetic pathway of 3a and 3b. Reaction conditions: (i) K2CO3, DMF, rt, 48 h. | ||
The 1H NMR spectrum of both of the 3a and 3b showed the disappearance of the OH protons at around 8.6 ppm, which proved that the starting material 1 had been converted to phthalonitriles 3a and 3b. In addition, the IR spectra of the compounds 3a and 3b displayed absorptions at 2230 and 2232 cm−1 respectively corresponding to the nitrile stretching frequencies.
The cyclotetramerization reaction of the phthalonitrile derivatives to ZnPc-1 and ZnPc-2 was accomplished with anhydrous Zn(OAc)2 in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and n-hexanol. Similarly, employing InCl3 gave the corresponding Pc derivatives, InPc-1 and InPc-2. The synthetic pathway and structures of the Pc compounds (ZnPc-1 and ZnPc-2, InPc-1 and InPc-2) were given in Scheme 2. The newly synthesized Pc derivatives were characterized by FT-IR, 1H and 13C NMR, UV-vis, MALDI-TOF mass spectrometry (Fig. S1–S28†) and elemental analysis. The IR spectra clearly indicated formation of the Pc derivatives with disappearing of nitrile stretching bands at around 2230 cm−1. In the FT-IR spectra, aromatic C–H stretching vibrations belonging to substituted group were observed around 3050 cm−1 while aliphatic C–H stretching vibrations were observed between 2900–2850 cm−1 for the Pcs. The 1H NMR spectra of the Pc compounds displayed the aromatic protons resonances belonging to the HDACi group overlapping with Pc ring protons, while the chemical shifts of aliphatic protons of the compounds at around 5.8 and 2.2 ppm were observed (Fig. S9, S14, S19 and S24†). A common feature of the 1H NMR spectra of all of the Pc derivatives was the broad absorptions when compared with that of the phthalonitrile derivatives, probably caused by the aggregation of the Pc which is frequently encountered at the concentrations used for NMR measurements. The signals of aromatic protons are considerably broadened and multipled due to the formation of an isomeric mixture (Fig. S10, S15, S20 and S25†). The MALDI-TOF spectra of the Pc compounds showed molecular ion peaks supported the proposed molecular formula for these compounds (Fig. S11, S16, S21 and S26†).
 | ||
| Scheme 2 Synthetic pathway of ZnPc1, InPc-1, ZnPc-2 and InPc-2 and 3b. Reaction conditions: (i) DBU, n-hexanol, the corresponding metal salt, 18 h. | ||
Phthalocyanines are known for their unique two strong absorption Q band and Soret (B) bands, including both ultraviolet and visible regions. The electronic absorption spectra of the Pc derivatives (ZnPc-1, ZnPc-2, InPc-1 and InPc-2) were studied in DMSO. In the UV-vis spectrum, Q-bands were observed at 706, 672, 722 and 690 nm corresponding to studied Pcs (ZnPc-1, ZnPc-2, InPc-1 and InPc-2) respectively (Fig. S13, S18, S23 and S28†). On the other hand, Soret bands for compounds were observed between 350–360 nm. The InPc derivatives (InPc-1 and InPc-2) showed a shift of the Q bands to higher wavelengths in comparison with their Zn derivatives (ZnPc-1, ZnPc-2) because of central metal ion effect. When compared to substitution position of the Pcs, non-peripheral derivatives (ZnPc-1 and InPc-1) displayed red shifting of Q bands as expected. All spectral data of the compounds (3a, 3b, ZnPc-1, ZnPc-2, InPc-1 and InPc-2) were given in ESI.†
Photochemical properties
The singlet oxygen quantum yield (ΦΔ) is described as the number of molecules of singlet oxygen generated per number of photons absorbed by the sensitizer. A number of different techniques for the determination of ΦΔ measurement of efficiency have been developed over the five decades.42–44 In our work, we had applied a direct method in DMF. Singlet-oxygen phosphorescence spectra for all compounds excited with a xenon-arc source at their respective absorption maxima were recorded by a near-IR-sensitive detector. Singlet oxygen phosphorescence spectra of Pc derivatives in DMF at equal absorbances (0.23) were obtained to directly determine ΦΔ. Table S1† shows ΦΔ values of all the Pc complexes in DMF (Fig. S29–S32†). ΦΔ for ZnPc-1 and InPc-1 derivatives were obtained at same value (0.57). Although peripherally 3-HPT bearing Pc derivative (ZnPc-2) shown the highest value (0.73), InPc-2 gave the lowest value (0.50) (Table S1†).Dark and light cytotoxicity
None of the compounds tested showed toxicity after incubation for 24 hours under dark conditions, even at high concentrations (p > 0.05) (Fig. S33–S35†). When considering ZnPc-HDACis; IC50 values of ZnPc-1 on HUVECs, MCF-7 and MDA-MB-231 were reported as 72.1, 65 and 63 μM; whereas calculated as 24.9, 16.8 and 24 μM, respectively for ZnPc-2 (Fig. S34†). IC50 values for InPc-HDACis were calculated as 84.3, 67.4 and 56.5 μM for InPc-1; 80.7, 56.4 and 48.4 μM for InPc-2 on HUVECs, MCF-7 and MDA-MB-231 cells, respectively (Fig. S35†). IC50 doses for the positive control in this study, tetrakis-(4,7,10-trioxaundecan-1-sulfanyl)phthalocyaninato zinc (ZnPc-C) was calculated as 89.5, 63.5 and 82.4 μM for HUVECs, MCF-7 cells and MDA-MB-231 cells, respectively (Fig. S33†).Evaluation of apoptosis and viability
Apoptosis is a tightly regulated cell death mechanism that is essential for the maintenance of normal cellular homeostasis by either eliminating undesired or potentially harmful cells. Dysregulations in apoptotic pathways are related to various pathological conditions including cancer.45 Apoptosis is considered as the main cell death pathway in both PDT and HDAC inhibition.32 Here, both ZnPc-HDACi derivatives decreased viability significantly for all cell lines compared to control groups while viability rates of all cell lines were significantly higher for ZnPc-1 treated cells compared to ZnPc-2 treatment (p < 0.0001) (Fig. 1b). ZnPc-1 did not induce apoptosis significantly (p > 0.05) while ZnPc-2 increased apoptotic cell rate in all cells lines (p < 0.0001). Comparisons between cell lines indicated MCF-7 cells had highest late apoptotic cell population (p < 0.0001) as well as HUVECs had significantly lower apoptosis rates compared to both MCF-7 and MDA-MB-231 cells (Fig. 1d). ZnPc-1 treatment did not alter late apoptotic cell ratios between cell lines (p > 0.05). However, in contrast to ZnPc-2 which did not induce early apoptosis, ZnPc-1 significantly increased early apoptosis in all cell lines, most significantly in MCF-7 cells which had higher early apoptotic cell rates compared to MDA-MB-231 cells (p < 0.05) (Fig. 1c). Comparisons between treatment and control groups revealed ZnPc-1 treatment did not alter necrotic cell population in all cell lines (p < 0.05), however, ZnPc-2 induced necrosis significantly on HUVECs and MCF-7 cells but not MDA-MB-231 cells (p < 0.0001 and p < 0.01, respectively) (Fig. 1e). In conclusion, while both ZnPc-HDACi derivatives decreased viability compared to the control groups, ZnPc-1 was reported to be less effective in terms of inducing apoptosis.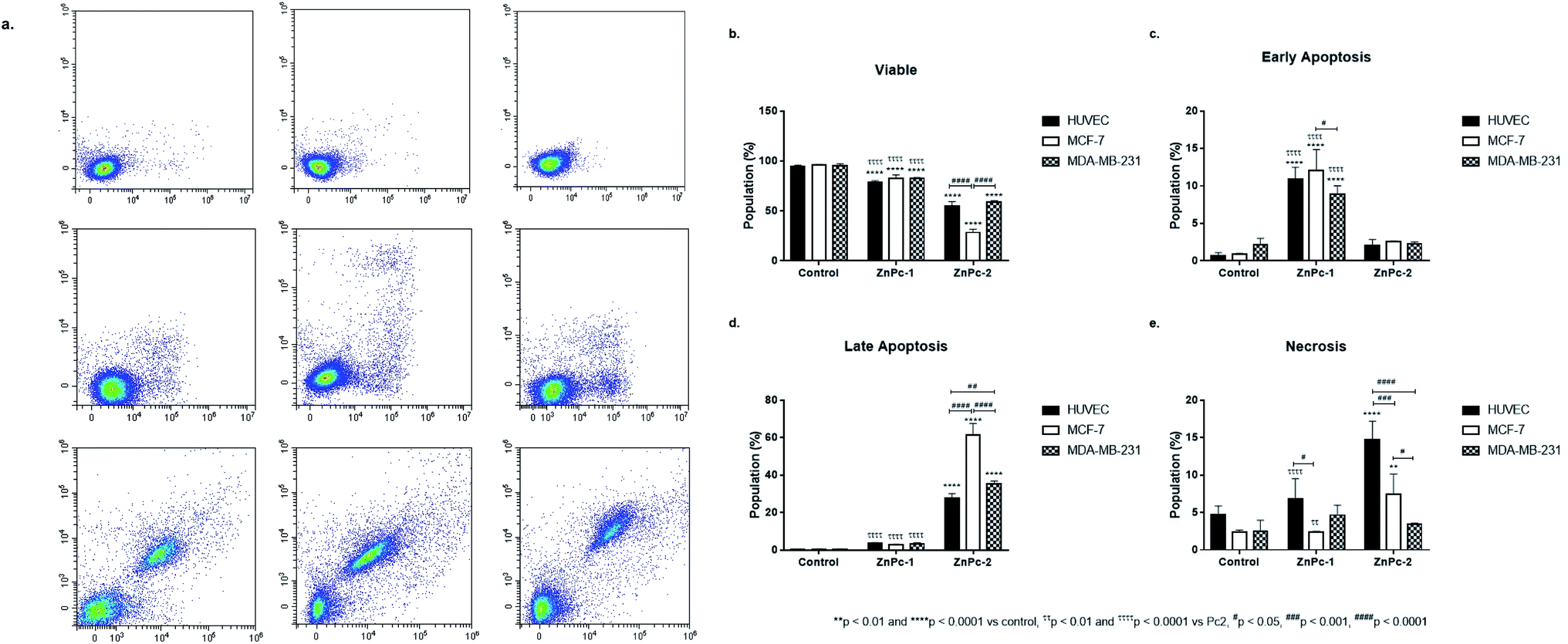 | ||
| Fig. 1 Both ZnPc-1 and ZnPc-2 treatment decreases all cell lines' viability while promoting apoptosis. (a) Representative flow cytometry histograms. Comparisons between two different ZnPc derivatives in terms of (b) viability, (c) early apoptosis, (d) late apoptosis and (e) necrosis revealed that ZnPc-1 treatment increased early apoptosis rates more effectively in all cell lines compared to ZnPc-2; on the other hand, late apoptotic cell populations in all cell lines were significantly higher compared to ZnPc-1 treated counterparts, suggesting non-peripheral and peripheral substitution of HDACi residues may induce different mechanisms of action. * denotes difference between control and treatment groups. # denotes difference between cell lines within the same treatment groups. τ indicates difference between either peripherally or non-peripheral HDACi substituted phthalocyanines bearing zinc as core metal. | ||
When considering InPc-HDACis, both compounds decreased viability significantly for all cell lines compared to control groups (p < 0.0001) while InPc-1 treated MCF-7 cells had significantly higher viability percentage compared to their InPc-2 treated counterparts (p < 0.0001) (Fig. 2b). The decrease in viability rates were accompanied by an increase in both early (Fig. 2c) and late apoptosis ratios (Fig. 2d) after InPc-1 and InPc-2 treatments (p < 0.0001). While no differences were observed between InPc-1 and InPc-2 groups in terms of early apoptosis (Fig. 2c), InPc-1 treated HUVECs as well as MCF-7 cells had significantly lower late apoptosis rates compared to InPc-2 treated counterparts (p < 0.001 and p < 0.0001, respectively).
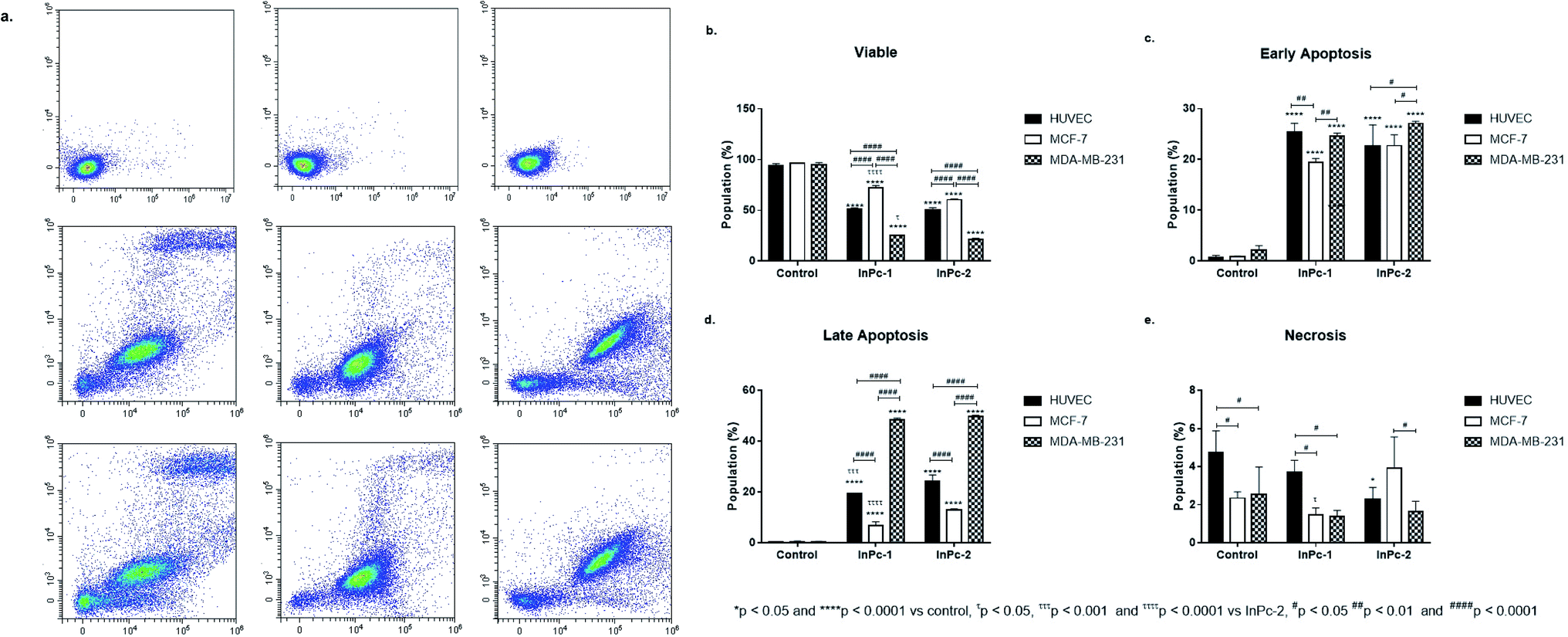 | ||
| Fig. 2 Both InPc-1 and InPc-2 treatments decreases all cell lines' viability while promoting apoptosis. (a) Representative flow cytometry histograms. Comparisons between two different InPc derivatives in terms of (b) viability, (c) early apoptosis, (d) late apoptosis and (e) necrosis. InPc-1 and InPc-2 induces both early and late apoptosis in all cell lines. While substation position of HDACi residues have no effect on early apoptosis rates, InPc-1 treatment led significantly higher viability accompanied with lower late apoptosis rates in MCF-7 cells compared to InPc-2 treatment. * denotes difference between control and treatment groups, # denotes difference between cell lines within the same treatment groups and τ indicates difference between either peripheral or non-peripheral HDACi substituted phthalocyanines bearing indium as core metal. | ||
DNA content analysis
DNA damage is a well-known to initiator of programmed cell death.46,47 PDT applications lead to DNA damage, either targeting DNA directly or promoting ROS formation and exerting indirect effects.48 Yet, the main cytotoxic product of PDT, 1O2 is thought to be not the main inducer of DNA damage due to its short lifespan and limited range unless it is generated close to DNA strands.48 On the other hand, HDAC inhibition is shown to repress DNA repair proteins and increase cellular ROS, which result in DNA damage.49 Former studies indicated that both Pcs and HDACis are shown to induce cell cycle arrest.50–59 When considering the distinct natures of various PS and HDACis, their differential effects on cell cycle progression would be attributed to their unique mechanism of actions, and the properties of the cells investigated. In our study, ZnPc-HDACis did not induce cell cycle arrest on HUVECs while ZnPc-1 led to a small but significant decrease in G0/G1 phase (p < 0.05) (Fig. 3b–d). In terms of cancer cell lines, ZnPc-1 led to G2/M-phase arrest in MCF-7 and G0/G1-phase arrest MDA-MB-231 (p < 0.0001) cells, which is accompanied by significant decrease of S phase in both cell lines (p < 0.0001). ZnPc-2 induced G2/M-phase arrest in both cancer cell lines, while in contrary to ZnPc-1, it decreased G0/G1 phase in MDA-MB-231 cells. This finding reveals that direct nuclear damage caused by PDT may induce G2/M phase arrest as when considering the nuclear localization of ZnPc-2, and Ps targeting non-nuclear cellular compartments may tend to lead G1/G0 or S phase arrests. In compliance with this hypothesis, both InPc-1 and InPc-2 increased G0/G1 phase (p < 0.01) in MDA-MB-231 cells and decreased G2/M phase (p < 0.0001) without altering S phase (p > 0.05), indicating a G0/G1 phase arrest (Fig. 3e–g). Similarly, both InPc-1 and InPc-2 derivatives increased SubG1 (p < 0.01) and S phases (p < 0.0001) accompanied by the decrease of G2/M phase (p < 0.0001) in MCF-7 cells, suggesting an S-phase arrest. When considering healthy cells, InPc-HDACis increased SubG1 phase (p < 0.01) while decreasing G2/M phase significantly in HUVECs (p < 0.05 for InPc-1 and p < 0.01 for InPc-2, respectively) while no differences between G0/G1 and S phases compared to control was observed (p > 0.05). Altogether, these data suggest when considering breast cancer cells, both ZnPc-HDACis and InPc-HDACis do not lead to cell cycle arrest on healthy endothelial cells (though InPc-HDACis increase the hypodiploidic cell population); ZnPc-HDACis induce cell cycle arrest at G2/M phase and the position of the HDACi moieties may alter the efficacy of the treatment while InPc-HDACis tend to lead accumulation in G0/G1 or S phases according to the cell line, and the position of HDACi moieties do not have a major impact on cell cycle arrest.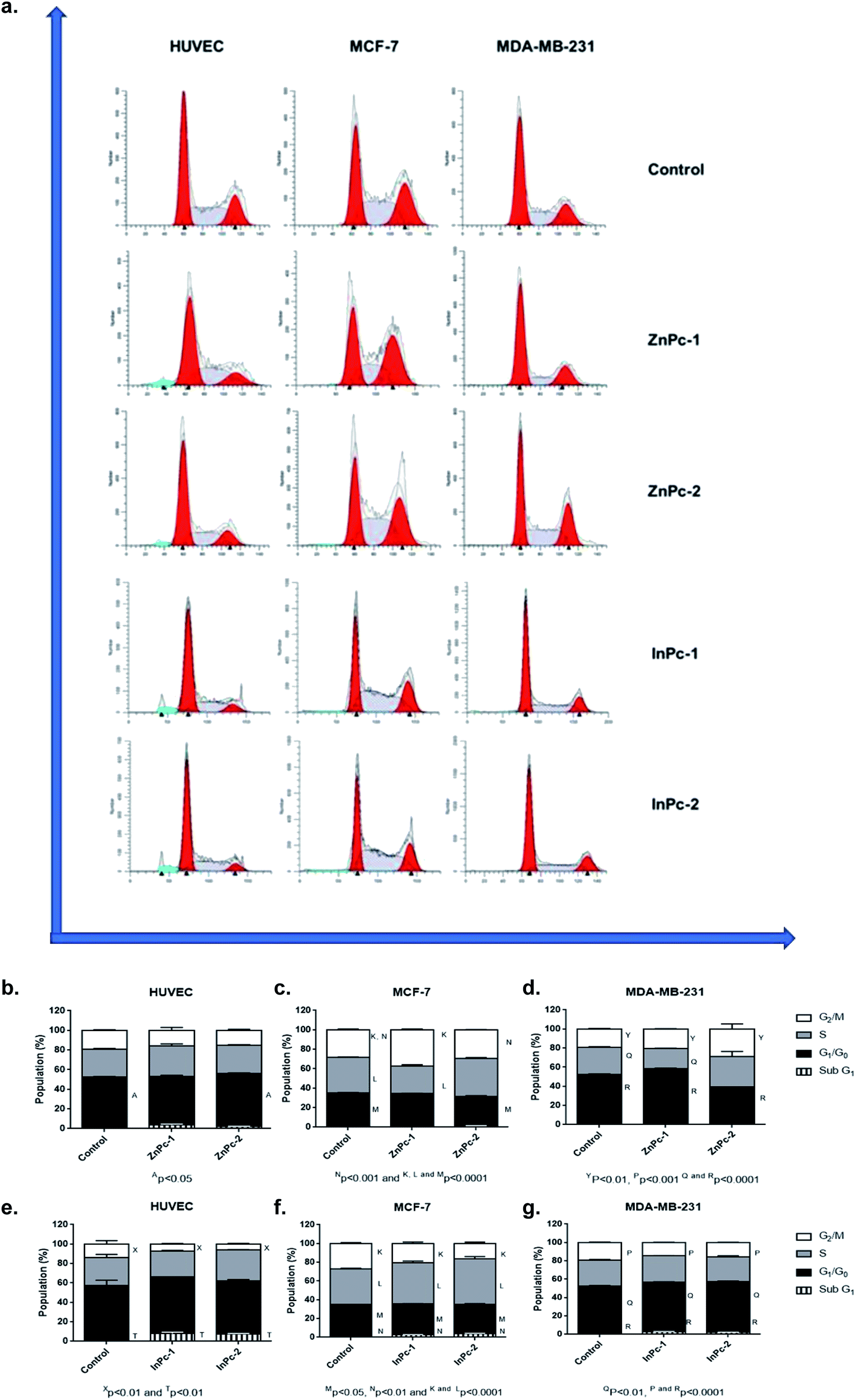 | ||
| Fig. 3 DNA content analysis upon treatment with ZnPc-HDACi and InPc-HDACi derivatives. (a) Representative histograms of DNA content. (b) Both ZnPc-1 and ZnPc-2 treatments did not lead to cell cycle arrest in HUVECs. (c) Both ZnPc-1 and ZnPc-2 increased G2/M phase in MCF-7 cells. (d) ZnPc-1 and ZnPc-2 induced G0/G1 and G2/M phase arrests in MDA-MB-231 cells, respectively. (e) Both InPc-1 and InPc-2 treatments decreased G2/M phase in HUVECs while increasing SubG1 phases. (f) Both InPc-1 and InPc-2 treatments decreased G2/M phase while inducing S-phase arrest in MCF-7 cells. (g) Both InPc-1 and InPc-2 treatments decreased G2/M phase while inducing G0/G1 phase arrest in MDA-MB-231 cells. | ||
Evaluation of HDAC6 and HDAC8 protein levels
Both ZnPc-1 (p < 0.001, p < 0.0001 and p < 0.01 for HUVEC, MCF-7 and MDA-MB-231 cell lines, respectively) and ZnPc-2 (p < 0.001 for all cell lines) treatments significantly decreased HDAC6 levels; both compounds also decreased HDAC8 levels on MDA-MB-231 cells (p < 0.05), but not on MCF-7 cells or HUVECs (Fig. 4b and c).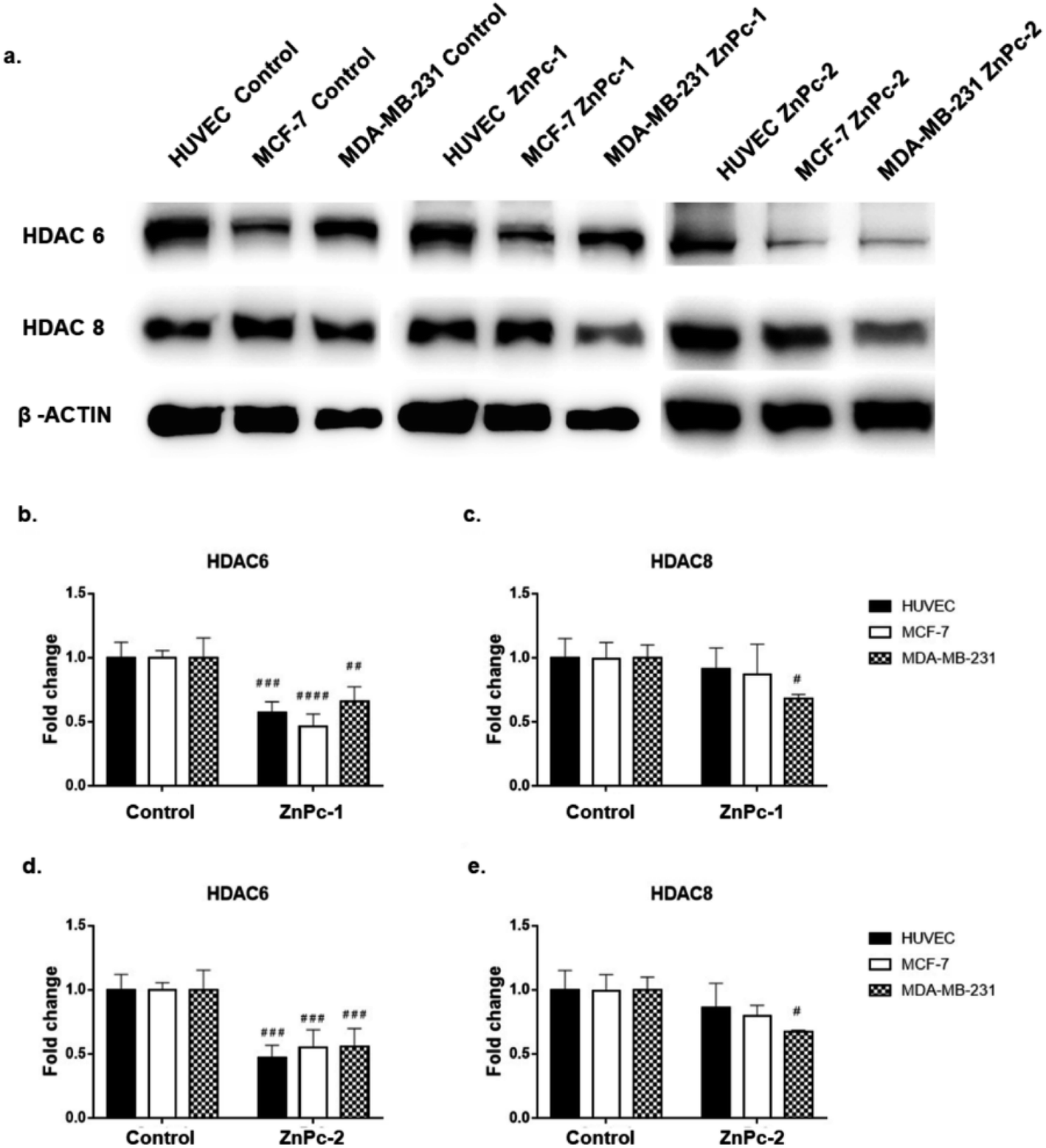 | ||
| Fig. 4 Both ZnPc-1 and ZnPc-2 successfully decrease HDAC6 levels in all cell lines while HDAC8 levels were only decreased in MDA-MB-231 cells. (a) Representative blot image of ZnPc-1 and ZnPc-2 treated HUVECs, MCF-7 and MDA-MB-231 cells. (b) Quantification of HDAC6 upon ZnPc-1 treatment. (c) Quantification of HDAC8 upon ZnPc-1 treatment. (d) Quantification of HDAC6 upon ZnPc-2 treatment. (e) Quantification of HDAC8 upon ZnPc-2 treatment. Significance level was considered as p < 0.05. * denotes significance between cell lines upon treatment whereas # denotes significance between treatment and control groups. | ||
Similar to ZnPc-HDACis, both InPc-1 (p < 0.001 for all cell lines) and InPc-2 (p < 0.05, p < 0.001 and p < 0.01 for HUVECs, MCF-7 and MDA-MB-231 cells, respectively) significantly decreased HDAC6 levels on all cell lines. InPc-1 did not alter HDAC8 protein levels (p > 0.05) in contrast to InPc-2 which significantly decreased HDAC8 levels in both HUVECs (p < 0.05) and MDA-MB-231 cells (p < 0.01). When considering 3-HPT has an IC50(nM) = 306 ± 69 for HDAC6 and IC50(nM) = 3105 ± 1649 for HDAC8,36 our results indicating successful HDAC6 inhibition in addition to a relatively weak effect on HDAC8 is compatible with the literature (Fig. 5).
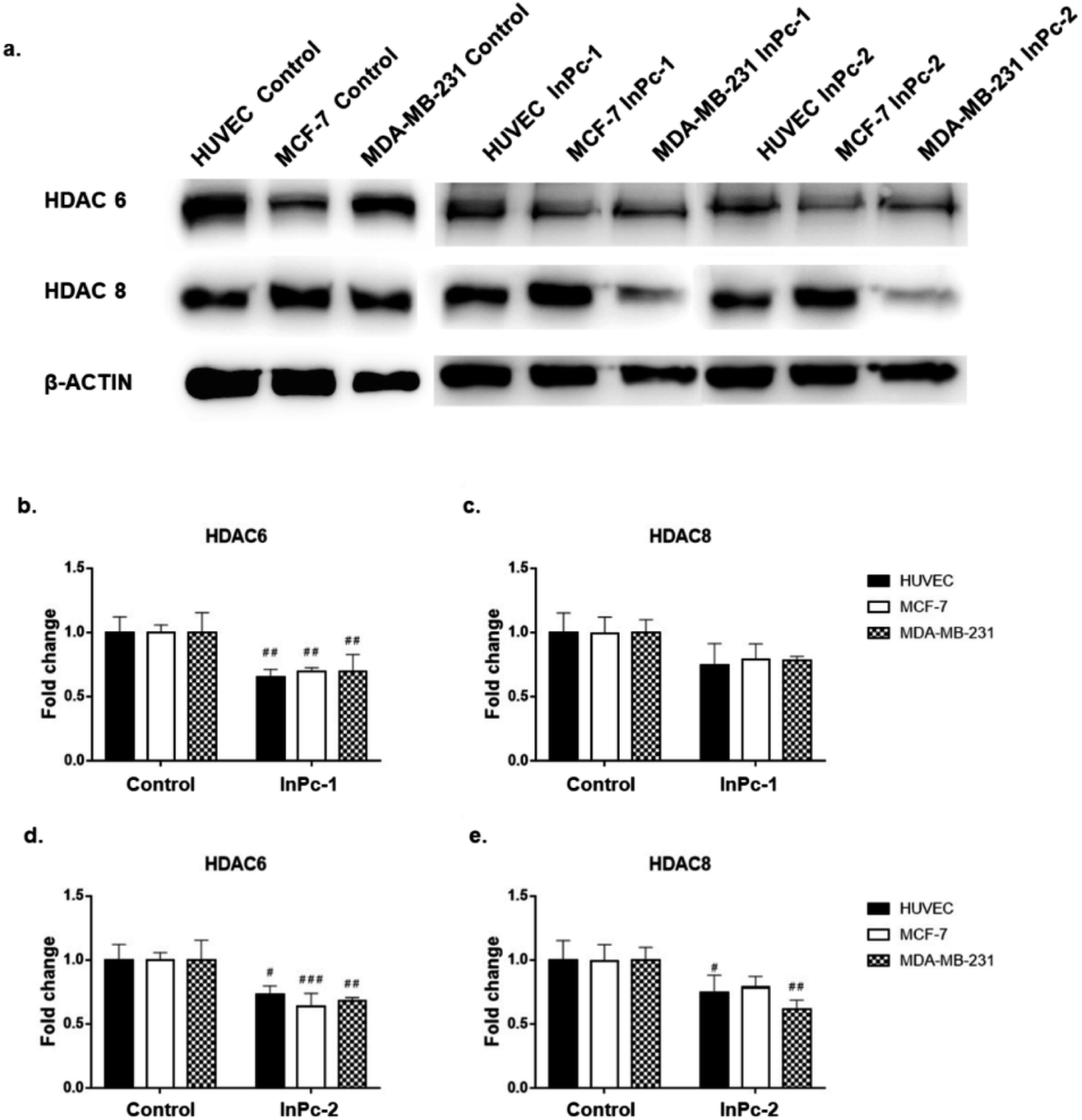 | ||
| Fig. 5 Both InPc-1 and InPc-2 successfully decreased HDAC6 levels in all cell lines while HDAC8 levels were only decreased in HUVECs and MDA-MB-231 cells treated with InPc-2. (a) Representative blot images of InPc-1 and InPc-2 treated HUVECs, MCF-7 and MDA-MB-231 cells. (b) Quantification of HDAC6 upon InPc-1 treatment. (c) Quantification of HDAC8 upon InPc-1 treatment. (d) Quantification of HDAC6 upon InPc-2 treatment. (e) Quantification of HDAC8 upon InPc-2 treatment. | ||
In summary, both ZnPc-HDACis and InPc-HDACis decreased HDAC6 levels in cell lines tested, confirming the 3-HPT moieties' success on HDAC downregulation. Along with HDAC6, ZnPc-HDACis were only shown to downregulate HDAC8 in MDA-MB-231 cells. The comparisons between InPc-HDACis revealed that InPc-2 decreased HDAC8 levels in HUVECs and MDA-MB-231 cells, yet InPc-1 remained ineffective. When considering non-peripheral Pc-HDACis; low penetration of ZnPc-1 due to steric effect was further confirmed with the higher efficacy of InPc-1 on downregulating HDAC8 levels in MDA-MB-231 cells. ZnPc-2 was reported to be more effective in terms of downregulating HDAC6 levels in HUVECs, and MDA-MB-231 cells compared to InPc-2 which may suggest a synergistic effect between PDT and HDAC inhibition when peripheral Pc-HDACis were compared. In contrast to assays evaluating the combinatorial anti-cancer activity of compounds, HDAC6 and HDAC8 protein levels were not expected to be influenced by PDT as a previous study, combining a PDT agent and a HDACi has indicated that HDAC activities are PDT-independent.33 The HDACi moiety, 3-HPT, should interact with its respective targets to exert its' activity. Thus, membrane or mitochondrial localization of Pc-HDACis may decrease their ability to inhibit HDACs. However, our HDAC evaluation strategy is limited to the semi-quantitative analysis of the protein levels and does not measure proteins' enzymatic activity. All compounds may still have disruptive effects on HDAC8 and HDAC6 proteins' deacetylating properties while preserving the proteins' integrity, which is not evaluated as a part of this study.
Visualization of cellular localization and autophagy
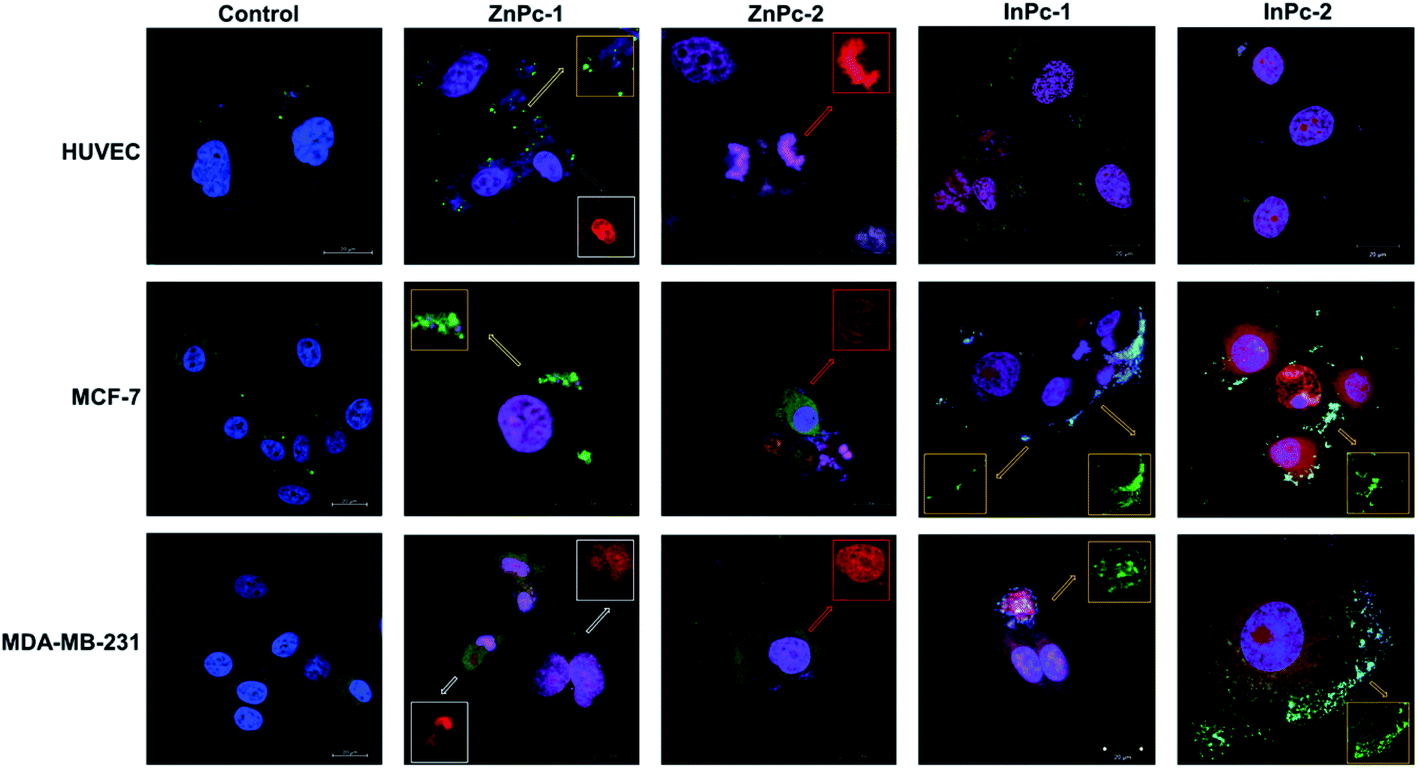
Cellular localization is an important factor that determines the anti-cancer effect of this dual system; as along with 1O2 generation, the HDACi moieties should interact with their respective targets to successfully downregulate respective proteins. For this study, the target proteins (HDAC6 and HDAC8) can be found both in nucleus and cytoplasm.60,61 Confocal imaging revealed that ZnPc-1 is either localizes to nucleoli or distributed homogeneous (which indicates cell membrane and/or cytoplasmic localization) whereas ZnPc-2 is localized into nucleus (Fig. 6). This homogenous staining of ZnPc-1 may explain significantly lower apoptosis rates upon treatment in contrast to the peripheral ZnPc-2. As suggested by the mitochondrial staining, ZnPc-1 disrupted mitochondrial network in HUVECs and MCF-7 cells whereas ZnPc-2 treatment impaired mitochondrial structure in HUVECs and MDA-MB-231 cells but not MCF-7 cells. On the contrary, in addition to homogenous staining (shown in white squares), both InPc-HDACis are localized to the mitochondrial network (shown in pale blue squares); which is not observed in ZnPc-1 (membrane/cytoplasmic) and ZnPc-2 (nuclear). Neither InPc-1 nor InPc-2 altered mitochondrial network in HUVECs while decreasing the signal in MDA-MB-231 cells. InPc-1 treated MCF-7 cells' mitochondrial network structure resembled control cells while InPc-2 treatment reduced the signal, similar to MDA-MB-231 cells. On the other hand, InPc-1 and InPc-2 were the only compounds showing mitochondrial localization, due to the absence of a mitochondria-targeting sequence of the compounds, this overlap suggests that the InPc-HDACis can target mitochondria themselves. In3+ is known to induce mitochondrial permeability transition by disrupting proton channels located in the inner mitochondrial membrane, leading to mitochondrial oxidative stress, cytochrome c release and eventually apoptosis.62,63 Yet, their induction of speckle formation (thus disrupting the mitochondrial network) was less prominent compared to ZnPc-HDACis.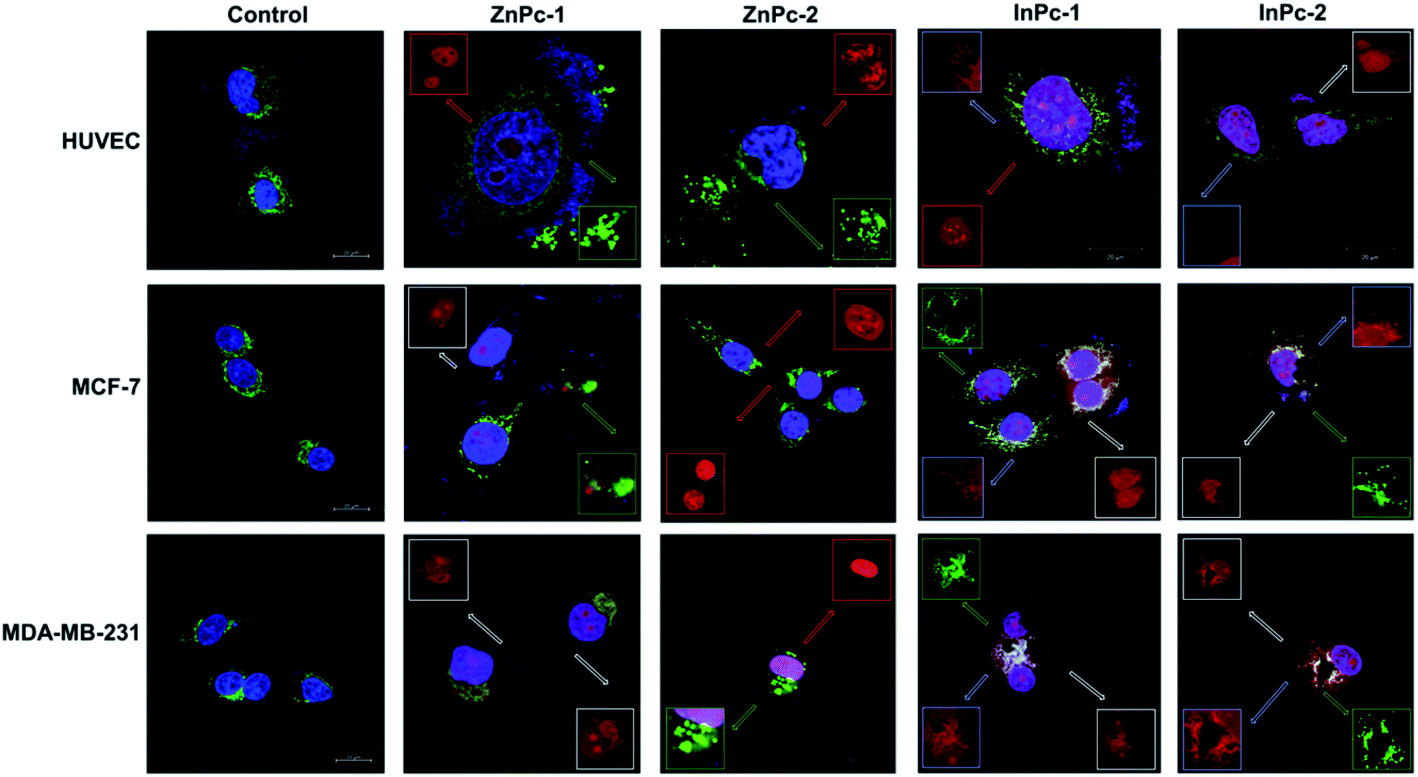 | ||
| Fig. 6 Micrographs of untreated and ZnPc-1, ZnPc-2, InPc-1 and InPc-2 treated cells indicating cellular localization. Cells were stained with anti-mitochondria antibody and counterstained with DAPI. Red squares indicate deposits in nuclei and nucleoli whereas white squares indicate homogenous staining that may indicate predominant membrane/cytoplasmic localization. Disrupted mitochondria are shown in green squares. Scale bars indicate 20 μm. All images were taken at 63X zoom with immersion oil. | ||
Role of autophagy upon PDT is controversial as it may either lead to treatment resistance by promoting recycling damaged organelles, or initiate death signals.64 Activation of apoptosis along with autophagy is reported to be crucial for an enhanced cell death response as MCF-7 cells were shown to develop PDT resistance when the autophagic response is inhibited.65,66 In conclusion, it is still unclear how exactly autophagy affects the outcome of PDT.67 Induction of autophagy is reported to be localization independent in PDT, and apoptosis was also observed in cells already undergoing autophagy.67 We investigated the link between localization of Pc-HDACis and the mode of cell death and revealed that both the core metal of the Pc derivative as well as the position of the HDACi moiety can influence the compounds' localization, and in turn, the effectivity of the treatment. In our study, untreated cells exhibited homogenous LC3A/B staining pattern throughout the cytoplasm. ZnPc-1 treatment induced autophagosome formation that is characterized with green puncta, indicated with yellow squares, and observed in HUVECs and MCF-7 cells but not MDA-MB-231 cells. In addition, ZnPc-1 did not localize into autophagosomes and accumulated on either cell membrane or in nucleus which is also indicated above; suggesting ZnPc-1 treatment may induce autophagosome formation but do not directly interact with autophagic lysosomes (Fig. 7). On the other hand, ZnPc-2 treatment did not lead to autophagosome formation yet localized mainly in the nucleus as shown in Fig. 7. InPc-1 and InPc-2 treatment induced autophagy in both cancer cell lines, but not in HUVECs which is indicated in Fig. 7. Similar to ZnPc-1, neither InPc-1 nor InPc-2 did not localize to the autophagosomes, but the cyan colour observed in merged photos suggest that autophagosomes contain nuclear material, which is not present upon ZnPc-1 or ZnPc-2 treatment. Induction of autophagy upon DNA damage is suggested by various studies.68,69 However, the origin of the nuclear material (genomic DNA, or mitochondrial DNA) inside the autophagosomes remains unknown.
 | ||
| Fig. 7 Micrographs of untreated and ZnPc-1, ZnPc-2, InPc-1 and InPc-2 treated cells indicating autophagosome formation. Cells were stained with rabbit polyclonal anti-LC3A/B antibody followed by donkey anti-rabbit Alexa Fluor 488 secondary antibody and counterstained with DAPI. Puncta formations are indicated in yellow squares while red squares and white squares indicate nuclear and cytoplasmic/membrane localization, respectively. Scale bars indicate 20 μm. All images were taken at 63X zoom with immersion oil. | ||
Evaluation of mitochondrial activity
As 1O2 formation depends on the availability of oxygen, mitochondria are important organelles in PDT due to their high oxygen levels.70 A previous study even suggested that low levels of singlet oxygen produced in the mitochondria are two times more toxic than membrane targeting, and thrice more effective than nuclear targeting.71 We evaluated mitROS accumulation by DHR123 staining. Both ZnPc-1 (p < 0.001 for HUVECs, p < 0.0001 for MCF-7 and MDA-MB-231 cells) and ZnPc-2 (p < 0.0001 for all cell lines) treatments significantly increased DHR-123 MFI in all cell lines (Fig. 8b). Comparisons between ZnPc-HDACis indicated ZnPc-2 treatment significantly increased DHR-123 MFI in all cell lines compared to ZnPc-1 treatment (p < 0.0001). A similar trend for InPc-1 (p < 0.0001 for all cell lines) and InPc-2 (p < 0.001 for HUVECs, p < 0.0001 for MCF-7 and MDA-MB-231 cells) was observed when treatment and control groups were compared (Fig. 8c). Contrary to ZnPc-HDACis, higher DHR-123 MFI values were observed for InPc-1 treated HUVECs as well as MCF-7 cells (p < 0.0001 and p < 0.05, respectively) in comparison with peripheral InPc-2. Comparisons between non-peripheral Pcs indicated InPc-1 increased DHR-123 MFI in all cell lines compared to its zinc counterpart ZnPc-1 (p < 0.0001 for all cell lines) (Fig. 8d). A similar phenomenon was observed when MCF-7 and MDA-MB-231 cells were compared after ZnPc-2 and InPc-2 treatments as peripheral substituted InPc derivative led to significantly increased MFI values in MCF-7 and MDA-MB-231 cells (p < 0.0001 and p < 0.001, respectively), yet no difference between HUVECs was observed (p > 0.05.) (Fig. 8e).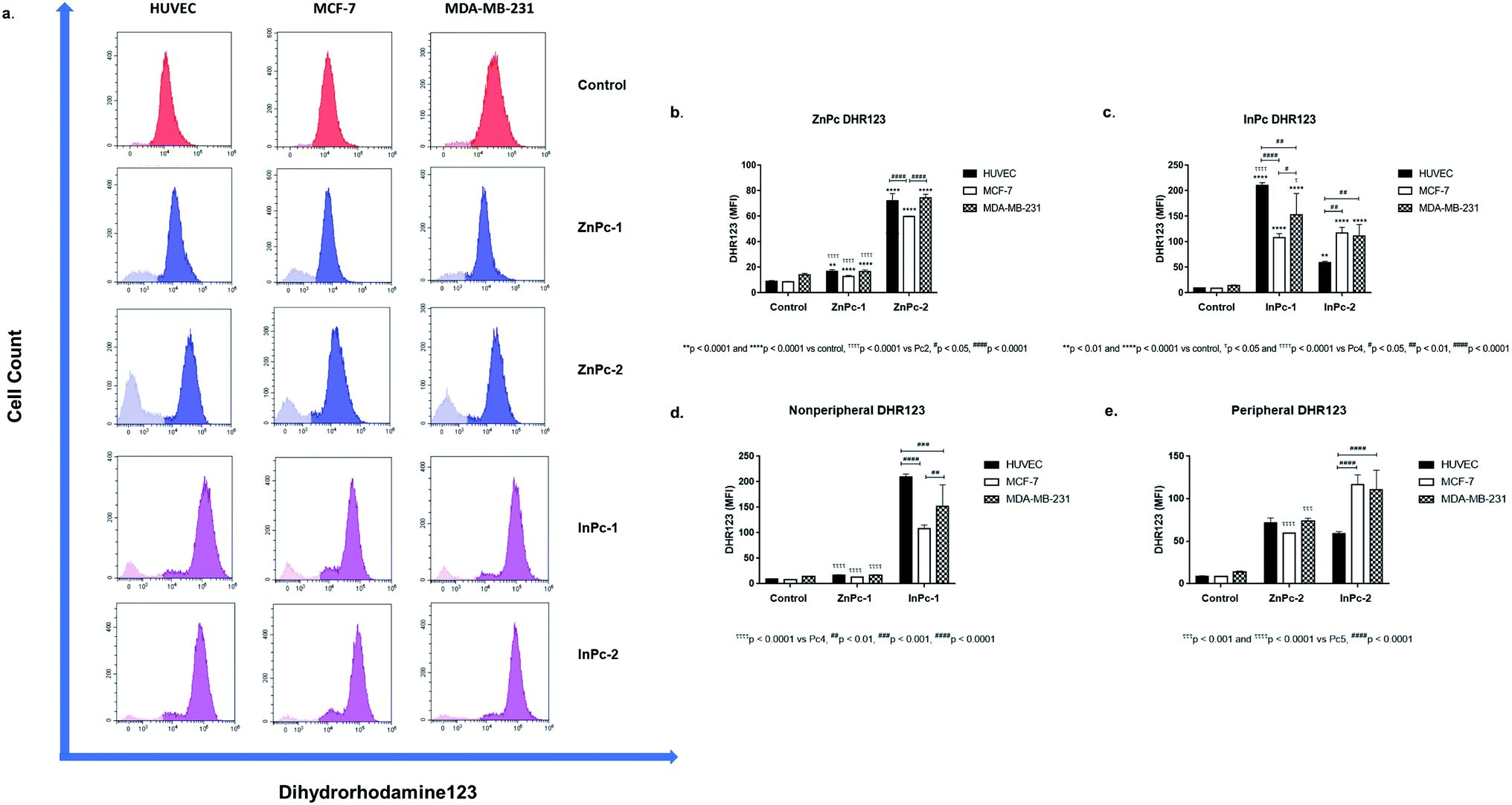 | ||
| Fig. 8 Both Zn and InPc-HDACi derivatives increased dihydrorhodamine-123 mean fluorescence intensity in all cell lines. (a) Representative flow cytometry histograms. (b) Bar graphics indicating comparisons between cell lines and DHR-123 MFI values for ZnPc-1 and ZnPc-2 treatments. (c) Bar graphics indicating comparisons between cell lines and DHR-123 MFI values for InPc-1 and InPc-2 treatments. (d) Comparisons between ZnPc-1 and InPc-1 treatments in terms of mitROS accumulation for evaluating Zn or In as core metals as core metals. (e) Comparisons between ZnPc-2 and InPc-2 treatments in terms of mitROS accumulation for evaluating Zn or In as core metals. | ||
In summary, InPc-HDACis which target mitochondria along with homogenous localization resulted in higher mitROS accumulation compared to ZnPc-HDACis (yet, ZnPc-2 was as effective as InPc-2 on HUVECs in terms of mitROS induction). Here, the mechanism behind the ZnPc-HDAis′ disruptive effect on the mitochondrial network without targeting the mitochondria (observed in the confocal microscopy as green speckles), and how its' integrity is preserved after mitochondria localizing InPc-HDACi treatments can be questioned. This result may be an outcome of the general cellular damage induced by 1O2 formation as even if the PS localizes to a different site of the cell (such as ZnPc-2 targeting nucleus), the resulting signalling cascades may alter mitochondrial network. Another possibility is the partial localization of ZnPc-HDACis to mitochondria-associated membranes (MAMs), the junctions where ER and mitochondria are physically connected.72 Several proteins involve in cellular homeostasis reside at MAMs including the ones take part in programmed cell death mechanisms and tumorigenesis. Yet, this hypothesis should be validated with nucleus-ER-mitochondria-compound quadruple staining along with the analysis of mitROS accumulation.
Evaluation of CD44, CXCR4 and CCR7 levels by flow cytometry
Leading cause of the cancer related deaths is the migration of tumour cells to form secondary lesions in distant organs. Recent studies indicate that chemokine receptor family belonging to G protein-coupled receptors play important roles in this process.73 While preliminary studies suggested CXCR4 and CCR7 expression facilitates migration of tumour cells towards the chemokine gradient, today it is known that both chemokine receptors also plays role in cellular growth, endothelial adhesion and extravagation.74 Upon binding to its ligand CXCL12, CXCR4 induces various signalling pathways that eventually induce gene expression, cellular motility, survival, and proliferation.75 CXCR4 has been confirmed to form homo- or hetero-dimers76 which activates JAK/STAT signalling pathway that promotes chemotactic responses.75 Disruption of CXCL12/CXCR4 interaction significantly impairs breast cancer cells' migration towards regional lymph nodes.77 Along with CXCR4, CCR7 is also shown to be highly expressed in certain malignancies including breast cancer.77 CD44, a large protein family that regulates cell-ECM interactions can contribute to cancer progression including cell growth, angiogenesis, cell migration and invasion,78 in addition, it also modulates CXCR4-CXCL12 signalling.79In our study, ZnPc-1 did not alter CXCR4 protein levels in cell lines (p > 0.05) while decreasing CD44 and CCR7 protein levels in HUVECs and MDA-MB-231 cells (p < 0.001 and p < 0.0001, respectively) (Fig. 9a). ZnPc-2 led to a significant decrease in CD44 levels in all cell lines (p < 0.0001) while increasing CXCR4 levels (p < 0.0001 for HUVECs and MCF-7 cells, p < 0.01 MDA-MB-231) (Fig. 9b). In addition, ZnPc-2 treatment did not alter CCR7 protein levels in MCF-7 cells while decreasing it both in HUVECs and MDA-MB-231 cells (p < 0.0001). On the other hand, CXCR4 protein levels were increased upon ZnPc-2 treatment in MCF-7 cells at a significantly higher rate than both HUVECs and MDA-MB-213 cells (p < 0.001, Fig. 9b). Representative flow cytometry histograms regarding ZnPc-1 and ZnPc-2 are given in Fig. S36 and S37,† respectively. InPc-1 significantly reduced CD44 protein levels in all cell lines (p < 0.0001); similarly, CCR7 levels were decreased significantly in MCF-7 and MDA-MB-231 cells (p < 0.0001 and p < 0.001, respectively) while HUVECs remain unaffected (p > 0.05). However, InPc-1 significantly increased CXCR4 levels in HUVECs (p < 0.0001) and MCF-7 cells (p < 0.01), but not in MDA-MB-231 cells (Fig. 9c). InPc-1 treatment increased CXCR4 levels in HUVECs significantly compared to MCF-7 cells (p < 0.05) while decreasing CXCR4 levels significantly in MDA-MB-231 cells, though this alteration was not significant. InPc-2 altered CD44 and CXCR4 protein levels a similar manner with InPc-1 in HUVECs (p < 0.001) while also decreasing CCR7 levels compared to control (p < 0.0001) (Fig. 9d). Also, in MCF-7 cells, InPc-2 acted in a similar way with its non-peripheral counterpart by decreasing CD44 (p < 0.0001) and increasing CXCR4 (p < 0.0001) levels; yet, the decrease in CCR7 levels was not significant (p > 0.05). In MDA-MB-231 cells, InPc-2 slightly but significantly decreased both CD44 and CCR7 protein levels (p < 0.0001 and p < 0.001, respectively), but did not alter CXCR4 levels. Representative flow cytometry histograms regarding InPc-1 and InPc-2 are given in Fig. S38 and S39,† respectively.
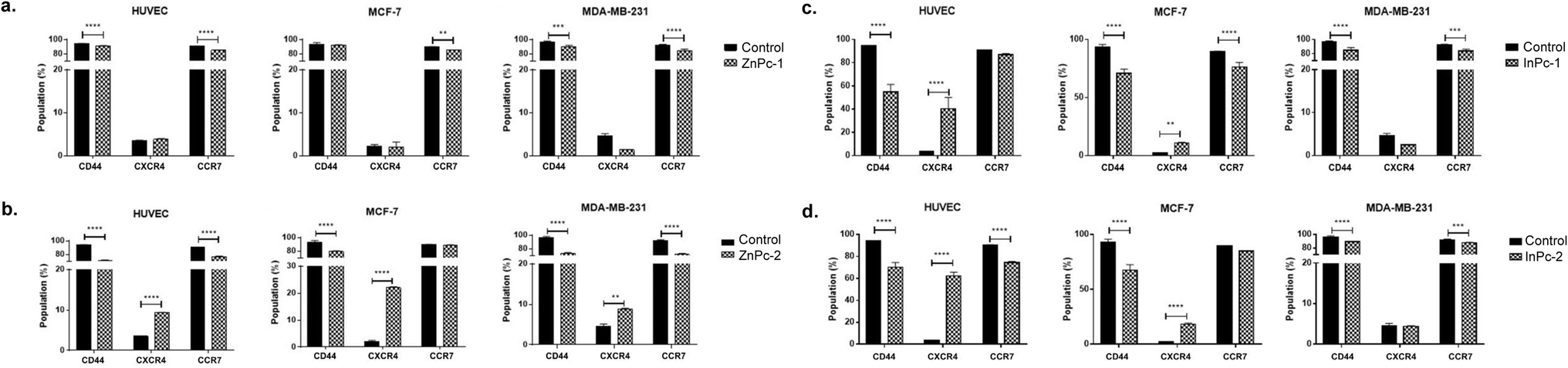 | ||
| Fig. 9 (a) ZnPc-1 decreased CCR7 protein levels in all cell lines and CD44 in HUVECs as well as MDA-MB-231 cells while increasing CXCR4 in HUVECs and MCF7 cells. (b) ZnPc-2 decreased CD44 protein levels in all cell lines while increasing CXCR4 in HUVECs and MCF7 cells. (c) InPc-1 decreased CD44 protein levels in all cell lines and CCR7 in cancer cell lines while increasing CXCR4 in HUVECs and MCF7 cells. (d) InPc-2 decreased CD44 protein levels in all cell lines and CCR7 HUVECs as well as MDA-MB-231 cells while increasing CXCR4 in HUVECs and MCF7 cells. * denotes significance between control and treatment groups. | ||
In our study, all Pc-HDACis are shown to downregulate either CD44 or CCR7 protein levels, or both, suggesting that PDT along with HDAC inhibition may have an inhibitory effect on migration. While the effect of PDT on tumour cells' CCR7 expression is not evaluated previously, the response of immune cells to PDT in cancer, especially dendritic cells is well studied,80,81 suggesting PDT promotes normal function of dendritic cells to possess anti-cancer functions compared to their untreated, tumour-educated, immunosuppressive counterparts, further underlining the importance of the presence of immune system while evaluating the efficacy of PDT in cancer treatment. However, treatment-induced alterations in CXCR4 protein levels were not in line between compounds as ZnPc-2 increased CXCR4 levels in all cell lines, especially in MCF-7 while ZnPc-1 remained ineffective in terms of CXCR4 expression. In a similar manner, InPc-1 and InPc-2 also induced CXCR4 protein expression in HUVECs and MCF-7 cells, but not MDA-MB-231 cells. Ierano et al. hypothesized that HDACi treatment would result in decreased migration capacity due to HDACs role in cellular motility, and revealed that treatment of different cell lines (including breast, colon, lung and renal cancer) with various HDACis lead to CXCR4 mRNA expression along with disrupting CXCL12-mediated signalling cascades to prevent metastasis.82 According to this study, increased CXCR4 levels upon Pc-HDACi treatment is an expected outcome and should be evaluated together with the other components of CXCL12 signalling pathway.
Conclusions
Due to its selectivity and specificity, PDT stands out as an attractive alternative for cancer treatment.3 Besides killing malignant cells by promoting ROS formation, PDT can also destroys the tumour-associated vasculature and modulate immune system to promote anti-tumoural activity.6,7 Today, the majority of approved PDT protocols target superficial skin and luminal organ lesions while interstitial and intra-operative approaches are under investigation for treating solid tumours including breast tumours.83Studies regarding combinatorial administration of HDACi along with PDT agents are restricted while none of them involve a Pc derivative for photodynamic action. Ye et al. synthesized four cyclometalated Ir(III)-HDACi complexes and investigated their anticancer efficacy on non-small cell lung cancer cell line A549 as well as its cisplatin resistant counterpart and reported that along with inhibiting HDAC activity, compounds induce mitochondrial damage and ROS production to lead apoptosis.32 Evaluated on ovarian cancer cell line A2780, suberoyl-bis-hydroxamic acid (SubH)-conjugated photosensitive platinum(IV) complex showed higher toxicity than its' biologically inactive ligand substituted counterpart or cisplatin, indicating HDACi moieties are able to induce distinct cell death pathways than PDT or cisplatin alone.33 Even in case of PDT resistance, HDACi pre-treatment was reported to increase the impact of PDT by promoting epigenetic alterations.31 Liu et al. combined chlorin-6 (Ce6) and vorinostat to target hypoxia signalling pathway and reported enhanced anti-cancer activity along with decreased vascular endothelial growth factor and hypoxia inducible factor 1 alpha (HIF-1α) levels.84 We have previously reported an 3-HPT substituted SiPc derivative and evaluated its' anti-cancer efficacy on MCF-7 and MDA-MB-231 cells as well as HUVECs.12 According to our results, SiPc-HDACi had shown the greatest efficiency on the TNBC cell line MDA-MB-231 as the lowest viable cell population among with highest apoptosis rate and mitROS accumulation was observed in this cell line. In our current study, we explored biological effects of novel 3-HPT substituted Zn and In Pc derivatives that combines chemotherapeutic action of the HDACi targeting HDAC6 and HDAC8 with PDT. Comparisons between ZnPc-HDACis revealed that peripheral substitution of HDACi moieties increase cellular penetration of the ZnPc-2 to promote a more prominent anti-cancer efficacy compared to non-peripheral ZnPc-1, indicating a possible steric effect for non-peripheral HDACi substitution. Such effect was not observed between InPc-1 and InPc-2, in addition, both compounds showed a distinct localization pattern that also targets the mitochondrial network to induce mitROS accumulation more efficiently compared to their Zn counterparts. ZnPc-HDACis and InPc-HDACis also led to cell cycle arrest in different phases of cell cycle, which may be attributed to their cellular localization. ZnPc-2 promoted G2/M phase arrest in both cancer cell lines while ZnPc-1, too, led to G2/M accumulation in MCF-7 cells but not in MDA-MB-231 cells which ended up with G0/G1 phase arrest. This finding suggests that direct nuclear damage caused by the PS may induce G2/M phase arrest while PS targeting non-nuclear cellular compartments may tend to lead G1/G0 or S phase arrests. In compliance with this phenomenon, localizing to cytoplasm and mitochondria, InPc-1 and InPc-2 treatments induced accumulation in S and G0/G1 phases in MCF-7 and MDA-MB-231 cells, respectively. In addition, DNA content upon treatment with both ZnPcs and InPcs did not lead to cell cycle arrest in HUVECs, which may indicate that healthy cells can cope with 1O2-induced DNA damage to a greater extent in comparison with cancer cells.
It is clear that more research is required to develop new PDT agents targeting tumour cells to increase treatment efficiency. For this purpose, differences between various Pc derivatives' cellular impacts including their target programmed cell death pathways and localizations should be well documented to determine right PS for the photodynamic action according to the respective cancer type. Different positions for introduced HDACis (peripheral vs. non-peripheral and/or asymmetrical vs. symmetrical) may be evaluated to achieve optimum cellular penetration and various HDACis may be considered according to the targeted cancers' transcription profiles. Moreover, due to the oxygen dependent nature of PDT, in vivo models that can accurately reflect the outcomes of the treatment in humans to be employed for a complete evaluation of PDT drug candidates is crucial. Such evaluations would also provide insight to the compounds' effects on different cell types of the tumour, immune system, tumour vasculature, many other components play role in cancer progression, and cannot be represented in vitro.
Author contributions
A. G. Gürek, G. Yanıkkaya Demirel and D. Atilla contributed to the conception and design of the research. B. Aru performed all biological experiments. A. Günay and D. Atilla, synthesized and characterized ZnPc-1, ZnPc-2, InPc-1, InPc-2, and ZnPc-C compounds. B. Aru prepared the figures and drafted the manuscript. A. G. Gürek, G. Yanıkkaya Demirel and D. Atilla edited and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank The Scientific and Technological Research Council of Turkey (TUBITAK) for financial support (project no. 118Z693).References
- J. Dang, H. He, D. Chen and L. Yin, Biomater. Sci., 2017, 5, 1500–1511 RSC.
- P.-C. Lo, M. S. Rodríguez-Morgade, R. K. Pandey, D. K. P. Ng, T. Torres and F. Dumoulin, Chem. Soc. Rev., 2020, 49, 1041–1056 RSC.
- X. Shi, C. Y. Zhang, J. Gao and Z. Wang, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2019, 11, e1560 Search PubMed.
- J. J. Schuitmaker, P. Baas, H. L. van Leengoed, F. W. van der Meulen, W. M. Star and N. van Zandwijk, J. Photochem. Photobiol., B, 1996, 34, 3–12 CrossRef CAS.
- A. Juzeniene and J. Moan, Photodiagn. Photodyn. Ther., 2007, 4, 3–11 CrossRef CAS PubMed.
- P. Agostinis, K. Berg, K. A. Cengel, T. H. Foster, A. W. Girotti, S. O. Gollnick, S. M. Hahn, M. R. Hamblin, A. Juzeniene, D. Kessel, M. Korbelik, J. Moan, P. Mroz, D. Nowis, J. Piette, B. C. Wilson and J. Golab, Ca-Cancer J. Clin., 2011, 61, 250–281 CrossRef PubMed.
- V. Mashayekhi, C. O. Hoog and S. Oliveira, J. Porphyrins Phthalocyanines, 2019, 23, 1229–1240 CrossRef CAS PubMed.
- S. B. Brown, E. A. Brown and I. Walker, Lancet Oncol., 2004, 5, 497–508 CrossRef CAS PubMed.
- M. A. MacCormack, Semin. Cutaneous Med. Surg., 2008, 27, 52–62 CrossRef CAS PubMed.
- L. V. Zhorina, B. C. Elena, A. V. Agronskaya and G. G. Maria, presented in part at Proc. SPIE 2100, June,1994.
- Y. Zhang and J. F. Lovell, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2017, 9(1), e1420 Search PubMed.
- B. Aru, A. Günay, E. Şenkuytu, G. Yanıkkaya Demirel, A. G. Gürek and D. Atilla, ACS Omega, 2020, 5, 25854–25867 CrossRef CAS PubMed.
- S. Alpugan, D. Topkaya, D. Atilla, V. Ahsen, J. H. Niazi and F. Dumoulin, J. Porphyrins Phthalocyanines, 2017, 21, 887–892 CrossRef CAS.
- S. Minucci and P. G. Pelicci, Nat. Rev. Cancer, 2006, 6, 38–51 CrossRef CAS PubMed.
- E. Seto and M. Yoshida, Cold Spring Harbor Perspect. Biol., 2014, 6, a018713 CrossRef PubMed.
- Y. Li and E. Seto, Cold Spring Harbor Perspect. Med., 2016, 6, a026831 CrossRef PubMed.
- T. Eckschlager, J. Plch, M. Stiborova and J. Hrabeta, Int. J. Mol. Sci., 2017, 18, 1414 CrossRef PubMed.
- J. A. Halsall and B. M. Turner, BioEssays, 2016, 38, 1102–1110 CrossRef CAS PubMed.
- S. E. Bates, N. Engl. J. Med., 2020, 383, 650–663 CrossRef CAS PubMed.
- B. E. Gryder, Q. H. Sodji and A. K. Oyelere, Future Med. Chem., 2012, 4, 505–524 CrossRef CAS PubMed.
- F. M. Robertson, K. Chu, K. M. Boley, Z. Ye, H. Liu, M. C. Wright, R. Moraes, X. Zhang, T. L. Green, S. H. Barsky, C. Heise and M. Cristofanilli, J. Exp. Ther. Oncol., 2013, 10, 219–233 CAS.
- J. Shen, C. Huang, L. Jiang, F. Gao, Z. Wang, Y. Zhang, J. Bai, H. Zhou and Q. Chen, Biochem. Pharmacol., 2007, 73, 1901–1909 CrossRef CAS PubMed.
- U. Lassen, L. R. Molife, M. Sorensen, S. A. Engelholm, L. Vidal, R. Sinha, R. T. Penson, P. Buhl-Jensen, E. Crowley, J. Tjornelund, P. Knoblauch and J. S. de Bono, Br. J. Cancer, 2010, 103, 12–17 CrossRef CAS PubMed.
- M. P. Coleman, M. Quaresma, F. Berrino, J. M. Lutz, R. De Angelis, R. Capocaccia, P. Baili, B. Rachet, G. Gatta, T. Hakulinen, A. Micheli, M. Sant, H. K. Weir, J. M. Elwood, H. Tsukuma, S. Koifman, G. A. E Silva, S. Francisci, M. Santaquilani, A. Verdecchia, H. H. Storm and J. L. Young, Lancet Oncol., 2008, 9, 730–756 CrossRef PubMed.
- C. E. DeSantis, J. Ma, M. M. Gaudet, L. A. Newman, K. D. Miller, A. Goding Sauer, A. Jemal and R. L. Siegel, Ca-Cancer J. Clin., 2019, 69, 438–451 CrossRef PubMed.
- J. Y. S. Tsang and G. M. Tse, Adv. Anat. Pathol., 2020, 27, 27–35 CrossRef CAS PubMed.
- D. Banik, S. Noonepalle, M. Hadley, E. Palmer, M. Gracia-Hernandez, C. Zevallos-Delgado, N. Manhas, H. Simonyan, C. N. Young, A. Popratiloff, K. B. Chiappinelli, R. Fernandes, E. M. Sotomayor and A. Villagra, Cancer Res., 2020, 80, 3649–3662 CrossRef CAS PubMed.
- P. An, F. Chen, Z. Li, Y. Ling, Y. Peng, H. Zhang, J. Li, Z. Chen and H. Wang, Oncogene, 2020, 39, 4956–4969 CrossRef CAS PubMed.
- V. R. Pidugu, N. S. Yarla, A. Bishayee, A. M. Kalle and A. K. Satya, Apoptosis, 2017, 22, 1394–1403 CrossRef PubMed.
- G. Rahmani, S. Sameri, N. Abbasi, M. Abdi and R. Najafi, Pathol., Res. Pract., 2021, 220, 153396 CrossRef CAS PubMed.
- A. Halaburková, R. Jendželovský, J. Kovaľ, Z. Herceg, P. Fedoročko and A. Ghantous, Clin. Epigenet., 2017, 9, 62 CrossRef PubMed.
- R. R. Ye, C. P. Tan, L. He, M. H. Chen, L. N. Ji and Z. W. Mao, Chem. Commun., 2014, 50, 10945–10948 RSC.
- J. Kasparkova, H. Kostrhunova, O. Novakova, R. Křikavová, J. Vančo, Z. Trávníček and V. Brabec, Angew. Chem., 2015, 54, 14478–14482 CrossRef CAS PubMed.
- R. D. George and A. W. Snow, J. Heterocycl. Chem., 1995, 32, 495–498 CrossRef CAS.
- J. G. Young and W. Onyebuagu, J. Org. Chem., 1990, 55, 2155–2159 CrossRef CAS.
- V. Patil, Q. H. Sodji, J. R. Kornacki, M. Mrksich and A. K. Oyelere, J. Med. Chem., 2013, 56, 3492–3506 CrossRef CAS PubMed.
- D. Atilla, N. Saydan, M. Durmuş, A. G. Gürek, T. Khan, A. Rück, H. Walt, T. Nyokong and V. Ahsen, J. Photochem. Photobiol., A, 2007, 186, 298–307 CrossRef CAS.
- H. A. Byth, B. I. McHunu, I. A. Dubery and L. Bornman, Phytochem. Anal., 2001, 12, 340–346 CrossRef CAS PubMed.
- J. D. Gelles and J. E. Chipuk, Cell Death Dis., 2016, 7, e2493 CrossRef CAS PubMed.
- Z. Darzynkiewicz, Current protocols in cytometry, 2011, ch. 7, unit 7.2 Search PubMed.
- C. Sobreira, M. Davidson, M. P. King and A. F. Miranda, J. Histochem. Cytochem., 1996, 44, 571–579 CrossRef CAS PubMed.
- M. Niedre, M. S. Patterson and B. C. Wilson, Photochem. Photobiol., 2002, 75, 382–391 CrossRef CAS PubMed.
- R. Schmidt, C. Tanielian, R. Dunsbach and C. Wolff, J. Photochem. Photobiol., A, 1994, 79, 11–17 CrossRef CAS.
- E. Önal, Ö. Tüncel, M. Albakour, G. G. Çelik, A. G. Gürek and S. Özçelik, RSC Adv., 2021, 11, 6188–6200 RSC.
- S. Elmore, Toxicol. Pathol., 2007, 35, 495–516 CrossRef CAS PubMed.
- M. Redza-Dutordoir and D. A. Averill-Bates, Biochim. Biophys. Acta, Mol. Cell Res., 2016, 1863, 2977–2992 CrossRef CAS PubMed.
- A. G. Eliopoulos, S. Havaki and V. G. Gorgoulis, Front. Genet., 2016, 7, 204 Search PubMed.
- A. El-Hussein, M. Harith and H. Abrahamse, Int. J. Photoenergy, 2012, 281068 Search PubMed.
- J. H. Lee, M. L. Choy, L. Ngo, S. S. Foster and P. A. Marks, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14639–14644 CrossRef CAS PubMed.
- S. L. Haywood-Small, D. I. Vernon, J. Griffiths, J. Schofield and S. B. Brown, Biochem. Biophys. Res. Commun., 2006, 339, 569–576 CrossRef CAS PubMed.
- N. Ahmad, D. K. Feyes, R. Agarwal and H. Mukhtar, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6977–6982 CrossRef CAS PubMed.
- C. Xia, Y. Wang, W. Chen, W. Yu, B. Wang and T. Li, Molecules, 2011, 16, 1389–1401 CrossRef CAS PubMed.
- J. Schmidt, W. Kuzyniak, J. Berkholz, G. Steinemann, R. Ogbodu, B. Hoffmann, G. Nouailles, A. G. Gürek, B. Nitzsche and M. Höpfner, Int. J. Mol. Med., 2018, 42, 534–546 CAS.
- J. Shao, J. Xue, Y. Dai, H. Liu, N. Chen, L. Jia and J. Huang, Eur. J. Cancer, 2012, 48, 2086–2096 CrossRef CAS PubMed.
- P. Finzer, C. Kuntzen, U. Soto, H. zur Hausen and F. Rösl, Oncogene, 2001, 20, 4768–4776 CrossRef CAS PubMed.
- E. Bernhart, N. Stuendl, H. Kaltenegger, C. Windpassinger, N. Donohue, A. Leithner and B. Lohberger, Oncotarget, 2017, 8, 77254–77267 CrossRef PubMed.
- I. Hrgovic, M. Doll, J. Kleemann, X. F. Wang, N. Zoeller, A. Pinter, S. Kippenberger, R. Kaufmann and M. Meissner, BMC Cancer, 2016, 16, 763 CrossRef PubMed.
- S. Roy, K. Packman, R. Jeffrey and M. Tenniswood, Cell Death Differ., 2005, 12, 482–491 CrossRef CAS PubMed.
- Z. Dong, Y. Yang, S. Liu, J. Lu, B. Huang and Y. Zhang, Oncotarget, 2018, 9, 512–523 CrossRef PubMed.
- A. Chakrabarti, I. Oehme, O. Witt, G. Oliveira, W. Sippl, C. Romier, R. J. Pierce and M. Jung, Trends Pharmacol. Sci., 2015, 36, 481–492 CrossRef CAS PubMed.
- Y. Liu, L. Peng, E. Seto, S. Huang and Y. Qiu, J. Biol. Chem., 2012, 287, 29168–29174 CrossRef CAS PubMed.
- L. Yuan, J. Zhang, Y. Liu, J. Zhao, F. Jiang and Y. Liu, J. Inorg. Biochem., 2017, 177, 17–26 CrossRef CAS PubMed.
- P. K. Tsai, S. W. Wu, C. Y. Chiang, M. W. Lee, H. Y. Chen, W. Y. Chen, C. J. Chen, S. F. Yang, C. B. Yeh and Y. H. Kuan, Ecotoxicol. Environ. Saf., 2020, 193, 110348 CrossRef CAS PubMed.
- I. Mfouo-Tynga and H. Abrahamse, Int. J. Mol. Sci., 2015, 16, 10228–10241 CrossRef CAS PubMed.
- L. Y. Xue, S. M. Chiu and N. L. Oleinick, Autophagy, 2010, 6, 248–255 CrossRef CAS PubMed.
- A. D. Garg, H. Maes, E. Romano and P. Agostinis, Photochem. Photobiol. Sci., 2015, 14, 1410–1424 CrossRef CAS PubMed.
- P. Mroz, A. Yaroslavsky, G. B. Kharkwal and M. R. Hamblin, Cancers, 2011, 3, 2516–2539 CrossRef CAS PubMed.
- N. I. Orlotti, G. Cimino-Reale, E. Borghini, M. Pennati, C. Sissi, F. Perrone, M. Palumbo, M. G. Daidone, M. Folini and N. Zaffaroni, Autophagy, 2012, 8, 1185–1196 CrossRef CAS PubMed.
- V. V. Eapen and J. E. Haber, Autophagy, 2013, 9, 440–441 CrossRef CAS PubMed.
- S. M. Mahalingam, J. D. Ordaz and P. S. Low, ACS Omega, 2018, 3, 6066–6074 CrossRef CAS PubMed.
- N. Rubio, S. P. Fleury and R. W. Redmond, Photochem. Photobiol. Sci., 2009, 8, 457–464 CrossRef CAS PubMed.
- S. Missiroli, S. Patergnani, N. Caroccia, G. Pedriali, M. Perrone, M. Previati, M. R. Wieckowski and C. Giorgi, Cell Death Dis., 2018, 9, 329 CrossRef PubMed.
- M. Kochetkova, S. Kumar and S. R. McColl, Cell Death Differ., 2009, 16, 664–673 CrossRef CAS PubMed.
- T. Kakinuma and S. T. Hwang, J. Leukocyte Biol., 2006, 79, 639–651 CrossRef CAS PubMed.
- S. Chatterjee, B. Behnam Azad and S. Nimmagadda, Adv. Cancer Res., 2014, 124, 31–82 CrossRef CAS PubMed.
- B. Ge, J. Lao, J. Li, Y. Chen, Y. Song and F. Huang, Sci. Rep., 2017, 7, 16873 CrossRef PubMed.
- A. Müller, B. Homey, H. Soto, N. Ge, D. Catron, M. E. Buchanan, T. McClanahan, E. Murphy, W. Yuan, S. N. Wagner, J. L. Barrera, A. Mohar, E. Verástegui and A. Zlotnik, Nature, 2001, 410, 50–56 CrossRef PubMed.
- J. M. Louderbough and J. A. Schroeder, Mol. Cancer Res., 2011, 9, 1573–1586 CrossRef CAS PubMed.
- K. Fuchs, A. Hippe, A. Schmaus, B. Homey, J. P. Sleeman and V. Orian-Rousseau, Cell Death Dis., 2013, 4, e819 CrossRef CAS PubMed.
- N. Trempolec, B. Doix, C. Degavre, D. Brusa, C. Bouzin, O. Riant and O. Feron, Cancers, 2020, 12(3), 545 CrossRef CAS PubMed.
- F. Zhang, Y. Zhu, G. Fan and S. Hu, Oncol. Lett., 2018, 16, 5034–5040 CrossRef PubMed.
- C. Ierano, A. Basseville, K. K. To, Z. Zhan, R. W. Robey, J. Wilkerson, S. E. Bates and S. Scala, Cancer Biol. Ther., 2013, 14, 175–183 CrossRef CAS PubMed.
- A. Master, M. Livingston and A. Sen Gupta, J. Controlled Release, 2013, 168, 88–102 CrossRef CAS PubMed.
- N. Liu, H. Liu, H. Chen, G. Wang, H. Teng and Y. Chang, Colloids Surf., B, 2020, 188, 110753 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05404j |
| This journal is © The Royal Society of Chemistry 2021 |